
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of an oligonucleotide with a nicotinamide mononucleotide residue and its molecular recognition in DNA helices†
A.
Göckel
and
C.
Richert
*
Institute for Organic Chemistry, University of Stuttgart, 70569 Stuttgart, Germany. Fax: +49 (0) 711 608 64321; Tel: +49 (0) 711 608 64311E-mail: lehrstuhl-2@oc.uni-stuttgart.de
First published on 4th September 2015
Abstract
Nicotinamide adenine dinucleotide (NAD) is a pivotal redox cofactor of primary metabolism. Its redox reactivity is based on the nicotinamide mononucleotide (NMN) moiety. We investigated whether NMN+ can engage in pairing interactions, when incorporated into an oligonucleotide. Here we describe the incorporation of NMN+ at the 3′-terminus of an oligodeoxynucleotide via a phosphoramidate coupling in solution. The stability of duplexes and triplexes with the NMN+-containing strand was measured in UV-melting curves. While the melting points of duplexes with different bases facing the nicotinamide were similar, triplex stabilities varied greatly between different base combinations, suggesting specific pairing. The most stable triplexes were found when a guanine and an adenine were facing the NMN+ residue. Their triplex melting points were higher than those of the corresponding triplexes with a thymidine residue at the same position. These results show that NMN+ residues can be recognized selectively in DNA helices and are thus compatible with the molecular recognition in nucleic acids.
Introduction
Nicotinamide adenine dinucleotide (NAD) is a pivotal cofactor in the cell. It consists of nicotinamide mononucleotide (NMN) and adenosine monophosphate, linked by a pyrophosphate bond. The reduced form of NAD+, NADH, is generated during glycolysis and the citric acid cycle, and is consumed in the respiratory chain.1 Further, NADH and its oxidized form, NAD+, are coenzymes for a host of oxidoreductase enzymes. Also, NAD+ is a substrate for the ADP-ribose transfer to enzymes.2 Moreover, NAD+ serves as a precursor of cyclic ADP-ribose, a second messenger.3 Bacterial DNA ligases consume the NAD+ when joining two DNA strands.4 Nicotinamide residues are also found as a cap for regulatory RNAs.5 Additionally, it is becoming increasingly clear that NAD+ and its phosphorylated form, nicotinamide adenine dinucleotide phosphate (NADP), have roles in signal transduction and in cellular defence systems.6 On the extracellular level NAD+ is assumed to act as a neurotransmitter.7It is not surprising that NAD+ is conserved in all kingdoms of life. Apparently, it originated early in evolution. Structurally, NAD+ consists of two ribonucleotides. This points to a possible origin in the so-called ‘RNA world’.8,9 If so, it is likely that nature first used NAD+ in biomolecular complexes that formed, directed by base pairing. But while the adenosine monophosphate portion of NAD+ is well known to engage in hydrogen bonding interactions underlying duplex and triplex formation,10 it is not clear whether the NMN+ residue is able to engage in base pairing with other nucleobases in a duplex or triplex context (Fig. 1). If it does, this would be most useful for the design of novel functional DNA nanostructures11–13 with tailor-made active sites.
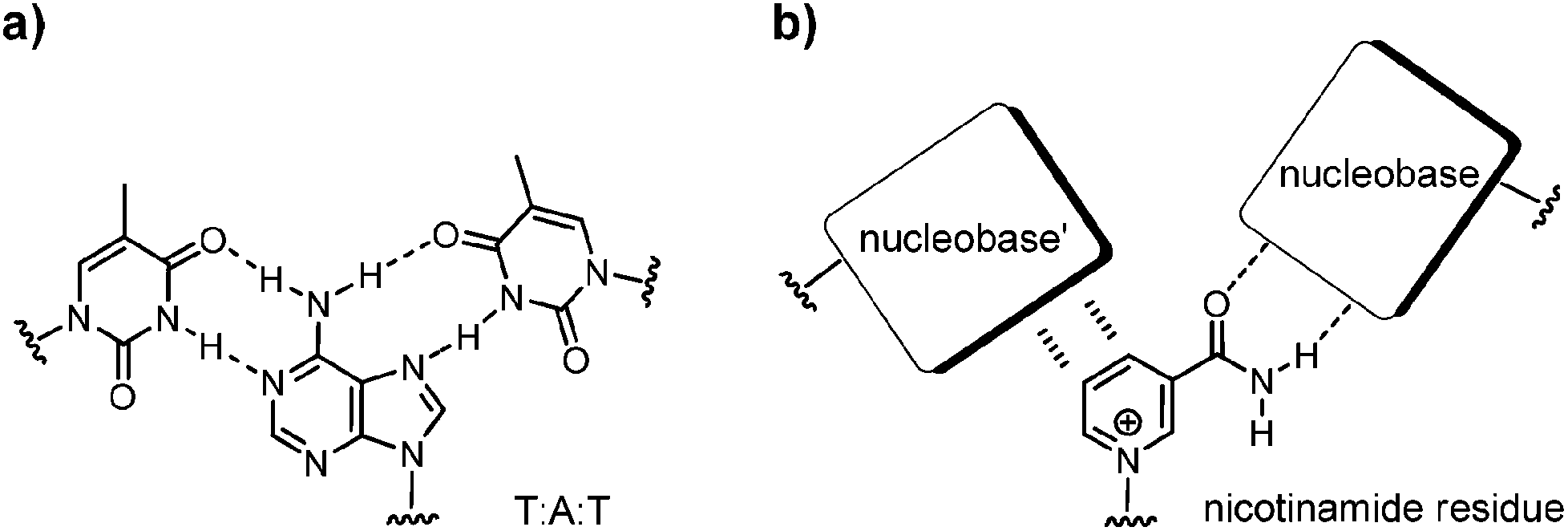 | ||
| Fig. 1 (a) Base pairing as found in a triplex, with a combination of Watson–Crick and Hoogsteen pairings; (b) nicotinamide residue of NAD+ and possible pairing interactions with nucleobases in the context of a DNA triplex. | ||
We therefore sought a methodology for incorporating nicotinamide mononucleotide residues in oligonucleotides. Liu and Orgel had previously attempted to incorporate NAD+ residues at the terminus of primers terminating in a ribonucleoside. Enzymatic extension of oligoribonucleotides and untemplated oligomerization was achieved, using di- or triphosphates of pyridine nucleosides.14 Attempts to induce in vitro transcription or enzyme-free primer extension were unsuccessful, though. The authors noted that NMN+ should, in principle, be able to undergo base pairing, but because both enzymatic and enzyme-free template-directed incorporation of NMN+ failed, they concluded that NMN+ does not act like an ‘informational unit of a polymer’.
More recently, Mutschler and Holliger described a ribozyme that catalyzes addition of nicotinamide adenine dinucleotide 2′,3′-cyclic phosphate to the 5′-terminus of RNA strands through a nucleotidyl transferase activity,15 but the resulting constructs contained the NMN+ residue in the pyrophosphate-linked form, not as part of a continuous phosphodiester chain. Here we report a methodology for incorporating NMN+ residues at the 3′-terminus of DNA strands via a phosphoramidate linkage, together with binding data that show what base combinations best accommodate the NMN+ residue in a double helix or triplex. The challenge to link the labile NMN+ substructure to a synthetic oligonucleotide sequence was met by converting the NMN+ residue into an active ester and subsequent coupling to the aminoterminal oligonucleotide in aqueous solution.
Results
The chemical synthesis of oligodeoxynucleotides with an NAD+ residue is complicated due to the lability of the pyridine nucleotides. In the cationic, oxidized form, lability to a base is a significant problem, while the reduced form reacts quickly under acidic conditions.16,17 This was confirmed in exploratory reactions involving NADH, NAD+, and their pyrophosphate cleavage products. The labilities precluded a synthetic approach involving conventional chain assembly by solid-phase DNA synthesis, which employs both acidic and strongly basic conditions. Further, it was not immediately clear how a phosphoramidite, H-phosphonate, or a phosphodiester building block could be generated, starting from commercially available starting materials. This prompted us to study the conversion of fully deprotected NMN+ under conditions established for enzyme-free primer extension.18,19Liu and Orgel had reported the synthesis of a methylimidazolide of NMN+ using a redox condensation in DMSO, but no characterization of the product was given.14 This was also true for the oligomerization products of their enzymatically catalyzed conversions that gave electrophoretic bands contaminated with broad smears that the authors attributed to the partial loss of the nicotinamide from the nucleotide under the basic conditions employed. When we prepared the methylimidazolide of NMN+, using conditions optimized for ribonucleotides,20 we observed the expected signal by ESI mass spectrometric analysis of the precipitated product (Fig. S1, ESI†). Attempts to bring about a coupling reaction with a primer were unsuccessful, though, both with an RNA primer, and with a more reactive DNA primer with a 3′-terminal, 3′-amino-2′,3′-dideoxynucleoside. The latter gives a phosphoramidate linkage that is isoelectronic and largely isosteric to natural phosphodiester linkages.21 The aminoterminal primer had been prepared by solid-phase synthesis, starting from controlled pore glass loaded with the aminonucleoside.22
Scheme 1 shows the successful synthetic approach. It starts with the activation of NMN+ to the oxyazabenzotriazolide (OAt ester) 1, using hydroxyazabenzotriazolide (HOAt) and N-ethyl-N′-dimethylaminopropyl carbodiimide (EDC) as the condensation agent in aqueous solution at pH 5.5.19,23 The formation of the active ester was confirmed by 31P-NMR and UV-Vis spectroscopy (Fig. S2 and S3, ESI†). An aliquot of the solution of 1 was then added to a solution of aminoterminal strand 2 in primer extension buffer containing MgCl2 and NaCl at pH 8.9.24 The concentration of the buffer was increased to 0.5 M to ensure a proper pH shift from that of the activation mixture and thus high-yielding reactions. Typically, 0.1 M OAt ester 1 was used, and the reaction was allowed to proceed for 48 h at 20 °C. As expected, the isolation and purification of NMN+-containing oligonucleotide 3 proved far from trivial. While gel permeation chromatography or affinity chromatography with an immobilized complementary strand to 3 and subsequent dialysis gave unsatisfactory results, anion exchange HPLC, followed by desalting on a C4-phase gave a solution showing pure 3 (Fig. 2). This solution was evaporated to dryness without decomposition by using a gentle stream of nitrogen.
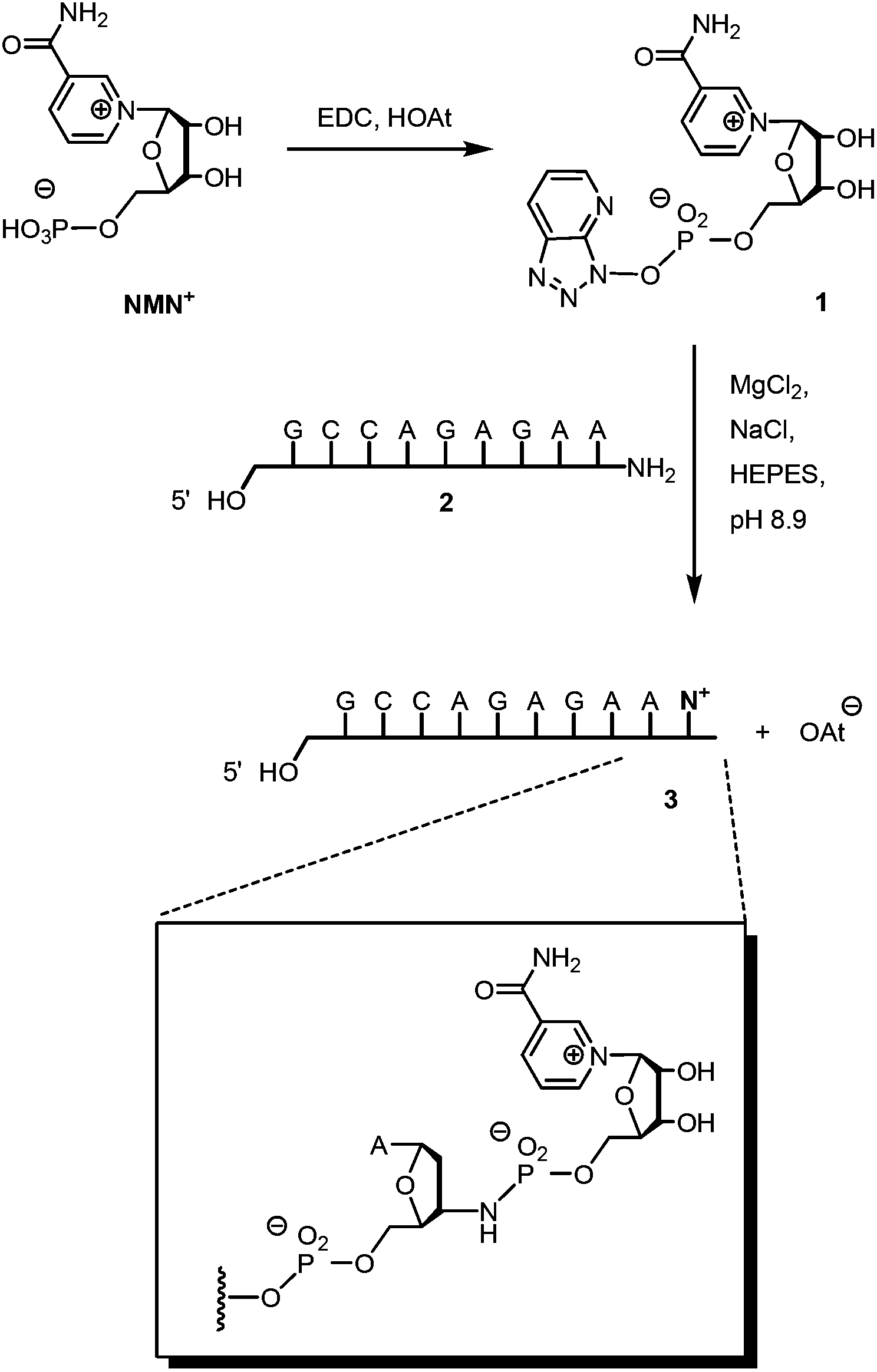 | ||
| Scheme 1 Synthesis of oligonucleotide 3 featuring a 3′-terminal nicotinamide mononucleotide residue, connected via a phosphoramidate linkage. | ||
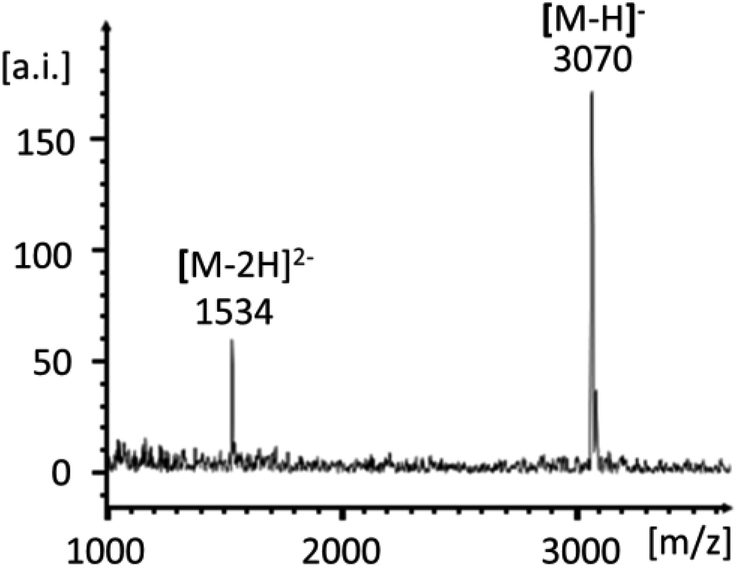 | ||
| Fig. 2 MALDI-TOF mass spectrum of NMN+-terminated oligonucleotide 3. | ||
With NMN+-containing oligonucleotide 3 in hand, we then proceeded toward studying the molecular recognition of the nicotinamide mononucleotide in duplex and triplex contexts. For this, the four-strand triplex shown in Fig. 3 was designed. Besides 3 or unmodified T-terminated control strand 4, it features a long strand (5a–t) engaging in Watson–Crick base pairing, a triplex-forming oligonucleotide (TFO, 6a–t) that bridges the two duplexes, and the purine-rich central strands (3/4 and 7).
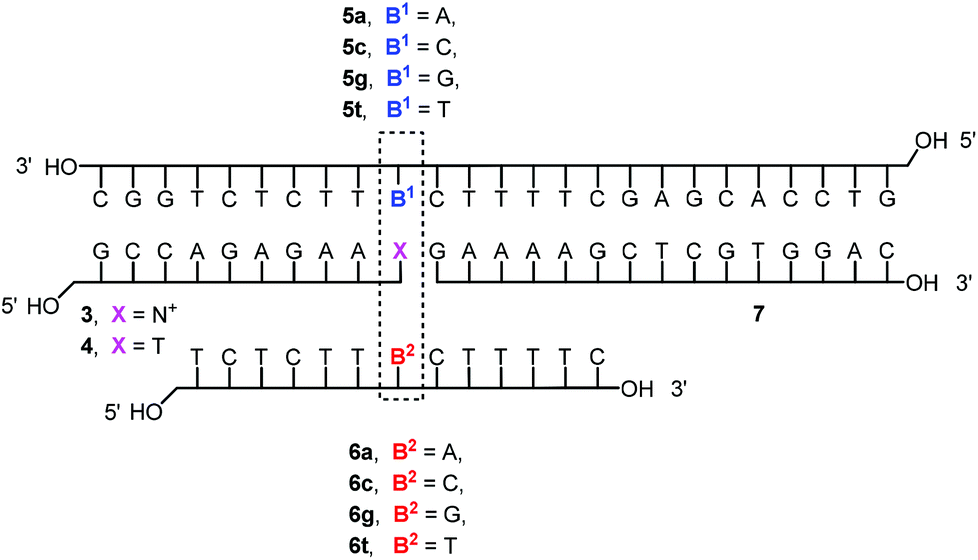 | ||
| Fig. 3 Sequences of DNA triplexes studied. | ||
Three subsequent melting transitions were expected for the triplexes (Fig. 4a). The first releases the TFO from the duplexes, the second involves the dissociation of the short purine-rich strand, and the third transition leads to the dissociation of the remaining long duplex. Fig. 4b shows that all three transitions are indeed observable experimentally. This allowed us to screen for pairing interactions with the nicotinamide residue, both in a duplex and in a triplex environment, by offering each of the four different nucleobases (A/C/G/T) in either of the two pairing strands (5 and 6) and measuring the thermal stability of the resulting complexes. The third melting transition (Tm3, Fig. 4a) should be largely unaffected by the sixteen possible base combinations of 5a–t and 6a–t, thus serving as an internal control. This was indeed the case, with a Tm3 of 66 ± 1 °C in all cases.
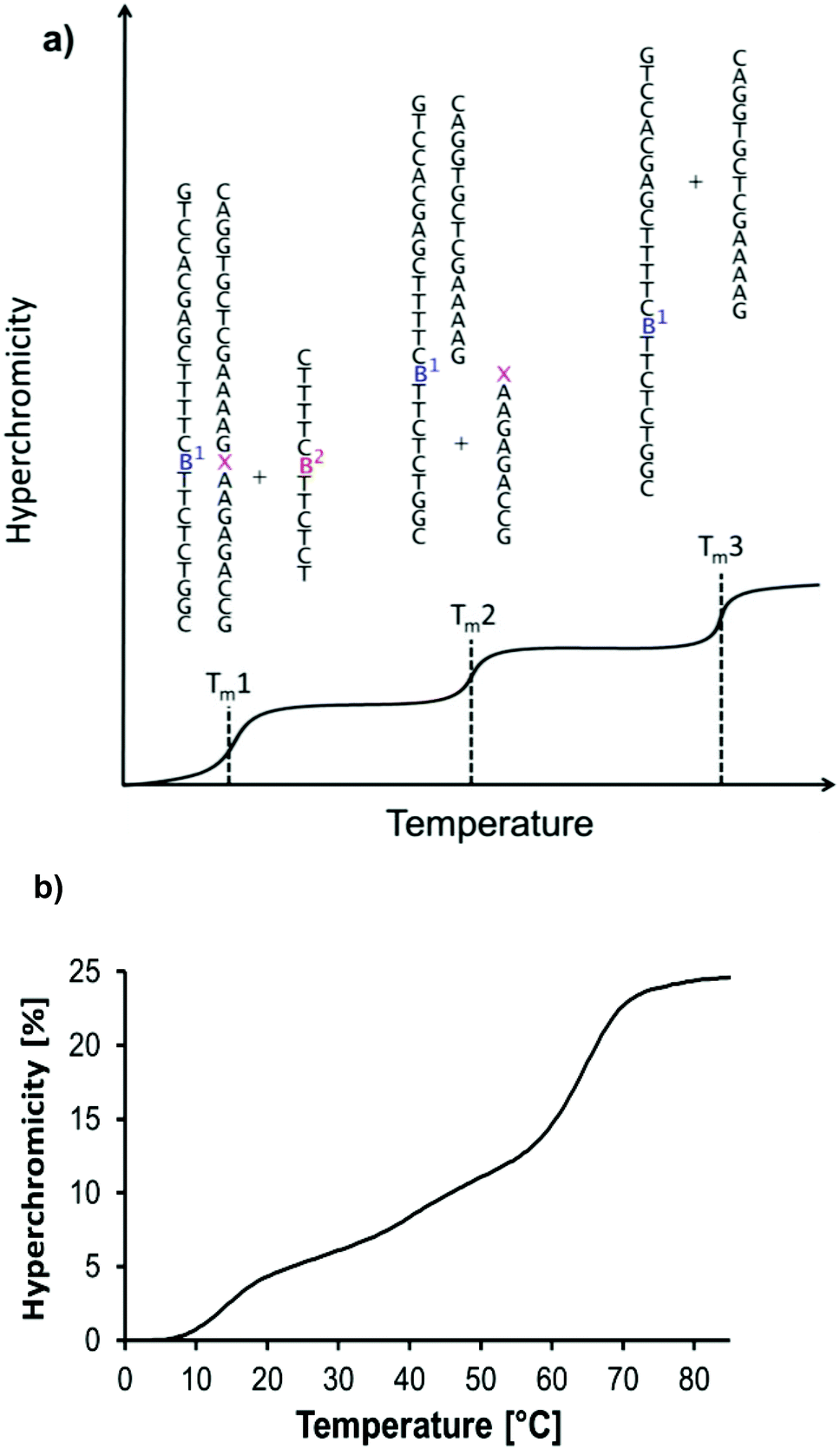 | ||
| Fig. 4 UV-melting transitions in a four-strand triple helical complex. (a) Cartoon of a melting curve for the triplex shown in Fig. 3, containing a non-natural residue (artists rendition). Three transitions are expected, based on the release of the triplex-forming oligonucleotide, the short, and the long central strands. (b) Experimental melting curve at λdet 260 nm showing the expected transitions for the triplex formed by oligonucleotides 5t, 3, 6a, and 7 at 1 μM strand concentration, each; 10 mM phosphate buffer pH 7.0, 1 M NaCl, and 10 mM MgCl2. | ||
Table 1 lists the Tm2 and Tm1 values determined in our screening process. We measured at least two melting curves for each base combination and analyzed the transitions with two different methods (first derivative of numerical values, or graphical determination of Tm as the point at which 50% of the hyperchromicity is reached). The duplex melting points for the thermal release of 3 were very similar for each of the four possible bases in 5a–t, ranging from 39.4 to 42.5 °C. This suggested that the NMN+ residue does not engage in highly selective pairing in a duplex context.
| Partner strands | Triplet |
T
m2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (duplex) [°C] b (duplex) [°C] |
T m1b (triplex) [°C] | Hyperchromicityb (triplex) [%] |
|---|---|---|---|---|
|
a Conditions: 1 μM oligonucleotides, each; 10 mM phosphate buffer, pH = 7.0, 1 M NaCl, and 10 mM MgCl2. Melting points are averages of at least two measurements. The melting points of all duplex-to-single strand transitions of the remaining long duplexes (5a–t:7) were near-identical with Tm3 values of 66 ± 1 °C.
b ± One standard deviation whenever ≥ three data points were available.
c Melting transition not sigmoidal.
|
||||
| 5a, 6a |
A:N+:A |
40.6 ± 0.5 | 14.3 ± 0.8 | 6.9 ± 0.8 |
| 5a, 6c |
A:N+:C |
40.7 ± 0.3 | 10.4 ± 0.5 | 6.8 ± 0.3 |
| 5a, 6g |
A:N+:G |
41.7 ± 0.3 | 19.9 ± 0.1 | 7.6 ± 0.2 |
| 5a, 6t |
A:N+:T |
40.7 ± 0.3 | 12.7 ± 0.4 | 6.6 ± 0.9 |
| 5c, 6a |
C:N+:A |
40.9 ± 0.9 | 14.0 ± 0.5 | 5.2 ± 0.7 |
| 5c, 6c |
C:N+:C |
41.9 ± 0.4 | 14.2 | 6.5 |
| 5c, 6g |
C:N+:G |
42.5 ± 0.4 | <10c | <5c |
| 5c, 6t |
C:N+:T |
41.9 ± 0.3 | 12.8 | 6.0 ± 0.9 |
| 5g, 6a |
G:N+:A |
40.3 ± 0.3 | 18.5 ± 0.2 | 10.3 ± 0.1 |
| 5g, 6c |
G:N+:C |
39.4 ± 0.4 | 11.5 ± 0.6 | 6.4 ± 1.0 |
| 5g, 6g |
G:N+:G |
40.1 ± 0.2 | 14.5 ± 0.2 | 8.0 ± 0.0 |
| 5g, 6t |
G:N+:T |
40.4 ± 0.3 | 12.7 | 5.7 ± 0.2 |
| 5t, 6a |
T:N+:A |
42.1 ± 0.1 | 15.6 ± 0.6 | 5.0 ± 0.6 |
| 5t, 6c |
T:N+:C |
40.6 ± 0.0 | (10.2 ± 0.7)c | <5.0c |
| 5t, 6g |
T:N+:G |
41.7 ± 0.3 | 11.5 | 5.0 |
| 5t, 6t |
T:N+:T |
41.4 ± 0.5 | (10.1)c | 5.0c |
However, triplex stabilities varied greatly with the nucleobases in the pairing strands (5/6). Here, the lowest melting point was below 10 °C, and the highest Tm1 was just below 20 °C. The triplexes of 3 with 5a/6g and 5g/6a were significantly more stable than other triplexes. This was confirmed by the melting points obtained by the graphical data analysis mode (Table S1, ESI†), where Tm1's of 19.5 ± 0.2 °C and 19.6 ± 0.4 °C were obtained. Fig. 5 shows how much the triplex-to-duplex transition is shifted for a stabilized versus a non-stabilized triplex.
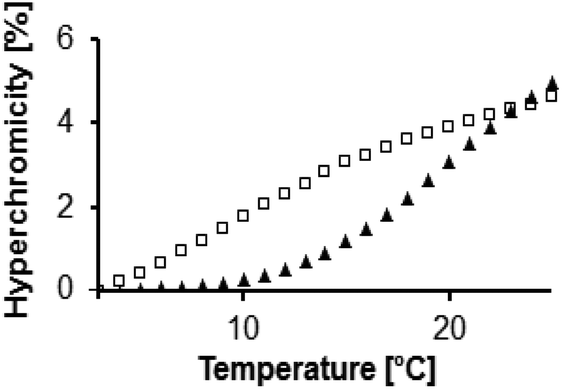 | ||
| Fig. 5 Expansion of melting curves showing the triplex-to-duplex melting curves for a weakly stabilized triplex (squares, strands 5t, 3, 6c and 7) and a strongly stabilized triplex (triangles, strands 5a, 3, 6g and 7). | ||
To exclude that the high stability of the triplexes with adenine, guanine and NMN+ is due to non-specific or non-triplet interactions between the two purines, we then measured a series of melting curves with control strand 4 replacing 3 in the triplex. We chose seven sequences, focusing on the base combination with a purine in at least one of the pairing positions. The thymine residue of 4 is similar in size and shape to the nicotinamide residue of 3 but offers very different hydrogen bonding functionalities. Table 2 lists the results from these experiments.
| Partner strands | Triplet | T m2 (duplex) [°C] |
T
m1b (triplex) [°C] |
Hyperchromicity (triplex) [%] |
|---|---|---|---|---|
|
a Conditions: 1 μM oligonucleotides, each, 10 mM phosphate buffer, pH = 7.0, 1 M NaCl, 10 mM MgCl2; melting points are averages of at least two measurements. All duplex-to-single strand transitions of the remaining long duplex (5a–t:7) were within 0.5 °C of 65.1 °C.
b ±1 standard deviation when ≥three data points were available.
|
||||
| 5a, 6a |
A:T:A |
50.4 | 12.4 ± 1.0 | 6.0 |
| 5a, 6c |
A:T:C |
51.9 | <10 | <5 |
| 5a, 6g |
A:T:G |
51.3 | 15.8 ± 0.4 | 6.5 |
| 5a, 6t |
A:T:T |
51.1 | <10 | <5 |
| 5c, 6a |
C:T:A |
41.5 | 15.8 ± 0.6 | 7.5 |
| 5g, 6a |
G:T:A |
42.8 | 17.3 ± 0.9 | 7.5 |
| 5t, 6a |
T:T:A |
43.3 | 15.3 ± 0.8 | 7.0 |
As expected, the thymidine residue of 4 pairs sequence-specifically with the A of 5a, following the base-pairing rules of Watson and Crick. Mismatched bases in 5 (C, G or T) lead to a depression in Tm2 of 7.1–8.9 °C (Table 2). However, neither of the four possible bases of 6a–t gave melting points as high as those found for triplexes with 3 and the combinations of A and G as pairing partners. This was confirmed by the results of the graphical data analysis method, as detailed in Table S2 of the ESI.† Here, the melting points of the most stable triplexes with 4 gave Tm1's of 15.9 ± 0.3 and 16.8 ± 0.1 °C.
Discussion
Thus far, it was assumed that nicotinamide mononucleotide residues do not act as base pairing partners when incorporated into nucleic acid chains.14 Our results now show that a combination of adenine and guanine as pairing partners for an NMN+ residue gives specific and surprisingly stable triplexes (Table 1). It is therefore interesting to ask what the interactions may be that lead to these stabilized triplexes. Two different hydrogen bonding patterns present themselves as likely interaction modes with the NMN+ at the center position of a triplet. One of these patterns features two hydrogen bonds with guanine on the Watson–Crick side of the pairing (Fig. 6a). It involves the so-called cis-conformation of the NMN+ that places the carbonyl group of the amide moiety towards the C2 carbon of the pyridinium ring.25 Computational work and crystal structures of nicotinamides show that this is the preferred conformation of free NAD+.26,27 The second pairing pattern is shown in Fig. 6b. It involves the trans-conformation of the amide of the NMN which has been described as the main conformation of NAD+ in complexes with proteins.28 Both conformers are close enough in energy to make it likely that they are populated to a significant extent.27,28 In the second pairing arrangement, adenine is found on the Watson–Crick side of the triplet, whereas guanine is the base facing the nicotinamide in the triplex-forming oligonucleotide. | ||
| Fig. 6 Possible hydrogen bonding interactions between the nicotinamide residue (N+) and adenine (A) and guanine (G) as pairing partners. (a) Pairing of the cis-conformation of the nicotinamide, and (b) pairing of the trans-conformation of the nicotinamide. In either case, two hydrogen bonds can be formed with the pairing partner in the Watson–Crick-pairing strand of the triplex, whereas the base of the triplex-forming strand engages in no more than a single hydrogen bond. Alternative pairing arrangements may exist for conformers of N+ that are resulting from a 180° rotation about the glycosidic bond. | ||
We note that a single hydrogen bond may not provide a decisive directing force and that shape complementarity and stacking interactions with neighboring bases in the same strand may be important factors for pairing. They cannot be predicted as easily as hydrogen bonding, though. Elucidating them will require a detailed three-dimensional structure, to be obtained e.g. by X-ray crystallography or NMR and restrained molecular dynamics. However, it is reasonable to assume that the interior of a stack of triplets provides an environment, in which a combination of stacking, classical hydrophobic effect, and hydrogen bonding allows for such triplet pairings between aromatic rings, as shown in Fig. 6. Thus, such fairly open pairing arrangements that may not compete well with hydrogen bonding with the solvent in the more exposed situation of duplexes may now be sufficiently stable.
Conclusions
Independent of what the finer structural details of the pairing interactions are at the site of the NMN+ in the triplex, it is now clear that a preferred sequence contexts exist for accommodating NMN+ in a helical DNA structure. This is important as NMN+ is the reactive part of NAD+, a pivotal redox cofactor in the metabolism of cells. Together with the synthetic methodology to covalently incorporate nicotinamide mononucleotide residues into oligonucleotides, our results may thus help to design new functional DNA structures, including structures with catalytic activity that rely on NMN+ residues as prosthetic groups.Experimental
General
Nicotinamide mononucleotide was obtained from Sigma-Aldrich (Deisenhofen, Germany). Both HOAt and EDC were purchased from Carbolution (Saarbrücken, Germany). All chemicals used for automated solid-phase DNA synthesis were from Proligo (Hamburg, Germany), including the phosphoramidite building blocks of deoxynucleosides, DCI activator (0.25 M 4,5-dicyanoimidazol in acetonitrile), amidite diluent (acetonitrile, ≤90 ppm water), tetrazole activator solution (0.45 M in abs. acetonitrile), cap B (THF/pyridine/1-methylimidazole 80:10:10 v/v/v), cap A (THF/tert-butylphenoxyacetic anhydride 100:5 v/v), oxidizer solution (0.1 M iodine in THF/pyridine/water 77:21:2, v/v/v), and deblock solution (trichloroacetic acid in CH2Cl, 3:97 v/v). Controlled-pore glass loaded with 5′-protected 3′-amino-2′,3′-dideoxyadenosine, linked with a hexafluoroglutaric acid linker (pore size 1000 Å, 25 μmol g−1 loading) was prepared as reported.22 Yields of oligonucleotides were determined by UV-absorption at 260 nm. MALDI-TOF mass spectra were acquired on a Bruker Reflex IV spectrometer in linear negative mode using 0.3 M 2,4,6-trihydroxyacetophenone monohydrate in ethanol as a matrix and 0.1 M diammonium citrate in water as a co-matrix solution.
:1, v/v). Oligonucleotide 2 eluted at 10% acetonitrile; overall yield 10% (based on the loading of the support), MALDI-TOF MS, calc. for C88H109N42O47P8 [M − H]− 2753.5 (average mass), found 2752.7 (maximum of unresolved isotope pattern).
UV-melting curves
The UV-melting curves were measured on a Lambda 25 spectrophotometer (Perkin Elmer) with Peltier-based temperature control, using Suprasil precision microcuvettes (Hellma, Müllheim, Germany) and sample volumes of 80 μL, with a protective layer of liquid paraffin (50 μL) to avoid evaporation. Control experiments without the protective layer showed identical melting points. Absorption was measured at 260 nm in the temperature range of 3–85 °C with a heating or cooling rate of 1 °C min−1. Melting points are extrema of the first derivative of heating or cooling curves, determined numerically, using UV-WinLab software. In some instances, even though the overall appearance of a curve was similar to other melting curves of the same sample, the transition was not sigmoidal enough to allow for unambiguous determination of the lowest melting point. If so, the number of available data points was sometimes insufficient to calculate an accurate standard deviation and an average value is being reported instead. Melting points, determined graphically as the temperature at which 50% of the hyperchromicity of the triplex-to-duplex transition was reached, are given in Tables S1 and S2 in the ESI.†Acknowledgements
The authors thank Dr S. Tschierlei and S. Vollmer for discussions and a review of the manuscript. Support by Stiftung Energieforschung Baden-Württemberg, grant no. A 308 14 to C. R. and DFG, grant no. RI 1063/13-1 is gratefully acknowledged.Notes and references
- M. Friedkin and A. Lehninger, J. Biol. Chem., 1949, 178, 611–623 CAS
.
- M. Ziegler, Eur. J. Biochem., 2000, 267, 1550–1564 CrossRef CAS
- A. Guse, Curr. Med. Chem., 2004, 11, 847–855 CrossRef CAS
- A. Wilkinson, J. Day and R. Bowater, Mol. Microbiol., 2001, 40, 1241–1248 CrossRef CAS
- H. Cahová, M.-L. Winz, K. Höfer, G. Nübel and A. Jäschke, Nature, 2015, 519, 374–377 CrossRef PubMed
- N. Pollak, C. Dölle and M. Ziegler, Biochem. J., 2007, 402, 205–218 CrossRef CAS PubMed
- V. Mutafova-Yambolieva, S. J. Hwang, X. Hao, H. Chen, M. X. Zhu, J. D. Wood, S. M. Ward and K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16359–16364 CrossRef CAS PubMed
- N. Lehman, WIREs RNA, 2010, 1, 202–213 CrossRef CAS PubMed
-
N. Raffaelli, Nicotinamide coenzyme synthesis: A case of ribonucleotide emergence or a byproduct of the RNA World? in Origins of Life: The Primal Self-Organization, ed. R. Ege, D.-H. Lankenau and A. Y. Mulkidjanian, Springer, Berlin, 2011, pp. 185–208 Search PubMed
- C. Kröner, A. Göckel, W. Liu and C. Richert, Chem. – Eur. J., 2013, 19, 15879–15887 CrossRef PubMed
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed
- N. V. Voigt, T. Tørring, A. Rotaru, M. F. Jacobsen, J. B. Ravnsbaek, R. Subramani, W. Mamdouh, J. Kjems, A. Mokhir, F. Besenbacher and K. V. Gothelf, Nat. Nanotechnol., 2010, 5, 200–203 CrossRef CAS PubMed
- B. Saccà and C. M. Niemeyer, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef PubMed
- R. Liu and L. E. Orgel, Nucleic Acids Res., 1995, 23, 3742–3749 CrossRef CAS PubMed
- H. Mutschler and P. Holliger, J. Am. Chem. Soc., 2014, 136, 5193–5196 CrossRef CAS PubMed
- N. O. Kaplan, S. P. Colowick and C. C. Barnes, J. Biol. Chem., 1951, 191, 461–472 CAS
- O. H. Lowry, J. V. Passoneau and M. K. Rock, J. Biol. Chem., 1961, 236, 2756–2759 CAS
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99–123 CrossRef CAS PubMed
- J. A. R. Stütz, E. Kervio, C. Deck and C. Richert, Chem. Biodiversity, 2007, 4, 784–802 Search PubMed
- C. Deck, M. Jauker and C. Richert, Nat. Chem., 2011, 3, 603–608 CrossRef CAS PubMed
- V. Tereshko, S. Gryaznov and M. Egli, J. Am. Chem. Soc., 1998, 120, 269–283 CrossRef CAS
- R. Eisenhuth and C. Richert, J. Org. Chem., 2009, 74, 26–37 CrossRef CAS PubMed
- E. Kervio, A. Hochgesand, U. E. Steiner and C. Richert, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12074–12079 CrossRef CAS PubMed
- P. Hagenbuch, E. Kervio, A. Hochgesand, U. Plutowski and C. Richert, Angew. Chem., Int. Ed., 2005, 44, 6588–6592 CrossRef CAS PubMed
- U. Berg and N. Åström, Acta Chem. Scand., 1995, 49, 599–608 CrossRef CAS
- H. Li and B. M. Goldstein, J. Med. Chem., 1992, 35, 3560–3567 CrossRef CAS
- G. De Luca, T. Marino, T. Mineva, N. Russo and M. Toscano, J. Mol. Struct., 2000, 501–502, 215–220 CrossRef CAS
- V. I. Polshakov, R. R. Biekofsky, B. Birdsall and J. Feeney, J. Mol. Struct., 2002, 602–603, 257–267 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Spectra, representative UV-melting curves and tables with melting points obtained by graphical analysis. See DOI: 10.1039/c5ob01714a |
| This journal is © The Royal Society of Chemistry 2015 |