
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and applications of secondary amine derivatives of (+)-dehydroabietylamine in chiral molecular recognition†
Tiina
Laaksonen
,
Sami
Heikkinen
and
Kristiina
Wähälä
*
Department of Chemistry, University of Helsinki, A.I. Virtasen aukio 1, P.O. Box 55, FI-00014 Helsinki, Finland. E-mail: kristiina.wahala@helsinki.fi
First published on 24th August 2015
Abstract
(+)-Dehydroabietylamine (1a), the novel derivatives (2a–6a) and their NTf2 salts (1b–6b) were tested as chiral NMR solvating agents for the resolution of enantiomers of the model compound Mosher's acid (7) and its n-Bu4N salt (8). Best enantiomeric discrimination of 7 was obtained using bisdehydroabietylamino-N1,N2-ethane-1,2-diamine (6a), and of 8 using N-(dehydroabietyl)-2-(dehydroabietylamino)ethanaminium bis((trifluoromethyl)-sulfonyl)-amide (6b). For the maximal resolution of enantiomers of 8, 1.0 eq. of 6b were needed. However, 0.5 eq. of 6a sufficed for the maximal resolution of enantiomers of 7. Enantiomeric excess studies were successfully conducted using 6a and 6b. The capability of 6a and 6b to recognize the enantiomers of various α-substituted carboxylic acids and their n-Bu4N salts were examined. Best resolutions were observed for aliphatic and aromatic carboxylic acids bearing an electronegative α-substituent. Now the ee studies on such non-aromatic carboxylic acids are also feasible.
Introduction
Analytical enantiomeric purity determinations are of great interest particularly in organic, medicinal and biological chemistry, where stereocontrol is important. Compared to commonly used techniques such as HPLC, NMR is more versatile, and the development of more sensitive instruments has made it a potential competitor for the traditional methods in chiral recognition. As NMR can provide fast and easy enantiomeric excess (ee) measurements (up to 94–99% ee),1 it is the ideal tool for quick ee determinations when developing new asymmetric synthesis or when studying the efficacy of a new chiral catalyst. Also, if the enantiomeric resolution cannot be efficiently performed with traditional methods, NMR can be used as an alternative.2In NMR, two general methods to investigate the enantiomeric purity of a compound may be applied. One is to use an enantiomerically pure chiral derivatising agent to produce two diastereomers. However, this method is time consuming and also may cause concerns of kinetic resolution and racemization. The second, more convenient and faster method is provided by chiral solvating agents (CSAs) where the resolving ability is based on the supramolecular complexation between two enantiomers of a chiral guest and a chiral host.3,4 Since complex formation is strongly dependent on interactions between a host and a guest, CSAs may contain hydrogen bond acceptor and donor groups (such as –NH2, –OH, –COOH), aromatic functionality for π–π stacking, and ionic and dipolic groups for ion–ion, dipole–dipole and ion–dipole interactions.4,5 However, the functional groups of the host and guest are not solely responsible for an efficient enantiomeric discrimination, but also the deuterated solvent used, concentration, molar ratio of host and guest, temperature and the anion (if present) of the chiral host6 have an effect on the resolution.3 The non-ionic and ionic CSAs are often derived from chiral natural compounds such as amino acids, menthol or mandelic acid. These compounds are generally not only chiral but also contain suitable functionalities for complex formation.
In this study we have used the readily available softwood resin derivative (+)-dehydroabietylamine7 (1a, Scheme 1) as a starting material to create novel CSAs for the enantiomeric resolution of racemic carboxylic acids by NMR. As only few of the reported amine based CSAs are ionic, and it being thought advisable to see if ionic functionalities might provide better resolution, protonated forms of our amines were also tested.
 | ||
| Scheme 1 Preparation of (+)-dehydroabietylamine based secondary amines and their corresponding NTf2 salts. (DAB = (+)-dehydroabietylamine, BrR = BrEt, Br(CH2)2OH, Br(CH2CH2O)2Me, Br(CH2)2Br). | ||
Results and discussion
Five secondary amine derivatives 2a–6a of 1a were prepared and converted along with 1a to bis(trifluoromethane)-sulfonimide (NTf2) salts (1b–6b) to give CSAs with ionic functionality. NTf2 was chosen for a counter anion as it has been found to provide better enantiomeric resolution with an ionic CSA.7,8 Compounds 3a–6a were readily prepared by an expedient one step reaction with suitable alkyl bromides in a microwave reactor (Scheme 1). Compound 2a was synthesized in two steps by the conversion of 1a to formamide followed by reduction. Physical data of the compounds are listed in Table 1, showing that when the amines are converted to NTf2 salts the melting points increase and the optical rotations decrease.| No. | R | Anion | mp (°C) | [α]22D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
[M]22Db |
|---|---|---|---|---|---|
| a c = 1.0, CHCl2. b Molar rotation calculated from [α]22D × M/100, where M is the molar mass. c DAB = (+)-Dehydroabietylamine. | |||||
| 1a | — | — | 44.2 | +44.35 | +126.60 |
| 1b | — | NTf2 | 197.2 | +17.11 | +96.96 |
| 2a | Me | — | Liquid at rt. | +50.64 | +151.65 |
| 2b | Me | NTf2 | 180.3 | +10.86 | +63.06 |
| 3a | Et | — | Liquid at rt. | +49.47 | +155.10 |
| 3b | Et | NTf2 | 180.9 | +9.80 | +58.35 |
| 4a | (CH2)2OH | — | 74.0 | +42.05 | +138.57 |
| 4b | (CH2)2OH | NTf2 | 160.7 | +7.03 | +42.92 |
| 5a | (CH2CH2O)2Me | — | Liquid at rt. | +34.07 | +132.05 |
| 5b | (CH2CH2O)2Me | NTf2 | Viscous liquid at rt. | +7.03 | +47.00 |
| 6a | (CH2)2DABc | — | 63.8 | +43.32 | +258.58 |
| 6b | (CH2)2DABc | NTf2 | 237.3 | +13.54 | +118.93 |
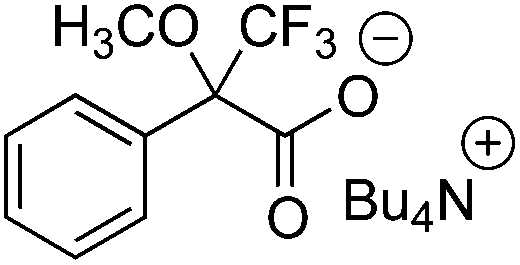
The ability of compounds 1–6a and b to recognize the chirality of ionic and non-ionic racemic carboxylic acids was examined using Mosher's acid 7 and its n-Bu4N salt 8. The effect of concentration of CSA was also investigated, as according to literature, the magnitude of non-equivalence (Δδ) increases when the concentration of host is higher than that of the guest.3,4 CDCl3 was chosen as solvent for the experiments because it is known that polar solvents can solvate ions and protic solvents may interfere in hydrogen bond formation which are important for complex formation.9 NMR experiments were performed by taking 0.5 mL (1.0 eq., 22.0 mM) of a stock solution containing 7 or 8 (guest) and dissolved CSA (host) (1.0 or 2.0 eq.). Both 1H and 19F NMR spectra were recorded (Table 2). Results indicate that 1–6a and b form a diastereometric salt pair with the model compound (7 or 8) as no resolution was detected between a non-ionic CSA (1a–6a) and 8, or an ionic CSA (1b–5b) and 7. This is most probably caused by the lack of suitable interactions in forming a salt pair. However, the ionic 6b resolved the non-ionic 7 presumably because the former has a non-ionic amine group to be protonated by 7 and is therefore able to resolve the enantiomers of 7. Due to the poor solubility of 6b in CDCl3 the resolution of 7 under 2:1 conditions could not be determined. No significant increase in the chemical shift difference between R and S enantiomers (Δδ) was detected when the concentration of host was doubled. In some cases the increase of concentration even led to a decrease in Δδ. This was especially notable in the case of compounds 6a and 6b. Among the non-ionic CSAs, 1a, 2a and 6a resolved the enantiomers of 7 highly efficiently. Especially 6a worked exceptionally well (host:guest ratio 1:1) both in 1H NMR (0.14 ppm, 71.8 Hz) and in 19F NMR (0.045 ppm, 21.2 Hz). The extent of resolution decreased both in 1H NMR and in 19F NMR when the molar ratio was increased to 2:1. For the resolution of 8 the corresponding NTf2 salts 1b, 2b and 6b gave best results. In this case the highest resolution was obtained with 6b both in 1H NMR (0.16 ppm, 81.1 Hz) and in 19F NMR (0.076 ppm, 35.8 Hz). As in the case of 6a, an increase in concentration lowered Δδ in 1H NMR; however, in 19F NMR Δδ was increased to 0.32 ppm (149.9 Hz).
| CSA | Host:Guest |
|
|
||
|---|---|---|---|---|---|
| 7 (ppm, (Hz)) | 8 (ppm, (Hz)) | ||||
| 1H | 19F | 1H | 19F | ||
| a Could not be measured since 6b was not soluble in CDCl3 at high concentrations. nd (no resolution was detected). | |||||
| 1a | 1:1 |
0.024 (12.1) | 0.074 (34.6) | nd | nd |
| 2:1 |
0.022 (10.9) | 0.064 (30.3) | nd | nd | |
| 1b | 1:1 |
nd | nd | 0.014 (6.8) | 0.051 (23.9) |
| 2:1 |
nd | nd | 0.016 (8.0) | 0.059 (27.7) | |
| 2a | 1:1 |
0.037 (18.7) | nd | nd | nd |
| 2:1 |
0.04 (20.0) | 0.029 (13.5) | nd | nd | |
| 2b | 1:1 |
nd | nd | 0.033 (16.6) | 0.019 (8.7) |
| 2:1 |
nd | nd | 0.025 (12.7) | nd | |
| 3a | 1:1 |
0.013 (6.7) | nd | nd | nd |
| 2:1 |
0.016 (7.8) | nd | nd | nd | |
| 3b | 1:1 |
nd | nd | 0.014 (6.9) | nd |
| 2:1 |
nd | nd | 0.0074 (3.7) | nd | |
| 4a | 1:1 |
0.0059 (3.0) | 0.017 (8.0) | nd | nd |
| 2:1 |
0.0092 (4.6) | 0.0066 (3.1) | nd | nd | |
| 4b | 1:1 |
nd | nd | 0.0051 (2.5) | nd |
| 2:1 |
nd | nd | 0.0068 (3.4) | nd | |
| 5a | 1:1 |
0.015 (7.5) | nd | nd | nd |
| 2:1 |
0.013 (6.3) | nd | nd | nd | |
| 5b | 1:1 |
nd | nd | 0.0082 (4.1) | nd |
| 2:1 |
nd | nd | 0.014 (6.8) | nd | |
| 6a | 1:1 |
0.14 (71.8) | 0.045 (21.2) | nd | nd |
| 2:1 |
0.027 (12.3) | 0.028 (13.3) | nd | nd | |
| 6b | 1:1 |
0.023 (11.3) | 0.035 (16.6) | 0.16 (81.1) | 0.076 (35.8) |
| 2:1 |
—a | —a | 0.038 (22.2) | 0.32 (149.9) | |

As compounds 6a and 6b gave the best results, their enantiomeric discrimination ability in NMR was further investigated. To find out how much guest is needed for maximum resolution and to obtain information about the composition of complex (e.g. 2:1 vs. 1:1 complex), the guest 7 (0.5 mL, 2.0 mM) was titrated with host 6a (46.6 mM). Due to the poor solubility of 6b in CDCl3, titration was performed in an opposite manner compared to 6a (i.e., titrating a 2.0 mM solution of host 6b with a 46.6 mM solution of guest 8) (Fig. 1). Titration results from both 1H and 19F NMR spectra indicate that the maximal resolution with host 6a occurs at the point where the molar ratio of host and guest was 0.5:1 (0.200 ppm, 99.9 Hz in 1H NMR and 0.088 ppm, 41.2 Hz, in 19F NMR), and with 6b at the molar ratio 1:1 (0.204 ppm, 101.8 Hz in 1H NMR and 0.093 ppm, 43.7 Hz in 19F NMR) (see ESI 3.1 and 3.2†). Commercially available CSAs are often expensive, and the ability of 6a to resolve enantiomers at the host:guest molar ratio 0.5:1 is a clear improvement as a minimum of 1.0 eq. (and in some cases an excess up to 24 eq.) of host is needed to obtain a maximal resolution.3 Since Δδ between the enantiomers of 7 (and of 8) decreased in 1H NMR when the concentration of 6a (or 6b) increased, it is assumed that the supramolecular complexation pattern changes when the concentration of host increases.
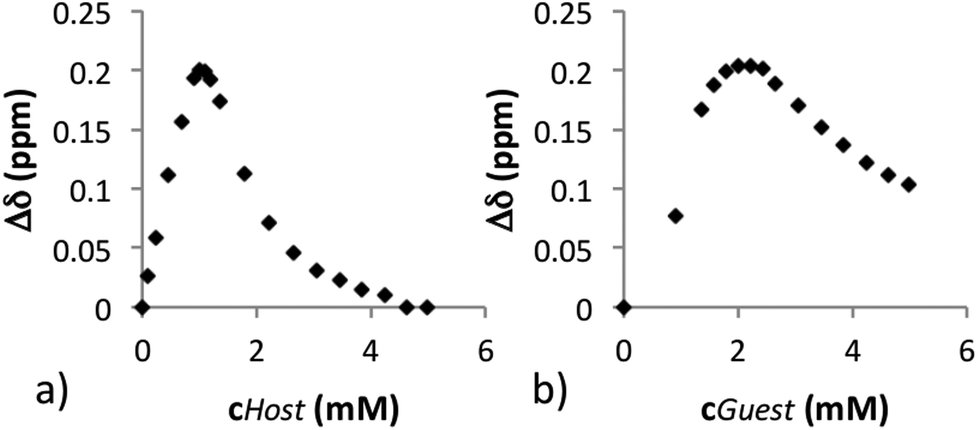 | ||
| Fig. 1 (a) The chiral resolution of enantiomers of 7 with 6a (1H NMR) and (b) chiral resolution of enantiomers of 8 with 6b (1H NMR). | ||
The suitability of CSAs 6a and 6b for enantiomeric excess (ee) NMR measurements was tested with 7 and 8. Both 6a and 6b can be used to detect the enantiomeric composition of samples with excellent reliability (Fig. 2a and b; see ESI 4.1 and 4.2†).
 | ||
| Fig. 2 Validation of enantiomeric composition of (a) 7 in the presence of 6a (expected ee%: (A) 80, (B) 75, (C) 70, (D) 60, (E) 50) and (b) 8 in the presence of 6b (expected ee%: (F) 50, (G) 58, (H) 67, (I) 75, (J) 79) by 1H NMR (500 MHz) in CDCl3 at 27 °C. (The peak of S enantiomer is wider due to strong binding with 6a, see ESI 3.1.1.†) | ||
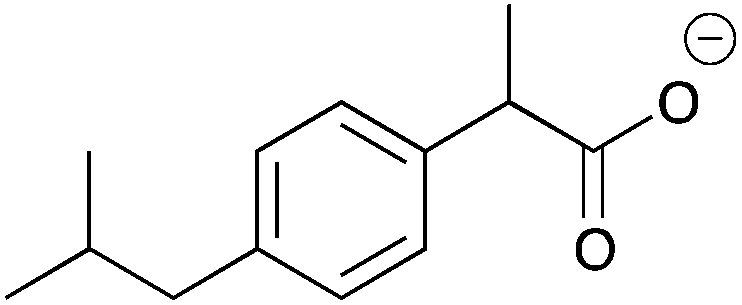
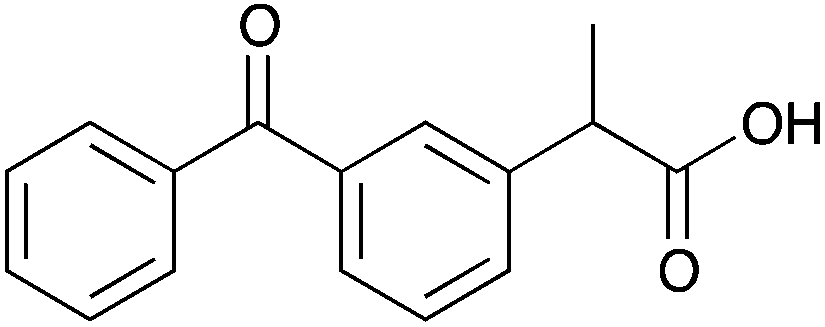
The resolution of racemic α-substituted carboxylic acids or their n-Bu4N salts by 6a and 6b, respectively, is presented in Table 3 and ESI 5.1 and 5.2.† CSAs 6a and 6b discriminate best carboxylic acids having an electronegative atom (e.g. O, N, Br) at the α-position (11–15a and b). Such aromatic or non-aromatic carboxylic acids were discriminated equally well. This is a major improvement as it has been suggested that the presence of an aromatic ring is necessary for good signal separation.10 In any case the resolution of non-aromatic carboxylic acids, especially using amine based CSAs, has been largely neglected.4,9,11
| Cmpd.a | Racemic carboxylic acid | Δδ | Cmpd.b | n-Bu4N salt of racemic carboxylic acid | Δδ | ||||
|---|---|---|---|---|---|---|---|---|---|
| ppm | Hz | ppm | Hz | ||||||
|
a 11.0 μL of a 46.6 mM 6a solution was added to 0.5 mL of a 2.0 mM solution of the analyte studied, to give an 0.5:1 host:guest molar ratio.
b 22.5 μL of a 46.6 mM solution of the analyte studied was added to 0.5 mL of a 2.0 mM solution of 6b, to give a 1:1 host:guest molar ratio.
c Peak overlapped with host peaks; nd (no resolution was detected).
|
|||||||||
| 9a |

|
Me | 0.0020 | 1.1 | 9b |
|
Me | 0.0099 | 5.0 |
| H | 0.0052 | 2.6 | H | 0.0053 | 2.6 | ||||
| CH2 | 0.0062 | 3.1 | CH2 | —c | —c | ||||
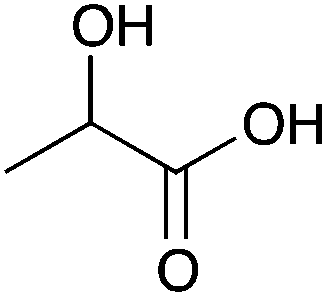
| 10a |
|
Me | 0.014 | 7.2 | 10b |

|
Me | 0.01 | 5.1 |
| H | nd | nd | H | nd | nd | ||||
| 11a |

|
H | 0.084 | 42.1 | 11b |
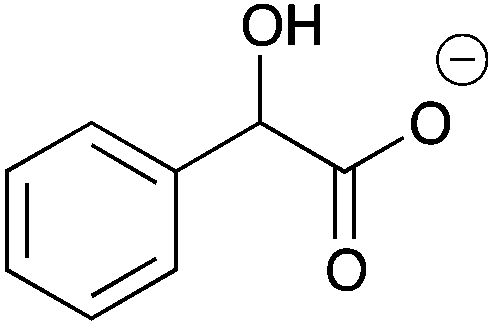
|
H | 0.10 | 50.8 |
| 12a |
|
H | 0.014 | 6.9 | 12b |

|
H | 0.038 | 19.1 |
| Me | 0.0062 | 3.1 | Me | —c | —c | ||||
| 13a |

|
H | 0.01 | 5.1 | 13b |
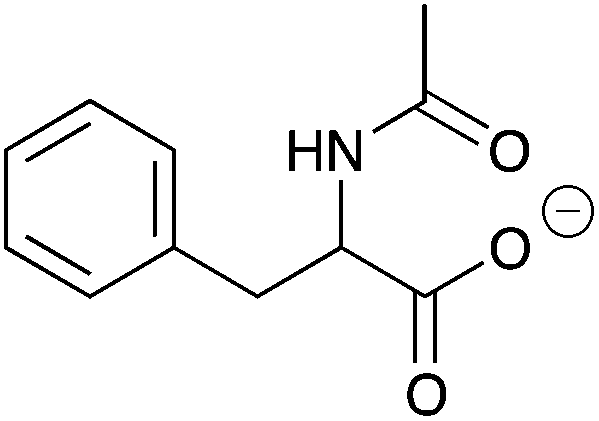
|
H | nd | nd |
| NH | 0.052 | 26.1 | NH | 0.051 | 25.5 | ||||
| Me | 0.033 | 16.3 | Me | 0.033 | 16.5 | ||||
| 14a |

|
H | 0.014 | 6.1 | 14b |
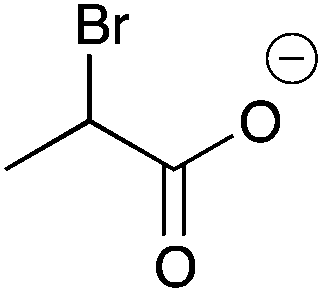
|
H | 0.016 | 8.2 |
| Me | 0.077 | 38.7 | Me | —c | —c | ||||
| 15a |

|
H | 0.016 | 8.2 | 15b |

|
H | 0.017 | 8.4 |
| NH | 0.088 | 43.8 | NH | 0.072 | 36.2 | ||||
The carboxylic acids 9a and b, 10a and b were discriminated by the corresponding host only moderately (1–7 Hz). This may be due to the lack of suitable interactions between the host and guest. This, however, is not a problem for ee determination as certain specialized NMR experiments are now available and can be used when the multiplet resolution needs improvement or when the overlapping of CSA and substrate is an issue.12 Compound 6b resolves the enantiomers of 9–12b better than its non-ionic form 6a resolves those of compounds 9–12a, indicating that stronger interaction can be obtained between the ionic CSA and ionic substrate. This can be used to advantage if the resolution of non-ionic CSA and substrate is not sufficient. CSA 6a resolves the enantiomers of 13–15a better than the corresponding ionic CSA 6b those of 13–15b. Interestingly 6a resolved the prochiral CH2 hydrogens in compound 9a. Since CSAs are usually able to resolve α-substituted carboxylic acids at the chiral α-site13 only, this is an additional advantage.
Conclusions
A number of derivatives (2a–6a) of (+)-dehydroabietylamine (1a) and their NTf2 salts (1b–6b) were prepared for use in chiral molecular recognition studies, the syntheses being carried out by highly expedient microwave techniques. The ability of the CSAs 1–6a and b to resolve racemic 7 and 8 was examined by 1H and 19F NMR. 6a showed excellent discrimination ability for 7 and its corresponding NTf2 salt 6b for 8. Optimum conditions for enantiomeric discrimination with 6a and 6b were determined by titration. 6b gives best results at a 1:1 host:guest molar ratio whereas 6a gave best resolution at a 0.5:1 host:guest molar ratio. This is a useful result since usually at least 1.0 eq. of CSA are needed for maximum resolution. 6a and 6b are highly useful in ee determination as well. In resolving various α-substituted racemic carboxylic acids using 6a or 6b, best results were given by acids bearing an electronegative α-substituent. In general, acids bearing or lacking an aromatic moiety performed equally well. For carboxylates, somewhat better results were obtained when ionic CSA 6b was used for the resolution than when using 6a for non-ionic substrates. In future, the applicability of compounds 6a and 6b in resolution by NMR will be further investigated, and the development of new (+)-dehydroabietylamine based CSAs are being continued.
Experimental
Materials and methods
All reagents and solvents were obtained from commercial suppliers and were used without further purification unless otherwise stated. Dehydroabietylamine was purchased (Sigma Aldrich) as 60% grade and purified by a method described in the literature14 with slight modifications (see below). Flash chromatography was performed on 40–63 mesh silica gel. Microwave oven reactions were performed using the CEM Focused Microwave™ Synthesis System (Model Discover). Melting points were determined on a digital melting point apparatus (Büchi B 545). Optical rotations were determined on a digital polarimeter (JASCO DIP-1000) at 22 °C in CHCl3 or MeOH as solvent. Exact masses were obtained using high-resolution mass spectrometry (Bruker MicroTOF LC) with electrospray ionisation (ESI).Synthesis of chiral solvating agents
General procedure for the preparation of secondary amines under microwave irradiation
The mixture was filtered and solvent evaporated. The crude product was dissolved in Et2O, dried over MgSO4 and filtered. The solvent was evaporated and the product dried under vacuum, and purified by column chromatography (1:9 MeOH:DCM). Yield 0.60 g, 42.0%; colourless liquid; calc. for C21H34N [M + H]+ 300.2686, found 300.2690; [α]22D +50.6360 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ ppm 0.951 (s, 3H, CH3), 1.223 (s, 3H, CH3), 1.229 (d, J = 6.90 Hz, 6H, 2 × CH3), 1.406 (m, 1H, CHH), 1.431 (m, 2H, CH2), 1.543 (dd, J = −12.27, 2.54 Hz, 1H, CH), 1.771 (m, 2H, CH2), 1.789 (m, 2H, CH2), 2.334 (d, J = −11.72 Hz, 1H, CHH), 2.433 (s, 3H, CH3), 2.457 (d, J = −11.72 Hz, 1H, CHH), 2.826 (sep J = 6.90 Hz, 1H, CH), 2.893 (m, 2H, CH2), 6.885 (d, J = 2.11 Hz, 1H, CHAr), 6.986 (dd, J = 8.20, 2.11 Hz, 1H, CHAr), 7.172 (d, J = 8.20 Hz, 1H, CHAr); 13C NMR (300 MHz, CDCl3) δ ppm 19.008 (CH3), 19.091 (CH2), 19.218 (CH2), 24.146 (2 × CH3), 25.409 (CH3), 30.361 (CH2), 33.590 (CH), 36.520 (CH2), 37.094 (C), 37.580 (C), 37.879 (CH3), 38.664 (CH2), 46.061 (CH), 64.881 (CH2), 123.902 (CHAr), 124.355 (CHAr), 126.896 (CHAr), 134.908 (CAr), 145.582 (CAr), 147.694 (CAr).
General procedure for preparation of secondary amines under microwave radiation
(+)-Dehydroabietylamine (1.0 eq.), 1-bromoethane (1.05 eq.) and Na2CO3 (0.6 eq.) were added to a microwave tube with isopropanol (in the case of 6a (+)-dehydroabietylamine (2.0 eq.), 1,2-dibromoethane (1.0 eq.) and Na2CO3 (1.0 eq.) were used). The reaction mixture was microwave irradiated (110 W at 110 °C) for 2 h. The solvent was evaporated and the residue triturated with ether, filtered and mixed with Et2O. The separated phases and organic phase was washed with water until neutral. The organic phase was dried over Na2SO4 and filtered, the solvent evaporated and the product dried under vacuum.:9 MeOH:DCM). Yield 0.78 g 74.3%; white solid; mp. 63.8 °C; calc. for C42H65N2 [M + H]+ 597.5142, found 597.5132; [α]22D +43.3160 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ ppm 0.914 (s, 6H, 2 × CH3), 1.204 (s, 6H, 2 × CH3), 1.229 (d, J = 7.02 Hz, 12H, 4 × CH3), 1.336 (m, 2H, 2 × CHH), 1.375 (m, 4H, 2 × CH2), 1.565 (dd, J = −12.25, 2.66 Hz, 2H, 2 × CH), 1.603 (m, 4H, 2 × CH2), 1.713 (m, 2H, 2 × CHH), 1.753 (m, 2H, 2 × CHH), 2.233 (dt, J = −12.83, 3.27 Hz, 2H, 2 × CHH), 2.319 (d, J = −11.80 Hz, 2H, 2 × CHH), 2.509 (d, J = −11.80 Hz, 2H, 2 × CHH), 2.696 (s, 4H, 2 × CH2), 2.824 (sep J = 7.02 Hz, 2H, 2 × CH), 2.876 (m, 4H, 2 × CH2), 6.872 (d, J = 1.85 Hz, 2H, 2 × CHAr), 6.983 (dd, J = 8.13, 1.85 Hz, 2H, 2 × CHAr), 7.158 (d, J = 8.13 Hz, 2H, 2 × CHAr); 13C NMR (500 MHz, CDCl3) δ ppm 18.983 (4 × CH2), 19.348 (2 × CH3), 24.138 (4 × CH3), 25.466 (2 × CH3), 30.459 (2 × CH2), 33.582 (2 × CH), 36.390 (2 × CH2), 37.167 (2 × C), 37.548 (2 × C), 38.583 (2 × CH2), 45.624 (2 × CH), 50.018 (CH2), 61.606 (2 × CH2), 123.910 (2 × CHAr), 124.412 (2 × CHAr), 126.888 (2 × CHAr), 134.883 (CAr), 145.541 (2 × CAr), 147.637 (2 × CAr).
General procedure of NTf2 salts
Primary amine 1a or secondary amine 2a–6a (1.0 eq.) was dissolved to DCM (0.5 mL). HNTf2 (1.0 eq.) was added at 0 °C. Reaction mixture was stirred for 1 h at rt. The layers were separated and the organic phase washed with water (3 × 2.0 mL). The organic solvent was evaporated and product dried in vacuum.Synthesis of guests
N-Acetylation of phenylalanine was performed according to literature.16 Preparation of [NBu4]+ salts was performed by adding tetrabutylammoniumhydroxide (1.0 M in MeOH, 1.0 eq.) to racemic acid (1.0 eq.) in MeOH. After stirring for 1–3 h, the solvent was evaporated and product was dried in vacuum.Acknowledgements
TL is grateful for the support of the University of Helsinki and The Finnish Society of Sciences and Letters.Notes and references
- (a) H. Bergmann, B. Grosch, S. Sitterberg and T. Bach, J. Org. Chem., 2004, 69, 970 CrossRef CAS PubMed; (b) K. S. Heo, M. H. Hyun, Y. J. Cho and J. J. Ryoo, Chirality, 2011, 23, 281 CrossRef CAS PubMed; (c) S. H. Grimm, L. Allmendinger, G. Hoefner and K. T. Wanner, Chirality, 2013, 25, 923 CrossRef CAS PubMed.
- M. Suzuki, J. R. Deschamps, A. E. Jacobson and K. C. Rice, Chirality, 2015, 27, 287 CrossRef CAS PubMed.
- (a) G. Uccello-Barretta and F. Balzano, Top. Curr. Chem., 2013, 341, 69 CrossRef CAS; (b) K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235 CrossRef CAS PubMed.
- N. Jain, M. B. Mandal and A. V. Bedekar, Tetrahedron, 2014, 70, 4343 CrossRef CAS PubMed.
- T. J. Wenzel, Discrimination of Chiral Compounds Using NMR Spectroscopy, John Wiley & Sons, Hoboken, New Jersey, 2007 Search PubMed.
- V. Kumar, C. E. Olsen, S. J. C. Schaeffer, V. S. Parmar and S. V. Malhotra, Org. Lett., 2007, 9, 3905 CrossRef CAS PubMed.
- First used by N. Guranatne et al. for the construction of an enantio-recognizing reagent: M. B. Foreiter, H. Q. N. Gunaratne, P. Nockemann, K. R. Seddon, P. J. Stevenson and D. F. Wassell, New J. Chem., 2013, 37, 515 RSC.
- (a) T. Heckel, A. Winkel and R. Wilhelm, Tetrahedron: Asymmetry, 2013, 24, 1127 CrossRef CAS PubMed; (b) B. Altava, D. S. Barbosa, M. Isabel Burguete, J. Escorihuela and S. V. Luis, Tetrahedron: Asymmetry, 2009, 20, 999 CrossRef CAS PubMed.
- L. Gonzalez, B. Altava, M. Bolte, M. I. Burguete, E. Garcia-Verdugo and S. V. Luis, Eur. J. Org. Chem., 2012, 4996 CrossRef CAS PubMed.
- K. Tanaka and N. Fukuda, Tetrahedron: Asymmetry, 2006, 17, 916 CrossRef PubMed.
- (a) A. R. Chaudhary, P. Yadav and A. V. Bedekar, Tetrahedron: Asymmetry, 2014, 25, 767 CrossRef CAS PubMed; (b) L. Liu, M. Ye, X. Hu, X. Yu, L. Zhang and X. Lei, Tetrahedron: Asymmetry, 2011, 22, 1667 CrossRef CAS PubMed; (c) M. Periasamy, M. Dalai and M. Padmaja, J. Chem. Sci., 2010, 122, 561 CrossRef CAS; (d) X. Yang, G. Wang, C. Zhong, X. Wu and E. Fu, Tetrahedron: Asymmetry, 2006, 17, 916 CrossRef CAS PubMed; (e) S. Tabassum, M. A. Gilani and R. Wilhelm, Tetrahedron: Asymmetry, 2011, 22, 1632 CrossRef CAS PubMed; (f) S. Satishkumar and M. Periasamy, Tetrahedron: Asymmetry, 2009, 20, 2257 CrossRef CAS PubMed; (g) S. Bozkurt, M. Durmaz, H. N. Naziroglu, M. Yilmaz and A. Sirit, Tetrahedron: Asymmetry, 2011, 22, 541 CrossRef CAS PubMed; (h) W. Wang, F. Ma, X. Shen and C. Zhang, Tetrahedron: Asymmetry, 2007, 18, 832 CrossRef CAS PubMed; (i) C. Pena, J. Gonzalez-Sabin, I. Alfonso, F. Rebolledo and V. Gotor, Tetrahedron, 2008, 64, 7709 CrossRef CAS PubMed.
- (a) M. Perez-Trujillo, L. Castanar, E. Monteagudo, L. T. Kuhn, P. Nolis, A. Virgili, R. T. Williamson and T. Parella, Chem. Commun., 2014, 50, 10214 RSC; (b) J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901 CrossRef CAS PubMed; (c) S. R. Chaudhari and N. Suryaprakash, Chem. Phys. Lett., 2013, 555, 286 CrossRef CAS PubMed; (d) N. Lokesh, S. R. Chaudhari and N. Suryaprakash, Org. Biomol. Chem., 2014, 12, 993 RSC.
- T. J. Wenzel, in Stereoselective Synthesis of Drugs and Natural Products, ed. V. Andrushko and N. Andryushko, John Wiley & Sons, Inc., Hoboken, New Jersey, 2014, pp. 1505–1528 Search PubMed.
- L. C. Cheney, US Pat, 2787637, 1957 Search PubMed.
- G. Su, L. Huo, W. Huang, H. Wang and Y. Pan, Chin. J. Struct. Chem., 2009, 28(6), 693–698 CAS.
- S. Stella and A. Chadha, Tetrahedron: Asymmetry, 2010, 21(4), 457–460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data, chiral recognition, titration, enantiomeric excess studies, separation of carboxylic acids. See DOI: 10.1039/c5ob01667c |
| This journal is © The Royal Society of Chemistry 2015 |