
Insights into the catalytic mechanism of synthetic glutathione peroxidase mimetics
Debasish
Bhowmick
and
Govindasamy
Mugesh
*
Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: mugesh@ipc.iisc.ernet.in; Fax: +91-80-2360 1552/2360 0683
First published on 7th September 2015
Abstract
Glutathione Peroxidase (GPx) is a key selenoenzyme that protects biomolecules from oxidative damage. Extensive research has been carried out to design and synthesize small organoselenium compounds as functional mimics of GPx. While the catalytic mechanism of the native enzyme itself is poorly understood, the synthetic mimics follow different catalytic pathways depending upon the structures and reactivities of various intermediates formed in the catalytic cycle. The steric as well as electronic environments around the selenium atom not only modulate the reactivity of these synthetic mimics towards peroxides and thiols, but also the catalytic mechanisms. The catalytic cycle of small GPx mimics is also dependent on the nature of peroxides and thiols used in the study. In this review, we discuss how the catalytic mechanism varies with the substituents attached to the selenium atom.
Introduction
Selenoenzymes are a class of proteins that contain a selenocysteine (Sec, U) residue in their active sites. The biological activities of these enzymes are interesting due to the unique redox properties of the selenium atom.1 Selenoenzymes act as antioxidants by maintaining the cellular redox balance or by controlling the thyroid hormone levels. To date, around 30 selenoproteins are known and few of these selenoproteins have been isolated and characterized biochemically.2 The major selenoenzymes discovered to date include formate dehydrogenases,3 hydrogenases,4 glycine reductase,5 iodothyronine deiodinases (ID),6 thioredoxin reductases (TrxR),7 selenophosphate synthetase,8 and glutathione peroxidase (GPx).9 Significant research efforts have been directed toward the synthesis of small molecule mimics of these selenoenzymes for better understanding of their catalytic activities and mechanisms. Although synthetic mimics are not known for many of these enzymes, synthetic compounds that mimic the function of GPx have been studied extensively.The mammalian GPx is an important antioxidant enzyme, which protects various biomolecules such as proteins, amino acids, lipids, DNA etc. from oxidative damage. GPx catalyzes the reduction of hydroperoxides using glutathione (GSH) as a cofactor.10 The GPx superfamily consists of four enzymes, cytosolic GPx (cGPx), phospholipid hydroperoxide GPx (PHGPx), plasma GPx (pGPx) and gastrointestinal GPx (giGPx).11 Although all of these enzymes contain the Sec residue in their active sites, their reactivity is highly dependent on the nature of peroxides and thiols.12
The proposed catalytic cycle of GPx contains three steps involving three active states of the enzyme. In the first step, the selenolate state (E-SeH) of the Sec residue reduces hydroperoxides to water (or alcohol) to form the oxidized selenenic acid (E-SeOH) state,9a,13 which subsequently reacts with one equivalent of GSH to generate the selenenyl sulfide (E-SeSG) state. Nucleophilic attack of a second equivalent of cellular GSH regenerates the active selenol species with elimination of the oxidized GSH (GSSG). Cleavage of the –Se–S– bond by GSH is the rate determining step in the overall process (Scheme 1).11
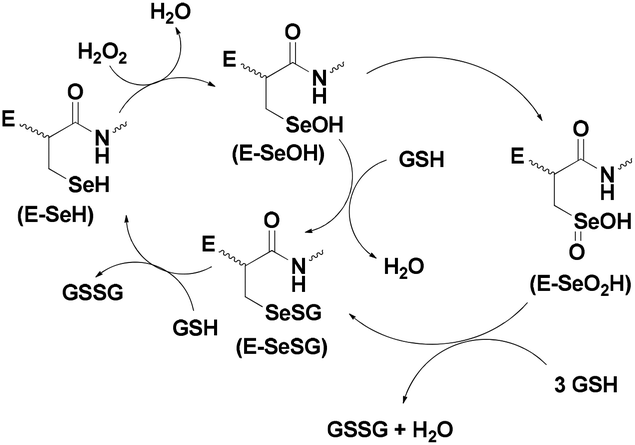 | ||
| Scheme 1 Proposed mechanism for the GPx-catalyzed reduction of H2O2. | ||
At higher concentrations of hydroperoxides, the selenium centre may undergo overoxidation to produce the corresponding seleninic acid (E-SeO2H), which may be converted to the selenenyl sulfide by reaction with excess GSH. However, the formation of the overoxidized seleninic acid derivative may reduce the catalytic activity, although it has not been demonstrated in vivo. The crystal structure of GPx (Fig. 1a) indicates that the Sec residue (Sec 45) remains very close to two other amino acid residues glutamine (Gln80) and tryptophan (Trp158) (Fig. 1b) forming a catalytic triad.14 These two amino acid residues are very important for the catalytic activity as they stabilize the selenolate moiety by hydrogen bonding interactions. Such interactions may also prevent the cleavage of the C–Se bond, which generally leads to toxicity of the organoselenium compounds.
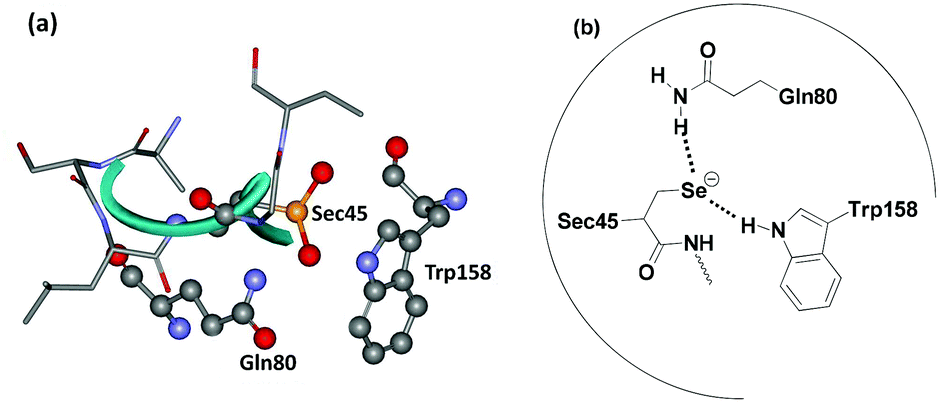 | ||
| Fig. 1 (a) Active site of glutathione peroxidase in seleninic acid form (PDB code 1GP1 figure) determined by X-ray crystallography. (b) Catalytic triad at the active site of GPx. | ||
Considerable attention has been directed toward the design and synthesis of small molecule organoselenium compounds that functionally mimic the GPx enzyme.15 Synthetic mimics were primarily synthesized to understand the chemistry at the active site of GPx. However, they may also be very useful as drugs for diseases related to oxidative stress. The synthetic mimics were designed with a heteroatom near the selenium centre considering the GPx catalytic triad in the active site. The majority of the synthetic mimics reported so far can be classified into two major categories on the basis of their structures. The first category of compounds contains the heteroatom directly bonded to the selenium centre. In the second category, the compounds do not have a direct selenium–heteroatom bond, but a heteroatom is placed near the selenium centre leading to weak selenium–heteroatom non-covalent interactions.15 Although these compounds mimic the function of the natural GPx enzyme, they may not necessarily follow a similar catalytic cycle, which is highly dependent on their structure and reactivity. Natural enzymes mostly use GSH as the thiol cofactor. However, synthetic mimics may use a thiol such as GSH or small aromatic/aliphatic thiols for their catalytic activities. The selenium atom in all the organoselenium compounds is responsible for the reduction of peroxides, but its reactivity towards thiol and peroxides depends on the electronic and steric environments around the selenium centre. Their catalytic mechanism is also dependent on the nature of peroxides and thiols employed in the study. Therefore, in this review, we provide an overview of the catalytic mechanism of different synthetic mimics.
Ebselen and related cyclic compounds
Ebselen (2-phenyl-1,2-benzisoselenazole-3)-(2H)-one (1) containing a direct Se–N bond (Fig. 2) was the earliest example of a synthetic GPx mimic. Ebselen exerts many biological functions both in vitro and in vivo systems.16 It is associated with many therapeutic properties such as the reduction of hydrogen peroxide (H2O2) and lipid peroxides, effective scavenging of highly reactive peroxynitrite and inhibition of a variety of free radical generating enzymes and enzymes involved in inflammatory diseases such as lipoxygenase and cyclooxygenase.17 Ebselen was found to be less toxic because of its stable isoselenazole moiety and it is under phase III clinical trial for the treatment of hearing loss, inflammation, stroke, neurodegenerative diseases, reperfusion injury, bipolar disorder etc.18 | ||
| Fig. 2 Chemical structures of the cyclic selenenyl amides 1–3. | ||
Although ebselen has been studied extensively as a GPx mimic, its catalytic cycle for the reduction of hydroperoxides by a thiol has been controversial for many years.19 The first proposed mechanism, shown in Scheme 2, involves selenol 5, selenenyl sulfide 4 and selenenic acid 6 as the intermediates. Although ebselen exhibits moderate catalytic activity using GSH as the cofactor, several studies have showed that it is a relatively inefficient catalyst in the presence of aromatic thiols such as thiophenol, or other related thiols such as benzyl thiol.20
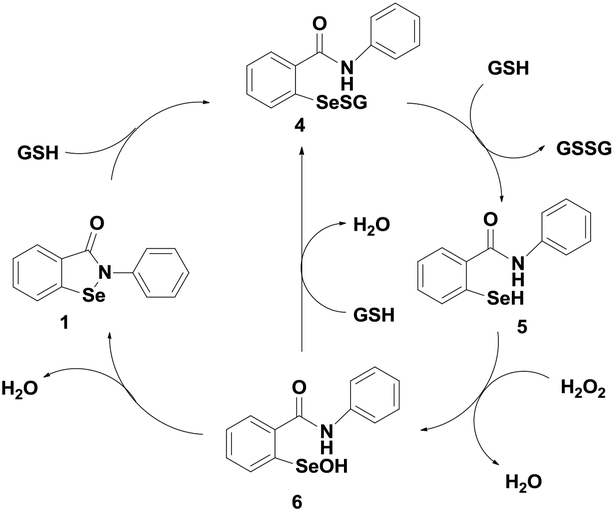 | ||
| Scheme 2 Initially proposed catalytic mechanism of ebselen. | ||
Detailed experimental and theoretical studies revealed that the poor activity of ebselen and related analogues is due to an extensive thiol exchange reaction, which is initiated by the strong Se⋯O non-covalent interactions in the selenenyl sulfide intermediate.20b As the thiol exchange reaction prevents the formation of the active selenol species, Mugesh and co-workers proposed a revised catalytic mechanism for the GPx-like activity of ebselen using PhSH based on the 77Se NMR experiments.20 According to this mechanism, the selenenyl sulfide 7 produces a diselenide intermediate 8 by disproportionation reaction instead of generating the corresponding selenol. The diselenide 8, which acts as the active catalyst in this mechanism, reduces peroxides and is oxidized to the corresponding selenenic acid 6 (Scheme 3).20d The disproportionation reaction of the selenenyl sulfide 7 to diselenide 8 was found to be the rate limiting step in the overall catalytic reaction. Therefore, the extent of thiol exchange reaction and the stability of the selenenyl sulfide intermediate in solution are highly dependent on the nature of the thiol used as the cofactor.
 | ||
| Scheme 3 A revised catalytic mechanism of ebselen (1). | ||
Reich et al. have reported the antioxidant activity of cyclic selenenyl amide 2 (Fig. 2).21 Based on the reactions of compound 2 with t-BuOOH (TBHP) and thiols, they have proposed a catalytic mechanism shown in Scheme 4. Similar to ebselen, the initial reaction of compound 2 with the thiol leads to the cleavage of the Se–N bond generating the corresponding selenenyl sulfide 10, which in the presence of excess thiol produces the active selenol species 11. Interestingly, selenol 11 reacts with peroxides to generate the diselenide 12 instead of producing the corresponding selenenic acid, which is observed for the native GPx enzymes (Scheme 1).22 It is noteworthy that both the catalytic cycles of ebselen and compound 2 involve the corresponding diselenide as the intermediates. However, the mechanism of formation of the diselenides is entirely different.
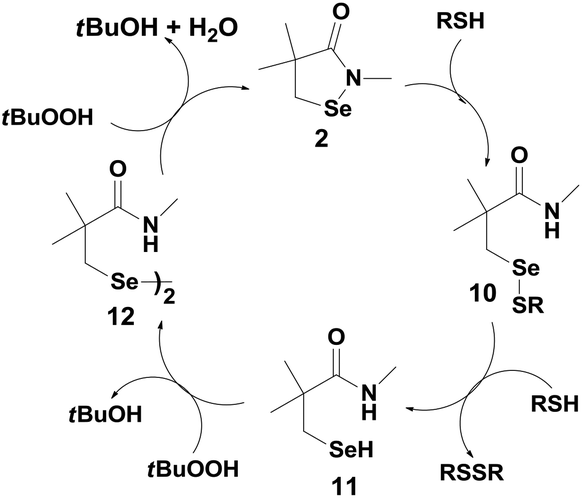 | ||
| Scheme 4 The proposed catalytic cycle of selenenyl amide 2. | ||
Back and Dyck have synthesized a camphor-based selenenyl amide 3 (Fig. 2), which exhibits GPx-like activity by reducing t-BuOOH using benzyl thiol (BnSH) as the cofactor.23 The catalytic mechanism that is followed by 3 is significantly different from that of 1 and 2. The first nucleophilic attack of thiol at the selenium centre cleaves the Se–N bond, generating the corresponding selenenyl sulfide 13, which forms a catalytic cycle with selenol 14 and selenenic acid 15 (Scheme 5). Thus, unlike other cyclic selenenyl amides, the catalytic mechanism of 3 resembles the catalytic cycle of the native GPx enzymes. Compound 3 acts as a pro-catalyst as it is not directly involved in the catalytic cycle. In the entire cycle, the conversion of selenenyl sulfide to selenol is the rate determining step.
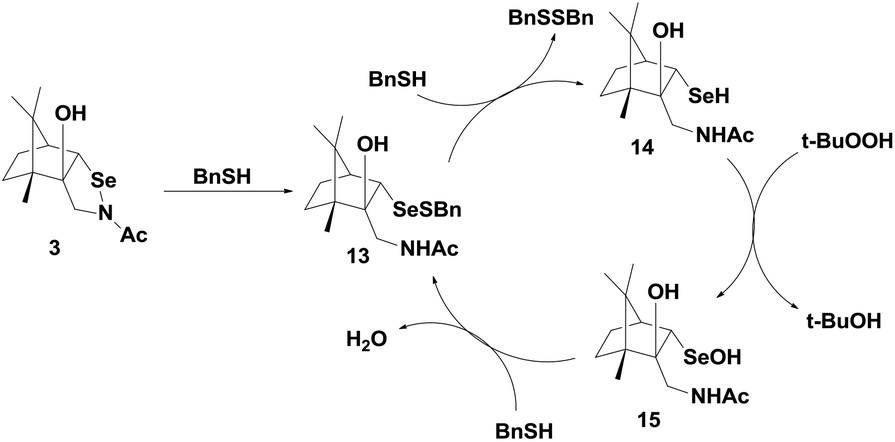 | ||
| Scheme 5 Proposed mechanism for the GPx activity of the camphor-derived selenenamide 3. | ||
Mugesh et al. reported a series of peptide-containing ebselen analogues (Fig. 3),24 whose activities and catalytic mechanisms are highly dependent on the nature of the peptide chains attached to the nitrogen atom. For example, the catalytic cycle for compound 16 is different from that of compounds 17–18. A catalytic mechanism for compound 16 was proposed using 77Se NMR. Interestingly, it was observed that the mechanism is significantly different from that of ebselen. In contrast to ebselen, compound 16 reacts with the thiol to generate the selenol intermediate, which is the active species in the catalytic cycle. Thus, the reactivity of selenenyl sulfide derived from 16 with thiol is different from that of compound 7, which does not produce any selenol in the cycle.20d In contrast, the peptide-based ebselen analogues having Phe residues such as compounds 17–18 follow a mechanism identical to that of ebselen (Scheme 3), involving the diselenide intermediates. Therefore, the catalytic mechanism of the peptide containing ebselen analogues was found to be dependent on the nature of the peptide chains.
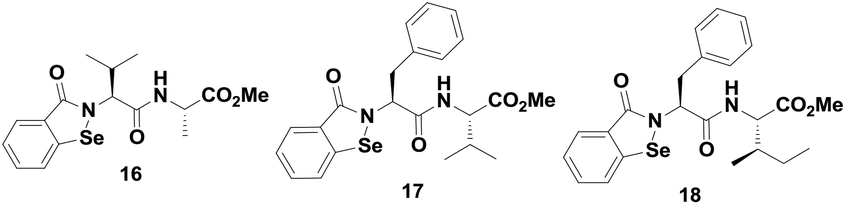 | ||
| Fig. 3 Chemical structures of peptides containing ebselen analogues 16–18. | ||
Singh and co-workers reported novel isoselenazolines 19–20 and isoselenazoline Se-oxides 21–22 (Fig. 4),25 which are stabilized by intramolecular Se⋯O interactions. These compounds exhibited higher GPx-like activities compared to that of ebselen that contains a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. Particularly, the selenoxides 21–22 are almost 3–4 times more active than ebselen. Based on the experimental studies, they have proposed a catalytic mechanism for compound 21.25
O group. Particularly, the selenoxides 21–22 are almost 3–4 times more active than ebselen. Based on the experimental studies, they have proposed a catalytic mechanism for compound 21.25
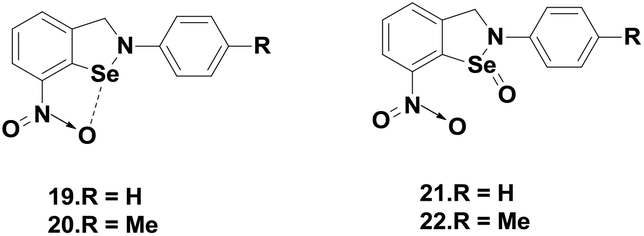 | ||
| Fig. 4 Chemical structures of isoselenazolines 19–20 and their oxides 21–22. | ||
Scheme 6 indicates that the selenenyl amide bond is cleaved by thiol (PhSH) to produce the selenenyl sulfide intermediate 23, which acts as a true catalyst in the catalytic mechanism involving compounds 24 and 25. Although this pathway is different from that of ebselen, the formation of the diselenide 26 from the corresponding selenenyl sulfide by disproportionation reaction is quite similar to that observed during the reduction of peroxides by ebselen.
 | ||
| Scheme 6 Proposed catalytic mechanism for the GPx-like activity of compound 21. | ||
Recently, Mugesh and coworkers have synthesized similar types of isoselenazole compounds 28–31 (Fig. 5),26 which contain a methoxy substituent in the ortho-position and alkyl substituents on the nitrogen atom. Interestingly, it was observed that compounds 28–31 display excellent glutathione peroxidase activity both in vitro and inside human cells. A comparison of the catalytic activity shows that all the isoselenazole compounds exert very high activities as compared to ebselen. Interestingly, these compounds also mimic the peroxiredoxins in human cells by using cellular thioredoxin as reducing agents.
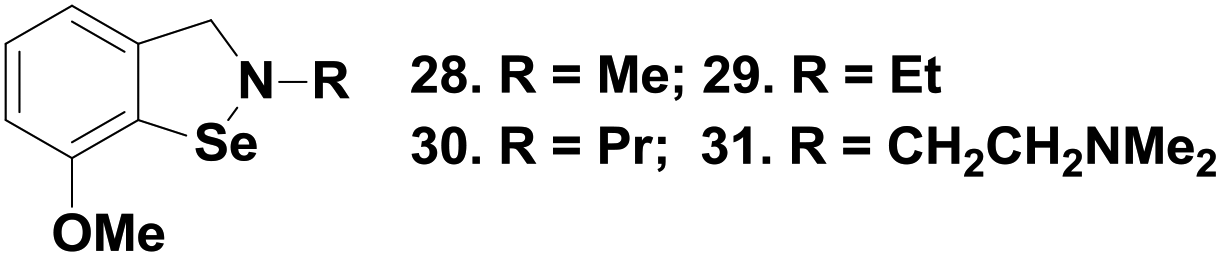 | ||
| Fig. 5 Chemical structures of isoselenazoles 28–31. | ||
Although these isoselenazoles are structurally similar to compounds 19–20, the reactivity towards thiol is significantly different. It has been previously observed that the selenium centre in cyclic selenenyl amide compounds readily undergoes reductive cleavage by the thiol. The amide moiety makes the selenium centre highly electrophilic facilitating the nucleophilic attack of the thiol. On the other hand, compounds 19–20, which lack the carbonyl group, also react with thiol to generate the corresponding selenenyl sulfides. It has been shown that the presence of intramolecular secondary Se⋯O interactions with the nitro group enhances the reactivity of the Se–N bond towards cleavage by thiol.25 In contrast, isoselenazoles 28–31 reacted very slowly with thiol to cleave the Se–N bond as the selenium centre is not activated. Thus, the catalytic mechanism of 31 involves the oxidation of selenium with peroxides generating the selenoxide intermediates 32, which subsequently undergoes rapid reaction with an excess amount of PhSH to produce the corresponding selenenyl sulfide 33. Compound 33 then follows a catalytic cycle similar to the native enzyme involving the selenol 34 and the selenenic acid 35 (Scheme 7). It is noticed that, unlike compound 26, the diselenide 36 is produced from the selenol intermediate after auto-oxidation. Although isoselenazoles 19 and 28 maintain a similar catalytic cycle which involves selenenyl sulfide, selenol and selenenic acids as the intermediates, the reactivity towards thiol and peroxides is significantly different due to the different electronic environments around the selenium atom.
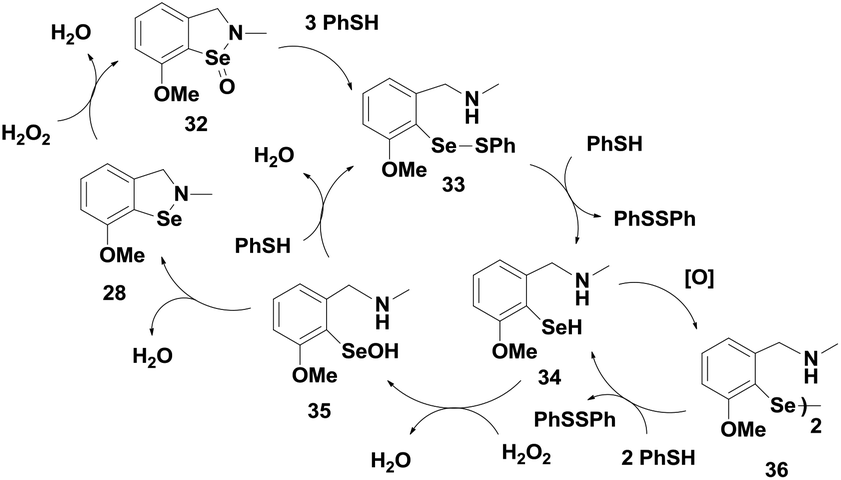 | ||
| Scheme 7 Catalytic mechanism for the GPx-like activity of compound 28. | ||
Selenides and seleninate esters
Back and Moussa showed that a number of allyl selenides 37–40 (Fig. 6) exhibit very good GPx-like activity in the presence of t-BuOOH as the substrate and benzyl thiol as the cofactor.27 These selenides act as pro-catalysts. Compound 37 having three spacer carbon atoms between selenium and the alcohol moiety was found to exhibit very high antioxidant activity. However, compound 43 with an amide moiety also shows good catalytic activity. | ||
| Fig. 6 Chemical structures of allyl selenides 37–40. | ||
The allyl selenide 37 reacts readily with TBHP to produce selenoxide 41, which undergoes a [2,3] sigmatropic rearrangement to generate the seleninate ester 42. In the presence of excess peroxide, the selenium undergoes overoxidation to produce an unstable intermediate 43, which after rapid cyclization generates the cyclic seleninate ester 44 (Scheme 8).
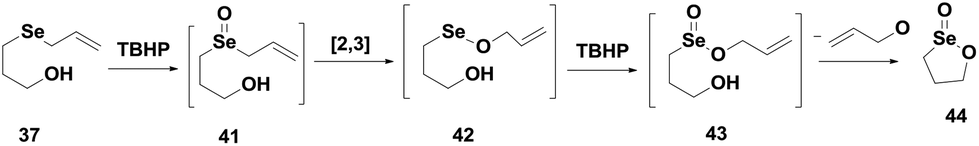 | ||
| Scheme 8 Reaction of compound 37 with peroxide to produce the cyclic compound 44. | ||
The catalytic mechanism (Scheme 9) suggests that compound 44 first reacts with BnSH to produce the thioseleninate 45, which undergoes further thiolysis reaction to afford the selenenic acid 46 and disulfide BnSSBn. The selenenic acid 46 reconverts to the seleninate ester 44 by oxidative cyclization in the presence of TBHP. However, in the presence of excess thiol, compound 46 reacts with another BnSH molecule to produce the selenenyl sulfide 47, which after prolonged reaction with peroxide regenerates the seleninate ester 44.
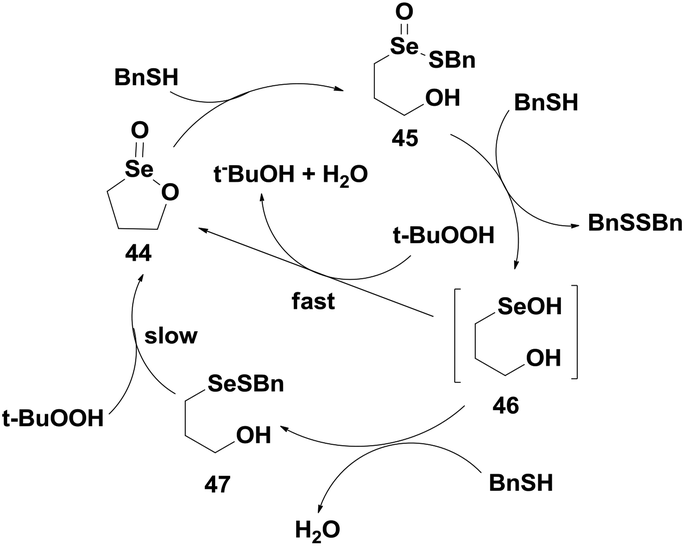 | ||
| Scheme 9 Proposed catalytic mechanism of the cyclic seleninate ester 44. | ||
Subsequently, Back et al. and Singh et al. have synthesized a series of aromatic analogues 48–51 (Fig. 7) of cyclic seleninate ester 44. Back et al. showed that the aromatic analogue 48 exhibits less catalytic activity compared to that of 44.28 However, the introduction of electron donating substituents such as the methoxy group (49) can enhance the activity considerably.29 Although the catalytic activities differ significantly, Back et al. observed that the catalytic cycles involving the cyclic ester, thioseleninate and the selenenic acid intermediates remain unaltered.28–30 The formation of selenenyl sulfide was observed in excess thiol concentration as a deactivation pathway. However, the cyclic seleninate ester is regenerated from the selenenyl sulfide in the presence of TBHP either via the thioseleninate intermediate or the corresponding diselenide. Interestingly, Back and co-workers could not observe any diselenide for the reduction of TBHP with BnSH, which suggests that disproportionation of selenenyl sulfide is not the major pathway in the regeneration of seleninate esters.
 | ||
| Fig. 7 Chemical structures of cyclic seleninate esters 48–51. | ||
In contrast, Singh et al. have shown that the catalytic mechanism of the cyclic seleninate ester 51 for the reduction of TBHP using PhSH is different when the thiol concentration is high.31 Compound 51 follows a catalytic mechanism similar to that of 48. However, in the presence of excess thiol, the selenenyl sulfide, generated from the corresponding selenenic acid in the deactivation pathway, regenerates the seleninate ester 51via formation of the diselenide intermediate, which was not observed by Back and co-workers.28–30 This might be due to the ortho-nitro substituent that stabilizes the selenenyl sulfide intermediate by Se⋯O non-covalent interactions leading to the formation of the diselenide by disproportionation reaction.
Back and co-workers have also reported a highly efficient GPx mimic di-(3-hydroxy-propyl)selenide 52, which exhibits almost 15 times higher activity than ebselen.32 It follows an interesting catalytic mechanism that involves a novel spirodioxaselenanonane 54 as an intermediate. Unlike the monoselenide 37, compound 52 directly takes part in the catalytic cycle and reduces peroxides. Initial oxidation by t-BuOOH produces a transient selenoxide 53, which undergoes spontaneous cyclization to generate the cyclic spirodioxaselenanonane 54. Selenide 52 is subsequently regenerated upon reaction with two equivalents of BnSH (Scheme 10). This was the first report of a catalytic mechanism involving a cyclic selenanonane as the intermediate.
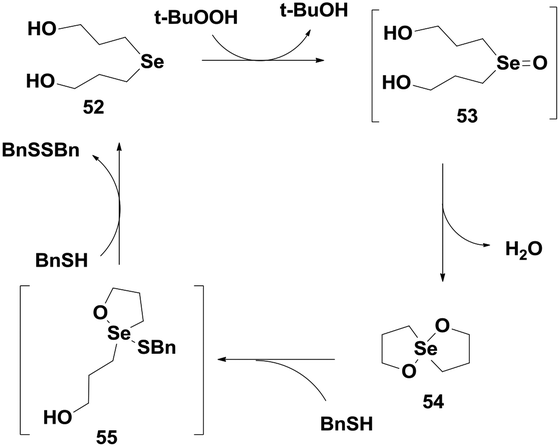 | ||
| Scheme 10 Catalytic cycle of selenide 52 for the reduction of TBHP. | ||
Singh and co-workers have reported a stable cyclic selenenate ester 56 (Fig. 8) that exhibits almost 300 times higher GPx-like activity as compared to that of diphenyl diselenide (PhSeSePh) in the presence of H2O2 as the substrate and PhSH as the cofactor.33 Unlike compound 44, the selenenate ester 56 first reacts with peroxides, generating the corresponding cyclic seleninate and thereby acting as the active intermediate in the catalytic cycle.
 | ||
| Fig. 8 Chemical structure of the selenenate ester 56. | ||
Excellent catalytic activities of compounds 44 and 56 also indicate that compounds containing intramolecular Se–O bonds are equally effective GPx mimics as the commonly studied Se–N derivatives. This interaction is also very important for the stabilization and isolation of catalytic intermediates.
Back and co-workers have shown the catalytic activity of some monoselenides 57–58 (Fig. 9).30 These monoselenides generally exhibit their antioxidant properties by Se(II)/Se(IV) redox cycle in the presence of peroxides and thiol. However, Braga, Detty and co-workers studied the GPx-like activities of a series of monoselenides 59–60 and their oxides 61–62 (Fig. 10) that follow a mechanism involving a hydroxy perhydroxy selenane intermediate.34
 | ||
| Fig. 9 Chemical structures of the monoselenides 57–58. | ||
 | ||
| Fig. 10 Chemical structures of the monoselenides 59–60 and their oxides 61–62. | ||
In the presence of peroxides, selenoxide 61 is converted to the hydroxy perhydroxy selenane 64via hydroxyselenonium 63. Compound 64 is a kinetically better oxidizing agent than the selenoxide 61. Compound 64 then reacts with two equivalents of PhSH to produce PhSSPh and 63avia an intermediate 65 restarting the catalytic cycle with H2O2 (Scheme 11). The catalytic activity of the selenoxide is little affected by electronic demands at the Se centre, however, selenoxides containing chelating groups, especially amino groups, were found to be better catalysts.
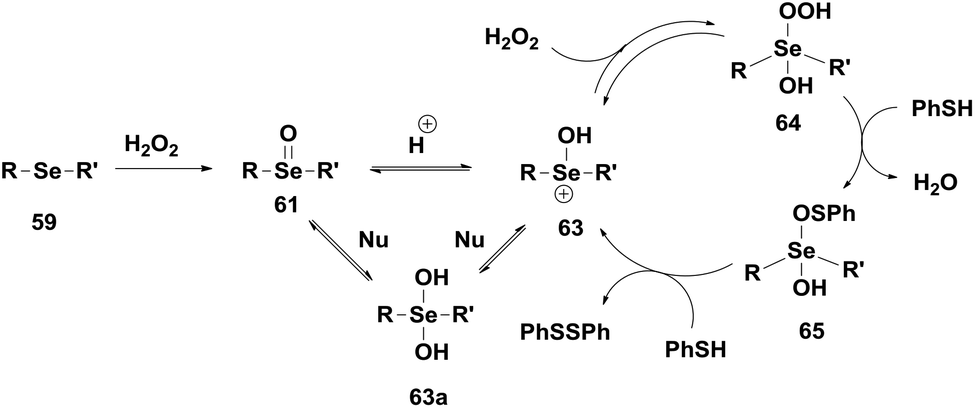 | ||
| Scheme 11 Proposed catalytic mechanism for the GPx-like activity of 59 and 61. | ||
Catalytic mechanism of diaryl diselenides
Diaryl diselenides are another class of GPx mimics that exhibit excellent catalytic activities for the reduction of peroxides. It was observed that diphenyl diselenide (PhSeSePh) exhibits two times higher activity than that of ebselen. The reductive cleavage of the diselenide bond by the thiol generates the corresponding selenol intermediate (PhSeH) that reduces peroxides. Subsequently, Spector and co-workers35 have reported diaryl diselenides 66–67 (Fig. 11), which contain a tert-amino moiety near the selenium centre. These diselenides were found to be more active than ebselen. They proposed that the selenium and the nitrogen atoms are involved in an intramolecular non-covalent interaction, which modulates the catalytic activity of diaryl diselenides. | ||
| Fig. 11 Chemical structures of the amine-based diselenides 66–68. | ||
Iwaoka and Tomoda have studied the catalytic mechanism of diselenide 68 for the reduction of H2O2 by PhSH using 77Se NMR spectroscopy.36 They have proposed a catalytic cycle (Scheme 12) based on the experimental findings. It is observed that the non-bonded Se⋯N interaction plays an important role in the catalytic cycle. The initial nucleophilic attack of the PhSH produces a mixture of selenenyl sulfide 71 and selenolate ion 69, which is generated from the corresponding selenol after deprotonation by the amino moiety. 69 and 71 form a catalytic cycle with selenenic acid 70. The diselenides actually act as pro-catalysts as they do not directly take part in the catalytic cycle. However, they follow a catalytic cycle similar to that of the native GPx enzyme for reducing peroxides.
 | ||
| Scheme 12 Catalytic cycle of diselenide 75 in the presence of H2O2 and PhSH. | ||
Based on the mechanistic observations, Iwaoka and Tomoda36 proposed the following possible roles of the amino group in the catalytic cycle: (i) the amino group can generate kinetically more reactive and more nucleophilic selenolate anions (E-Se−) from the selenol intermediate (E-SeH) after deprotonation. (ii) The non-covalent Se⋯N interaction prevents the selenenic acid moiety from overoxidation in the presence of excess peroxides. (iii) The Se⋯N interaction facilitates the nucleophilic attack of thiol at the sulfur center of the selenenyl sulfide intermediate allowing effective regeneration of the selenol species.
Although the tert-amine-based diselenides are significantly more active than ebselen, detailed experimental investigations indicate that their catalytic efficiency may be reduced considerably by other deactivation pathways. In the selenenic acid intermediate, the selenium centre undergoes overoxidation to produce the corresponding seleninic or selenonic acids, which reduces the catalytic activity significantly at lower concentrations of the thiol. On the other hand, in the selenenyl sulfide intermediate, the nucleophilic attack of the thiol predominantly takes place at the selenium centre due to the thiol exchange reaction (Scheme 13), which prevents the regeneration of active selenol species. Although the Se⋯N interactions in selenenyl sulfides derived from tert-amine-based diselenide are much fewer than Se⋯O interactions in 7, this Se⋯N interaction is sufficient to cause significant thiol exchange reactions.35 In this context, Singh and co-workers reported redox-active diferrocenyl diselenides 72–73 (Fig. 12), which exert a higher catalytic activity than that of diselenides 66–67.37
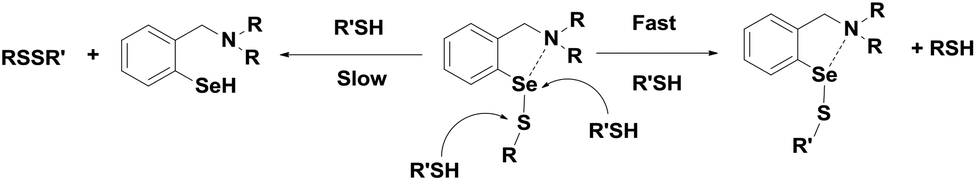 | ||
| Scheme 13 Thiol exchange reaction in the selenenyl sulfide intermediate. | ||
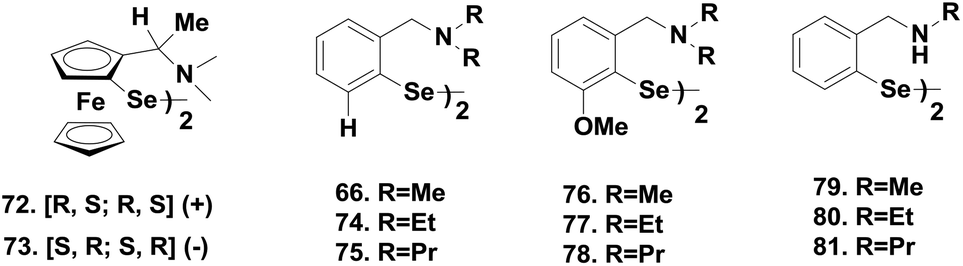 | ||
| Fig. 12 Chemical structures of the amine-based diselenides 66, 72–81. | ||
Experimental evidence shows that the catalytic cycle is identical to that of compound 68. However, the higher activity is ascribed to the negligible interaction between the nitrogen and selenium atoms in the selenenyl sulfide intermediate in the presence of a ferrocenyl moiety. Subsequently Mugesh and coworkers have reported novel amine-based diselenides 76–78 by substituting the ortho-hydrogen in compounds 66, 74–75 with the methoxy substituent (Fig. 12).38 Interestingly, a dramatic increase in the activity for the reduction of H2O2 by PhSH was observed. Particularly, compound 76 showed almost one order of magnitude higher catalytic activity than that of the parent diselenide 66. They proposed a catalytic mechanism for compounds 76–78 based on experimental and theoretical investigations. The catalytic mechanism involving selenol, selenenic acid and selenenyl sulfide intermediates was unaltered. However, it was noticed that the methoxy group protects the selenium centre in selenenic acid from overoxidation. Moreover, it reduces the thiol exchange reaction significantly in the selenenyl sulfide intermediate by reducing the Se⋯N interaction. 77Se NMR studies show that selenenyl sulfide derived from the diselenide 66 requires a large excess of PhSH to overcome the thiol exchange reaction.38,39 However, one equivalent of PhSH is sufficient for the complete conversion of selenenyl sulfide derived from 76 to the corresponding selenol. Rapid formation of the active selenol species is responsible for the very high catalytic activities of diselenides 76–78. Mugesh and co-workers also reported diselenides 79–81 by substituting the tert-amino moiety in compounds 66, 74–75 with sec-amine (Fig. 12).40 Introduction of the sec-amino group increases the activity considerably more than that of compounds 66, 74–75 due to their higher basicity and solubility. However, the catalytic cycle for the reduction of H2O2 using PhSH was unaffected.
As the catalytic cycle of ebselen involves the diselenide 8 as the key intermediate that reduces peroxides, Mugesh and co-workers synthesized and explored the catalytic activity of a series of diaryl diselenides 82–89 containing the amide moiety (Fig. 13).41sec-Amide-based diselenides 82–85 exhibit almost similar activities compared to that of ebselen, however, the activity is increased 10–20 times in the presence of the tert-amide moiety.
 | ||
| Fig. 13 Chemical structures of the amide-based diselenides 82–92. | ||
Amide-based diselenides were found to be much lower in activity than the corresponding amine-based diselenides. The strong Se⋯O interaction in the selenenyl sulfide intermediate induces an extensive thiol exchange reaction that reduces the catalytic activity. Moreover, experimental studies show that the reactivity of these diselenides towards thiol is completely different from that of ebselen, which reacts rapidly with the thiol to cleave the Se–N bond. In contrast, diselenides 82–89 undergo a very slow reaction with thiol to cleave the –Se–Se– bond. Therefore, the oxidative cleavage of the –Se–Se– bond by peroxides was found to be the first step in the catalytic cycle of the amide-based diselenides for the reduction of peroxides. The catalytic mechanisms were similar for both the sec- and tert-amide-based diselenides. Scheme 14 indicates that 82 and 86 first react with H2O2 to produce a mixture of the corresponding selenenic acids 87–88 and seleninic acids 89–90, which are readily converted to the respective selenenyl sulfides 91–92 by excess thiol (Scheme 14). Formation of 93 was observed from 87 after water elimination, which was not possible for 88 due to the absence of the hydrogen atom. Regeneration of diselenides by disproportionation is the rate determining step in the overall catalytic cycle of amide-based diselenides. Extensive thiol exchange reaction in the selenenyl sulfide intermediate prevents the formation of selenol species. Interestingly, introduction of an additional amino moiety (90) changes the catalytic mechanism completely. The thiol cofactor is deprotonated by the additional amino group generating the more nucleophilic thiolate anion, which leads to the rapid cleavage of the diselenide bond. Therefore, compound 90 follows a catalytic cycle that involves selenol as the active intermediate.42 Braga and co-workers also reported sec-amide based diselenides 91–92, which exhibit higher catalytic activities than ebselen.43
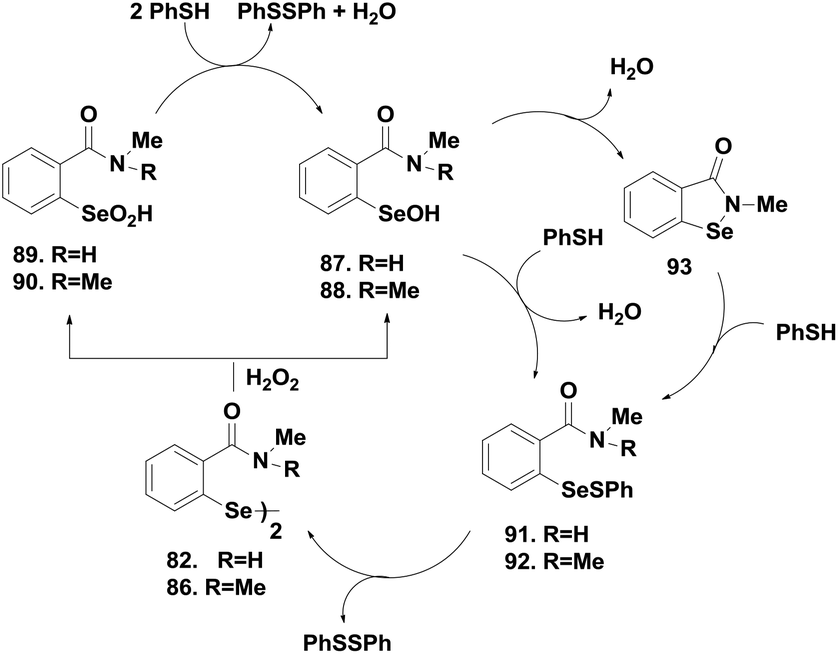 | ||
| Scheme 14 Catalytic mechanism of the amide-based diselenides 89 and 93. | ||
The excellent antioxidant activities of amine-based diaryl diselenides led many researchers to design and synthesize diaryl diselenides that contain oxygen as the heteroatom near the selenium centre. Wirth and co-workers reported a series of oxygen containing diaryl diselenides 94–98 (Fig. 14).44 Compound 94 showed the highest catalytic activity compared to that of compounds 95–97. However, these diselenides were found to be much less active than the corresponding amine-based diselenides despite having non-covalent Se⋯O interactions. However, they follow similar catalytic cycles for peroxide reduction.
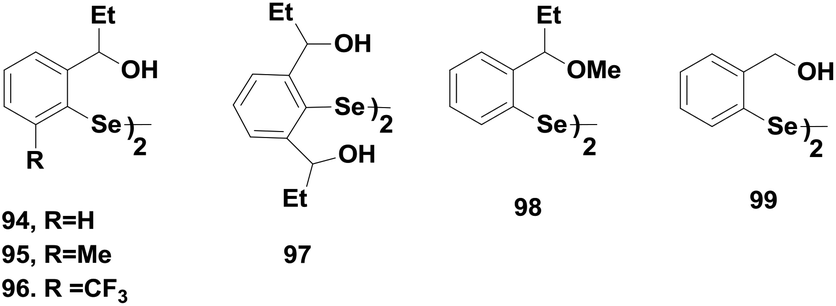 | ||
| Fig. 14 Chemical structures of the oxygen containing diselenides 94–99. | ||
Singh and co-workers have studied the catalytic activity of diselenide 99 in the presence of TBHP and PhSH.31a Diselenide 99 reacts with two equivalents of PhSH to produce the corresponding selenol, which forms a catalytic cycle with the corresponding selenenic acid and selenenyl sulfide intermediates. However, the selenenic acid or other overoxidized compounds could not be detected due to rapid conversion of the selenol to the selenenyl sulfide even at higher peroxide concentrations. The selenenyl sulfide was found to be the most stable intermediate in the catalytic cycle.
Spiroselenuranes
Spirodioxyselenurane as a GPx mimic was first described by Back and co-workers. Compound 54 exhibits excellent GPx-like activity for the reduction of TBHP using BnSH as the cofactor.32 In contrast, the corresponding aromatic analogues 100 exhibit lower antioxidant activity than 54 and the activity is further decreased in the presence of carbonyl moieties in compound 101 (Fig. 15).28,29 However, the activity of compound 100 is enhanced considerably in the presence of electron-donating substituents such as the methoxy group on the aromatic ring.29 The catalytic cycle of spirodioxyselenurane 54 for the reduction of peroxides is shown in Scheme 10. The aromatic derivatives 100–101 also follow a similar mechanism for the reduction of TBHP in the presence of BnSH. Although the GPx-like activities of spirodioxyselenuranes are studied extensively, the corresponding diazaselenuranes as GPx mimics are less explored. Mugesh and co-workers synthesized the first stable spirodiazaselenurane 102 (Fig. 15) and studied its antioxidant activity in the presence of H2O2 and BnSH.45 Compound 102 was found to be less active as a catalyst than ebselen. However, the activity is enhanced when an electron donating substituent is attached to the aromatic ring (103).46 Based on the 77Se NMR studies, they proposed a catalytic cycle for compound 102 that is shown in Scheme 15. It is observed that cleavage of the Se–N bonds by BnSH produces the selenide 104, which is oxidized to the selenoxide 105 by peroxides forming a catalytic cycle. However, in excess thiol concentrations, the selenoxide 105 may react with BnSH to afford the intermediate 106, which is converted to the selenide 104 upon reaction with a second equivalent of BnSH. Formation of the selenide is a crucial step as it is the active intermediate in the cycle. On the other hand, stability of the Se–N bonds highly depends on the nature of substituents on the nitrogen atoms. Phenyl substituents stabilize the Se–N bonds significantly, however, the stability is decreased considerably in the presence of heterocyclic, benzylic or aliphatic substituents leading to the rapid formation of the selenide intermediates.47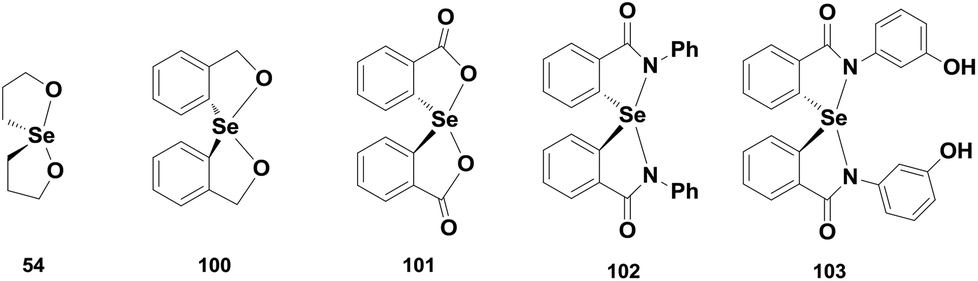 | ||
| Fig. 15 Chemical structures of the spiro compounds 54, 100–103. | ||
 | ||
| Scheme 15 Catalytic mechanism of spirodiazaselenurane 102. | ||
Conclusion
In this review, we have discussed how the catalytic mechanism of GPx mimics is altered depending on the structures of organoselenium compounds and the reactivity of various intermediates toward thiols and peroxides. A detailed analysis of the catalytic mechanisms reveals that synthetic GPx mimics can be classified into different categories based on their structures and compounds from each category follow a characteristic catalytic mechanism for the reduction of peroxides by thiols. However, the mechanisms are also altered between the same types of compounds having different substituents. Furthermore, the nature of the thiol cofactor can alter the catalytic mechanism.Acknowledgements
We acknowledge the Science and Engineering Research Board, New Delhi, for financial support.Notes and references
- H. Sies, in Oxidative Stress, ed. H. Sies, Academic Press, London, 1985, pp. 1–8 CrossRef CAS PubMed; H. Sies, Angew. Chem., 1986, 98, 1061–1075 ( Angew. Chem. Int. Ed. , 1986 , 25 , 1058–1071 ) CrossRef CAS PubMed.
- T. C. Stadtman, Annu. Rev. Biochem., 1996, 65, 83–100 CrossRef CAS PubMed; M. Birringer, S. Pilawa and L. Flohé, Nat. Prod. Rep., 2002, 19, 693–718 RSC; L. Johansson, G. Gafvelin and E. S. J. Arnér, Biochim. Biophys. Acta, 2005, 1726, 1–13 CrossRef PubMed; L. V. Papp, J. Lu, A. Holmgren and K. K. Khanna, Antioxid. Redox Signaling, 2007, 9, 775–806 CrossRef PubMed.
- J. C. Boyington, V. N. Gladyshev, S. V. Khangulov, T. C. Stadtman and P. D. Sun, Science, 1997, 275, 1305–1308 CrossRef CAS.
- R. Wilting, S. Schorling, B. C. Persson and A. Böck, J. Mol. Biol., 1977, 266, 637–641 CrossRef CAS PubMed; E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef; M. Pfeiffer, R. Bingemann and A. Klein, Eur. J. Biochem., 1998, 256, 447–452 Search PubMed.
- M. Wagner, D. Sonntag, R. Grimm, A. Pich, C. Eckerskorn, B. Söhling and J. R. Andreesen, Eur. J. Biochem., 1999, 260, 38–49 CrossRef CAS.
- D. Behne, A. Kyriakopoulos, H. Meinhold and J. Köhrle, Biochem. Biophys. Res. Commun., 1990, 173, 1143–1149 CrossRef CAS; J. C. Davey, K. B. Becker, M. J. Schneider, G. L. Germain and V. A. Galton, J. Biol. Chem., 1995, 270, 26786–26789 CrossRef PubMed; J. R. Arthur, F. Nicol and G. J. Beckett, Biochem. J., 1990, 272, 537–540 CrossRef; W. Croteau, S. K. Whittemore, M. J. Schneider and D. L. Germain, J. Biol. Chem., 1995, 270, 16569–16575 CrossRef PubMed.
- A. Lescure, D. Gautheret, P. Carbon and A. Krol, J. Biol. Chem., 1999, 274, 38147–38154 CrossRef CAS PubMed; T. Tamura and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1006–1011 CrossRef; S. Watabe, Y. Makino, K. Ogawa, T. Hiroi, Y. Yamamoto and S. Y. Takahashi, Eur. J. Biochem., 1999, 264, 74–84 CrossRef; S. R. Lee, J. R. Kim, K. S. Kwon, H. W. Yoon, R. L. Leveine, A. Ginsburg and S. G. Rhee, J. Biol. Chem., 1999, 274, 4722–4734 CrossRef PubMed.
- D. Mustacich and G. Powis, Biochem. J., 2000, 346, 1–8 CrossRef CAS.
- (a) L. Flohé, E. A. Günzler and H. H. Schock, FEBS Lett., 1973, 32, 132–134 CrossRef; (b) F. Ursini, M. Maiorino, M. Valente, L. Ferri and C. Gregolin, Biochim. Biophys. Acta, 1982, 710, 197–211 CrossRef CAS; (c) K. Takahasi, N. Avissar, J. Whittin and H. Cohen, Arch. Biochem. Biophys., 1987, 256, 677–686 CrossRef; (d) F.-F. Chu, J. H. Doroshow and R. S. Esworthy, J. Biol. Chem., 1993, 268, 2571–2576 CAS; (e) L. Flohé, J. R. Andreesen, R. Brigelius-Flohé, M. Maiorino and F. Ursini, IUBMB Life, 2000, 49, 411–420 CrossRef PubMed.
- J. T. Rotruck, A. L. Pope, H. E. Ganther, A. B. Swanson, D. G. Hafeman and W. G. Hoekstra, Science, 1973, 179, 588–590 Search PubMed; O. Epp, R. Ladenstein and A. Wendel, Eur. J. Biochem., 1983, 133, 51–69 CrossRef CAS PubMed; Selenium in Biology and Human Health, ed. R. F. Burk, Springer, New York, NY, 1994 CrossRef; G. Mugesh and W.-W. du Mont, Chem. – Eur. J., 2001, 7, 1365–1370 CrossRef; C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., 2003, 115, 4890–4907 ( Angew. Chem., Int. Ed. , 2003 , 42 , 4742–4758 ) CrossRef PubMed; G. Roy, B. K. Sarma, P. P. Phadnis and G. Mugesh, J. Chem. Sci., 2005, 117, 287–303 CrossRef.
- R. J. Hondal, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 5140–5141 CrossRef CAS; R. Quaderer, A. Sewing and D. Hilvert, Helv. Chim. Acta, 2001, 84, 1197–1206 CrossRef; M. Maiorino, K.-D. Aumann, R. Brigelius-Flohé, R. D. Doria, L. V. Heuvel, J. McCarthy, A. Rovery, F. Ursini and L. Flohé, Biol. Chem. Hoppe-Seyler, 1995, 376, 651–660 CrossRef; C. Rocher, J.-L. Lalanne and J. Chaudiére, Eur. J. Biochem., 1992, 205, 955–960 CrossRef PubMed; K. R. Maddipati and L. J. Marnett, J. Biol. Chem., 1987, 262, 17398–17403 Search PubMed; R. Brigelius-Flohé, Free Radicals Biol. Med., 1999, 27, 951–965 CrossRef.
- M. Maiorino, C. Grigolin and F. Ursini, Methods Enzymol., 1990, 186, 448–457 CAS; M. Björnstedt, J. Xue, W. Huang, B. Åkesson and A. Holmgren, J. Biol. Chem., 1994, 269, 29382–29384 Search PubMed.
- L. Flohé, in Free radicals in biology, ed. W. A. Pryor, Academic Press, New York, 1982, vol. 5, p. 223 Search PubMed.
- O. Epp, R. Ladenstein and A. Wendel, Eur. J. Biochem., 1983, 133, 51–69 CrossRef CAS PubMed; F. Ursini, M. Maiorino, R. Brigelius-Flohé, K.-D. Aumann, A. Roveri, D. Schomburg and L. Flohé, Methods Enzymol., 1995, 252, 38–53 Search PubMed.
- G. Mugesh and H. B. Singh, Chem. Soc. Rev., 2000, 29, 347–357 RSC; G. Mugesh, W.-W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2179 CrossRef CAS PubMed.
- A. Müller, E. Cadenas, P. Graf and H. Sies, Biochem. Pharmacol., 1984, 33, 3235–3239 CrossRef CAS; A. Wendel, M. Fausel, H. Safayhi, G. Tiegs and R. Otter, Biochem. Pharmacol., 1984, 33, 3241–3245 CrossRef; H. Sies, Angew. Chem., Int. Ed. Engl., 1986, 25, 1058–1071 CrossRef PubMed.
- A. Zembowicz, R. J. Hatchett, W. Radziszewski and R. J. Gryglewski, J. Pharmacol. Exp. Ther., 1993, 267, 1112–1118 Search PubMed; R. Hattori, R. Inoue, K. Sase, H. Eizawa, K. Kosuga, T. Aoyama, H. Masayasu, C. Kawai, S. Sasayama and Y. Yui, Eur. J. Pharmacol., 1994, 267, 1–6 CrossRef CAS; T. Nikawa, G. Schuch, G. Wagner and H. Sies, Biochem. Pharmacol., 1994, 47, 1007–1012 CrossRef.
- M. Parnham and H. Sies, Expert Opin. Invest. Drugs, 2000, 9, 607–619 CrossRef CAS PubMed; T. Yamaguchi, K. Sano, K. Takakura, I. Saito, Y. Shinohara, T. Asano and H. Yasuhara, Stroke, 1998, 29, 12–17 CrossRef PubMed; K. Jonathan, P. Carol, T. Huy, G. Rende and L. D. Eric, Hearing Res., 2007, 226, 44–51 CrossRef PubMed; N. Singh, A. C. Halliday, J. M. Thomas, O. V. Kuznetsova, R. Baldwin, E. C. Y. Woon, P. K. Aley, I. Antoniadou, T. Sharp, S. R. Vasudevan and G. C. Churchill, Nat. Commun., 2013, 4, 1332 CrossRef PubMed.
- I. M. Bell and D. Hilvert, Biochemistry, 1993, 32, 13969–13973 CrossRef CAS; H. Fisher and N. Dereu, Bull. Soc. Chim. Belg., 1987, 96, 757–768 CrossRef PubMed.
- (a) J. L. Kice and D. W. Purkiss, J. Org. Chem., 1987, 52, 3448–3451 CrossRef CAS; (b) B. K. Sarma and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 11477–11485 CrossRef CAS PubMed; (c) K. P. Bhabak and G. Mugesh, Chem. – Eur. J., 2007, 13, 4594–4601 CrossRef CAS PubMed; (d) B. K. Sarma and G. Mugesh, Chem. – Eur. J., 2008, 14, 10603–10614 CrossRef CAS PubMed; (e) K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1419 CrossRef CAS PubMed; (f) M. Elsherbini, W. S. Hamama, H. H. Zoorob, D. Bhowmick, G. Mugesh and T. Wirth, Heteroat. Chem., 2014, 25, 320–325 CrossRef CAS PubMed.
- H. J. Reich and C. P. Jasperse, J. Am. Chem. Soc., 1987, 109, 5549–5551 CrossRef CAS.
- G. Mugesh and W.-W. du Mont, Chem. – Eur. J., 2001, 7, 1365–1370 CrossRef CAS; C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef PubMed.
- T. G. Back and B. P. Dyck, J. Am. Chem. Soc., 1997, 119, 2079–2083 CrossRef CAS.
- K. Satheeshkumar and G. Mugesh, Chem. – Eur. J., 2011, 17, 4849–4857 CrossRef CAS PubMed.
- V. P. Singh, H. B. Singh and R. J. Butcher, Eur. J. Org. Chem., 2011, 5485–5497 CrossRef CAS PubMed.
- D. Bhowmick, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2015, 54, 8449–8453 CrossRef CAS PubMed.
- T. G. Back and Z. Moussa, J. Am. Chem. Soc., 2002, 124, 12104–12105 CrossRef CAS PubMed.
- T. G. Back, D. Kuzma and M. Parvez, J. Org. Chem., 2005, 70, 9230–9236 CrossRef CAS PubMed.
- D. J. Press, E. A. Mercier, D. Kuzma and T. G. Back, J. Org. Chem., 2008, 73, 4252–4255 CrossRef CAS PubMed.
- N. M. R. McNeil, M. C. Matz and T. G. Back, J. Org. Chem., 2013, 78, 10369–10382 CrossRef CAS PubMed.
- (a) S. K. Tripathi, U. Patel, D. Roy, R. B. Sunoj, H. B. Singh, G. Wolmershäuser and R. J. Butcher, J. Org. Chem., 2005, 70, 9237–9247 CrossRef CAS PubMed; (b) V. P. Singh, H. B. Singh and R. J. Butcher, Chem. – Asian J., 2011, 6, 1431–1442 CrossRef CAS PubMed.
- T. G. Back, Z. Moussa and M. Parvez, Angew. Chem., Int. Ed., 2004, 43, 1268–1270 CrossRef CAS PubMed.
- S. S. Zade, H. B. Singh and R. J. Butcher, Angew. Chem., Int. Ed., 2004, 43, 4513–4515 CrossRef CAS PubMed.
- V. Nascimento, E. E. Alberto, D. W. Tondo, D. Dambrowski, M. R. Detty, F. Nome and A. L. Braga, J. Am. Chem. Soc., 2012, 134, 138–141 CrossRef CAS PubMed.
- S. R. Wilson, P. A. Zucker, R.-R. C. Huang and A. Spector, J. Am. Chem. Soc., 1989, 111, 5936–5939 CrossRef CAS.
- M. Iwaoka and S. Tomoda, J. Am. Chem. Soc., 1994, 116, 2557–2561 CrossRef CAS.
- G. Mugesh, A. Panda, H. B. Singh, N. S. Punekar and R. J. Butcher, J. Am. Chem. Soc., 2001, 123, 839–850 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Chem. – Eur. J., 2008, 14, 8640–8651 CrossRef CAS PubMed.
- D. Bhowmick and G. Mugesh, Tetrahedron, 2012, 68, 10550–10560 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Chem. – Eur. J., 2009, 15, 9846–9854 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Chem. – Asian J., 2009, 4, 974–983 CrossRef CAS PubMed.
- D. Bhowmick and G. Mugesh, Org. Biomol. Chem., 2015, 13, 9072–9082 CAS.
- V. Nascimento, N. L. Ferreira, R. F. S. Canto, K. L. Schott, E. P. Waczuk, L. Sancineto, C. Santi, J. B. T. Rocha and A. L. Braga, Eur. J. Med. Chem., 2014, 87, 131–139 CrossRef CAS PubMed.
- T. Wirth, Molecules, 1998, 3, 164–166 CrossRef CAS PubMed.
- B. K. Sarma, D. Manna, M. Minoura and G. Mugesh, J. Am. Chem. Soc., 2010, 132, 5364–5374 CrossRef CAS PubMed.
- D. S. Lamani, D. Bhowmick and G. Mugesh, Org. Biomol. Chem., 2012, 10, 7933–7943 CAS.
- D. S. Lamani, D. Bhowmick and G. Mugesh, Molecules, 2015, 20, 12959–12978 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |