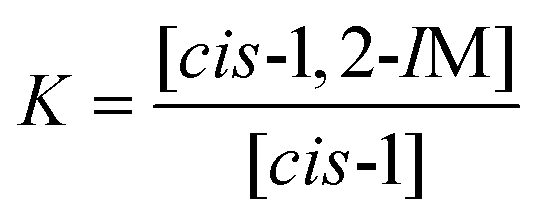DOI:
10.1039/C5OB01630D
(Paper)
Org. Biomol. Chem., 2015,
13, 10505-10510
Regioselective synthesis of fullerene multiadducts via tether-directed 1,3-dipolar cycloaddition†
Received
5th August 2015
, Accepted 28th August 2015
First published on 4th September 2015
Abstract
The regioselective synthesis of fullerene multiadducts was achieved from commercially available reagents in one pot over two steps. The configuration of the isolated regioisomers was determined using various NMR methods, UV-vis spectroscopy and electrochemical analysis with the structure of one isomer confirmed by single crystal X-ray analysis. Interesting variation in regioselectivity was observed when different amino acid reagents were used in the reactions. Theoretical calculations and additional experiments, such as deuterium exchange, led to a proposed mechanism for the regioselective product formation.
Introduction
Regiochemistry of fullerenes can be complex given the large number of carbon centres that can participate in reactions.1,2 An effective strategy is to control the regiochemistry using a tether between two reactive head groups.3,4 This is known as the tether-directed remote functionalization approach which was first demonstrated by Breslow et al.5 in mimicking highly selective enzymatic reactions on steroid compounds. This tether approach has been demonstrated in regio- and stereoselective fullerene functionalizations.6,7 Tethers reported include simple arenes, porphyrins, crown ethers as well as chiral Tröger base units.2 These previous studies investigated the scope of the tether-directed functionalization strategy but did not look into the application of the materials produced.
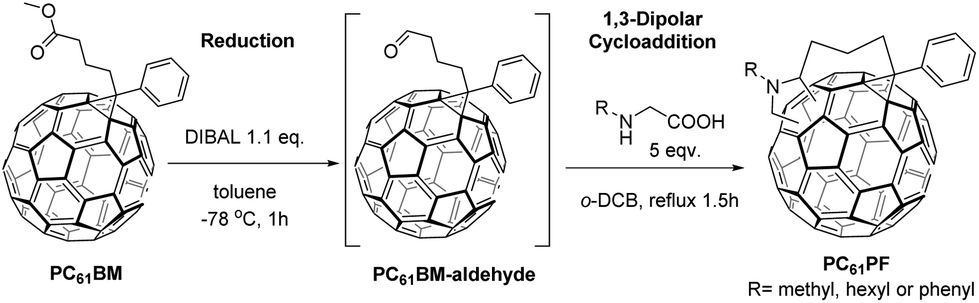
A central research theme in our group has been the development of organic electronic materials with particular emphasis on thin film solar cells. Bisadducts of fullerenes have been widely incorporated in solar cell devices but isomeric mixtures are commonly used. There has only been a few attempts to isolate single bisadduct isomers and investigate their performance in devices.8,9 In one case, a single isomer of indene-C70 bisadduct (IC70BA) was obtained by careful chromatographic separation.10 This single isomer showed significant device performance improvement compared to samples containing isomeric mixtures. However, chromatographic isolation of bisadduct isomers is extremely inefficient in terms of product yield and time required for the processing. There has been a few recent studies on the use of tether-directed functionalization to produce fullerene materials for solar cell applications.11–13 In particular, our group has developed a one-pot synthesis of a single C60 bisadduct isomer from commercially available phenyl-C61-butyric acid methyl ester (PC61BM).14 With the relatively short propyl tether and the azomethine ylide reactive head group, one major product was isolated and subsequently identified as the cis-1 C60 bisadduct derivative, N-methyl PC61PF (Fig. 1). This pure isomer material showed enhancement in performance compared to the isomer mixtures and the original PC61BM material.
 |
| | Fig. 1 Chemical structure representation of bisadduct derivatives discussed in this work and an illustration of possible C60 bisadduct regioisomer configurations. | |
This positive outcome gave us incentive to develop a series of related compounds with different substitution on the pyrrolidine nitrogen (Fig. 1). Interestingly, the substituent variations had significant effects on the regioselectivity of the 1,3-dipolar cycloaddition (Fig. 2). Different products were isolated when N-hexylglycine and N-phenylglycine was used instead of N-methylglycine. In a large proportion of examples, 1,3-dipolar cycloaddition to fullerenes occurred at the [6,6] bonds, namely the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds between two six-membered rings. This can result in 8 possible regioisomers for C60 fullerene bisadducts (Fig. 1). With the appropriate tether length, one can limit the 1,3-dipolar cycloaddition to the cis-hemisphere only. The cis-1 configuration was the most favoured in the case of the N-methyl PC61PF compound. Therefore, it was intriguing to observe regiochemistry variations for the N-hexyl and N-phenyl compounds even though the propyl tether was unchanged.
C bonds between two six-membered rings. This can result in 8 possible regioisomers for C60 fullerene bisadducts (Fig. 1). With the appropriate tether length, one can limit the 1,3-dipolar cycloaddition to the cis-hemisphere only. The cis-1 configuration was the most favoured in the case of the N-methyl PC61PF compound. Therefore, it was intriguing to observe regiochemistry variations for the N-hexyl and N-phenyl compounds even though the propyl tether was unchanged.
 |
| | Fig. 2 One-pot two-step synthesis of PC61PF derivatives. | |
In this study, our efforts in the elucidation of the regioisomer configuration of the isolated products are presented. Through computations and experiments, reaction mechanisms for the product outcomes are proposed. It is noteworthy that these tether-directed reaction resulted in fused ring structures, containing 3, 5, 6 and 7-membered rings, rarely observed in the literature.
Results and discussion
Synthesis and configuration identification
Two new C60 bisadducts, N-hexyl PC61PF and N-phenyl PC61PF, were synthesized and fully characterized (Fig. 1). As with the previously reported N-methyl PC61PF, the hexyl and phenyl analogues were synthesized from PC61BM in a one-pot-two-step reaction (Fig. 2). PC61BM was treated with the reducing reagent, diisobutylaluminium hydride (DIBAL), to give the aldehyde intermediate. Without isolation, the aldehyde intermediate was heated with N-hexylglycine or N-phenylglycine in 1,2-dichlorobenzene (o-DCB) at 180 °C to form the N-hexyl and N-phenyl PC61PF respectively. It was apparent immediately from thin layer chromatography analysis that the N-hexylglycine and N-phenylglycine gave different product distributions.
The crude product of N-hexyl PC61PF was purified by silica chromatography and two major fractions were isolated, N-hexyl PC61PF-1 and N-hexyl PC61PF-2, in yields of 9% and 14% respectively. The purity of the two hexyl samples were examined by HPLC analysis with Cosmosil Buckyprep-D column and found to be 95% and 99% for fraction 1 and 2 respectively (Fig. S22†). According to the 1H NMR spectrum, both fractions contained three distinct proton resonances characteristic to the pyrrolidine moiety, confirming the formation of the fulleropyrrolidine unit. However, the N-hexyl PC61PF-2 fraction showed two additional singlet resonance at 5.65 and 5.45 ppm compared to N-hexyl PC61PF-1. This observation corresponded to the mass spectrometry data where N-hexyl PC61PF-2 was two mass units heavier than N-hexyl PC61PF-1.
Full 1H NMR assignment of the two fractions was achieved using two-dimensional NMR analysis techniques shown in Fig. 3 and 4. The heteronuclear multiple-bond correlation (HMBC) spectrum of N-hexyl PC61PF-1 showed the C15 carbon coupled with both H5 and H2b protons (Fig. 3). This coupling was observed for the N-methyl PC61PF compound with known cis-1 regioisomer configuration. This strongly indicated cis-1 configuration for the N-hexyl PC61PF-1 fraction. With two extra proton signals, the HMBC spectrum of N-hexyl PC61PF-2 was more difficult to analyse (Fig. 4). There were clear HMBC coupling signals for H18 to C12 and C13 as well as H17 to C5 and C14, but no coupling with C15 and C16. This suggested that C17 and C18 were next to C13 and C14 but not directly adjacent to C15 and C16. In addition, H17 and H18 did not couple to each other. In light of these NMR experiments, a possible configuration of N-hexyl PC61PF-2 was shown in Fig. 4 with the pyrrolidine ring and the two additional protons occupied both the cis-1 and cis-2 position, designated as cis-1,2 hence forth.
 |
| | Fig. 3
1H, 13C and HMBC NMR spectrum of N-hexyl PC61PF-1 with resonances assigned for the key atoms in discussion. | |
 |
| | Fig. 4
1H, 13C and HMBC NMR spectrum of N-hexyl PC61PF-2 with resonances assigned for the key atoms in discussion. | |
Single crystals of N-hexyl PC61PF-2 were obtained by recrystallization from chloroform and subsequent X-ray crystallography experiment on the crystals revealed the relative configuration of the sample. The fused ring structure containing the pyrrolidine substituent was in agreement with the NMR assignments where the pyrrolidine ring and the two additional protons were in the cis-1,2 configuration (Fig. 4 and 5). Surprisingly, the crystallography data indicated the enantiomeric pairs in the crystal existed in a dimerized state (Fig. 5). The dimerization reaction probably proceeded through a photo-activated [2 + 2] cycloaddition driven by the close packing in the crystal structure.15 It is important to note that there was no evidence of the covalently linked dimer in mass spectrometry experiments prior to the crystallography analysis with the sample kept in ambient light conditions.
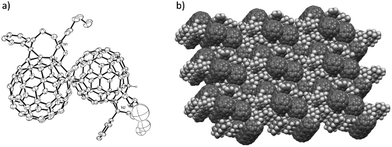 |
| | Fig. 5 Single crystal X-ray structure representation of N-hexyl PC61PF-2 dimer crystals grown from chloroform: (a) space filling model and (b) packing diagram in single crystal, where CHCl3 solvent molecules have been omitted for clarity. | |
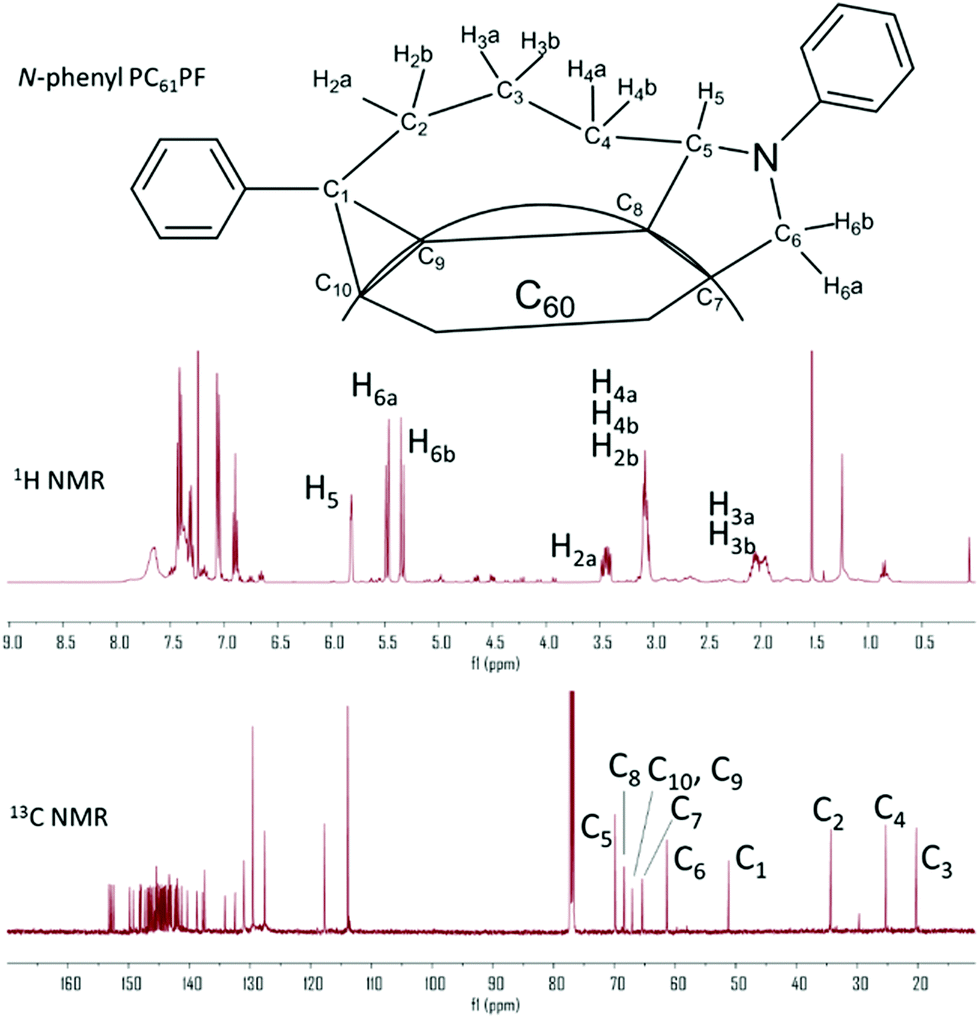
The crude product of N-phenyl PC61PF was purified by flash chromatography with one major fraction isolated in a yield of 14%. Some PC61BM stating material and aldehyde intermediate were recovered but the majority of reaction mixture consisted of insoluble material that could not be identified. The purity of the N-phenyl PC61PF sample was 77% by HPLC analysis (Fig. S22†). The three distinctive proton resonances of the pyrrolidine moiety were clearly observed at the chemical shift of 5.82, 5.47 and 5.35 ppm in the 1H NMR spectrum. As with the hexyl compounds, key structure assignments of N-phenyl PC61PF was achieved using two-dimensional NMR analysis (Fig. 6 and S13–15†). Due to overlapping resonances, it was not possible to identify the regioisomer configuration for N-phenyl PC61PF in NMR experiments.
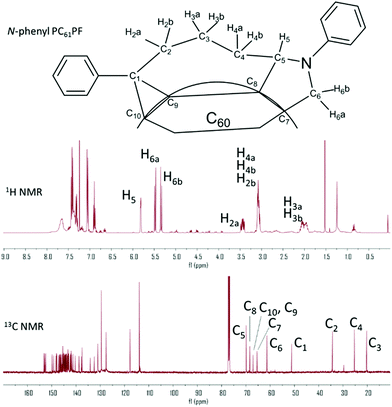 |
| | Fig. 6
1H and 13C spectrum of N-phenyl PC61PF with resonances assigned for the key atoms in discussion. | |
It is well-established that the isomeric configuration of fullerene bisadducts can be identified by comparing the UV-Vis spectrum of materials with known configuration.6 This is because the absorption bands of fullerene derivatives are closely related to the conjugated surface of the molecules. As shown in Fig. 7, the spectrum of N-hexyl PC61PF-1 and N-phenyl PC61PF contained a peak at 430 nm. Other C60 bisadducts of known cis-1 configuration, for example N-methyl PC61PF14 and C60-benzyne bisadduct,1 have similar spectral features. This provided support for the NMR analysis of the N-hexyl PC61PF-1 compound and hinted at the cis-1 configuration for N-phenyl PC61PF. Notably, the UV-vis spectrum of N-hexyl PC61PF-2 was different to that of N-hexyl PC61PF-1 and N-phenyl PC61PF with a peak at 442 nm (Fig. 7 and Table 1). Cyclic voltammetry was carried out to estimate the lowest unoccupied molecular orbital (LUMO) energy level of each PC61PF material (see ESI for details, Fig. S23†). The half-wave potential of the first reduction process (Ered1/2) were measured against the internal ferrocene standard and the LUMO energy can be calculated from this data (Table 1).16 The N-hexyl PC61PF-1 sample has higher electron affinity with ELUMO at −3.67 eV than N-hexyl PC61PF-2 with ELUMO at −3.58 eV. This corresponds to the increased substitution for N-hexyl PC61PF-2. The chemical structure of the isolated regioisomers, assigned using the characterisation data, are shown in Fig. 8. In the next section, calculations and discussion on the mechanism of product formation will provide support to the various characterisation techniques presented thus far.
 |
| | Fig. 7 UV-Vis spectrum of PC61PF compounds. | |
 |
| | Fig. 8 Chemical structures of N-hexyl PC61PF-1, N-hexyl PC61PF-2 and N-phenyl PC61PF. | |
Table 1 Summary of characterization data for PC61PF compounds
| |
Puritya (%) |
Configuration |
UV-vis λmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (nm) b (nm) |
E
red1/2c (eV) |
E
LUMOd (eV) |
|
Calculated by HPLC analysis.
Absorption coefficient (×103 M−1 cm−1) in brackets.
Half-wave reduction potential from cyclic voltammetry.
E
LUMO = −(Ered1/2 + 4.8) eV.
|
|
N-Hexyl PC61PF-1 |
95 |
cis-1 |
430 (5.8) |
−1.13 |
−3.67 |
|
N-Hexyl PC61PF-2 |
99 |
cis-1,2 |
442 (6.0) |
−1.22 |
−3.58 |
|
N-Phenyl PC61PF |
77 |
cis-1 |
431 (8.8) |
−1.25 |
−3.55 |
Density functional theory (DFT) calculations
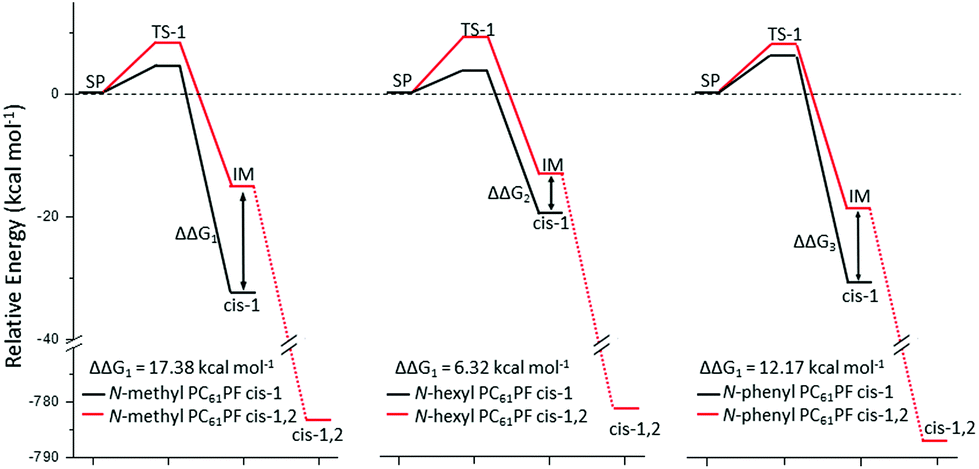
The unusual cis-1,2 configuration for N-hexyl PC61PF-2 was examined in DFT calculations to support our experimental observations. The proposed reaction pathway leading to the observed products is discussed below (Fig. 9). One can consider the azomethine ylide as the starting point of discussions. For N-hexyl PC61PF-1 and N-phenyl PC61PF, the cis-1 product can be formed by 1,3-dipolar cycloaddition in one step. On the other hand, the insertion of two additional protons for N-hexyl PC61PF-2 with cis-1,2 configuration probably involved at least two steps. In detail, the 1,3-dipolar cycloaddition proceeded first to form the pyrrolidine ring on the [5,6] bond followed by protonation to give the cis-1,2 product. Therefore, there must be an intermediate between the transition state of the cycloaddition step and the transition state of the protonation step. Using this model and applying DFT at B3LYP-D3 level17,18 with 6-311G basis set and polarization function (d,p), the relative energy of the azomethine ylide starting point, transition states, possible intermediates and final products were calculated (Fig. 10 and Table S1†).19,20
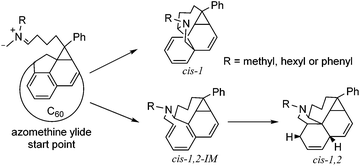 |
| | Fig. 9 Proposed reaction pathway for PC61PF products with cis-1 and cis-1,2 configuration. | |
 |
| | Fig. 10 Relative energy of starting point, transition states, intermediates and products of PC61PF compounds with cis-1 and cis-1,2 configuration from DFT calculations. | |
Since the product energy of PC61PFs in cis-1,2 configuration were much lower than both the intermediates cis-1,2-IM21,22 and the products of cis-1, the formation of the cis-1,2 product can be considered as irreversible (Fig. 10). On the other hand, an equilibrium may exist between cis-1 and cis-1,2-IM species. From the calculated Gibbs free energy difference (ΔΔG) values between cis-1 and cis-1,2-IM, relative equilibrium constant (K) values can be obtained using the equations:
| | 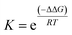 | (1) |
| | 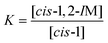 | (2) |
where
R is the gas constant and
T is the reaction temperature. With ΔΔ
G1 of
N-methyl PC
61PF larger than ΔΔ
G2 of
N-hexyl PC
61PF (
Fig. 10), the corresponding
K for the methyl species was much smaller (
K1 = 4.1 × 10
−9) than the hexyl species (
K2 = 8.9 × 10
−4). The consequence of this difference was that a much greater proportion of the hexyl species would exist in the
cis-1,2-IM form compared to the methyl at equilibrium, leading to a greater chance of formation of the
cis-1,2 product for the hexyl. This corresponded to the
N-hexyl PC
61PF-1 and
N-hexyl PC
61PF-2 compounds that were isolated. A steric argument can be invoked for the outcome of the calculated ΔΔ
G values. The significantly higher energy of the hexyl
cis-1 species can be attributed to the steric bulk of the hexyl group compared to the methyl group (Fig. S26
†). The steric effect of the hexyl moiety in
cis-1,2-IM configuration was not as significant resulting in similar energies for the hexyl and methyl
cis-1,2-IM species.
Although the phenyl group should have stronger steric effect than hexyl, cis-1,2 product was not observed for N-phenyl PC61PF. The DFT calculation revealed a relatively large ΔΔG3 for the phenyl compounds (Fig. 10). This meant that only a small concentration of cis-1,2-IM of N-phenyl PC61PF existed at equilibrium (K3 = 1.3 × 10−6), resulting in less cis-1,2 product compared to hexyl species. A possible reason could be that the phenyl group was able to stablize the cis-1 product by sharing the electron density with the pyrrolidine moiety. According to the DFT optimized molecular models, the orientation of the phenyl ring relative to the pyrrolidine ring was very different for the cis-1 and cis-1,2-IM species. Only the optimised geometry of cis-1 showed electronic stabilization effect of the phenyl group. In the cis-1,2-IM species, steric hinderance between the phenyl group and the H4 proton resulted in an orientation of the phenyl ring relative to the pyrrolidine ring that did not favor electronic stabilization (Fig. S27†).
Deuterium exchange experiments and proposed mechanism
To gain further insight into the reaction mechanism, deuterium exchange experiments were performed. The reactions to form the hexyl compounds were carried out as before except with the addition of deuterium oxide in the reaction mixture of the 1,3-dipolar cycloaddition step (see Experimental section for details). The 1H NMR spectrum showed that deuterium exchange occurred at two protons (H4a and H4b) in N-hexyl PC61PF-1 and four (H4a, H4b, H17 and H18) in N-hexyl PC61PF-2 (Fig. S16 and S17†). The exchanges at the H4a and H4b proton could be simply attributed to the keto–enol tautomerisation of the aldehyde starting material. The deuterium exchange at H17 and H18 for the N-hexyl PC61PF-2 material provided information on the source of those protons (Fig. 11). It was noteworthy that the percentage of deuterium exchange at H15 and H18 were both around 80% by integration. The following is a descirption of the proposed reaction mechanism for the formation of the cis-1 and cis-1,2 products taking into account all experimental data and calculations.
 |
| | Fig. 11 The proposed mechanism for the formation of PC61PF compounds in either cis-1 or cis-1,2 configuration. | |
The mechanism of formation of the cis-1 product proceeded via 1,3-dipolar cycloaddition of azomethine ylide to cis-1 bond of the C60 derivative (Fig. 11). This reaction, known as the Prato reation, is commonly observed in the literature but usually proceeds without regioselectivity.23,24 In order to achieve the cis-1,2 product outcome, the 1,3-dipolar cycloaddition would be required to occur at the [5,6] bond between the cis-1 and cis-2 positions (Fig. 11).21,22 This open cage intermediate cis-1,2-IM would then be converted to the close cage intermediate via a hydride transfer reaction. Further protonation would lead to the cis-1,2 product. It is feasible that the hydride source is from the fullerene aldehyde substrate leading to a fullerene amide by-product. This proposal is in agreement with the observed product yields. A reaction with deuterated DIBAL reagent was performed in an attempt to gather further evidence for the proposed mechanism (see ESI, Fig. S18†). In agreement with the azomethine ylide formation mechanism, the H5 proton on the pyrrolidine ring was deuterated. However, deuteration was not observed for either the H17 or H18 on the fullerene surface. It was found that the acidity of H17 and H18 was such that there was facile proton exchange under the reaction conditions (Fig. S19†). Attempts were made to identify intermediates and by-products from the reaction mixture using mass spectrometry. While some relevant fragments were observed, these experiments did not provide conclusive evidence for the proposed reaction mechanism.
Conclusions
The regioselective synthesis of three fullerene derivatives was presented. Starting from commercially available PC61BM, the multi-adducts were obtained via reduction of the methyl ester followed by 1,3-dipolar cycloaddition of the azomethine ylide in one pot. While the yields were moderate, intriguing regioselectivity variations were observed when different amino acid reagents were used. With a range of NMR methods, UV-Vis spectroscopy and electrochemical analysis, it was possible to confidently assign the configurations of the isolated regioisomers N-hexyl PC61PF-1 (cis-1), N-hexyl PC61PF-2 (cis-1,2) and N-phenyl PC61PF (cis-1). The relative configurations for N-hexyl PC61PF-2 was confirmed by single crystal X-ray analysis. Mechanism for the product formation was proposed with the aid of DFT calculations and deuterium exchange experiments.
Acknowledgements
This work was made possible by support from the Australian Renewable Energy Agency which funds the project grants within the Australian Centre for Advanced Photovoltaics. WWHW is supported by an Australian Research Council Future Fellowship (FT130100500). Responsibility for the views, information, or advice herein is not accepted by the Australian Government. We thank Dr Lars Goerigk for providing advice on the DFT calculations.
Notes and references
- Y. Nakamura, N. Takano, T. Nishimura, E. Yashima, M. Sato, T. Kudo and J. Nishimura, Org. Lett., 2001, 3, 1193–1196 CrossRef CAS PubMed.
- C. Thilgen and F. Diederich, C. R. Chim., 2006, 9, 868–880 CrossRef CAS PubMed.
- M. R. Cerón, M. Izquierdo, A. Aghabali, J. A. Valdez, K. B. Ghiassi, M. M. Olmstead, A. L. Balch, F. Wudl and L. Echegoyen, J. Am. Chem. Soc., 2015, 137, 7502–7508 CrossRef PubMed.
- L. Isaacs, R. F. Haldimann and F. Diederich, Angew. Chem., Int. Ed., 1994, 33, 2339–2342 CrossRef PubMed.
- R. Breslow, Acc. Chem. Res., 1980, 13, 170–177 CrossRef CAS.
- W. W. H. Wong and F. Diederich, Chem. – Eur. J., 2006, 12, 3463–3471 CrossRef CAS PubMed.
- Z. Zhou, D. I. Schuster and S. R. Wilson, J. Org. Chem., 2006, 71, 1545–1551 CrossRef CAS PubMed.
- X. Meng, G. Zhao, Q. Xu, Z. a. Tan, Z. Zhang, L. Jiang, C. Shu, C. Wang and Y. Li, Adv. Funct. Mater., 2014, 24, 158–163 CrossRef CAS PubMed.
- F. Zhao, X. Meng, Y. Feng, Z. Jin, Q. Zhou, H. Li, L. Jiang, J. Wang, Y. Li and C. Wang, J. Mater. Chem. A, 2015, 3, 14991–14995 CAS.
- W. W. H. Wong, J. Subbiah, J. M. White, H. Seyler, B. Zhang, D. J. Jones and A. B. Holmes, Chem. Mater., 2014, 26, 1686–1689 CrossRef CAS.
- M.-H. Liao, Y.-Y. Lai, Y.-Y. Lai, Y.-T. Chen, C.-E. Tsai, W.-W. Liang and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2014, 6, 996–1004 CAS.
- R. Tao, T. Umeyama, T. Higashino, T. Koganezawa and H. Imahori, Chem. Commun., 2015, 51, 8233–8236 RSC.
- R. Tao, T. Umeyama, T. Higashino, T. Koganezawa and H. Imahori, ACS Appl. Mater. Interfaces, 2015, 7, 16676–16685 CAS.
- B. Zhang, J. Subbiah, Y. Y. Lai, J. M. White, D. J. Jones and W. W. H. Wong, Chem. Commun., 2015, 51, 9837–9840 RSC.
- G.-W. Wang, K. Komatsu, Y. Murata and M. Shiro, Nature, 1997, 387, 583–586 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Osuna, M. Swart and M. Sola, J. Phys. Chem. A, 2011, 115, 3491–3496 CrossRef CAS PubMed.
- J.-C. Brodovitch, B. Addison-Jones, K. Ghandi, I. McKenzie and P. W. Percival, Phys. Chem. Chem. Phys., 2015, 17, 1755–1762 RSC.
- Z. Li and P. B. Shevlin, J. Am. Chem. Soc., 1997, 119, 1149–1150 CrossRef CAS.
- T. Grösser, M. Prato, V. Lucchini, A. Hirsch and F. Wudl, Angew. Chem., Int. Ed. Engl., 1995, 34, 1343–1345 CrossRef PubMed.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519–526 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis procedures, characterization and DFT calculations of the materials. Detailed experimental procedures, NMR spectra and other characterization data as well as details on the modelling and theoretical calculations. CCDC 1416759. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01630d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Wallace W. H.
Wong
*
and
Wallace W. H.
Wong
*