
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically synthesized dicarba H2 relaxin analogues retain strong RXFP1 receptor activity but show an unexpected loss of in vitro serum stability
Mohammed Akhter
Hossain
*abc,
Linda M.
Haugaard-Kedström
d,
K. Johan
Rosengren
d,
Ross A. D.
Bathgate
abe and
John D.
Wade
*abc
aFlorey Institute of Neuroscience and Mental Health, The University of Melbourne, Victoria 3010, Australia. E-mail: akhter.hossain@florey.edu.au; Tel: +61 3 8344 7330
bFlorey Department of Neuroscience and Mental Health, The University of Melbourne, Victoria 3010, Australia. E-mail: john.wade@florey.edu.au; Tel: +61 3 8344 7330
cSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia
dSchool of Biomedical Sciences, University of Queensland, Brisbane, QLD 4072, Australia
eDepartment of Biochemistry and Molecular Biology, The University of Melbourne, Victoria 3010, Australia
First published on 9th September 2015
Abstract
Peptides and proteins are now acknowledged as viable alternatives to small molecules as potential therapeutic agents. A primary limitation to their more widespread acceptance is their generally short in vivo half-lives due to serum enzyme susceptibility and rapid renal clearance. Numerous chemical approaches to address this concern have been undertaken in recent years. The replacement of disulfide bonds with non-reducible elements has been demonstrated to be one effective means by eliminating the deleterious effect of serum reductases. In particular, substitution with dicarba bonds via ring closure metathesis has been increasingly applied to many bioactive cystine-rich peptides. We used this approach for the replacement of the A-chain intramolecular disulfide bond of human relaxin 2 (H2 relaxin), an insulin-like peptide that has important regulatory roles in cardiovascular and connective tissue homeostasis that has led to successful Phase IIIa clinical trials for the treatment of acute heart failure. Use of efficient solid phase synthesis of the two peptide chains was followed by on-resin ring closure metathesis and formation of the dicarba bond within the A-chain and then by off-resin combination with the B-chain via sequential directed inter-chain disulfide bond formation. After purification and comprehensive chemical characterization, the two isomeric synthetic H2 relaxin analogues were shown to retain near-equipotent RXFP1 receptor binding and activation propensity. Unexpectedly, the in vitro serum stability of the analogues was greatly reduced compared with the native peptide. Circular dichroism spectroscopy studies showed subtle differences in the secondary structures between dicarba analogues and H2 relaxin suggesting that, although the overall fold is retained, it may be destabilized which could account for rapid degradation of dicarba analogues in serum. Caution is therefore recommended when using ring closure metathesis as a general approach to enhance peptide stability.
Introduction
Human relaxin-2 (H2 relaxin) is one of 10 members of insulin-relaxin superfamily and is structurally similar to insulin having two chains (A and B) linked by three disulfide bonds.1–3 Since its discovery in 1926,4 H2 relaxin has undergone several pre-clinical and clinical trials for the treatment of various conditions including cervical ripening, scleroderma, preeclampsia, fibromyalgia, orthodontics, and acute heart failure (AHF).2 It finally successfully completed a Phase IIIa clinical trial for the treatment of AHF.5,6 While a Phase IIIb clinical trial is in progress, H2 relaxin has been approved for sale in Russia for human use in clinical settings. However, like human insulin, H2 relaxin has a very short in vivo half-life.7 If injected into patients, it loses half its activity within 10 minutes7 due to degradation by blood enzymes as well as clearance by the kidney and liver. Thus, there is a need to develop a longer acting form of the peptide for extended therapeutic action in patients with AHF.To enhance the therapeutic potential of peptides,8 lipidation,9 PEGylation,10 PASylation,11 XTENylation12 or the attachments of fusion proteins13,14 are popular methods for increasing the molecular mass of target peptides or proteins and correspondingly slowing or preventing renal clearance thus extending the half-life.15 The additional in vivo lability of these biomolecules to exo- and endopeptidases has been addressed in several ways including the use of non-native amino acids (e.g.D-amino acids) within the hitherto-identified labile bonds.16 Another strategy is the introduction of cross-links that stabilize the folded state of the protein, such as cyclization,17 and have the potential to provide a “global protection” as proteases tend to target regions that are unstructured. This is evident from the fact that proteins that are disulfide-rich are often highly stable.18 However, disulfide bonds themselves provide another potential key target for increasing the in vitro and in vivo stability of proteins.19,20 Intracellular components such as the enzyme disulfide reductase, glutathione or other reducing conditions can rupture such bridges making the peptide unfold and become further susceptible to enzymatic cleavage. Therefore, substitution or replacement of disulfide bonds with stable isosteres such as diselenide, lactam, or dicarba bonds can result in a significant contribution to improved peptide half-life of peptides.19–21
Disulfide bonds are common structural motifs in many bioactive peptides including insulin/relaxin-like peptides where they play a critical role in maintaining the overall 3D-structure.22–25 In addition, disulfide bridges may also directly interact with cellular receptors.25 It has recently been shown for insulin-like peptide 3 (INSL3), an important germ cell maturation regulator, that the N-terminal inter-chain disulfide bridge may be directly involved in interacting with its RXFP2 receptor.26 Although there is thus far no evidence that a disulfide bridge of H2 relaxin is involved in direct interaction with its RXFP1 receptor, it is known that each of the three disulfide bonds are essential for maintenance of its tertiary structure.27,28 Furthermore, the formation of the intra-A-chain disulfide bond is a critical first step for in vitro and in vivo chain combination and folding.29 We were intrigued whether the A-chain disulfide bond is vulnerable to reductase activity and subsequent stability.
Optimum folding is very important for maintaining the full activity and in vitro and in vivo stability of any cystine-rich bioactive peptide. It is thus essential that replacement of the disulfide bond with a non-reducible bond does not alter native configuration. Otherwise, the resulting analogues may become inactive as was recently shown for insulin analogues where one of the two inter-chain disulfide bonds were replaced by a 1,2,3-triazole bond.30 Thus, in this study, we replaced the intra-A-chain disulfide bond of H2 relaxin with a dicarba bond, which more closely mimics the disulfide bond than a triazole bond (Fig. 1). Due to its unsaturated nature, the dicarba bond is more conformationally restrained than the native disulfide thus, in addition to being non-reducible, could potentially provide an increased stability of the overall fold. We report herein the chemical synthesis of dicarba isosteric analogues of H2 relaxin and show that it retains near-native in vitro activity. We also show that the intra-A-chain dicarba relaxin analogues (Fig. 1) unexpectedly exhibit reduced serum stability that is likely due to subtle changes in the structure as suggested by circular dichroism (CD) spectroscopy.
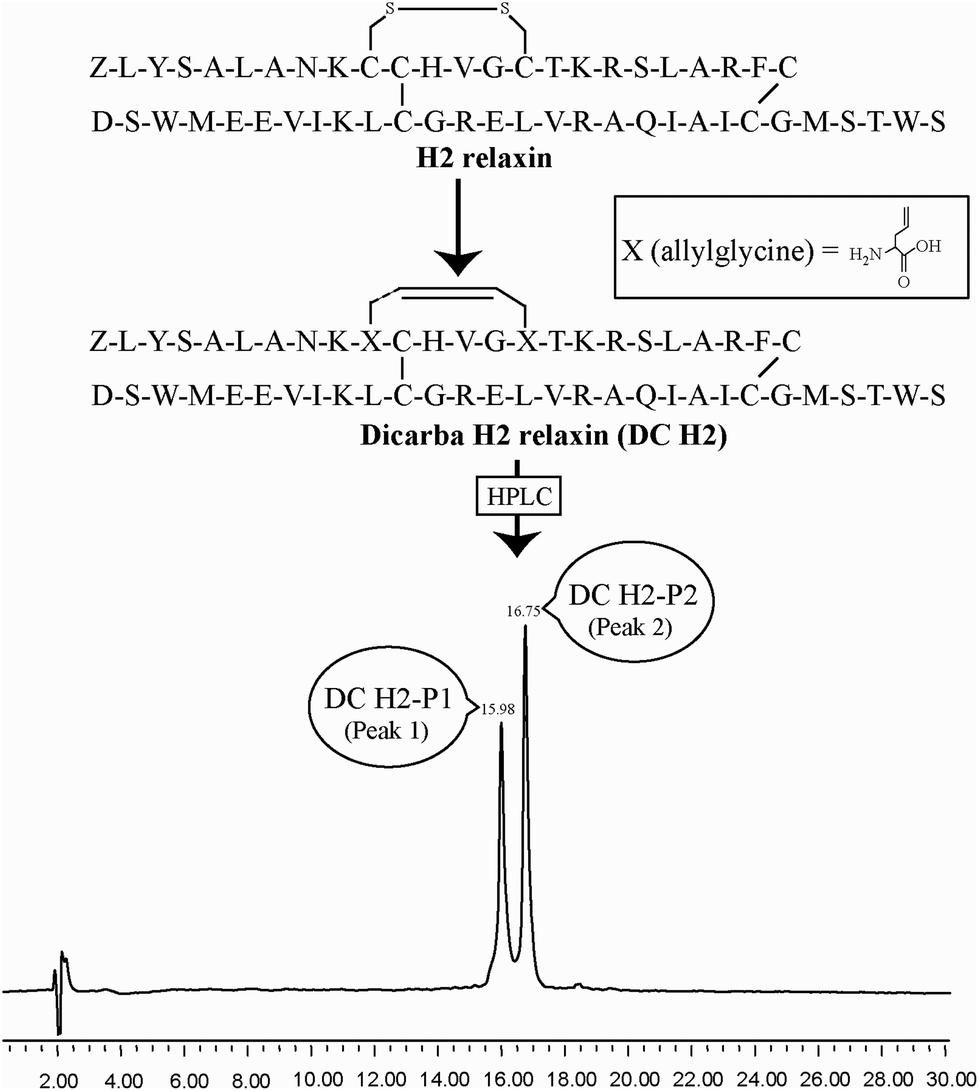 | ||
| Fig. 1 Primary structures and analytical RP-HPLC analyses of dicarba H2 relaxin peptides, DC H2-P1 (cis isoform) and DC H2-P2 (trans isoform). Z corresponds pyroglutamic acid. | ||
Results and discussion
Given the recent success with chemically assembling the related peptides, H3 relaxin and INSL3,31,32 with an intra-A-chain dicarba bond in place of a native disulfide bond and demonstration of retention of native RXFP3/2 receptor binding and activation.31,32 we undertook to employ a similar strategy for H2 relaxin and to determine its in vitro stability with the goal of improving its pharmacokinetics and subsequent potential clinical value. Briefly, the synthesis was started by separate solid phase assembly of the two chains (A-chain and B-chain) (Fig. 2).33 The A-chain contained a pair of allylGly (Hag) residues in place of the Cys residues at positions 10 and 15 that form an intra-chain cystine bond. On-resin microwave-assisted ring closing metathesis (RCM) of the A-chain using 2nd generation Grubb's catalyst was successfully achieved within 1 hour as indicated by a pilot cleavage and MALDI-TOF MS analyses (Fig. 2 and 3). The primary product after RCM of the A-chain showed two species corresponding to the cis and trans forms of the dicarba bond. Each of the A-chains (cis or trans forms) was activated with a good leaving group (S-pyridinyl; SPyr) and then combined separately with the purified B-chain by sequential disulfide bond formation using thiolysis and iodolysis respectively (Fig. 2 and 3). The two isomers of the dicarba H2 relaxin were isolated by RP-HPLC (Fig. 1) in good overall yield. The two peptides appeared as single species on analytical RP-HPLC (Fig. 1) and both had the correct molecular mass as measured by MALDI-TOF MS with MH+ values of 5927.79 and 5927.68 respectively (theoretical value MH+ 5926.03) (Fig. 3). We were previously unable to definitively differentiate between the two isomers of related H3 relaxin dicarba analogues using solution NMR spectroscopy.31 This was because the region around the dicarba bond did not adopt a single stable conformation in solution but rather experienced structural rearrangement that resulted in chemical shift averaging and consequently caused broadening and loss of signals. However, the bond lengths and angles for the dicarba bond were generated by energy minimization using ChemDraw 3D Ultra (CambridgeSoft, v.8.0) and compared with those for a disulfide bond that were taken from the CNS forcefield used for the NMR structure determination of H3 relaxin. The evidence strongly suggested that the RP-HPLC earlier-eluting isomer is the conformationally more compact cis form whereas the extended trans conformer is the later eluting isomer.31 This conclusion is supported by RP-HPLC thermodynamic principles where the peptide in the later eluting peak must have a more extended hydrophobic surface, which is what would be expected for a trans conformation. On this basis, we concluded that for the synthetic H2 relaxin dicarba isomers, the early-eluting RP-HPLC peak (Fig. 1, peak 1) corresponded to the cis form and the later eluting peak the trans isomer (Fig. 1; peak 2). We named these two isomers as DC H2-P1 and DC H2-P2, which correspond to peak 1 and peak 2 respectively (Fig. 1).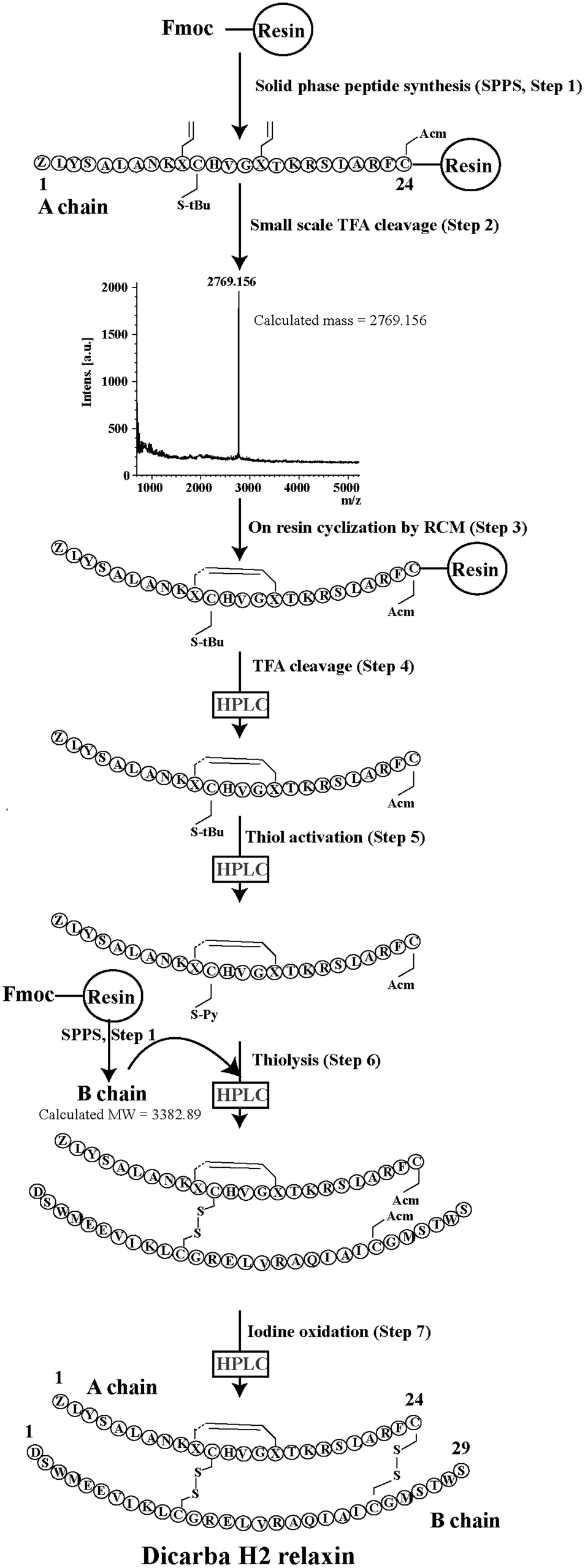 | ||
| Fig. 2 Schematic representation of the assembly of dicarba H2 relaxin analogues. | ||
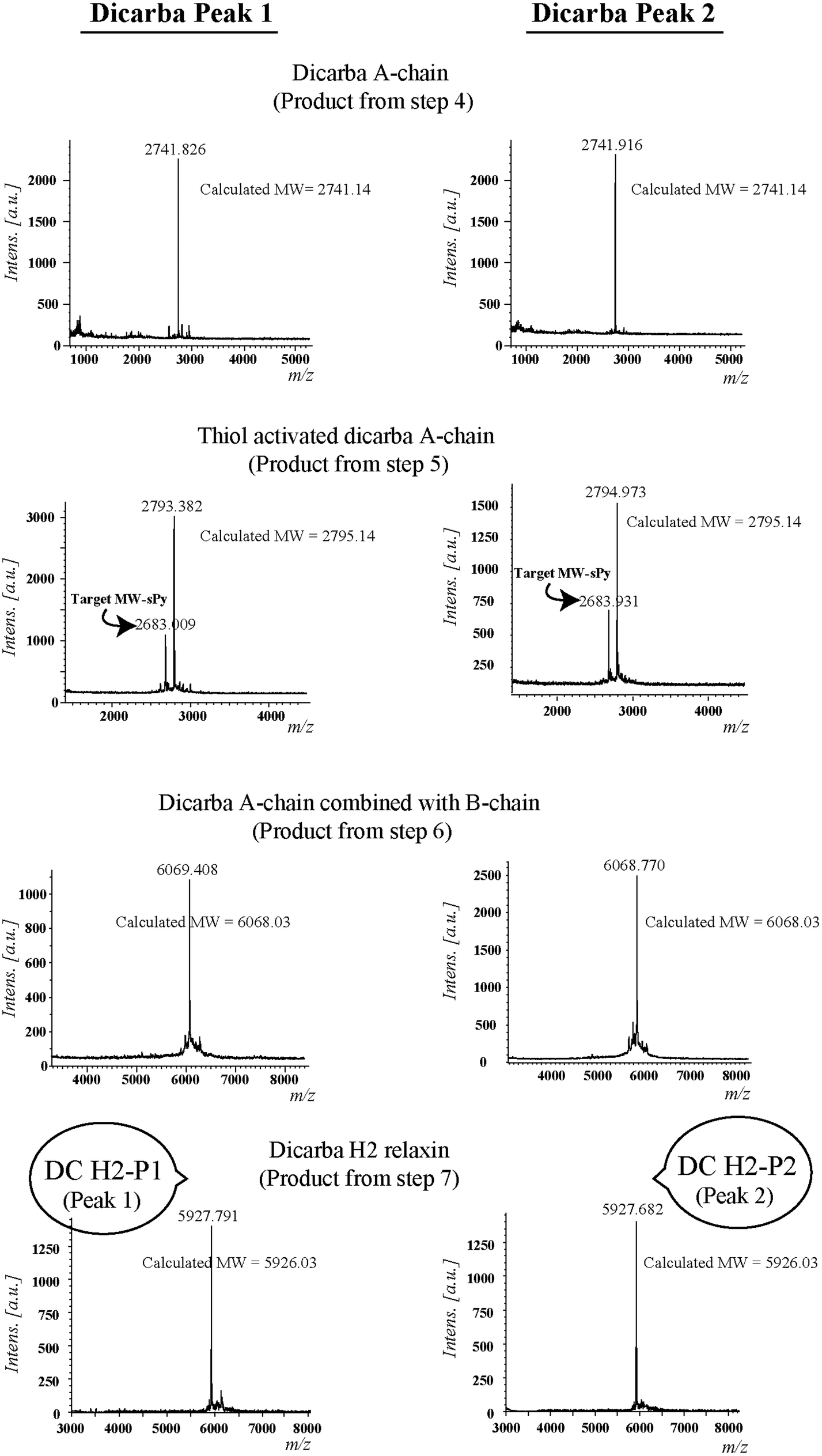 | ||
| Fig. 3 MALDI-TOF MS analysis of dicarba H2 relaxin and their intermediates. | ||
The two isomers, DC H2-P1 and DC H2-P2, were then subjected to in vitro binding and activity assays and were undertaken in comparison to the native recombinantly produced H2 relaxin. These assays were first carried out in HEK-293T cells stably expressing relaxin family peptide receptor 1 (RXFP1), the cognate receptor of H2 relaxin. The peptides were found to have high affinity and potency at the RXFP1 receptor (Fig. 4A and B; Table 1). Notably, the DC H2-P2 isomer of dicarba H2 relaxin was more potent than the DC H2-P1 isomer and equipotent to native H2 relaxin (Fig. 4A and B; Table 1).
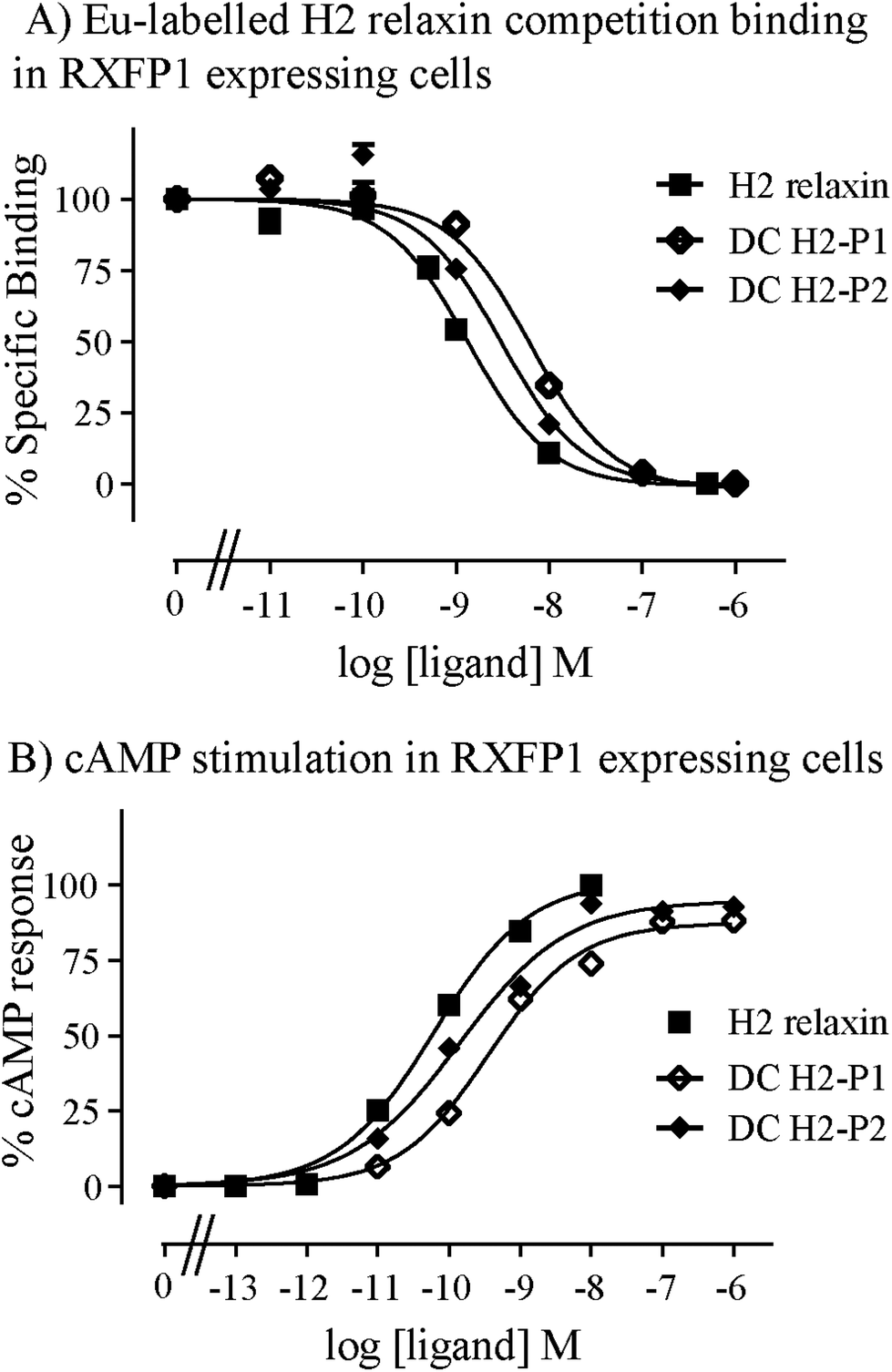 | ||
| Fig. 4 Activity of dicarba H2 relaxin analogues at RXFP1. (A) Competition binding of native H2 relaxin and dicarba H2 relaxin analogues in the presence of the competitive ligand Eu3+-labeled H2 relaxin tested in HEK-293T cells stably expressing RXFP1. (B) Effect of native H2 relaxin and dicarba H2 relaxin analogues on cAMP-related activity in HEK-293T cells expressing RXFP1 using a pCRE-galactosidase reporter gene system. Data are expressed as a percentage of specific binding or maximum relaxin stimulated cAMP response and are pooled data from at least three experiments performed in triplicate. | ||
| RXFP1 | RXFP2 | |||
|---|---|---|---|---|
| Ligand | Eu-H2 pIC50 | cAMP pEC50 | Eu-H2 pIC50 | cAMP pEC50 |
| *p < 0.05; **p < 0.01; ***p < 0.001 vs. H2 relaxin. | ||||
| H2 relaxin | 9.08 ± 0.07 (5) | 10.3 ± 0.04 (3) | 8.12 ± 0.22 (4) | 9.13 ± 0.06 (3) |
| DC H2-P1 | 8.27 ± 0.04 (3)* | 9.43 ± 0.10 (4)** | 7.28 ± 0.21 (4)* | 7.41 ± 0.15 (4)*** |
| DC H2-P2 | 8.59 ± 0.01 (3) | 9.86 ± 0.11 (4) | 7.63 ± 0.20 (4)* | 8.00 ± 0.17 (4)*** |
Both isomers were then tested on cells expressing RXFP2, the native receptor for related INSL3 which is also activated by H2 relaxin although to a much lesser extent than by INSL3. Interestingly, the peptides displayed significantly weaker binding and activation propensity suggesting that the introduction of the dicarba macrocycle adversely affects interaction of H2 relaxin to the RXFP2 receptor (Fig. 5A and B; Table 1).
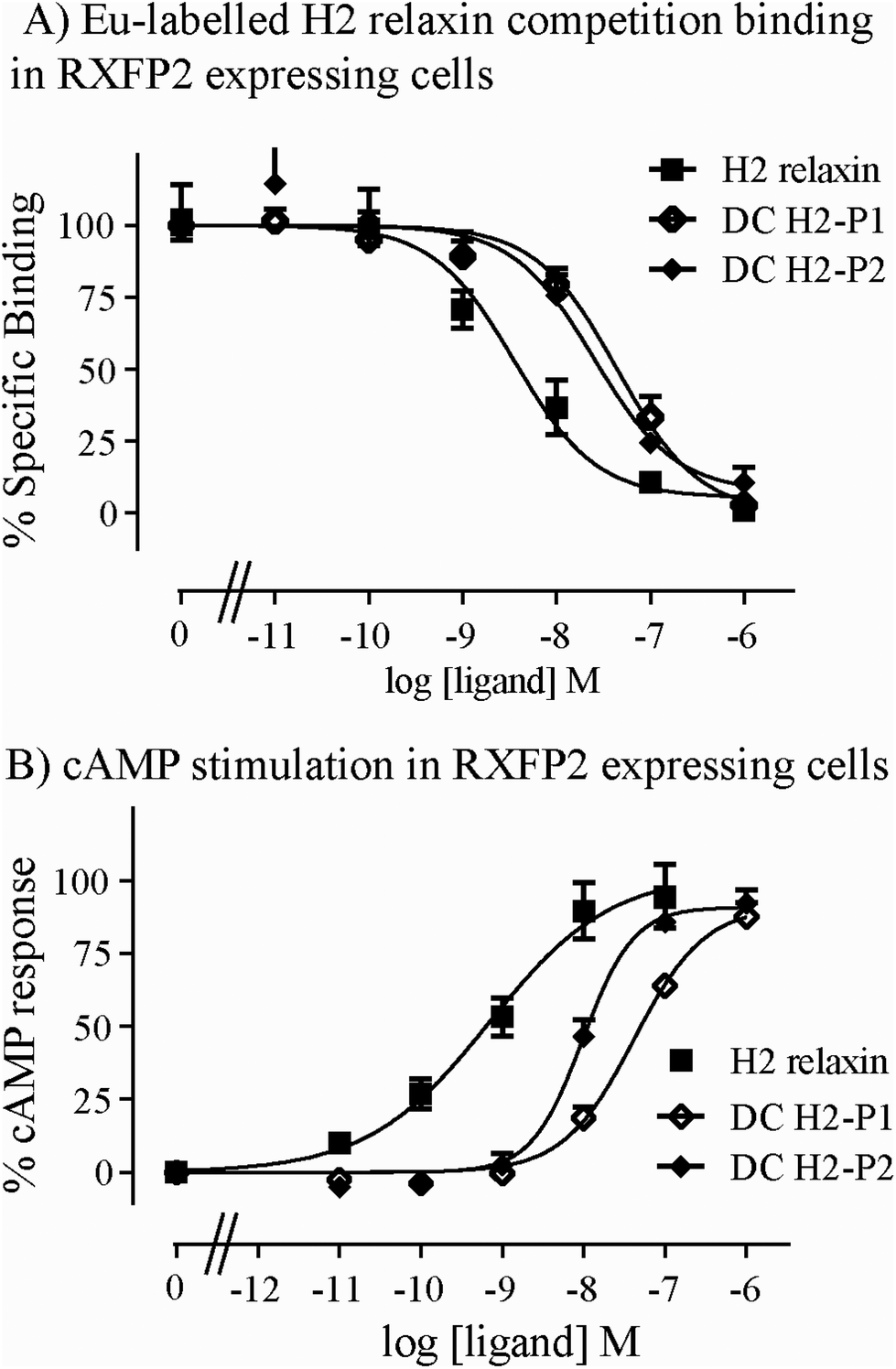 | ||
| Fig. 5 Activity of dicarba H2 relaxin analogues at RXFP2. (A) Competition binding of native human INSL3 and dicarba H2 relaxin analogues in the presence of the competitive ligand Eu3+-labeled H2 relaxin tested in HEK-293T cells stably expressing RXFP2. (B) Effect of native H2 relaxin and dicarba H2 relaxin analogues on cAMP-related activity in HEK-293T cells expressing RXFP2 using a pCRE-galactosidase reporter gene system. Data are expressed as a percentage of specific binding or maximum relaxin-stimulated cAMP response and are pooled data from at least three experiments performed in triplicate. | ||
To correlate the binding and activity data with the secondary structure of H2 relaxin, CD spectroscopic analyses were undertaken (Fig. 6). Consistent with potent binding and activity data, the DC H2-P1 and DC H2-P2 isomers were found to retain a high degree of α-helical conformation (with pronounced double minima at approximately 208 nm and 222 nm) along with some β-sheet and random coil structure (Fig. 6). The α-helical content of native H2 relaxin was found to be 40% compared with 32% and 36% for DC H2-P1 and DC H2-P2 isomer respectively.34 These values were calculated from the mean residue ellipticity (MRE) at 222 nm, the [θ]222 values for H2 relaxin, peaks 1 and 2 being −13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 808, −11014 and −12561, respectively. The difference between the MRE and helix content between these peptides suggested that there is a subtle variation in, or destabilization of, the secondary structure in the dicarba isomers compared to native H2 relaxin. This compromised structure, particularly for DC H2-P1, likely explains the reason that binding and cAMP were slightly lower at RXFP1 compared with native H2 relaxin. We have previously shown that some mutations at the A-chain of H2 relaxin disrupts the overall structure of H2 relaxin and such structural destabilization affects RXFP2 activity more than it affects RXFP1.35 The present study confirmed our previous observation that slightly less structured dicarba isomers exhibit significantly less activity at RXFP2 compared with RXFP1.35
808, −11014 and −12561, respectively. The difference between the MRE and helix content between these peptides suggested that there is a subtle variation in, or destabilization of, the secondary structure in the dicarba isomers compared to native H2 relaxin. This compromised structure, particularly for DC H2-P1, likely explains the reason that binding and cAMP were slightly lower at RXFP1 compared with native H2 relaxin. We have previously shown that some mutations at the A-chain of H2 relaxin disrupts the overall structure of H2 relaxin and such structural destabilization affects RXFP2 activity more than it affects RXFP1.35 The present study confirmed our previous observation that slightly less structured dicarba isomers exhibit significantly less activity at RXFP2 compared with RXFP1.35
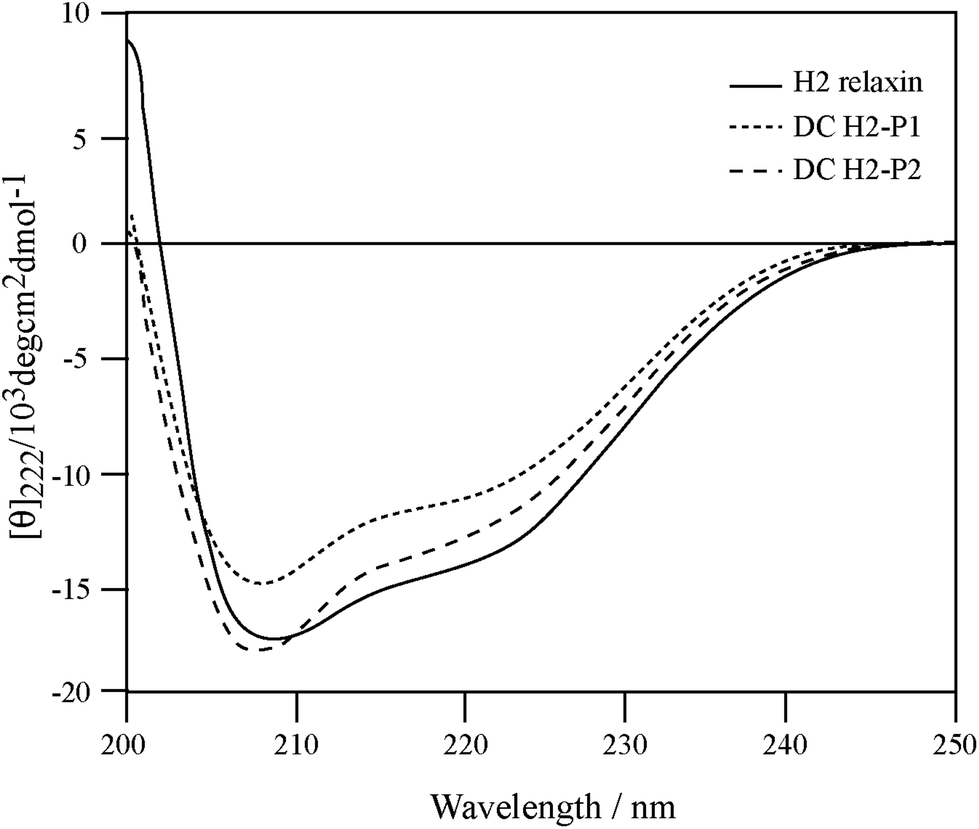 | ||
| Fig. 6 Secondary structure analysis of dicarba H2 relaxin isomers and H2 relaxin by CD spectroscopy. | ||
The in vitro serum stability of the dicarba H2 relaxin analogues was then examined. Under the conditions employed, the dicarba analogues of H2 relaxin were each significantly, and equally, less stable (t1/2 ∼1 h) than the native peptide (t1/2 ∼7 h) (Fig. 7). Specifically, over 40% of native H2 relaxin was still detected in serum after about 8 h, while the dicarba analogues were completely degraded by that time again suggesting that native H2 relaxin is more structured and thus more stable compared with dicarba H2 relaxin analogues. With complete resistance to reductases and isomerases endowed by the dicarba bridge, it is clear that, at least in this example, introduction of such an isosteric replacement for the disulfide bond has caused increased lability to other degrading enzymes. This suggests that the dicarba bond could not be fully structurally accommodated and that the resulting change in the structural stability of the peptides allowed enzymes access to amide bonds to be cleaved more easily compared with more compact structured native H2 relaxin peptide. There are several reports of the use of the dicarba bond resulting in a significant increase in resistance to proteases21,36 probably through, at least in part, preventing extended peptide conformation that the enzymes need to hydrolyze amide bonds. Locking in a well-ordered fold by a suitable restraint can indeed have dramatically beneficial effects. For example cyclization, of the alpha-conotoxin MII by introduction of a 6–7 residue linker sequence between the N and the C-termini prevents cleavage by the protease endoGluC despite the fact that the endoGluC cleavage site is on the opposite site of the molecule.17 However there are also reports of no beneficial effect of such macrocyclization on stability presumably because degradation occurs beyond the confines of the constraint,37,38 and the modification is insufficient to achieve a global stabilizing effect.
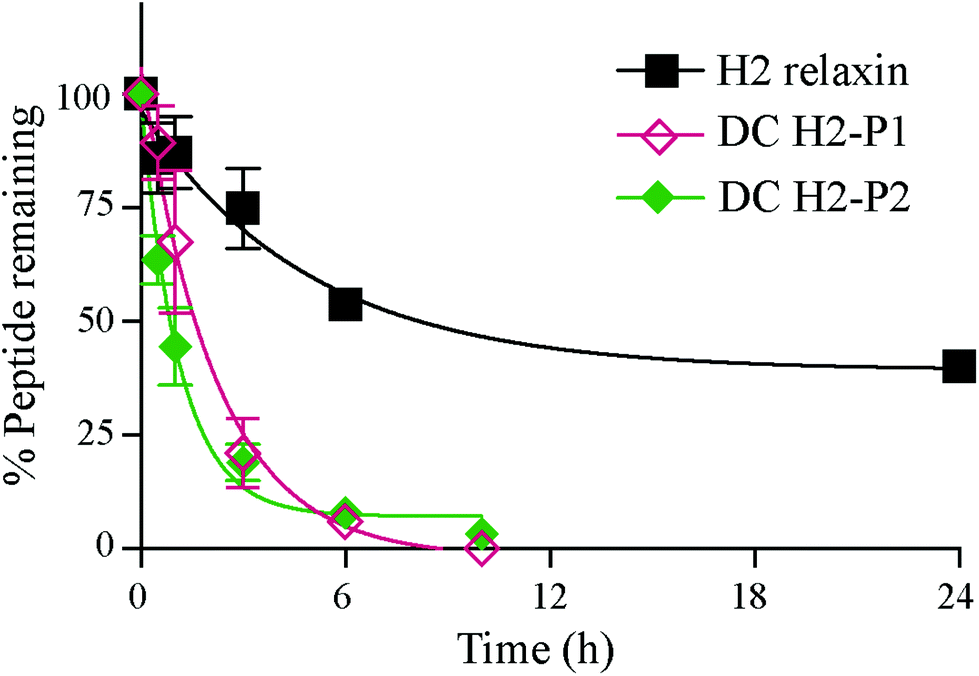 | ||
| Fig. 7 In vitro serum stability of H2 relaxin and dicarba H2 relaxin isomers. | ||
Conclusions
We have successfully chemically synthesized and characterized dicarba H2 relaxin analogues and showed that the analogues exhibit near-native RXFP1 activity and improved selectivity over RXFP2. Unexpectedly, the dicarba analogues exhibited poor serum stability due to subtle change in their structure which was confirmed by CD spectroscopy. Our observation of dramatically decreased stability is a critical one not hitherto reported elsewhere and that flags caution for the utility of such macrocyclization techniques.Materials and methods
9-Fluorenylmethoxycarbonyl (Fmoc) protected L-α-amino acids and 1-[bis(dimethylamino)methylene]-1H-benzotriazolium hexafluorophosphate 3-oxide (HBTU) were purchased from GL Biochem (Shanghai, China). Piperidine (PPD) and trifluoroacetic acid (TFA) were purchased from Auspep (Melbourne, Australia). Fmoc-PAL-PEG-PS resins with substitution of 0.18 mmol g−1 were purchased from Applied Biosystems Inc. (Melbourne, Australia). Dimethylformamide (DMF), methanol, diethyl ether, and dichloromethane (DCM) were purchased from Merck (Melbourne, Australia). 3,6-Dioxa-1,8-octanedithiol (DODT), triisopropylsilane (TIPS), diisopropylethylamine (DIPEA) and trifluoromethanesulfonic acid (TFMSA) were purchased from Sigma-Aldrich (Sydney, Australia). 2,2-Dipyridyl disulfide (DPDS) was purchased from Fluka (Switzerland). Acetonitrile was purchased from BDH Laboratory Supplies, (Poole, UK). All other reagents were from Sigma-Aldrich (Sydney, Australia). The tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydro-imidazol-2-ylidene](benzylidene)ruthenium(II) dichloride (2nd generation Grubbs’ catalyst) was supplied by Aldrich (Sydney, Australia).Solid-phase peptide synthesis (SPPS)
Regioselectively S-protected A- and B-chains of H2 relaxin were separately synthesized by the Fmoc solid-phase method by using microwave-assisted synthesis on a Liberty system (CEM Corporation, Charlotte, NC, USA)39 (Fig. 1, step 1). The following side chain protecting groups were used: Arg, Pbf; Asn and Gln, Trt; Asp and Glu, O-But; His, Trt; Lys, Boc; Ser and Thr, tBu. Selective cysteine S-protection was also employed: Cys(A11, B10), acetamidomethyl; Cys(A24), tert-butyl, and Cys(B22), trityl. For the subsequent formation of the dicarba bond, Fmoc-L-allylglycine (Hag) was used in positions 10 and 15 of the A-chain. A small amount of resin bound peptide was cleaved by TFA and analyzed by RP-HPLC and LALDI TOF MS which confirmed successful synthesis of linear A- and B-chains (Fig. 1, step 2).On-resin microwave-accelerated ring closing metathesis (RCM) of the A-chain
The RCM reaction carried out as previously described31,32 (Fig. 2, step 4). Briefly, a microwave reactor vessel was loaded with resin-bound A-chain peptide (0.55 g, 0.1 mmol), DCM (10 mL), 0.4 M LiCl in DMF (200 μL) and 2nd generation Grubbs’ catalyst (17 mg, 20 μmol, 20 mol%) in an inert environment. The system was sealed and the reaction mixture irradiated with 40 W of microwave energy and stirred at 100 °C for 1 h. The reaction mixture was cooled to room temperature, filtered through a fritted syringe and the resin washed with DCM (7 mL, 3 × 1 min) and MeOH (7 mL, 3 × 1 min) then left to dry in vacuo for 1 h. Post-metathesis, the resin-bound peptide was washed with DMF (5 × 1 min), DCM (3 × 1 min) and MeOH (3 × 1 min). After acid-mediated cleavage, RP-HPLC and mass spectral analysis showed the formation of the desired cyclic A-chain peptide as two isomers (Fig. 2 and 3; step 4).Regioselective disulfide bond formation
:4 v/v) were added and the mixture stirred for 1 h on ice. The peptide was then precipitated with ice-cold diethyl ether, and the pellet collected by centrifugation, washed 3 times with ice-cold diethyl ether, air-dried and subjected to RP-HPLC purification. The MALDI TOF MS analysis confirmed the formation of the desired SPyr activated A-chain isomers (Fig. 2 and 3: step 5).
Chemical characterization
The purity of each intermediate and the final dicarba H2 relaxin analogues was assessed by analytical RP-HPLC and MALDI-TOF mass spectrometry using a Bruker Autoflex II instrument (Bremen, Germany) in the linear mode at 19.5 kV. Peptides were also quantitated by amino acid analysis of a 24 hour vapour phase acid hydrolyzate followed by derivatization with AccuTag chemistry and resolution of the labeled residues using a Shimadzu microbore RP-HPLC system (Melbourne, Australia).CD spectroscopy
The secondary structural changes of the peptides were measured by recording their CD spectra on JASCO model J815 spectropolarimeter as previously described.42,43 The CD spectra were recorded in phosphate buffered saline (10 mM) with peptide concentrations made up to 0.1–0.2 μg μl−1. Helix content of peptide is directly proportional to mean residue ellipticity at 222 nm [θ]222. The [θ]222 value for each peptide was determined from the CD spectra that were measured at 25 °C. One hundred percent helicity was calculated by using the formula max[θ]222 = −40000 × [(1 − 2.5/n)] + (100 + T), where n = number of amino acid residues and T = temperature of the peptide solution in 25 °C.34 Percentage helicity was then calculated as 100 × [θ]222/max[θ]222.
Ligand binding assay
Human embryonic kidney (HEK-293T) cells stably transfected with RXFP1 or RXFP2 were cultured in RPMI 1640 medium (Sigma) supplemented with 10% fetal calf serum, 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin and 2 mM L-glutamine and plated into 96-well plates. The 96-well plates were pre-coated with poly-L-lysine for whole cell binding assays. Competition binding experiments were carried out using europium-labeled H2 relaxin44 in the absence or presence of increasing concentrations of unlabeled DC H2 analogues. Fluorescent measurements were recorded at an excitation wavelength of 340 nm and emission of 614 nm on a BMG PolarStar plate reader. All data were are presented as the mean ± S.E. of the percentage of the total specific binding of triplicate wells, repeated in at least three separate experiments, and curves were fitted using one-site binding curves in GraphPad Prism 4 (GraphPad Inc, San Diego, CA). Statistical differences in pKi values were analyzed using one-way analysis of variance coupled to Newman Keul's multiple comparison test for multiple group comparisons in GraphPad Prism 5.Functional cAMP assay
The evaluation of the ability of native H2 relaxin and dicarba relaxin analogues to stimulate a cAMP response in cells expressing the relaxin receptor RXFP1 was conducted using a cAMP reporter gene assay as described previously.45 HEK-293T cells co-transfected with either RXFP1 or RXFP2 and a pCRE β-galactosidase reporter plasmid were plated in 96-well plates. The co-transfected cells were incubated with increasing concentrations of H2 relaxin or relaxin analogues in RXFP1- or RXFP2-transfected cells. The amount of cAMP-driven β-galactosidase expression in each well was assessed with a colormetric assay measuring absorbance at 570 nm on a Benchmark Plus microplate spectrophotometer (BioRad). Ligand-induced cAMP stimulation was expressed as a percentage of maximal response of relaxin for RXFP1 and RXFP2 cells. Each data point was measured in triplicate and each experiment conducted independently at least three separate times. Statistical differences in pEC50 values were analyzed using one-way analysis of variance coupled to Newman Keul's multiple comparison test for multiple group comparisons in GraphPad Prism 5.In vitro serum stability
Human pooled male serum (Sigma-Aldrich) was pre-incubated for 15 min at 37 °C, before addition of 35 μg peptide to 665 μl serum. Samples were taken out at different time points, (0, 0.5, 1, 3, 6 and 10 h for H2 dicarba peptides and at 0, 0.5, 1, 3, 6 and 24 h for H2 relaxin. Each serum aliquot, 100 μl, was quenched with 900 μl 100 mM ammonium acetate, pH 3 and left on ice for 30 min. To separate the peptide from the serum components, Oasis HLB 3 cc 60 mg cartridges (Waters) were prepared by washing the column with 6 ml methanol, followed by 3 ml 70% acetonitrile, 1% formic acid and 3 ml 1% formic acid. The serum sample was then loaded onto the column and further washed with 3 ml 1% formic acid. The peptide was eluted with increasing concentrations of acetonitrile in 1% formic acid. The eluted sample was lyophilised and redissolved in 100 μl 1% formic acid and analysed on an API2000 (AB Sciex) LC-MS. The experiment was repeated three times for each time point and analysed using Prism 5. Statistical differences in t1/2 values were analyzed using one-way analysis of variance coupled to Tukey's multiple comparison test for multiple group comparisons in GraphPad Prism 6. There is a significance difference (p < 0.01) between the t1/2 values of H2 vs. DC P1 or DC P2 but no significant difference between the t1/2 values DC P1 vs. DC P2.Acknowledgements
We thank Mrs Sharon Layfield and Mrs Tania Ferraro for expert technical assistance and Associate Professor Andrea J. Robinson and Dr Bianca van Lierop (Monash University) for providing assistance with the RCM reaction. This work was supported by National Health & Medical Research Council (NHMRC) of Australia Grants (GNT1023321, GNT1023078, and GNT1065481) awarded to M. A. H., J. D. W, K. J. R, and R. A. D. B. Research at The Florey Institute of Neuroscience and Mental Health is supported by the Victorian Government Operational Infrastructure Support Program.References
- R. A. Bathgate, M. L. Halls, E. T. van der Westhuizen, G. E. Callander, M. Kocan and R. J. Summers, Physiol. Rev., 2013, 93, 405–480 CrossRef CAS PubMed.
- L. J. Chan, M. A. Hossain, C. S. Samuel, F. Separovic and J. D. Wade, Protein Pept. Lett., 2011, 18, 220–229 CrossRef CAS.
- F. Shabanpoor, F. Separovic and J. D. Wade, Vitam. Horm., 2009, 80, 1–31 CAS.
- F. L. Hisaw, Proc. Soc. Exp. Biol. Med., 1926, 23, 661–661 CrossRef.
- J. R. Teerlink, G. Cotter, B. A. Davison, G. M. Felker, G. Filippatos, B. H. Greenberg, P. Ponikowski, E. Unemori, A. A. Voors, K. F. Adams Jr., M. I. Dorobantu, L. R. Grinfeld, G. Jondeau, A. Marmor, J. Masip, P. S. Pang, K. Werdan, S. L. Teichman, A. Trapani, C. A. Bush, R. Saini, C. Schumacher, T. M. Severin and M. Metra, Lancet, 2013, 381, 29–39 CrossRef CAS.
- J. R. Teerlink, M. Metra, G. M. Felker, P. Ponikowski, A. A. Voors, B. D. Weatherley, A. Marmor, A. Katz, J. Grzybowski, E. Unemori, S. L. Teichman and G. Cotter, Lancet, 2009, 373, 1429–1439 CrossRef CAS.
- S. A. Chen, A. J. Perlman, N. Spanski, C. M. Peterson, S. W. Sanders, R. Jaffe, M. Martin, T. Yalcinkaya, R. C. Cefalo and N. C. Chescheir, et al. , Pharm. Res., 1993, 10, 834–838 CrossRef CAS.
- L. Otvos Jr. and J. D. Wade, Front. Chem., 2014, 2, 62 Search PubMed.
- S. Curry, P. Brick and N. P. Franks, Biochim. Biophys. Acta, 1999, 1441, 131–140 CrossRef CAS.
- C. Ginn, H. Khalili, R. Lever and S. Brocchini, Future Med. Chem., 2014, 6, 1829–1846 CrossRef CAS PubMed.
- M. Schlapschy, U. Binder, C. Borger, I. Theobald, K. Wachinger, S. Kisling, D. Haller and A. Skerra, Protein Eng. Des. Sel., 2013, 26, 489–501 CrossRef CAS PubMed.
- V. N. Podust, B. C. Sim, D. Kothari, L. Henthorn, C. Gu, C. W. Wang, B. McLaughlin and V. Schellenberger, Protein Eng. Des. Sel., 2013, 26, 743–753 CrossRef CAS PubMed.
- J. T. Sockolosky, S. Kivimae and F. C. Szoka, PLoS One, 2014, 9, e102566 Search PubMed.
- J. Seijsing, M. Lindborg, I. Hoiden-Guthenberg, H. Bonisch, E. Guneriusson, F. Y. Frejd, L. Abrahmsen, C. Ekblad, J. Lofblom, M. Uhlen and T. Graslund, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17110–17115 CrossRef CAS PubMed.
- R. E. Kontermann, Curr. Opin. Biotechnol., 2011, 22, 868–876 CrossRef CAS PubMed.
- M. Werle and A. Bernkop-Schnurch, Amino Acids, 2006, 30, 351–367 CrossRef CAS PubMed.
- R. J. Clark, H. Fischer, L. Dempster, N. L. Daly, K. J. Rosengren, S. T. Nevin, F. A. Meunier, D. J. Adams and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13767–13772 CrossRef CAS PubMed.
- M. L. Colgrave and D. J. Craik, Biochemistry, 2004, 43, 5965–5975 CrossRef CAS PubMed.
- K. Meinander, M. Pakkala, J. Weisell, U. H. Stenman, H. Koistinen, A. Narvanen and E. A. Wallen, ACS Med. Chem. Lett., 2014, 5, 162–165 CrossRef CAS PubMed.
- M. Muttenthaler, A. Andersson, A. D. de Araujo, Z. Dekan, R. J. Lewis and P. F. Alewood, J. Med. Chem., 2010, 53, 8585–8596 CrossRef CAS PubMed.
- S. Chhabra, A. Belgi, P. Bartels, B. J. van Lierop, S. D. Robinson, S. N. Kompella, A. Hung, B. P. Callaghan, D. J. Adams, A. J. Robinson and R. S. Norton, J. Med. Chem., 2014, 57, 9933–9944 CrossRef CAS PubMed.
- S. Kalra, N. Li, S. Seetharam, D. H. Alpers and B. Seetharam, Am. J. Physiol. Cell Physiol., 2003, 285, C150–C160 CrossRef CAS PubMed.
- A. Maemoto, X. Qu, K. J. Rosengren, H. Tanabe, A. Henschen-Edman, D. J. Craik and A. J. Ouellette, J. Biol. Chem., 2004, 279, 44188–44196 CrossRef CAS PubMed.
- N. R. Maiti and W. K. Surewicz, J. Biol. Chem., 2001, 276, 2427–2431 CrossRef CAS PubMed.
- N. A. Patil, J. Tailhades, R. A. Hughes, F. Separovic, J. D. Wade and M. A. Hossain, Int. J. Mol. Sci., 2015, 16, 1791–1805 CrossRef CAS PubMed.
- E. E. Büllesbach and C. Schwabe, Biochemistry, 2012, 51, 4198–4205 CrossRef PubMed.
- C. Eigenbrot, M. Randal, C. Quan, J. Burnier, L. O'Connell, E. Rinderknecht and A. A. Kossiakoff, J. Mol. Biol., 1991, 221, 15–21 CAS.
- L. M. Haugaard-Kedstrom, M. A. Hossain, N. L. Daly, R. A. Bathgate, E. Rinderknecht, J. D. Wade, D. J. Craik and K. J. Rosengren, ACS Chem. Biol., 2015, 10, 891–900 CrossRef CAS PubMed.
- J. G. Tang, Z. H. Wang, G. W. Tregear and J. D. Wade, Biochemistry, 2003, 42, 2731–2739 CrossRef CAS PubMed.
- G. M. Williams, K. Lee, X. Li, G. J. Cooper and M. A. Brimble, Org. Biomol. Chem., 2015, 13, 4059–4063 CAS.
- M. A. Hossain, K. J. Rosengren, S. Zhang, R. A. Bathgate, G. W. Tregear, B. J. van Lierop, A. J. Robinson and J. D. Wade, Org. Biomol. Chem., 2009, 7, 1547–1553 CAS.
- S. Zhang, R. A. Hughes, R. A. Bathgate, F. Shabanpoor, M. A. Hossain, F. Lin, B. van Lierop, A. J. Robinson and J. D. Wade, Peptides, 2010, 31, 1730–1736 CrossRef CAS PubMed.
- M. A. Hossain and J. D. Wade, Curr. Opin. Chem. Biol., 2014, 22, 47–55 CrossRef CAS PubMed.
- J. M. Scholtz, H. Qian, E. J. York, J. M. Stewart and R. L. Baldwin, Biopolymers, 1991, 31, 1463–1470 CrossRef CAS PubMed.
- L. J. Chan, K. J. Rosengren, S. L. Layfield, R. A. Bathgate, F. Separovic, C. S. Samuel, M. A. Hossain and J. D. Wade, J. Biol. Chem., 2012, 287, 41152–41164 CrossRef CAS PubMed.
- C. A. MacRaild, J. Illesinghe, B. J. van Lierop, A. L. Townsend, M. Chebib, B. G. Livett, A. J. Robinson and R. S. Norton, J. Med. Chem., 2009, 52, 755–762 CrossRef CAS PubMed.
- F. Giordanetto, J. D. Revell, L. Knerr, M. Hostettler, A. Paunovic, C. Priest, A. Janefeldt and A. Gill, ACS Med. Chem. Lett., 2013, 4, 1163–1168 CrossRef CAS PubMed.
- K. J. Rosengren, U. Goransson, L. Otvos Jr. and D. J. Craik, Biopolymers, 2004, 76, 446–458 CrossRef CAS PubMed.
- J. D. Wade, F. Lin, M. A. Hossain and R. M. Dawson, Amino Acids, 2012, 43, 2279–2283 CrossRef CAS PubMed.
- M. A. Hossain, R. A. Bathgate, C. K. Kong, F. Shabanpoor, S. Zhang, L. M. Haugaard-Jonsson, K. J. Rosengren, G. W. Tregear and J. D. Wade, ChemBioChem, 2008, 9, 1816–1822 CrossRef CAS PubMed.
- S. Zhang, F. Lin, M. Hossain, F. Shabanpoor, G. Tregear and J. Wade, Int. J. Pept. Res. Ther., 2008, 14, 301–305 CrossRef CAS.
- L. V. Najbar, D. J. Craik, J. D. Wade, D. Salvatore and M. J. McLeish, Biochemistry, 1997, 36, 11525–11533 CrossRef CAS PubMed.
- M. Hossain, F. Lin, S. Zhang, T. Ferraro, R. Bathgate, G. Tregear and J. Wade, Int. J. Pept. Res. Ther., 2006, 12, 211–215 CrossRef CAS.
- F. Shabanpoor, R. A. Bathgate, A. Belgi, L. J. Chan, V. B. Nair, J. D. Wade and M. A. Hossain, Biochem. Biophys. Res. Commun., 2012, 420, 253–256 CrossRef CAS PubMed.
- D. J. Scott, S. Layfield, Y. Yan, S. Sudo, A. J. Hsueh, G. W. Tregear and R. A. Bathgate, J. Biol. Chem., 2006, 281, 34942–34954 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |