
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of quinoxalines or quinolin-8-amines from N-propargyl aniline derivatives employing tin and indium chlorides†
Stefan
Aichhorn
a,
Markus
Himmelsbach
b and
Wolfgang
Schöfberger
*a
aInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria. E-mail: wolfgang.schoefberger@jku.at
bInstitute of Analytical Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria
First published on 12th August 2015
Abstract
Pyrazino compounds such as quinoxalines are 1,4-diazines with widespread occurrence in nature. Quinolin-8-amines are isomerically related and valuable scaffolds in organic synthesis. Herein, we present intramolecular main group metal Lewis acid catalyzed formal hydroamination as well as hydroarylation methodology using mono-propargylated aromatic ortho-diamines. The annulations can be conducted utilizing equal aerobic conditions with either stannic chloride or indium(III) chloride and represent primary examples for main group metal catalyzed 6-exo-dig and 6-endo-dig, respectively, cyclizations in such settings. Both types of reactions can also be utilized in a one-pot manner starting from ortho-nitro N-propargyl anilines using stoichiometric amounts SnCl2·2H2O or In powder. Mechanistic considerations are presented regarding the substituent-depending regioselectivity.
Introduction
Pyrazine motifs1 are widespread in biological organisms2 and integrated into versatile chemical structures3 also with pharmaceutical value.4 1,4-Diazines are commonly prepared from α-keto carbonyl moieties and 1,2-diamines through twofold imine condensation.5 The second conventional route is to initiate self-condensation of α-amino ketones, often derived from phenacyl halides.6 These syntheses appear to be quite straightforward cyclizations, but only a few supply a broader substituent scope.7 Also, routes via N-oxides have been utilized to synthesize polycyclic quinoxalines.8 On the other hand, propargylic amines and related structures found some use for the transition metal mediated construction of aromatic nitrogen heterocycles.9 Furthermore, quinoline-8-amines, which act as valuable directing groups,10 ligands for coordination chemistry11 as well as agents for various diseases,12 can be considered as structural isomers of methylquinoxalines. Functionalized quinoline platforms found broad interest with organic chemists over decades.13 In the course of attempting to synthesize N4-propargylquinolin-3,4-diamine 2 from its 3-nitro precursor 1, the reduction using stannous chloride dihydrate in ethanol gave a minor amount of 2, but a considerable fraction of the product mixture was found to consist of an unknown compound. The seemingly mismatching simplicity of its NMR spectra encouraged us to elucidate the structure. It could be verified by HMBC experiments as well as HR-MS that the resulting assembly appears to be 3-methylpyrazino[2,3-c]quinoline 3. To the best of our knowledge, such 3-alkylated pyrazino[2,3-c]quinoline scaffolds and the preparation of pyrazine or quinoxaline derivatives in this fashion have not been reported so far.Results and discussion
In order to exploit this observation towards more general application for aromatic heterocycles, we investigated a one-pot procedure for six-membered nitrogen motifs,14 starting from ortho-nitro N-propargyl aniline derivatives. Furthermore, we decided to examine whether next to tin also homologous indium species could be facilitated in this context, as indium powder is a reducing agent for nitro groups and In(III) has been demonstrated to activate alkynes.15 Performing initial screening reactions revealed that the use of methanol as solvent did lead to high quantity of an unidentified side product. It is noteworthy that it was possible to conduct the six-membered nitrogen ring formations in aqueous solution at 85 °C, although such reactions resulted in decreased yields. Farther, the same is true for employing toluene as solvent at 100 °C. Finally, we have developed two pragmatic protocols for the construction of pyrazines (Fig. 1). Both standard procedures are based on the use of isopropanol as solvent while stirring the reaction mixtures at reflux under aerobic conditions. To obtain the different 2-methylquinoxalines, the best yields were obtained employing 5.5 equivalents of SnCl2·2H2O. Moving to elementary indium, 3 equivalents in combination with hydrochloric acid gave similar results. With these optimized protocols in hand, we conducted one-pot syntheses of quinoxalines using starting materials 1, 4, and 13. With reaction times ranging from two to 20 hours, derivatives 3, 6, and 15 could be afforded in good yields from 71–82% (Fig. 1).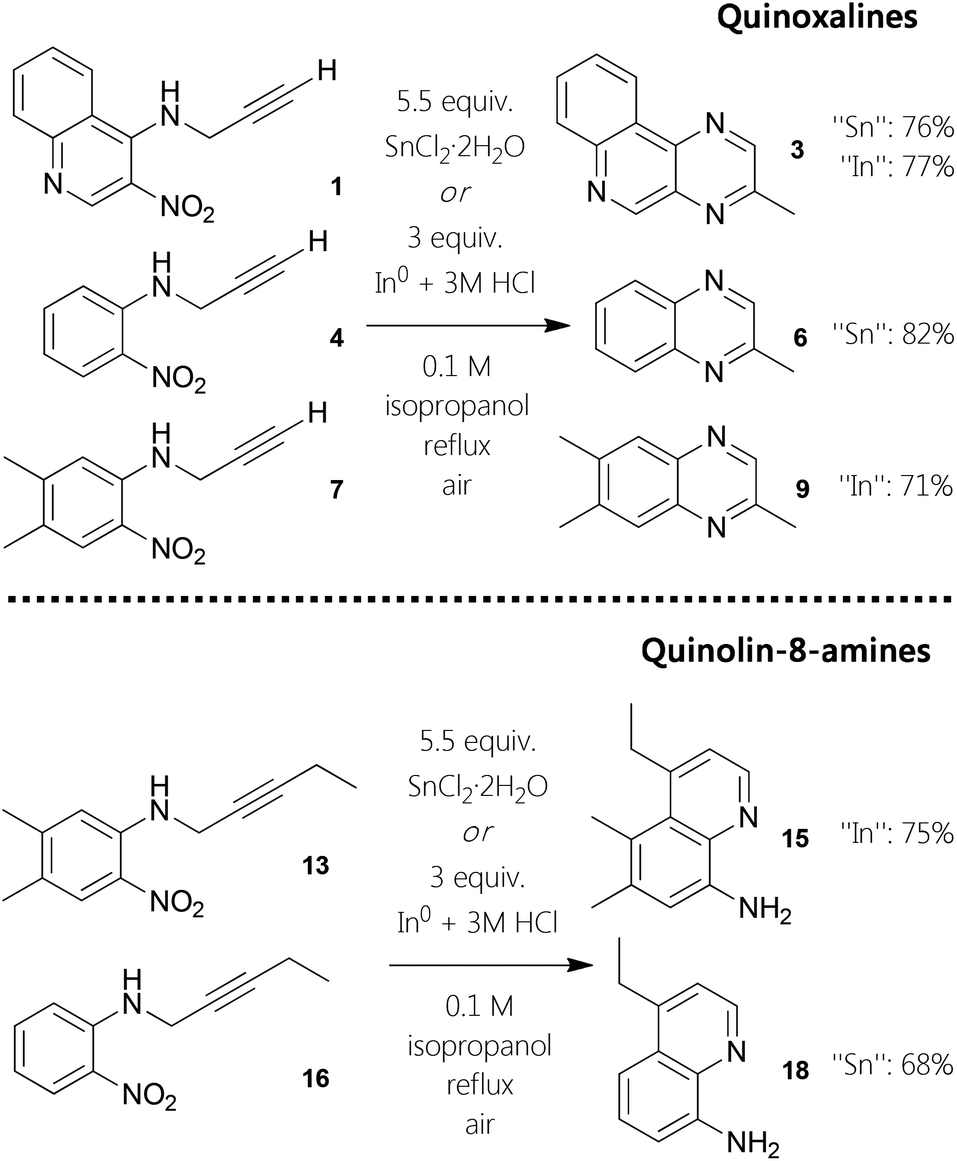 | ||
| Fig. 1 Substituent-dependent one-pot synthesis of either pyrazines or quinolin-8-amines. | ||
To explore a variant of this approach with related substrates, readily available anilides as masked anilines were chosen. The monofunctionalization of an anilide appears to be less susceptible compared to primary ortho-nitro anilines. An acetyl group as in anisyl-compound 10 can be cleaved under acidic conditions like present in the one-pot procedures. By subjecting the N-propargyl anilide to hot hydrochloric acid solution, quantitative deacetylation can be established. When a standard reductive one-pot cyclization-aromatization reaction is conducted with 10, almost exclusively N1-propargyl-C2-methyl-benzimidazole 12 is created. Under these circumstances, reduction of the nitro-group and subsequent condensation of the five-membered ring seems to be faster than the incorporation of the alkyne. In this fashion, functionalized benzimidazoles can be generated in good yield (Fig. 2). For the synthesis of quinoxalines, the protocol had to be adjusted by stirring the alkynyl–anilide in 3 M HCl at 90 °C first. Employing our developed standard protocol in the same pot (isopropanol and the respective reducing agent) gave the quinoxaline 11 in good yields.
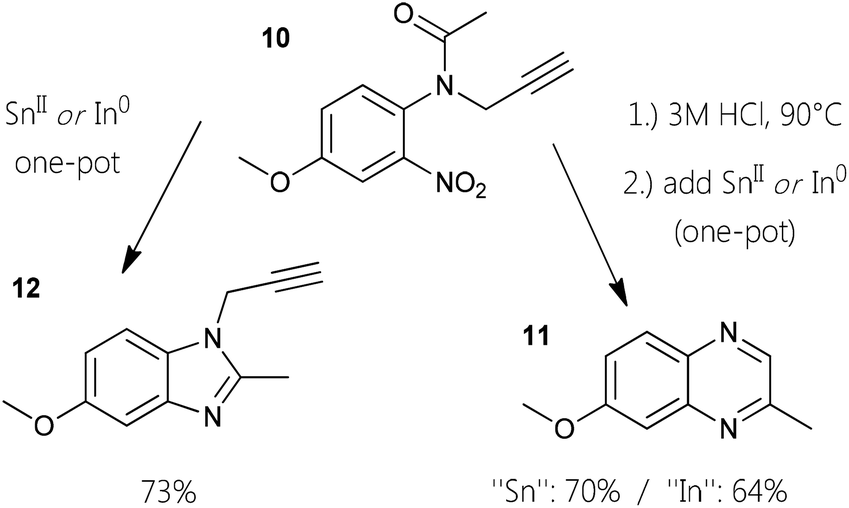 | ||
| Fig. 2 Alkynylanilides as substrates for either propargylic benz-imidazoles or quinoxalines. | ||
Soon, it became inevitable to examine whether only terminal β-γ-alkynylamines would serve as platform for such formal 6-exo-dig cyclizations.16 To access 3′-alkylated derivatives, the primary ortho-nitro arylamines were treated with sodium hydride and 1-bromo-2-pentyne, which gave the internal alkynes 13, 16, and 19. Those were subjected to reflux in isopropanol under the established aerobic one-pot conditions. At this point, it was surprising to observe complete consumption of educts without creation of pyrazine products.
While the reduced primary amines in ortho-position remained not further functionalized, we realized that indeed six-membered rings were synthesized from 13 and 16 (Fig. 1), but in contrast to previous intramolecular annulations, quinoline-8-amines 15 (75%) and 18 (68%) were afforded, formally in a 6-endo-dig fashion, exclusively. In case of 3-nitro-N-(pent-2-yn-1-yl)quinolin-4-amine, only reduction giving the corresponding 3,4-diamine occurred. These tendencies allow for a distinction between two different types of cascade reactions, which can be conducted employing identical conditions whilst generating unlike heterocycles. When a para-diazine compound is generated, the initial cyclization of the reduced intermediate can be considered as intramolecular hydroamination reaction. In contrast, the formation of quinolinamine structures follows a hydroarylation pathway prior to aromatization.
With protocols for specific six-membered rings in hand, we became interested in exploring the mechanistic aspects of this kind of annulation reactions. Preliminary experiments were conducted with β-γ-alkynyl-diamines like 2 or 14 exposed to oxygen in isopropanol at 82 °C. In the absence of any additives, no conversion was observed. The same was true for the addition of water or stannic chloride. By using less than one equivalent of SnCl2·2H2O or InCl3, product formation was achieved analogous to one-pot results. With these promising results, we were interested to demonstrate our synthetic procedures to obtain the six-membered rings in catalytic fashion with well-chosen substrates.
A study of literature reveals that there are a number of related syntheses starting from propargylamine derivatives towards either diaza-compounds (Fig. S1† – left column) or other six-membered rings (Fig. S1† – right column). Gold-catalysis sets the standards in the field due to the advantageous properties of Au-complexes concerning the interactions with C–C triple bonds.17 Regarding the hydroamination type reactions, a few older reports cover intramolecular conversions attaining quinoxalines similarly to our approach. However, copper and mercury based mediating reagents have to be utilized in stoichiometric fashion.18 In this sense, the tin(II) as well as indium(III) promoted cyclizations described here are the first main group metal catalyzed 6-exo-dig hydroamination. The situation with electrophilic aromatic substitution reactions is different as there are no reports on vicinal monoalkynylated diamino-substrates, but several transition metal catalyzed annulations giving quinoline derivatives.19 The best matching related ring formations are In(III) catalyzed syntheses of phenanthrenes and chromenes.20 Considering this, the Sn(II) and In(III) mediated cyclizations described here are the first examples of main group metal catalyzed 6-endo-dig hydroarylation to afford quinoline derivatives.
To provide starting materials for several screening reactions, the nitro precursors had to be reduced to the corresponding diamines in a controlled manner. Therefore, chemoselective transformations of 1, 4, 7, 13, and 16, respectively, were conducted employing sodium dithionite as reducing agent to prepare 2, 5, 8, 14, and 17 (Fig. 3). This proved to be a useful reaction procedure, since the reaction stopped at the ortho-diamine stages in reasonable yields.
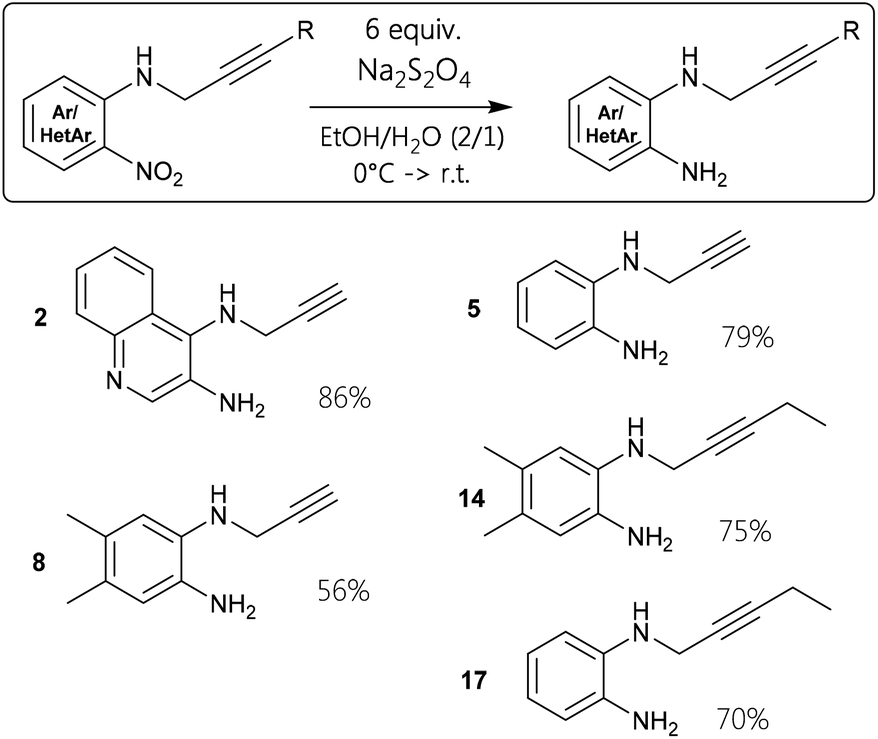 | ||
| Fig. 3 Dithionite as reducing agent to receive ortho-diamine compounds for catalytic annulation. | ||
Concerning the mediating metal species, tin(II) as well as indium(III) could be employed as catalysts (Table 1). In contrast to that, Sn(IV) salts and In(0) did not convert the substrates to the desired products. Soon we realized that no product is formed under strictly anaerobic conditions. On the other hand, atmospheric oxygen supply usually proves adequate and O2-bubbling is crucial only when it comes to syntheses in NMR tubes. It was observed that fast conversions are initiated above 70 °C, hence refluxing at 82 °C was chosen as general measure. Moreover, it turned out to be an important finding that a minimum amount of water is necessary for conversion of educts to proceed. With respect to one-pot procedures, which are not quenched in presence of an excess of water, it is noteworthy that water of crystallization from catalyst salts is sufficient for successful conversions.
Since a distinctly acidic regime is prevalent in a nitro-one-pot reaction mixture, intensely protic or Brønsted acidic conditions do not inhibit the intramolecular cyclization. In turn, our simple methodology demonstrated that product formation is promoted by Lewis acids, which result in a moderately acidic pH regime.
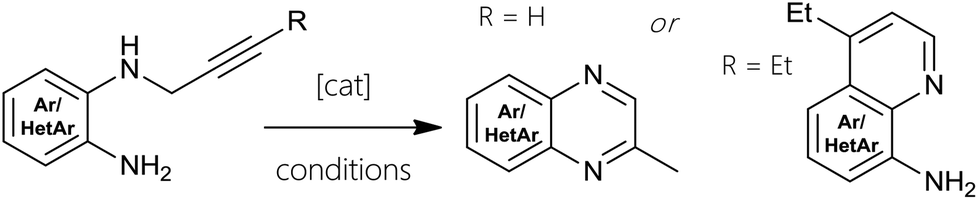
The overall tendency revealed that a well-working standard protocol consisted of refluxing the isopropanol solution of N1-β-γ-alkynyl ortho-diamine under air with stoichiometric water along with 25–50 mol% main group metal chloride. We reasoned that indium(III) is slightly more effective as catalyst than tin(II). In terms of catalyst loading, complete conversion could not be attained with less than 25 mol%. The best results were obtained with 0.5 equivalents loading, when syntheses could be completed within two to twelve hours. Decreasing the amount of SnCl2 or InCl3 would elongate reaction times up to two days. The highest yields with both catalyst systems were achieved by refluxing 2 overnight using 50 mol% metal chloride affording 3 in 91% (SnCl2·2H2O) and 93% (InCl3). Results of catalytic hydroamination and hydroarylation reactions are listed in Table 1 (also compare Fig. 1, 3 and 4).
 | ||
| Fig. 4 Sonogashira coupling for aryl–alkynes, subsequent cyclization, and literature comparison concerning In(III). | ||
A set of control experiments was conducted: we examined whether Brønsted acid could facilitate ring closure. Therefore, solutions of 5 and 8 were refluxed after the addition of tosylic acid, which only lead to the recovery of starting material both with 0.5 and 5 equivalents of TsOH·H2O, respectively. To study the behavior of the nitro-compounds, precursor 4 was reacted in combination with a non-reducing metal chloride. During both cases (catalytic and stoichiometric amounts of InCl3), no conversion of starting material 4 was observed utilizing our standard reaction protocol. The same was true for the N1-Boc-protected derivative of 5. Additionally, 2-(prop-2-yn-1-yloxy)aniline and 4-(pent-2-yn-1-yloxy)quinolin-3-amine were synthesized in order to study the probability of generating oxygen-heterocycles with our main group metal system. None of the alkynyloxy-compounds was converted to a cyclized scaffold after treatment with catalytic amounts of SnCl2·2H2O.
Further insight regarding the reactivity of the substrate-catalyst combinations could be achieved by subjecting simple N-(prop-2-yn-1-yl)-p-toluidine as well as N-(pent-2-yn-1-yl)-p-toluidine to standard catalytic conditions with both Sn(II) and In(III), respectively. After two days of stirring, the attempts with terminal alkynes just gave starting materials. On the other hand, reactions employing internal alkynes afforded small amounts of the hydroarylation product 4-ethyl-6-methylquinoline (“Sn”: 7%; “In”: 9%) besides educt.
Considering those tendencies, we have performed quantum mechanics calculations to determine the atomic charges and total charge densities (electrostatic potentials, ESPs) of compounds 5 and 17, respectively. By juxtaposing their relative charges concerning the propargylic moiety, there is a trend that the triple bond is much less polarized at the internal alkyne (Fig. S7 and S8†). In turn, the β-carbon of the terminal alkyne is clearly more electrophilic.
It is particularly remarkable that catalytic annulations of internal N1-β-γ-alkynyldiamines produce quinoline-8-amines equally exclusive as with the corresponding one-pot attempts. This indicates that the nature of the alkyne moiety decides about the type of ring closure. There does not seem to be a competition between rates of conversion for reduction vs. aromatic substitution reaction, which would have had influenced the given selectivity.
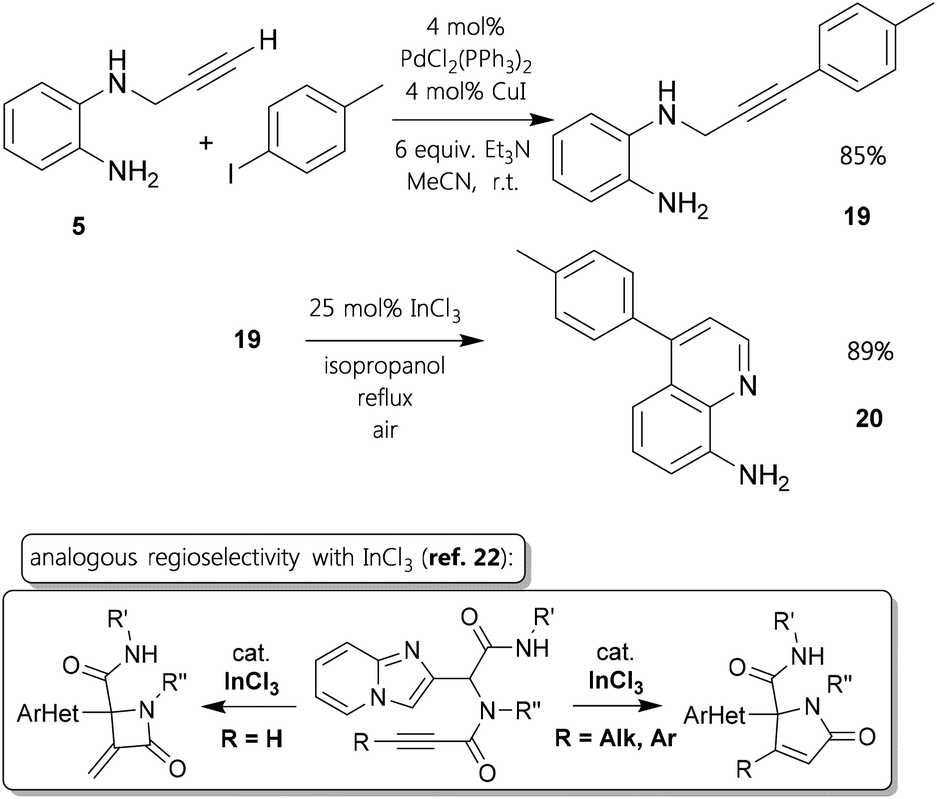
An important remaining aspect was, if aryl-substituents21 at the alkyne would favour an equal mode of ring closing as ethyl-alkynes. We did not succeed to functionalize ortho-nitro precursors like 4 employing Sonogashira cross-coupling methodology. However, we could generate that kind of internal alkynyl–amine by using the reduced compound 5. It was possible to afford derivative 19 in very good yield (Fig. 4). Subjecting it to standard conditions with 25 mol% of indium(III) chloride for 36 h, 4-p-tolylquinolin-8-amine 20 was synthesized in 89% yield. This result evidently demonstrates the tendency that in settings with ortho-diamino-scaffolds substituted triple bonds follow a hydroarylation pathway, whereas terminal alkynes undergo hydroamination. This distinction at ring closing reactions with propargylic amine-type substrates catalyzed by InCl3 was also described in a recent publication.22
As it could be confirmed that the unexpected ring closing reactions, depending on the given substrates, indeed produce either methylpyrazino- or quinoline-8-amino-compounds, the explored set of reactions can therefore be differentiated between 6-exo-dig and 6-endo-dig annulations, respectively.23 Under the given conditions, this would involve a Lewis acid (LA) complex to increase the electrophilic character of an alkyne. Metallic Lewis acids can bind weakly in a side-on manner to the triple bond or attach directly to an sp-carbon.24 Besides the more frequent reports on transition25 or main group26 metals and iodine27 to promote exo- and endo-dig-cyclizations,28 indium(III) species29 are able to promote such bond formations. The given reaction mixtures seem to be favouring intramolecular hydroaminations30 or hydroarylations.31 In order to examine mechanistic aspects of the annulation, syntheses were conducted in deuterated solvent and the mixtures analyzed by means of NMR and HR-MS.
The 1H NMR spectra of reactions in isopropanol-d8 reveal which hydrogen positions are affected during the procedure. The most striking spectroscopic difference to standard protic condition spectra is the low intensity of two signals: the aryl-singlet of the pyrazine ring as well as the methyl-peak show integrals of less than a third compared to their supposed values. This is true for every successfully tested experiment in deuterated reaction mixture, only the relative amounts of decreased peak area vary specifically with the employed metal salt type. The corresponding 13C NMR spectra exhibit split signals for those carbons, which hold primarily deuterium instead of protons. Hence, heteronuclear coupling of carbons with 2H nuclei is observable (1JC–D = 27 Hz). Furthermore, the aromatic peaks in the quinoxaline case appear to display differently overlapping multiplicities and to a minor extent reduced integrals, which can be explained by isotope exchange effects. ESI-mass spectra illustrated a range of product isomers with various deuteration grades. The influence of the alkyne-manipulation and cyclization on the initial aromatic body of the molecule can also be studied by a quinoxaline synthesis in deutero-solvent32 (Fig. 5): 1H and 13C NMR spectra indicate changes in peak patterns and intensities for the phenylene scaffold, mass spectra signify considerable amounts of isomers with more than four Ds (Fig. S2–5†). Most importantly, this experiment proves that the ortho-H position, which gives rise to the electrophilic aromatic substitution pathway, is also activated at substrates with terminal alkynes, but does not afford quinoline product in such cases.
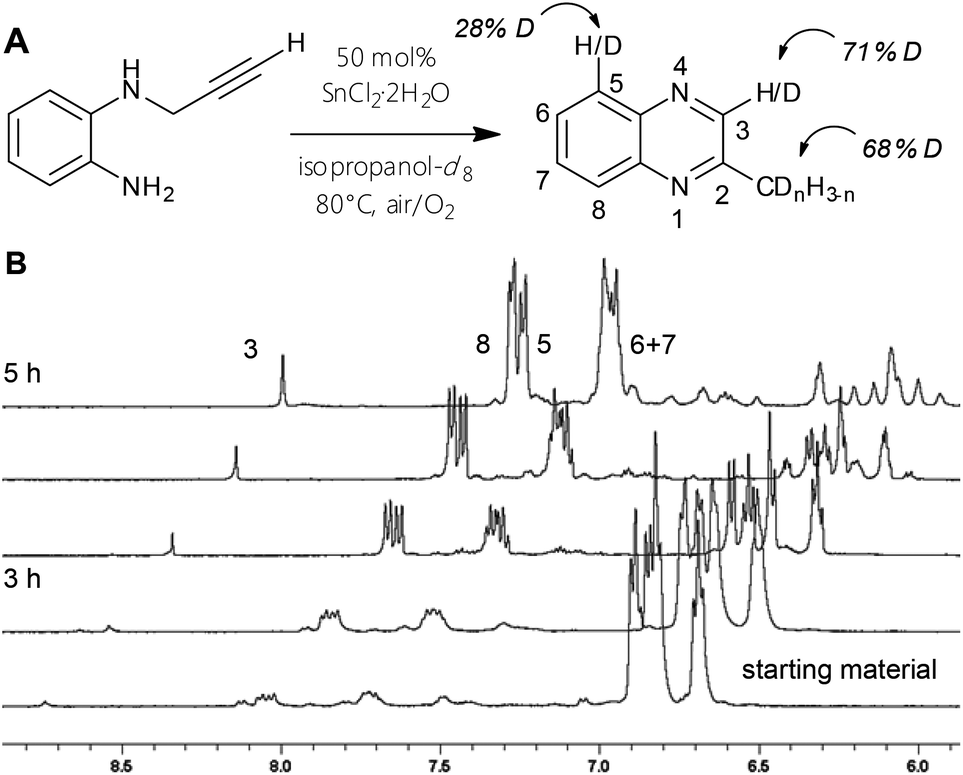 | ||
| Fig. 5 (A) Reaction of 5 to 6 to with 50 mol% SnCl2·2H2O in isopropanol-d8 to study the degree of activation on certain positions. (B) Time-course 1H NMR spectra of A). | ||
Comprehension of the ring closure might be facilitated by investigating certain screening reactions using 2 at reflux times of only 20 minutes along with catalytic amounts of Sn(II) and In(III) chlorides. After work-up, NMR spectra indicated approx. 50/50 mixtures of pyrazino-product and a species that was determined to be an intermediate. A similar observation was made during NMR-tube reactions of 14 with InCl3 (resembling the experiment depicted in Fig. 5). Considerable amounts of compound holding a CH2-group positioned between a double bond and an NH-group could be detected. HRMS and NMR measurements suggested dihydropyrazine/dihydroquinoline species (Scheme 1, structures III/VIII), which is likely to be a somehow stable structure prior to oxidation of the C–N-bond. We were not able to isolate these intermediates by column chromatography through silica, as the whole reduced species were converted into final product. The observation of those intermediates allows better understanding of the isotope distribution effects in deuterated media. A range of tautomeric equilibria might foster H/D exchange. Indium(III) and tin(II) chlorides lower the signal intensity of the methylene group (HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in products) down to values between 15–30%.
N in products) down to values between 15–30%.
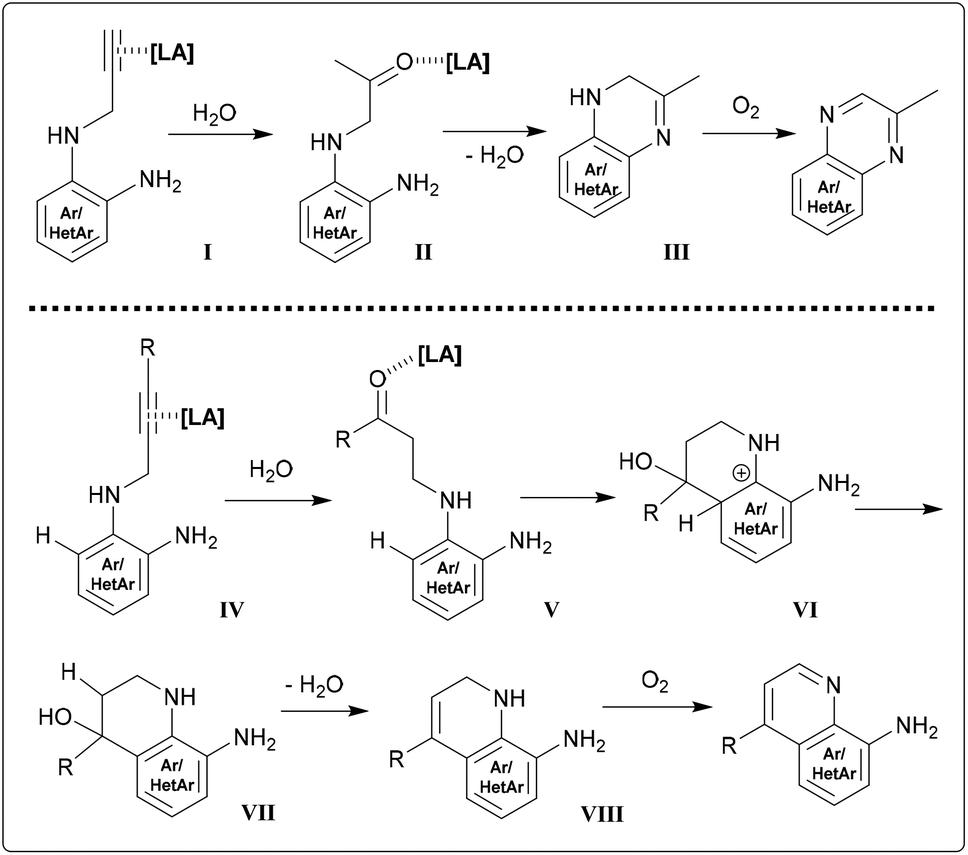 | ||
| Scheme 1 Proposed mechanisms of the annulation reactions. | ||
The need of molecular oxygen for the progress of the reaction can clearly be related to the oxidation of the 1,2-dihydro compounds (Scheme 1) to the final product. The gain of stability by reaching aromaticity explains the fast conversion of the cyclized skeleton. No evidence was found for the formation of an allene prior to ring closure.33 The prerequisite of water for the reactions to proceed might favour a hydration step prior to ring closure. Both tin and indium are known to be able to facilitate hydration reactions of alkynes to generate ketones.34 Those carbonyl intermediates could additionally be activated by Lewis acids. In a quinoxaline case, a structure like II may give rise to the condensation resulting in dihydro-compound III (Scheme 1 and Fig. S2†).
Applying this concept to a hydroarylation reaction, the hydration step (IV to V) would create the ketone at γ-position. This would then be attacked in an SEAr manner (V to VI), followed by rearomatization and water elimination (VI to VII to VIII). However, there is no evidence for such reactivity, although it would serve as credible explanation for the extensive deuteration also (Fig. S4 and S5†). For all those reasons mentioned, the course of the reactions might be covered by the sequence of steps depicted in Scheme 1.
What appears to be of special importance for the conception of a mechanism and the composition of the Lewis acid catalyst is the lack of reactivity in cases of nitro-precursors like 4 in combination with metal chlorides. The aromatic ortho-diamines are well suitable to act as bidentate ligand. As a consequence, we wish to emphasize that there could be deciding influence of a metal cation centred complex, involving an ortho-phenylene diamine type ligand,35 on mediating the cyclization steps.
Conclusions
We have demonstrated the utility of stannous chloride as well as indium powder under acidic conditions for intramolecular one-pot synthesis of either methylquinoxaline or quinolin-8-amine derivatives. Starting from ortho-nitro-N-propargylaniline compounds, selectivity is dependent on the substitution of the alkyne. It was shown that such aerobic annulation reactions are suitable to be conducted in catalytic fashion employing tin(II) and indium(III) chlorides as Lewis acids starting from ortho-diamino compounds. Those are the first examples of 6-exo-dig hydroaminations and 6-endo-dig hydroarylations with alkynylanilines, respectively, catalyzed by main group metals.Experimental part
General procedure for one-pot syntheses of quinoxalines or quinolin-8-amines
SnCl2·2H2O (5.5 equiv.) or indium powder (3 equiv. – in combination with hydrochloric acid solution (3 M; 15 equiv.)) were added to a stirred 0.15 M solution of ortho-nitro N-propargylaniline compound (1 equiv.) in 2-propanol at room temperature. The mixture was heated at reflux under air until judged completed as indicated by thin layer chromatography. The mixture was brought to room temperature, filtrated and the filtrate concentrated under reduced pressure. The residue was quenched with saturated NaHCO3 solution until pH 10. The aqueous solution was extracted with EtOAc three times, the combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (silica gel, heptanes/EtOAc = 3/1).General procedure for main group metal chloride catalyzed formation of quinoxalines or quinolin-8-amines
25–50 mol% of stannous chloride dihydrate or indium(III) chloride were added to a stirred 0.1 M solution of ortho-amino N-propargylaniline compound (1 equiv.) in 2-propanol at room temperature. Unless water of crystallization was present, 1 equiv. H2O was added. The mixture was heated at reflux under air until judged completed as indicated by thin layer chromatography. The mixture was concentrated under reduced pressure and the residue quenched with saturated NaHCO3 solution until pH 10. The aqueous solution was extracted with EtOAc three times, the combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (silica gel, heptanes/EtOAc = 3/1).General procedure for alkynylation of ortho-nitro anilines using NaH
Sodium hydride (95%, 1.1 equiv.) was added to a 0.2 M acetonitrile solution of 2-nitroaniline compound (1 equiv.). The mixture was stirred at 0 °C for 15 min. Then, 1-bromoalk-2-yne (1.1 equiv.) was added and the mixture was brought to room temperature. It was thoroughly stirred under N2 atmosphere for 40 h. The suspension was quenched with H2O and saturated NaHCO3 solution was added until pH 10. The resulting aqueous layer was extracted with DCM three times, the combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (silica gel, heptanes/EtOAc or MeOH/DCM).General procedure for the reduction of ortho-nitro N-alkynylanilines to diamines using Na2S2O4
To a stirred 0.1 M EtOH solution of 2-nitro-N-alk-2′-ynylaniline compound (1 equiv.) at 0 °C were added portions of sodium dithionite (85%, 6 equiv.) and H2O (half volume of EtOH) alternatingly. Then the mixture was allowed to warm to room temperature and stirred for 1 h. Then it was filtrated and the bulk of aqueous EtOH of the filtrate was evaporated in vacuo. Under reduced pressure, quick column chromatographic purification of the residue over a short pad of silica in a sintered glass filter was performed using a solvent gradient from DCM/MeOH (10/1) to pure MeOH. The polar fractions were collected and concentrated in vacuo.![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 2921, 2853, 1458, 1376, 951, 773 cm−1; HRMS (ESI): m/z calcd for C12H9N3: 196.0869 [M + H]+; found: 196.0870.
= 3194, 2112, 1519, 1489, 1439, 1276, 1198, 1151, 1034, 787 cm−1; HRMS (ESI): m/z calcd for C12H12N2O: 201.1022 [M + H]+; found: 201.1027.
= 2922, 2853, 1519, 1459, 1325, 1260, 1013, 797 cm−1; HRMS (ESI): m/z calcd for C13H16N2: 201.1386 [M + H]+; found: 201.1386.
= 3334, 3042, 1573, 1506, 1455, 1315, 1269, 738 cm−1; HRMS (ESI): m/z calcd for C11H14N2: 175.1230 [M + H]+; found: 175.1232.
= 3334, 2920, 1597, 1508, 1450, 1271, 816, 740 cm−1; HRMS (ESI): m/z calcd for C16H16N2: 237.1386 [M + H]+; found: 237.1384.
= 3447, 3319, 2360, 1618, 1502, 1358, 1282, 818, 761 cm−1; HRMS (ESI): m/z calcd for C16H14N2: 235.1230 [M + H]+; found: 235.1230.
= 2921, 2853, 1458, 1376, 951, 773 cm−1; HRMS (ESI): m/z calcd for C12H9N3: 196.0869 [M + H]+; found: 196.0870.
= 3194, 2112, 1519, 1489, 1439, 1276, 1198, 1151, 1034, 787 cm−1; HRMS (ESI): m/z calcd for C12H12N2O: 201.1022 [M + H]+; found: 201.1027.
= 2922, 2853, 1519, 1459, 1325, 1260, 1013, 797 cm−1; HRMS (ESI): m/z calcd for C13H16N2: 201.1386 [M + H]+; found: 201.1386.
= 3334, 3042, 1573, 1506, 1455, 1315, 1269, 738 cm−1; HRMS (ESI): m/z calcd for C11H14N2: 175.1230 [M + H]+; found: 175.1232.
= 3334, 2920, 1597, 1508, 1450, 1271, 816, 740 cm−1; HRMS (ESI): m/z calcd for C16H16N2: 237.1386 [M + H]+; found: 237.1384.
= 3447, 3319, 2360, 1618, 1502, 1358, 1282, 818, 761 cm−1; HRMS (ESI): m/z calcd for C16H14N2: 235.1230 [M + H]+; found: 235.1230.
Acknowledgements
WS kindly acknowledges the financial support of the project 836532 (“Prä-klinische Entwicklung einer Off-the-Shelf indvidualisierten Krebsimmuntherapie”) by the Austrian Research Promotion Agency (FFG). The NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”).Notes and references
- (a) I. J. Krems and P. E. Spoerri, Chem. Rev., 1947, 40, 279–358 CrossRef CAS; (b) W. Kaim, Angew. Chem., 1983, 95, 201–221 CrossRef CAS PubMed.
- (a) R. Müller and S. Rappert, Appl. Microbiol. Biotechnol., 2010, 85, 1315–1320 CrossRef PubMed; (b) P. Sjödin, H. Wallin, J. Alexander and M. Jägerstad, Carcinogenesis, 1989, 10, 1269–1275 CrossRef PubMed.
- (a) V. P. Mehta, A. Sharma, K. van Hecke, L. van Meervelt and E. V. Van der Eycken, J. Org. Chem., 2008, 73, 2382–2388 CrossRef CAS PubMed; (b) S. G. Modha, J. C. Trivedi, V. P. Mehta, D. S. Ermolat'ev and E. V. Van der Eycken, J. Org. Chem., 2011, 76, 846–856 CrossRef CAS PubMed; (c) A. Zaid, J.-S. Sun, C.-H. Nguyen, E. Bisagni, T. Garestier, D. S. Grierson and R. Zain, ChemBioChem, 2004, 5, 1550–1557 CrossRef CAS PubMed.
- (a) S. B. Ferreira and C. R. Kaiser, Expert Opin. Ther. Patents, 2012, 22, 1033–1051 CrossRef CAS PubMed; (b) J. Guillon, I. Forfar, M. Mamani-Matsuda, V. Desplat, M. Saliège, D. Thiolat, S. Massip, A. Tabourier, J.-M. Léger, B. Dufaure, G. Haumont, C. Jarry and D. Mossalayi, Bioorg. Med. Chem., 2007, 15, 194–210 CrossRef CAS PubMed.
- (a) F. Zhang, Y. Xi, Y. Lu, L. Wang, L. Liu, J. Li and Y. Zhao, Chem. Commun., 2014, 50, 5771–5773 RSC; (b) H. A. Oskooie, M. M. Heravi, K. Bakhtiari and S. Taheri, Monatsh. Chem. Chem. Mon., 2007, 138, 875–877 CrossRef CAS; (c) N. Xekoukoulotakis, C. Hadjiantoniou-Maroulis and A. Maroulis, Tetrahedron Lett., 2000, 41, 10299–10302 CrossRef CAS; (d) C. Qi, H. Jiang, L. Huang, Z. Chen and H. Chen, Synthesis, 2011, 387–396 CAS; (e) P. Ghosh and A. Mandal, Green Chem. Lett. Rev., 2012, 5, 127–134 CrossRef CAS PubMed.
- (a) F. Buron, A. Turck, N. Plé, L. Bischoff and F. Marsais, Tetrahedron Lett., 2007, 48, 4327–4330 CrossRef CAS PubMed; (b) G. Liu, G. Xu, J. Li, D. Ding and J. Sun, Org. Biomol. Chem., 2014, 12, 1387–1390 RSC; (c) Z. Chen, D. Ye, G. Xu, M. Ye and L. Liu, Org. Biomol. Chem., 2013, 11, 6699–6702 RSC.
- (a) C. Venkatesh, B. Singh, P. K. Mahata, H. Ila and H. Junjappa, Org. Lett., 2005, 7, 2169–2172 CrossRef CAS PubMed; (b) Z.-J. Yang, C.-Z. Liu, B.-L. Hu, C.-L. Deng and X.-G. Zhang, Chem. Commun., 2014, 50, 14554–14557 RSC.
- (a) F. Chen, X. Huang, X. Li, T. Shen, M. Zou and N. Jiao, Angew. Chem., Int. Ed., 2014, 126, 10663–10667 CrossRef PubMed; (b) K. K. D. Rao Viswanadham, M. Prathap Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517–13520 RSC.
- (a) D. D. Vachhani, V. P. Mehta, S. G. Modha, K. van Hecke, L. VanMeervelt and E. V. Van der Eycken, Adv. Synth. Catal., 2012, 354, 1593–1599 CrossRef CAS PubMed; (b) Q. Yang, T. Xu and Z. Yu, Org. Lett., 2014, 16, 6310–6313 CrossRef CAS PubMed; (c) D. D. Vachhani, S. G. Modha, A. Sharma and E. V. Van der Eycken, Tetrahedron, 2013, 69, 359–365 CrossRef CAS PubMed; (d) S. Jalal, K. Bera, S. Sarkar, K. Paul and U. Jana, Org. Biomol. Chem., 2014, 12, 1759–1770 RSC.
- (a) B. M. Monks, E. R. Fruchey and S. P. Cook, Angew. Chem., Int. Ed., 2014, 53, 11065–11069 CrossRef CAS PubMed; (b) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- C. E. Meyet and C. H. Larsen, J. Org. Chem., 2014, 79, 9835–9841 CrossRef CAS PubMed.
- A. Shi, T. A. Nguyen, S. K. Battina, S. Rana, D. J. Takemoto, P. K. Chiang and D. H. Hua, Bioorg. Med. Chem. Lett., 2008, 18, 3364–3368 CrossRef CAS PubMed.
- (a) M. Rehan, G. Hazra and P. Ghorai, Org. Lett., 2015, 17, 1668–1671 CrossRef CAS PubMed; (b) X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763–766 CrossRef CAS PubMed; (c) G. A. Salman, R. U. Nisa, V. O. Iaroshenko, J. Iqbal, K. Ayub and P. Langer, Org. Biomol. Chem., 2012, 10, 9464–9473 RSC.
- (a) G. S. Welmaker, J. A. Nelson, J. E. Sabalski, A. L. Sabb, J. R. Potoski, D. Graziano, M. Kagan, J. Coupet, J. Dunlop, H. Mazandarani, S. Rosenzweig-Lipson, S. Sukoff and Y. Zhang, Bioorg. Med. Chem. Lett., 2000, 10, 1991–1994 CrossRef CAS; (b) M. de F. Pereira and V. Thiéry, Org. Lett., 2012, 14, 4754–4757 CrossRef CAS PubMed.
- (a) N. Rajesh, R. Sarma and D. Prajapati, Synlett, 2014, 1448–1452 Search PubMed; (b) K. Surendra, W. Qiu and E. J. Corey, J. Am. Chem. Soc., 2011, 133, 9724–9726 CrossRef CAS PubMed; (c) Y. Zhang, P. Li, M. Wang and L. Wang, J. Org. Chem., 2009, 74, 4364–4367 CrossRef CAS PubMed.
- K. Gilmore and I. V. Alabugin, Chem. Rev., 2011, 111, 6513–6556 CrossRef CAS PubMed.
- (a) T. S. Symeonidis, I. N. Lykakis and K. E. Litinas, Tetrahedron, 2013, 69, 4612–4616 CrossRef CAS PubMed; (b) S. Basceken and M. Balci, J. Org. Chem., 2015, 80, 3806–3814 CrossRef CAS PubMed; (c) B. Alcaide, P. Almendros, J. M. Alonso, I. Fernández, G. Gómez-Campillos and M. R. Torres, Chem. Commun., 2014, 50, 4567–4570 RSC.
- (a) J. Barluenga, F. Aznar, R. Liz and M.-P. Cabal, Synthesis, 1985, 313–314 CrossRef CAS; (b) R. Mukhopadhyay and N. G. Kundu, Tetrahedron Lett., 2000, 41, 9927–9930 CrossRef CAS; (c) S. Knapp, J. Ziv and J. D. Rosen, Tetrahedron, 1989, 45, 1293–1298 CrossRef CAS.
- (a) H. Murase, K. Senda, M. Senoo, T. Hata and H. Urabe, Chem. – Eur. J., 2014, 20, 317–322 CrossRef CAS PubMed; (b) K. C. Majumdar, R. Kumar Nandi, S. Ganai and A. Taher, Synlett, 2011, 116–120 CrossRef CAS; (c) B. Roy, I. Ansary, S. Samanta and K. C. Majumdar, Tetrahedron Lett., 2012, 53, 5119–5122 CrossRef CAS PubMed.
- (a) Y. Kwon, H. Cho and S. Kim, Org. Lett., 2013, 15, 920–923 CrossRef CAS PubMed; (b) L. Alonso-Marañón, M. M. Martínez, L. A. Sarandeses and J. P. Sestelo, Org. Biomol. Chem., 2015, 13, 379–387 RSC.
- X. Zhang, T. Yao, M. A. Campo and R. C. Larock, Tetrahedron, 2010, 66, 1177 CrossRef CAS PubMed.
- Z. Li, U. K. Sharma, Z. Liu, N. Sharma, J. N. Harvey and E. V. Van der Eycken, Eur. J. Org. Chem., 2015, 3957–3962 CrossRef CAS PubMed.
- (a) M. J. Gainer, N. R. Bennett, Y. Takahashi and R. E. Looper, Angew. Chem., Int. Ed., 2011, 123, 710–713 CrossRef PubMed; (b) C. Shu, L. Li, C.-B. Chen, H.-C. Shen and L.-W. Ye, Chem. – Asian J., 2014, 9, 1525–1529 CrossRef CAS PubMed; (c) N. T. Patil, A. Nijamudheen and A. Datta, J. Org. Chem., 2012, 77, 6179–6185 CrossRef CAS PubMed.
- L. Li, G. Huang, Z. Chen, W. Liu, X. Wang, Y. Chen, L. Yang, W. Li and Y. Li, Eur. J. Org. Chem., 2012, 5564–5572 CrossRef CAS PubMed.
- (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–704 CrossRef PubMed; (b) D. Y. Li, H. J. Chen and P. N. Liu, Org. Lett., 2014, 16, 6176–6179 CrossRef CAS PubMed.
- F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104–114 RSC.
- N. M. Mishra, D. D. Vachhani, S. G. Modha and E. V. Van der Eycken, Eur. J. Org. Chem., 2013, 693–700 CrossRef CAS PubMed.
- (a) A. Kumar, Z. Li, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Chem. Commun., 2013, 49, 6803–6805 RSC; (b) J. M. French and S. T. Diver, J. Org. Chem., 2014, 79, 5569–5585 CrossRef CAS PubMed; (c) A.-L. Girard, T. Enomoto, S. Yokouchi, C. Tsukano and Y. Takemoto, Chem. – Asian J., 2011, 6, 1321–1324 CrossRef CAS PubMed.
- (a) R. Sarma and D. Prajapati, Chem. Commun., 2011, 47, 9525–9527 RSC; (b) J. Augé, N. Lubin-Germain and J. Uziel, Synthesis, 2007, 1739–1764 CrossRef.
- (a) K. Okuma, J.-i. Seto, K.-i. Sakaguchi, S. Ozaki, N. Nagahora and K. Shioji, Tetrahedron Lett., 2009, 50, 2943–2945 CrossRef CAS PubMed; (b) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407–1420 RSC; (c) A. Ranjan, R. Yerande, P. B. Wakchaure, S. G. Yerande and D. H. Dethe, Org. Lett., 2014, 16, 5788–5791 CrossRef CAS PubMed.
- (a) X.-Y. Liu, P. Ding, J.-S. Huang and C.-M. Che, Org. Lett., 2007, 9, 2645–2648 CrossRef CAS PubMed; (b) B. Michelet, J.-R. Colard-Itté, G. Thiery, R. Guillot, C. Bour and V. Gandon, Chem. Commun., 2015, 51, 7401–7404 RSC.
- (a) N. Yoshikai, S. Asako, T. Yamakawa, L. Ilies and E. Nakamura, Chem. – Asian J., 2011, 6, 3059–3065 CrossRef CAS PubMed; (b) W. Xie, J. Yang, B. Wang and B. Li, J. Org. Chem., 2014, 79, 8278–8287 CrossRef CAS PubMed.
- (a) A. Saito, S. Oda, H. Fukaya and Y. Hanzawa, J. Org. Chem., 2009, 74, 1517–1524 CrossRef CAS PubMed; (b) J. K. Vandavasi, W.-P. Hu, G. C. Senadi, S. S. K. Boominathan, H.-Y. Chen and J.-J. Wang, Eur. J. Org. Chem., 2014, 6219–6226 CrossRef CAS PubMed.
- (a) X. Jin, T. Oishi, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2011, 17, 1261–1267 CrossRef CAS PubMed; (b) E. Bosch and L. Jeffries, Tetrahedron Lett., 2001, 42, 8141–8142 CrossRef CAS; (c) M. B. T. Thuong, A. Mann and A. Wagner, Chem. Commun., 2012, 48, 434–436 RSC; (d) L. Hintermann and A. Labonne, Synthesis, 2007, 1121–1150 CrossRef CAS; (e) S. Liang, G. B. Hammond and B. Xu, Chem. Commun., 2015, 51, 903–906 RSC.
- M. Normand, V. Dorcet, E. Kirillov and J.-F. Carpentier, Organometallics, 2013, 32, 1694–1709 CrossRef CAS.
- S. Grivas, W. Tian, S. V. Meshkov, H. Lönnberg, M. Dahlqvist, M. M. Kady and S. B. Christensen, Acta Chem. Scand., 1992, 46, 1109–1113 CrossRef CAS.
- Y. Ichikawa, T. Nishimura and T. Hayashi, Organometallics, 2011, 30, 2342–2348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01532d |
| This journal is © The Royal Society of Chemistry 2015 |