
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total syntheses of natural products containing spirocarbocycles
Laura K.
Smith
* and
Ian R.
Baxendale
Department of Chemistry, Durham University, Stockton Road, Durham, DH1 3LE, UK. E-mail: l.k.smith@dur.ac.uk; Tel: +44 (0)191 334 2042
First published on 1st September 2015
Abstract
The structures of natural products from a variety of sources contain spirocycles, two rings that share a common atom. The spiro motif is finding increasing inclusion in drug candidates, and as a structural component in several promising classes of chiral ligands used in asymmetric synthesis. Total syntheses of products containing all-carbon spirocycles feature several common methods of ring closure which we examine in this review.
 Laura K. Smith | Laura K. Smith, PhD student, Department of Chemistry, Durham University, Stockton Road, Durham. DH1 3LE. Laura Smith attended Durham University, graduating in 2013 with an M. Chem (Hons) in Chemistry. Her fourth-year undergraduate research project was completed at Durham under the supervision of Prof. Patrick G. Steel on the subject of C–H activation and iridium-catalysed borylation of amino acids and peptides. She is currently reading for a PhD under the supervision of Prof. Ian R. Baxendale in the areas of asymmetric synthesis and catalysis. |
 Ian R. Baxendale | Ian R. Baxendale, Professor of Synthetic Chemistry, Department of Chemistry, Durham University, Stockton Road, Durham. DH1 3LE. Ian obtained his PhD from the University of Leicester (Prof. Pavel Kocovsky), before conducting postdoctoral studies at University of Cambridge (Prof. Steven V. Ley). In 2002 he was elected a Fellow and Dean of Sidney Sussex College and became the director of Natural Science teaching. In 2008 he was promoted to Senior Research Associate at Cambridge and in 2009 was awarded a Royal Society University Research Fellowship. In 2012 he moved to his current position as the Chair of Synthetic Chemistry at Durham. His research focuses on new enabling technologies including flow synthesis, automation and immobilised reagents and scavengers. |
1. Introduction
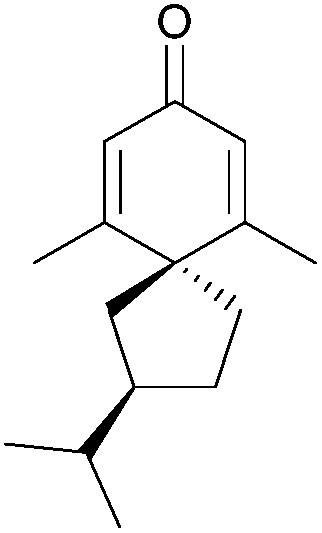
Natural products have been an invaluable source of medicines and other resources throughout history. In recent years, the search for novel compounds has moved from the land to the sea, concentrating on marine organisms rather than terrestrial species. There are several ways of classifying such compounds: by genus of their source, by physiological effect, or by a common structural element such as their carbon skeleton. This review will focus on natural products containing the spiro motif. Spiro compounds possess two or more rings which share only one common atom, with the rings not linked by a bridge.1Spiro-containing compounds have been isolated from a wide range of biological sources, from plants to frogs to marine sponges. The shift from earth to water in search of compounds is mirrored here. Selected examples of such compounds are shown below (Fig. 1). Spirocycles containing heteroatoms, such as the spirooxindoles, are well-known in nature, and there have been several reviews on these species.2,3 One of the earliest isolated spiro natural products was β-vetivone, extracted from vetiver oil in 1939 by Pfau and Plattner.4 However, for many years its structure was believed to be hydroazulenic (1.6) rather than a spiro[4.5]decane (1.7), an error which was only rectified by its total synthesis by Marshall et al. in 1968.5,6
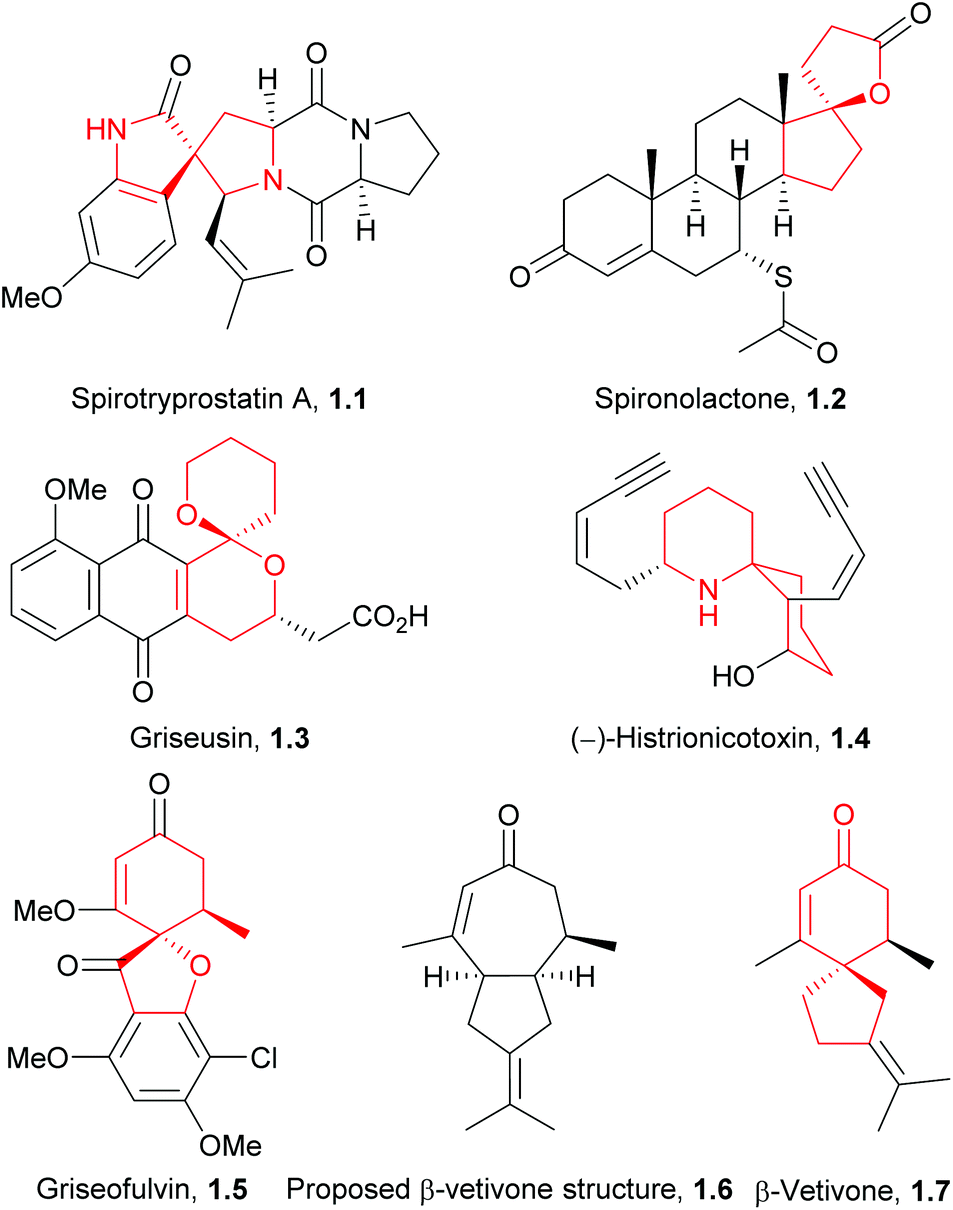 | ||
| Fig. 1 Natural products and drugs containing spirocycles. | ||
The spiro motif is becoming more prevalent as a template in drug discovery, and has been the subject of a recent review.7 Spiro rings of different sizes convey both increased three-dimensionality for potential improved activity, and novelty for patenting purposes. Two examples of marketed drugs containing spirocycles are spironolactone (1.2) and griseofulvin (1.5, Fig. 1). Both are on the World Health Organisation's list of essential medicines; spironolactone is a diuretic and anti-hypertensive drug, whilst griseofulvin is an anti-fungal agent.
An additional important application of spiro compounds is in asymmetric synthesis. Enantioselectivity in organic reactions can be achieved through the use of chiral ligands, such as BINOL (1.8) or cinchona alkaloids (1.9, Fig. 2). Some spirocycles, such as the spirobiindane SPINOL (1.10), belong to a class of privileged chiral ligands which show particular promise.8 The increased rigidity and restricted rotation at the spiro centre can assist in improving enantiomeric excess in asymmetric reactions.
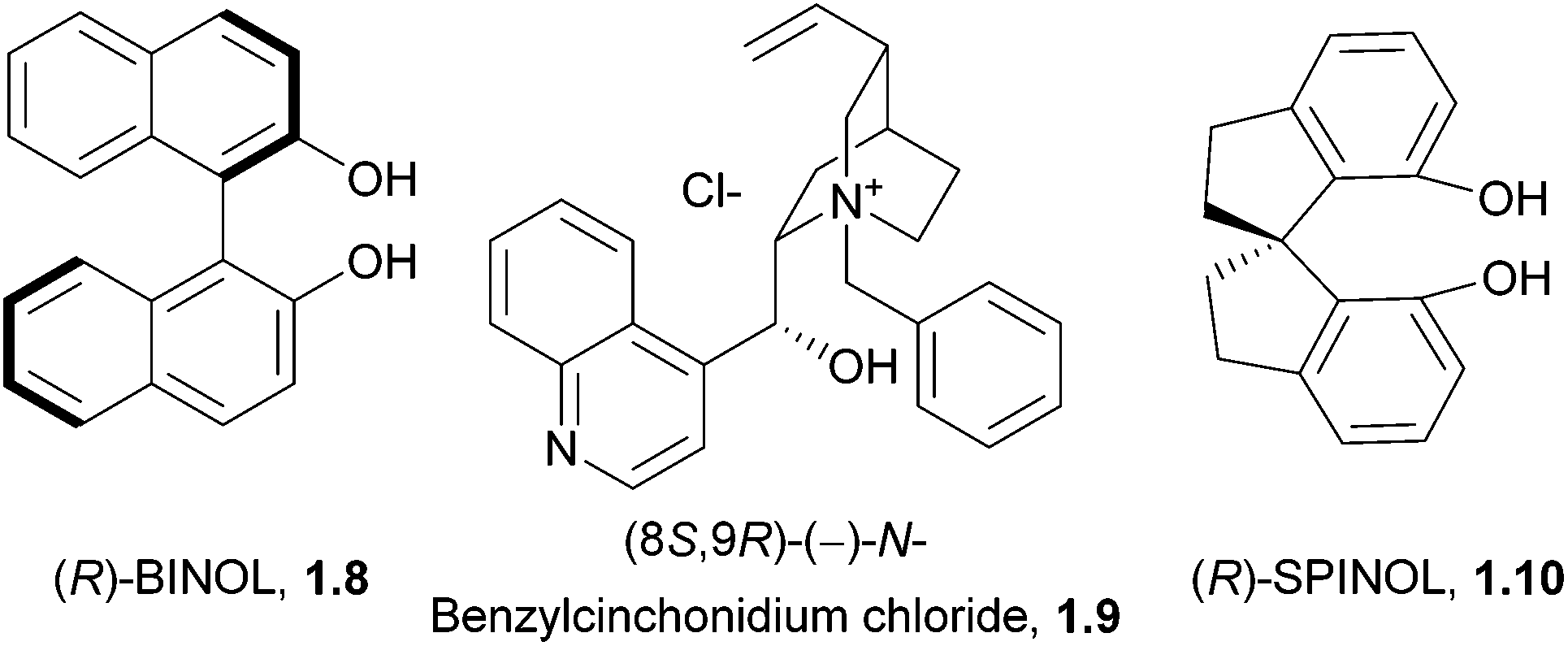 | ||
| Fig. 2 Selected privileged ligand classes. | ||
The asymmetric synthesis of quaternary carbon centres is extremely challenging,9 with the stereocontrolled preparation of products containing such spiroatoms10–12 conveying additional difficulty. Enantioselective syntheses of spirocyclic ligands utilise several construction methods, such as alkylation, radical cyclisation, rearrangements, cycloaddition reactions, cleavage of bridged ring systems and the use of transition metal catalysts.13 Several ring-closing strategies are common in the synthesis of spirocarbocycle-containing natural products, and these will be discussed in this review. Examination of the syntheses of these compounds will be first by reaction type of the spirocycle-forming step, followed by increasing ring size, from [2.5] through to [5.5] (Table 1). Not all ring sizes are represented in the natural world, nor have all spiro-containing natural products been the target of organic synthesis. Some reaction types have not been included in this article, for example the Wittig reaction.
| Natural product | Structure | Spiro | Section | Natural product | Structure | Spiro | Section |
|---|---|---|---|---|---|---|---|
| Illudin M |

|
[2.5] | 4 | Anhydro-β-rotunol (2005) |
|
[4.5] | 2 |
| Illudin S |
|
[2.5] | 4 | Axenol (2002, 2007) |

|
[4.5] | 4, 6 |
| (−)-Acutumine (synthesised in 2009) |

|
[4.4] | 7 | Axisonitrile-3 (2009) |

|
[4.5] | 6 |
| Ainsliadimer A (2010) |
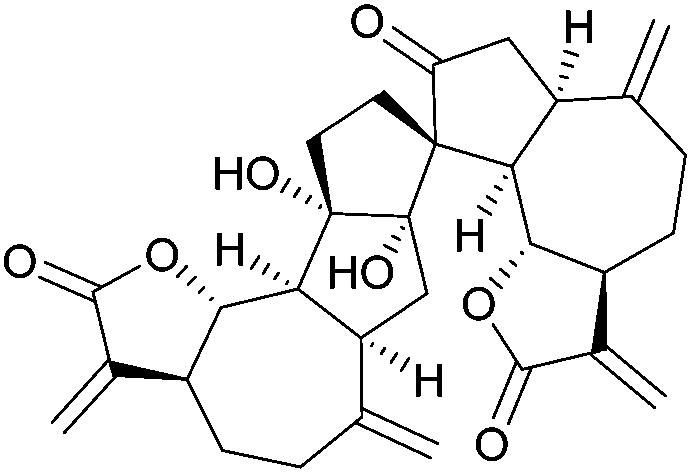
|
[4.4] | 5 | Bilinderone (2011) |
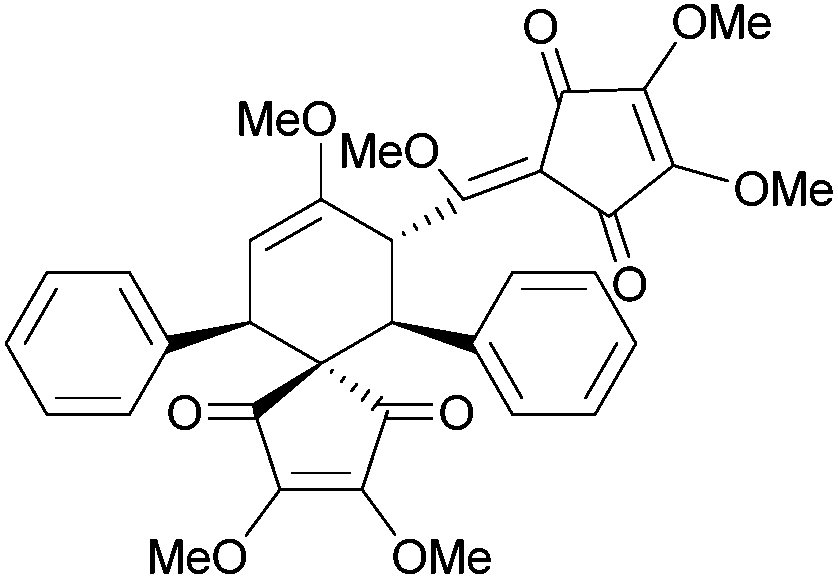
|
[4.5] | 7 |
| Vannusal A (R = Ac) |
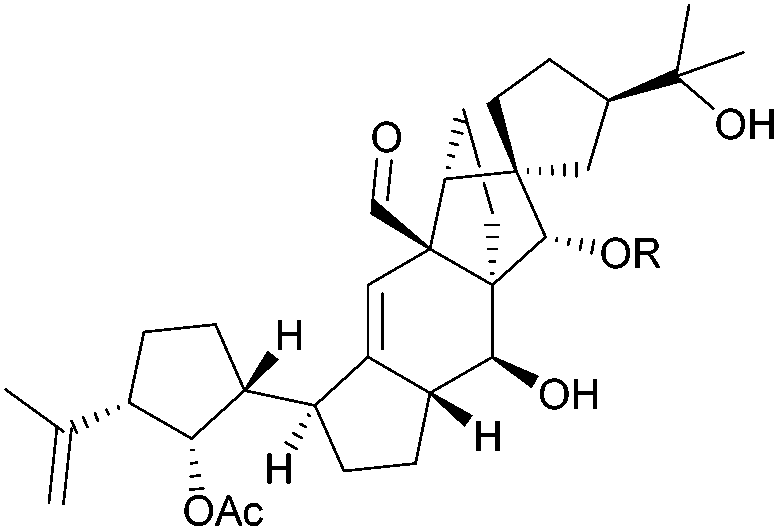
|
[4.4] | 2 | Cannabispirenone A: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, R1 = Me, R2 = H; B: CC, R1 = H, R2 = Me C, R1 = Me, R2 = H; B: CC, R1 = H, R2 = Me |

|
[4.5] | 2 |
| Vannusal B (R = H) | |||||||
| (2009) | |||||||
| Cannabispirone (C–C, R1 = Me, R2 = H) | |||||||
| (1981, 1982, 1984) | |||||||
| Acorenone (1978, 1982, 2011) |

|
[4.5] | 3, 4, 7 | Colletoic acid (2013) |

|
[4.5] | 5 |
| Acorenone B (1975, 1978) |
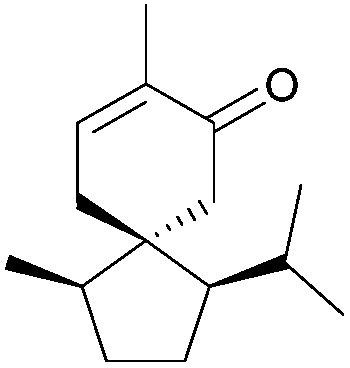
|
[4.5] | 2, 3 | Erythrodiene (1997, 1998) |
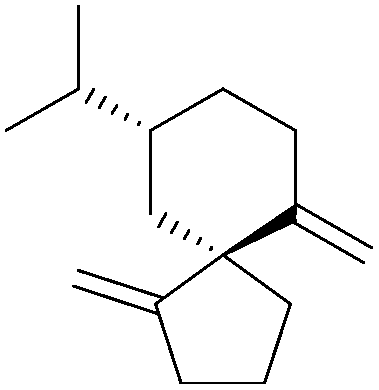
|
[4.5] | 2, 6 |
| Acorone (1975, 1977, 1978, 1996, 2007) |
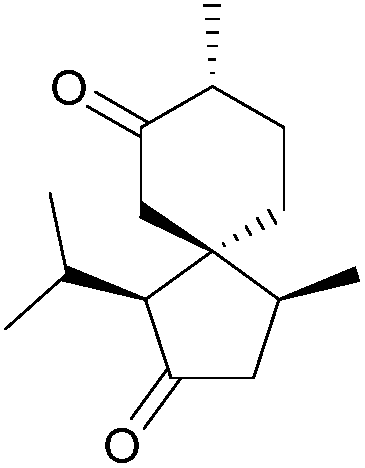
|
[4.5] | 2, 4, 5, 6 | Gleenol (2002, 2007, 2009) |
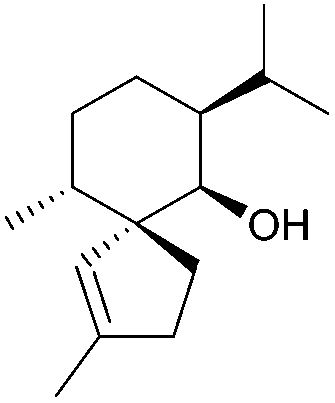
|
[4.5] | 4, 6 |
| Agarospirol (1970, 1975, 1980, 1995, 2006) |

|
[4.5] | 2, 3, 6, 7 | Gochnatiolide A (R = α-OH) |
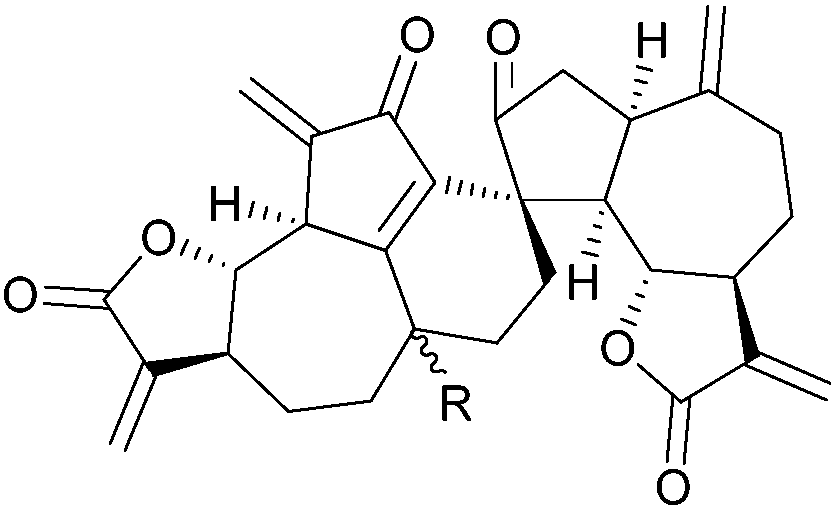
|
[4.5] | 5 |
| Gochnatiolide B (R = β-OH) | |||||||
| Gochnatiolide C (R = α-H) | |||||||
| (2013) | |||||||
| Ainsliadimer B (2013) |

|
[4.5] | 5 | Hinesene (2R,5S,10S) (2004) |
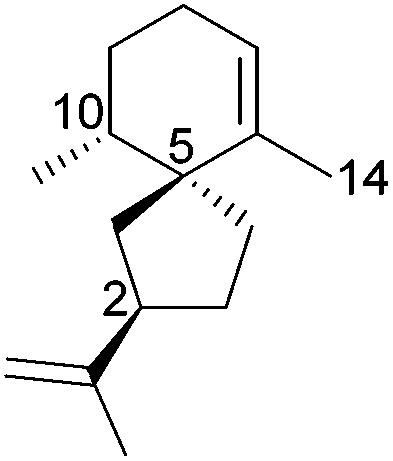
|
[4.5] | 3 |
| Hinesol (1970, 1973, 1980, 1995, 2004, 2006, 2007) |

|
[4.5] | 3, 6, 7 | Spirojatamol (1997, 1998) |
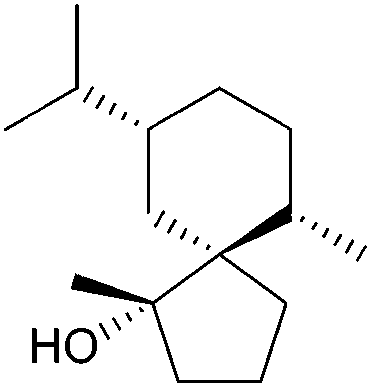
|
[4.5] | 2, 6 |
| Isoacorone (1975, 2000, 2007, 2008) |

|
[4.5] | 2, 4, 5, 6 | Spirolaurenone (1982) |

|
[4.5] | 3 |
| Lubimin (1984) |

|
[4.5] | 3 | Trichoacorenol (2011) |

|
[4.5] | 4 |
| Lubiminol (1996, 1998) |

|
[4.5] | 7 | (+)-Vitrenal (1982, 1986) |
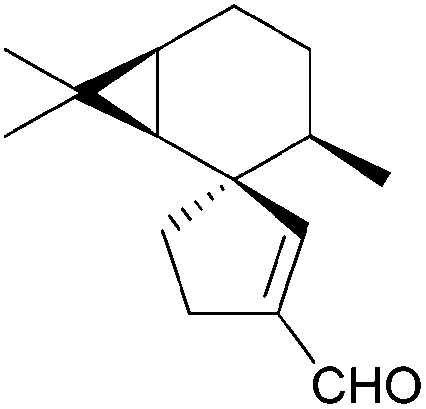
|
[4.5] | 2, 3 |
| Oxylubimin (1984) |
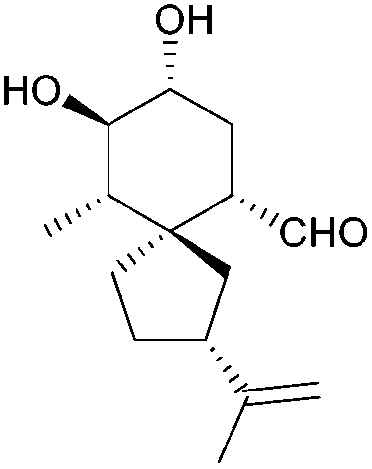
|
[4.5] | 3 | α-Acoradiene: |

|
[4.5] | 2, 4, 7 |
| L (1R,4S,5S) | |||||||
| R (1S,4R,5R) | |||||||
| (1983, 2001, 2004) | |||||||
| Premnaspirodiene (2R,5S,10R) (2004) |

|
[4.5] | 3 | α-Acorenol (2007) |

|
[4.5] | 4, 6 |
| Proaporphines: |

|
[4.5] | 7 | α-Vetispirene (1973, 1976, 1985, 2004, 2006, 2007) |

|
[4.5] | 2, 3, 6, 7 |
| Pronuciferine (R1 = Me, R2 = H) | |||||||
| Orientalinone (R1 = Me, R2 = OMe) | |||||||
| Kreysiginone (R1 = H, R2 = OMe) | |||||||
| (1971, 1972, 1978) | |||||||
| Sequosempervirin A (2007) |

|
[4.5] | 4 | β-Acoradiene (1975) |
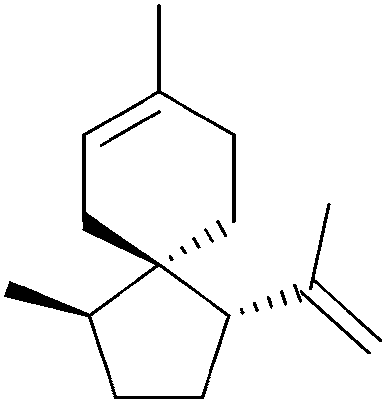
|
[4.5] | 5 |
| Solavetivone (1984) |
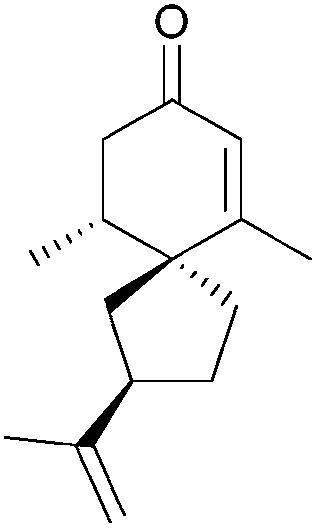
|
[4.5] | 3 | β-Acorenol (2007) |

|
[4.5] | 4, 6 |
| Spirocurcasone (2013, 2014) |

|
[4.5] | 3, 4 | β-Vetispirene (1973) |
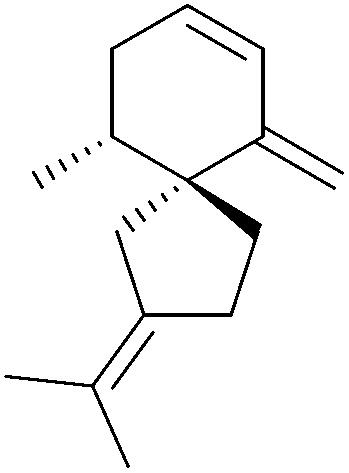
|
[4.5] | 3 |
| β-Vetivone (1968, 1973, 1984, 2006, 2007) |
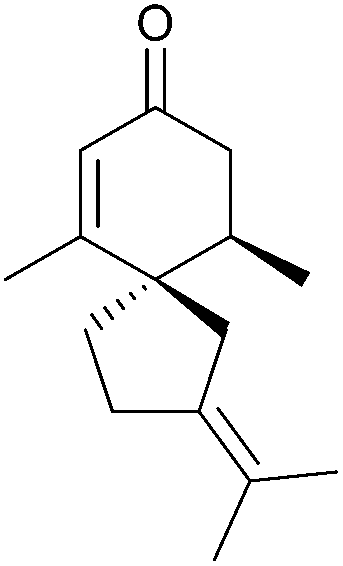
|
[4.5] | 2, 3, 6, 7 | Laurencenone B (2008) |

|
[5.5] | 4 |
| γ-Acoradiene (1973) |
|
[4.5] | 5 | Laurencenone C (2010) |
|
[5.5] | 2, 4 |
| δ-Acoradiene (1973) |
|
[4.5] | 5 | Majusculone (2007, 2011) |
|
[5.5] | 2, 3 |
| Linderaspirone A (2011) |
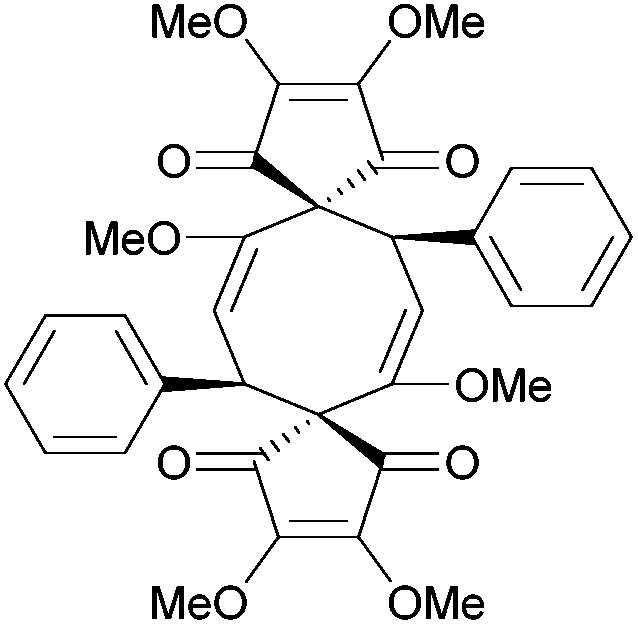
|
[4.3.4.3] | 7 | α-Chamigrene (1991, 2006) |
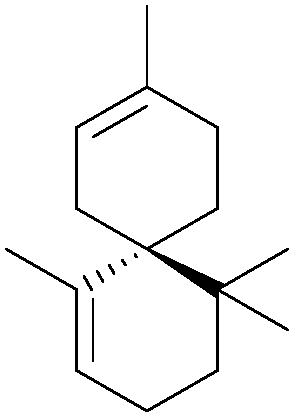
|
[5.5] | 3, 4 |
| Elatol (2008) |

|
[5.5] | 4 | β-Chamigrene (2006, 2008) |

|
[5.5] | 4 |
Some of the earliest-discovered and best-studied compounds such as the aforementioned β-vetivone have been synthesised by several of these methods. The field has matured significantly since the 1970s, when many of the structures of these spiro compounds were elucidated. Syntheses of these compounds span the last fifty years, and so not all of the original articles were published in English, but where relevant, have been translated and summarised here. In general, newer spirocyclisation methods tend to be both more efficient and are conducted enantioselectively. For example, Diels–Alder spirocyclisations of the acoranes were first attempted in 1975 and re-examined in 2013, where one isomer was selectively synthesised rather than the full range of four isomers (Scheme 1).
 | ||
| Scheme 1 Comparing the synthesis of acoranes by the Diels–Alder reaction. | ||
Reports from the last five years indicate that new spiro natural products are still being isolated and synthesised. Total syntheses of some compounds isolated many years ago were achieved using the less sophisticated methodologies available at the time, but there has been much improvement of these older methods by the application of more modern processing techniques and understanding. It may also be beneficial to apply some of the approaches recently developed in the enantioselective synthesis of chiral ligands to the synthesis of natural products. Several new natural product compounds have been recently identified that would benefit from an efficient preparative synthesis. Cyanthiwigin AC (Fig. 3), isolated from the marine sponge Myrmekioderma styx,14 is such an example. The cyanthiwigins exhibit a broad range of biological activity, including inhibition of human immunodeficiency virus and Mycobacterium tuberculosis, and cytotoxicity against human primary tumour cells.15 The first and only total synthesis of (+)-cyanthiwigin AC was achieved in 13 steps and 2% overall yield.16 Other compounds have also shown interesting preliminary biological activity ranging from anti-bacterial to anti-fouling activity but further investigation is hampered by the inability to isolate or synthesise sufficient quantities. The ritterazines are such a class of compounds: they are a family of 26 steroidal alkaloids of which 11 contain a spirocarbocycle in addition to the spiroketal shared by all ritterazines.17 They possess potent cytotoxic activity against apoptosis-resistant malignant cell lines but research has been limited by the small amounts of material successfully isolated from naturally occurring sources, so efforts have focussed on their total synthesis. There have been several published syntheses of the other 15 ritterazines and derivatives, but at the time of writing there has not yet been a published total synthesis of the spirocarbocycle-containing ritterazines.
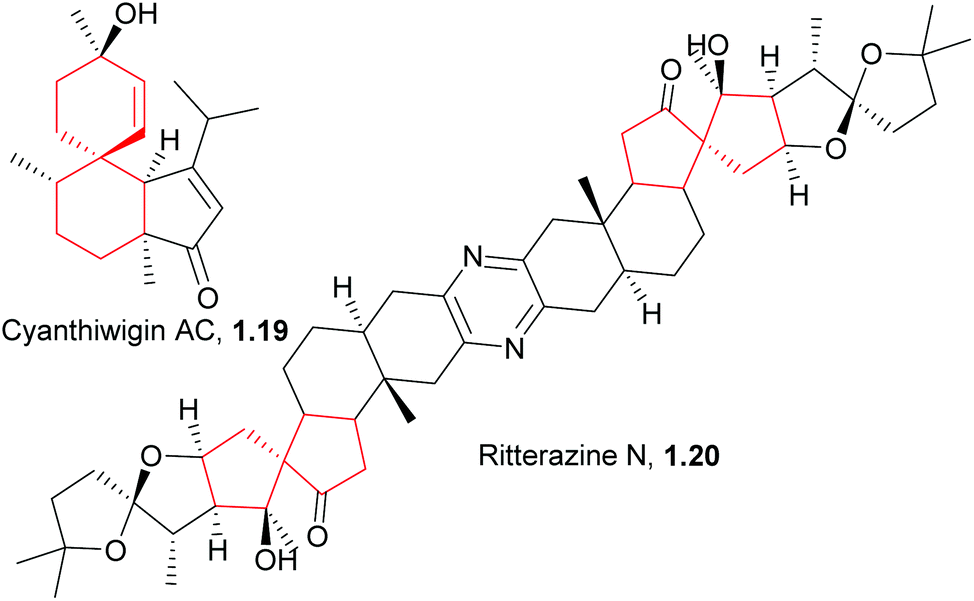 | ||
| Fig. 3 Structures of cyanthiwigin AC and ritterazine N. | ||
Taber et al. have come the closest to synthesising ritterazine N (1.20, Scheme 2) and related analogues18,19 by assembling a ritterazine N precursor. The final planned aldol cyclocondensation failed, but an alternative route via ozonolysis of an alkene followed by treatment with base successfully gave the related compound bis-18,18′-desmethylritterazine N (1.23).19,20
 | ||
| Scheme 2 Attempted syntheses of ritterazine N. Bis-18,18′-desmethylritterazine N was obtained as a single diastereomer in high yield. | ||
As this review aims to summarise the progress in the field, but also to identify areas for improvement, where natural products have been synthesised by more than one strategy, the methods will be compared.
2. Aldol reaction
The aldol reaction is a powerful carbon–carbon bond forming reaction.21 Stereochemical control is possible through the use of chiral aldehyde starting materials, or chiral auxiliaries. The asymmetric reaction can also be performed catalytically through the use of chiral Lewis acids.22 This approach has been particularly effective in the synthesis of various natural product spirocarbocycles as illustrated below.2.1 Vannusals A and B
Vannusals A and B are triterpenes with an unusual C30 backbone, comprising seven rings and thirteen chiral centres, three of which are quaternary. They were isolated from the tropical zooplankton Euplotes vannus, and their biosynthesis is thought to proceed from a squalene-type precursor (Fig. 4), via several possible routes.23 | ||
| Fig. 4 Proposed link between the structures of vannusals and squalene. | ||
The originally assigned structures for the vannusals were incorrect, and the true structures were only established following the total synthesis efforts of the Nicolaou group.24 The fused polycyclic carbon framework was assembled by an intramolecular spirocyclisation via a Mukaiyama-type aldol reaction25,26 (Scheme 3).
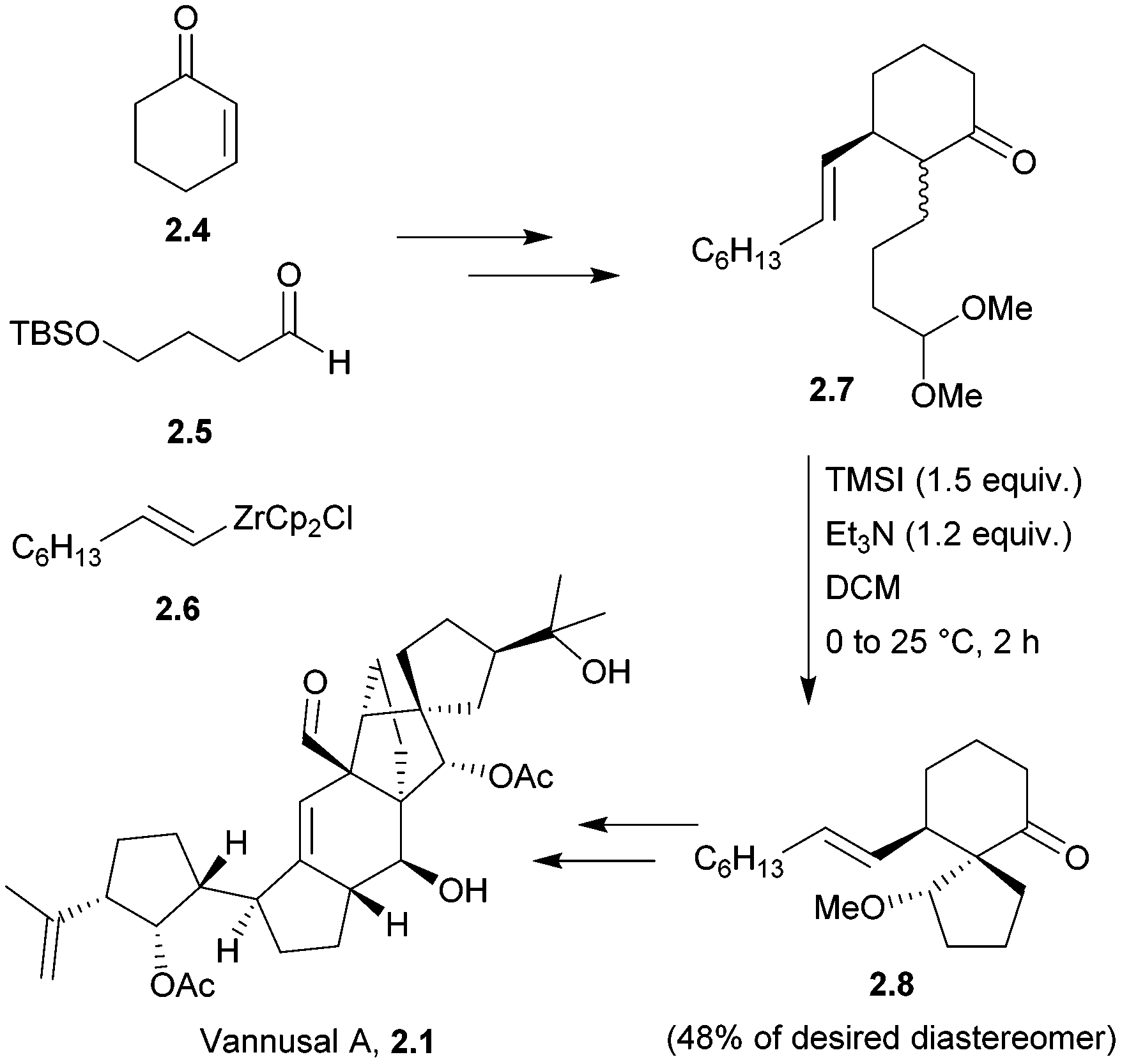 | ||
| Scheme 3 Mukaiyama aldol reaction in the synthesis of vannusal A. | ||
The above methodology was used in the total syntheses of the original incorrect vannusal B structure,27 as well as the resyntheses of the true vannusals A and B.24,28–30
2.2 Acoranes: α-acoradiene, acorone, isoacorone and acorenone B
The acoranes are a group of highly-oxygenated sesquiterpenes containing a spiro[4.5] core. They have been isolated from a variety of sources,31–33 including sweet flag Acorus calamus, the essential oils of the woods of Juniperus rigida and J. chinensis, and Australian sandalwood oil from Santalum spicatum.(±)-Acorone was synthesised in 1977 by the Dolby group, starting from 4-methyl-3-cyclohexene carboxaldehyde and using an intramolecular base-catalysed aldol cyclisation34 (NaOEt/EtOH). A similar method reported by Martin et al. in 1978 used an aldol cyclocondensation to also synthesise racemic acorone35 (Scheme 4).
 | ||
| Scheme 4 Base-catalysed cyclisation in the synthesis of (±)-acorone and (±)-isoacorone. No mention was made of the dr of intermediate 2.11. | ||
In addition, in 1996 Srikrishna et al. performed a formal total synthesis of (±)-acorone36 using an intramolecular aldol condensation of 1,4-cyclohexanedione. (±)-Acorone and (±)-isoacorone were later formally synthesised by the same group using a Claisen rearrangement-based methodology.37
Two enantiomers of the sesquiterpenoid α-acoradiene occur naturally: one is found in juniper wood, and the other is a pheromone of the broad-horned flour beetle. The structure of the beetle α-acoradiene was originally thought to be the (1R,4R,5S) enantiomer, which has been synthesised by ring-closing metathesis (see below). (1S,4R,5R)-α-Acoradiene was synthesised from (S)-(−)-pulegone in 10 steps and 16% overall yield38 (Scheme 5) by Mori et al. in 2004. The key step was the aldol condensation of a 1,5-diketone to form the 6-membered ring.
 | ||
| Scheme 5 Aldol condensation of 1,5-diketone 2.15 to form the spiro intermediate 2.16 in the synthesis of α-acoradiene. | ||
Acorenone B has been synthesised under basic conditions to form the spirocycle39 in high yield (Scheme 6) by Trost et al. in 1975.
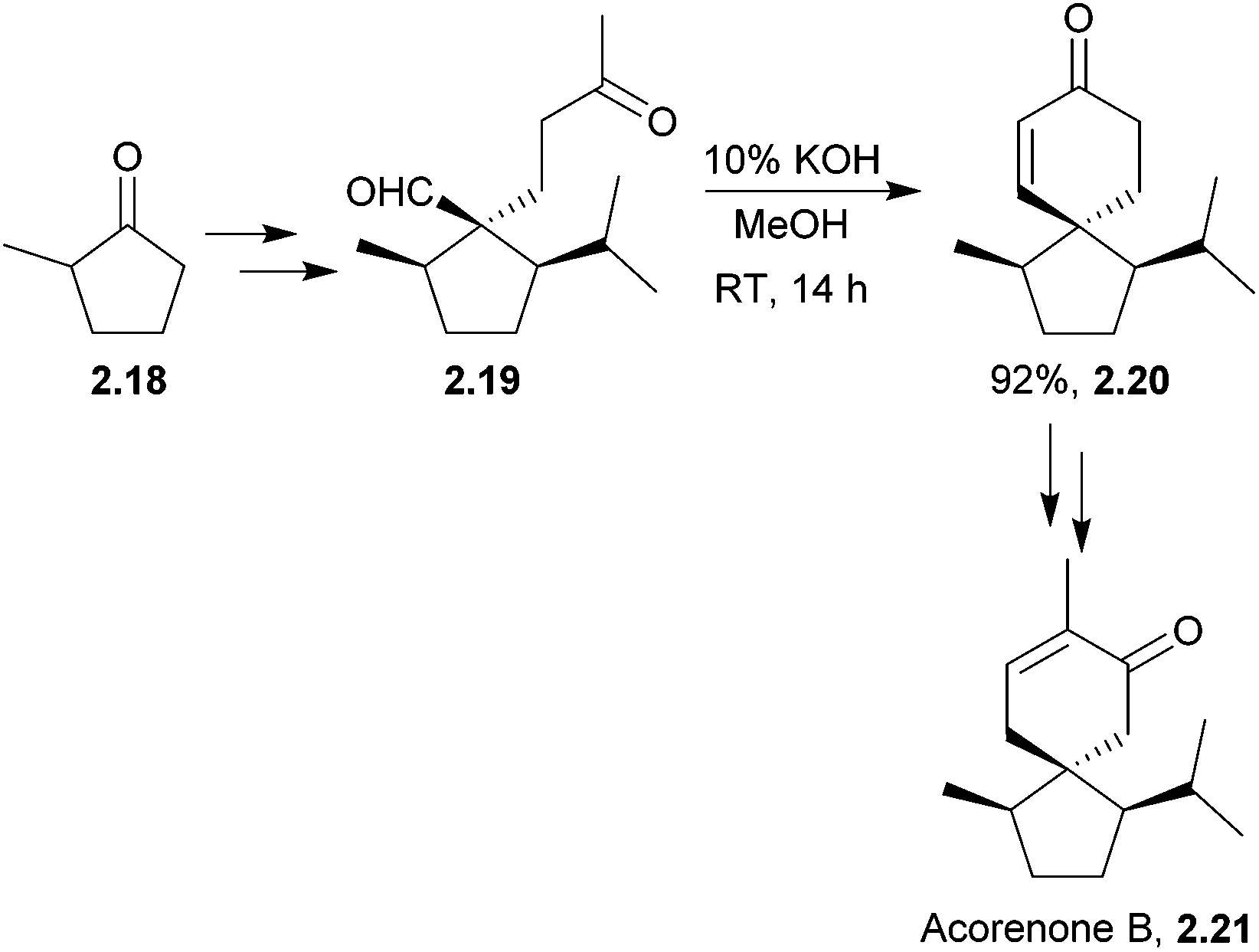 | ||
| Scheme 6 Aldol reaction in the synthesis of acorenone B. | ||
2.3 Spirovetivanes: agarospirol, β-vetivone, α-vetispirene and anhydro-β-rotunol
The spirovetivanes are sesquiterpenes with a spiro[4.5] core. They include metabolites and phytoalexins, compounds produced when a species is subjected to stress such as pathogenic attack.40 Vetiver oil from Vetiveria zizanioides contains several spirovetivanes, including β-vetivone,4 α-vetispirene and β-vetispirene.41 As stated above, for many years, the structure of β-vetivone was thought to be hydroazulenic but this was revised to the spiro[4.5]decane in 1968.5,6Following on from this work by Marshall and Johnson, Wenkert et al. used methanolic KOH to effect a basic aldol cyclisation of a key intermediate in the synthesis of β-vetivone.42 The reaction proceeded in 95% yield, as part of a formal total synthesis requiring 8 steps to reach the preliminary compound.
A tandem Michael-aldol condensation was used by Hutchins et al. in 1984 to synthesise racemic β-vetivone43 (Scheme 7) in a very short and efficient synthesis.
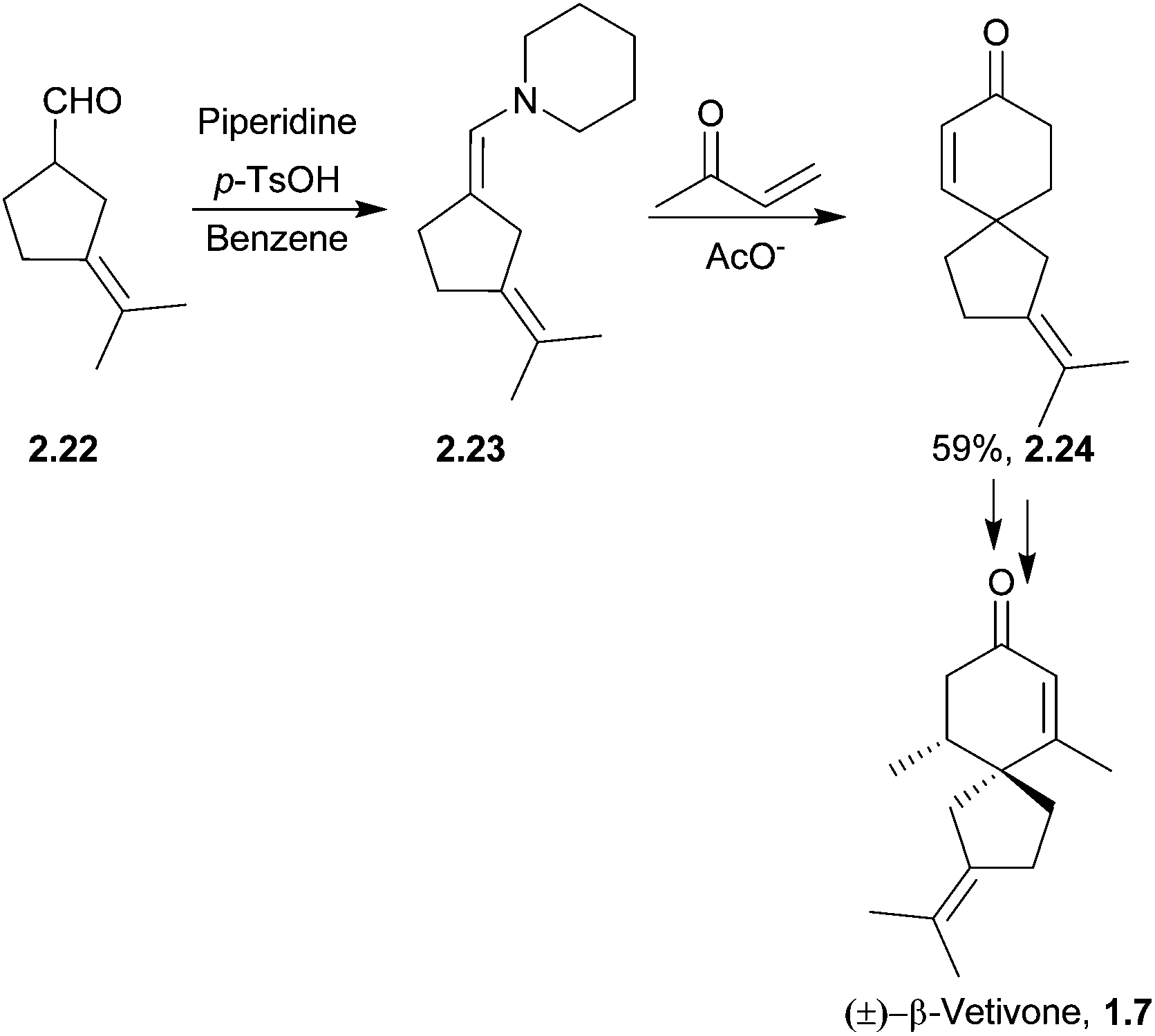 | ||
| Scheme 7 Tandem Michael-aldol condensation in the synthesis of β-vetivone. | ||
Starting from a spiro[5.5]acetal, (−)-agarospirol, a metabolite isolated from agarwood oil,44 was synthesised in 1975 via a 1,6-dicarbonyl and base to generate the α,β-unsaturated ketone.45
Ten years later, α-vetispirene was synthesised by Balme using a related intramolecular spirocondensation with a 1,5-dicarbonyl under basic conditions46 (Scheme 8).
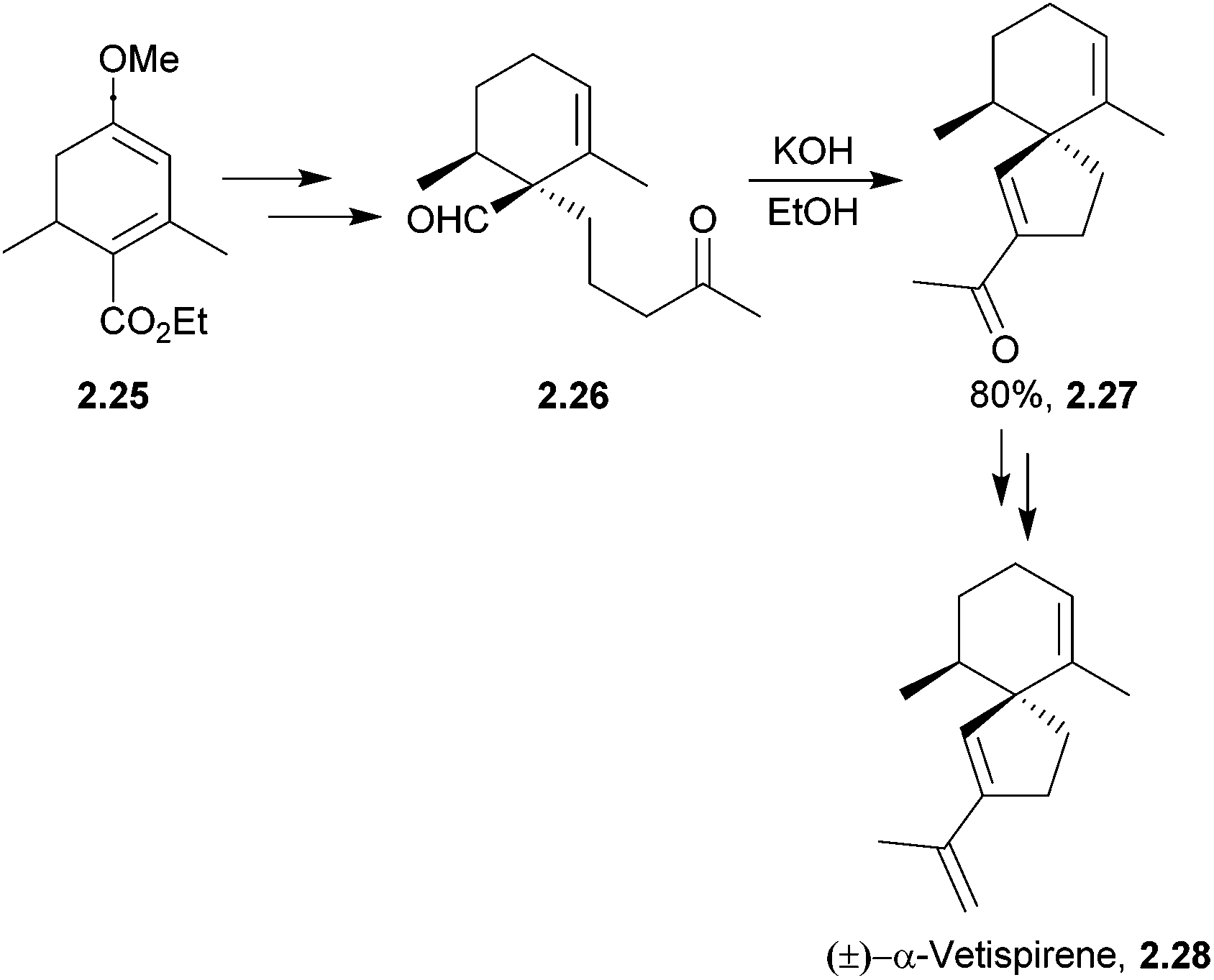 | ||
| Scheme 8 Basic aldol condensation in the synthesis of α-vetispirene. | ||
Anhydro-β-rotunol is a phytoalexin spirovetivadiene isolated from infected potato tubers.47 In 2005 Srikrishna et al. performed an enantioselective synthesis of (+)-anhydro-β-rotunol starting from (R)-carvone in 13 steps and 15% overall yield48 (Scheme 9). The spirocyclisation step involved a regioselective intramolecular aldol condensation of a keto-aldehyde, reminiscent of Balme's synthesis of α-vetispirene. Other conditions were attempted, but piperidine in AcOH was found to be the most successful.
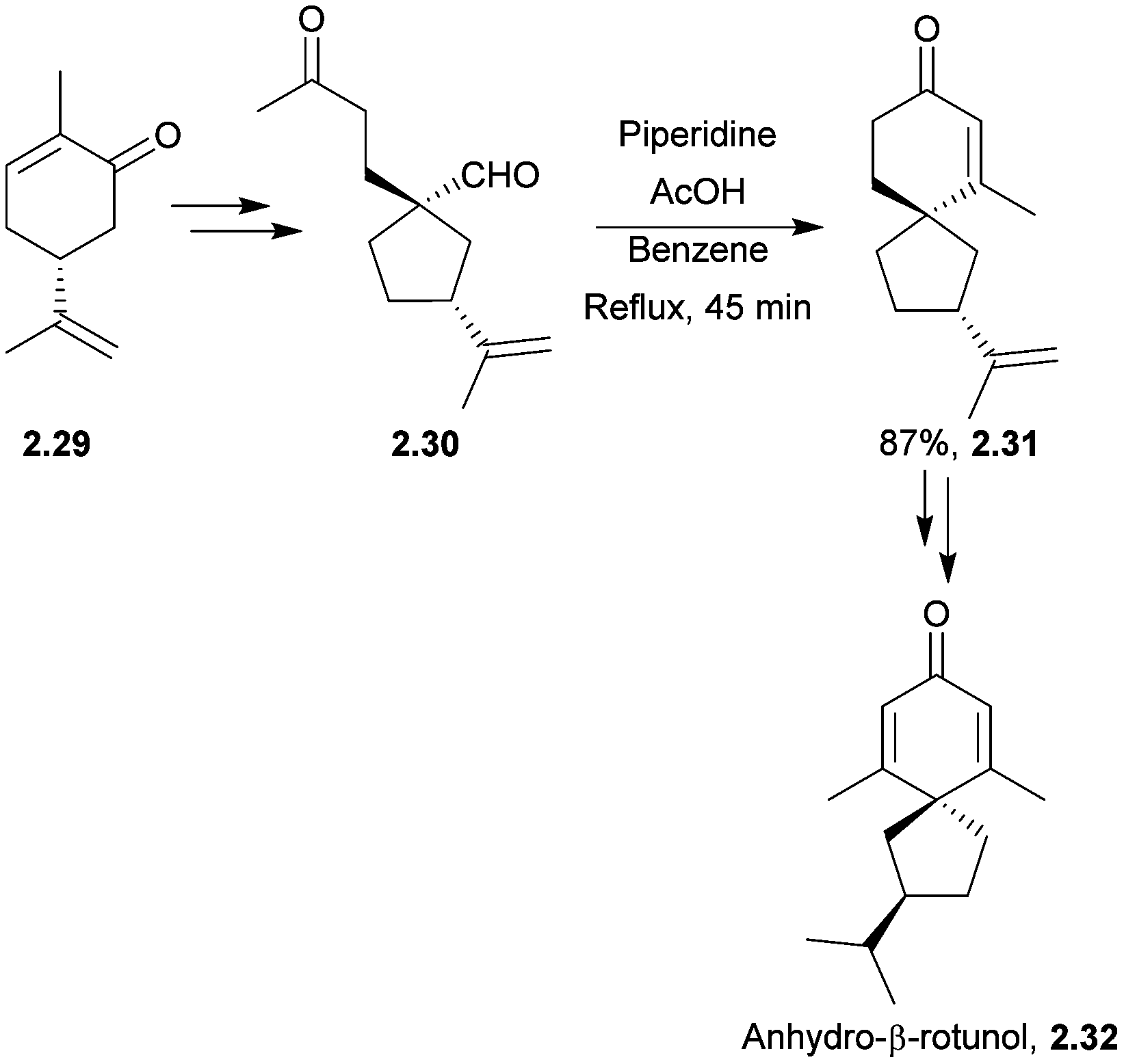 | ||
| Scheme 9 Aldol condensation in the synthesis of anhydro-β-rotunol. | ||
2.4 Erythrodiene and spirojatamol
Erythrodiene and spirojatamol are sesquiterpenes that share a common spirobicyclic [4.5]decane skeleton. (−)-Erythrodiene was isolated from a Caribbean encrusting coral, whereas (+)-spirojatamol was isolated from a Himalayan flowering plant. Erythrodiene and spirojatamol have been formally synthesised from N-cyclohexyl-4-isopropylcyclohexanimine49 by Ihara et al. in 1997. The key spirocyclisation step was a tandem intramolecular Michael-aldol reaction of a ketoacetal with Me3SiI/(Me3Si)2NH (Scheme 10). | ||
| Scheme 10 Aldol reaction during the synthesis of erythrodiene and spirojatamol. The diastereomers 2.35–2.38 were separable by column chromatography. | ||
Forsyth et al. have used spirocarbomercuration to synthesise erythrodiene and spirojatamol;50,51 more recently, Oppolzer et al. have used a Pd-catalysed allylzincation52 and Renaud et al. a thiophenol-mediated spirocyclisation.53
2.5 Vitrenal
(+)-Vitrenal is a sesquiterpenoid and a potent plant growth inhibitor.54 The unnatural enantiomer, (−)-vitrenal, has been synthesised from (+)-3-carene55 by Ito et al. in 1986. The key sequence is a one-pot deacetalisation-aldol condensation, which is preceded by a stereoselective Claisen rearrangement (Scheme 11). It showed weaker plant growth inhibition than the natural enantiomer. | ||
Scheme 11 One-pot deacetalisation-Aldol condensation in the synthesis of (−)-vitrenal. Intermediate 2.42 was present as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of geometrical isomers, which were only separable by gas chromatography. Both isomers gave the same acid (2.43). :1 mixture of geometrical isomers, which were only separable by gas chromatography. Both isomers gave the same acid (2.43). | ||
2.6 Cannabispirans: cannabispirenones A and B and cannabispirone
A variety of spiroindane products have been isolated from the marijuana plant, Cannabis sativa.56–62 They are structurally related to some synthetic oestrogenic-potentiating agents and so may show similar oestrogenic activity to marijuana.59 Crombie et al. used KOH to intramolecularly close the spiro ring of several cannabispirans63,64 (Scheme 12). | ||
| Scheme 12 Basic aldol reaction in the spirocyclisation of cannabispirenone A. | ||
Cannabispirone was synthesised by treating an aldehyde and methyl vinyl ketone with tBuOK65 (Scheme 13) by El-Feraly et al. in 1981.
 | ||
| Scheme 13 Spirocyclisation step in the synthesis of cannabispirone. | ||
Cannabispirenone A was asymmetrically synthesised by Natale et al. in 1984 from 3,5-dimethoxyaniline in 11 steps, using an enamine and methyl vinyl ketone followed by hydrolysis to form the spiro centre66 (Scheme 14).
 | ||
| Scheme 14 Aldol condensation in the synthesis of cannabispirenone A. | ||
Similarly, cannabispirenone B was synthesised by Novak et al. in 1982.67 Aldehyde 2.51 was transformed to the piperidine enamine, before it was reacted with methyl vinyl ketone followed by hydrolysis and aldol cyclisation. The increased difficulty in the synthesis of cannabispirenone B over A lay in the selective ether cleavage of the less sterically hindered aryl methoxy group. Previously this had only been possible using BBr3, which gave low yields and led to the formation of insoluble tars. Ten reagents were considered by the authors, of which only LiI in 2,4,6-trimethylpyridine was successful in 56% conversion and 82% yield.
2.7 Chamigrenes: majusculone and laurencenone C
The chamigrene family is a group of over 100 sesquiterpene natural products that contain a spiro[5.5]undecane core. Examples include majusculone and laurencenone C.68,69 Some chamigrenes are halogenated, and the group shows diverse biological activity.Zhu et al. synthesised racemic laurencenone C in 2010, and the key step was a base-catalysed aldol condensation70 (Scheme 15). The full sequence involved 11 steps with an overall yield of 17%.
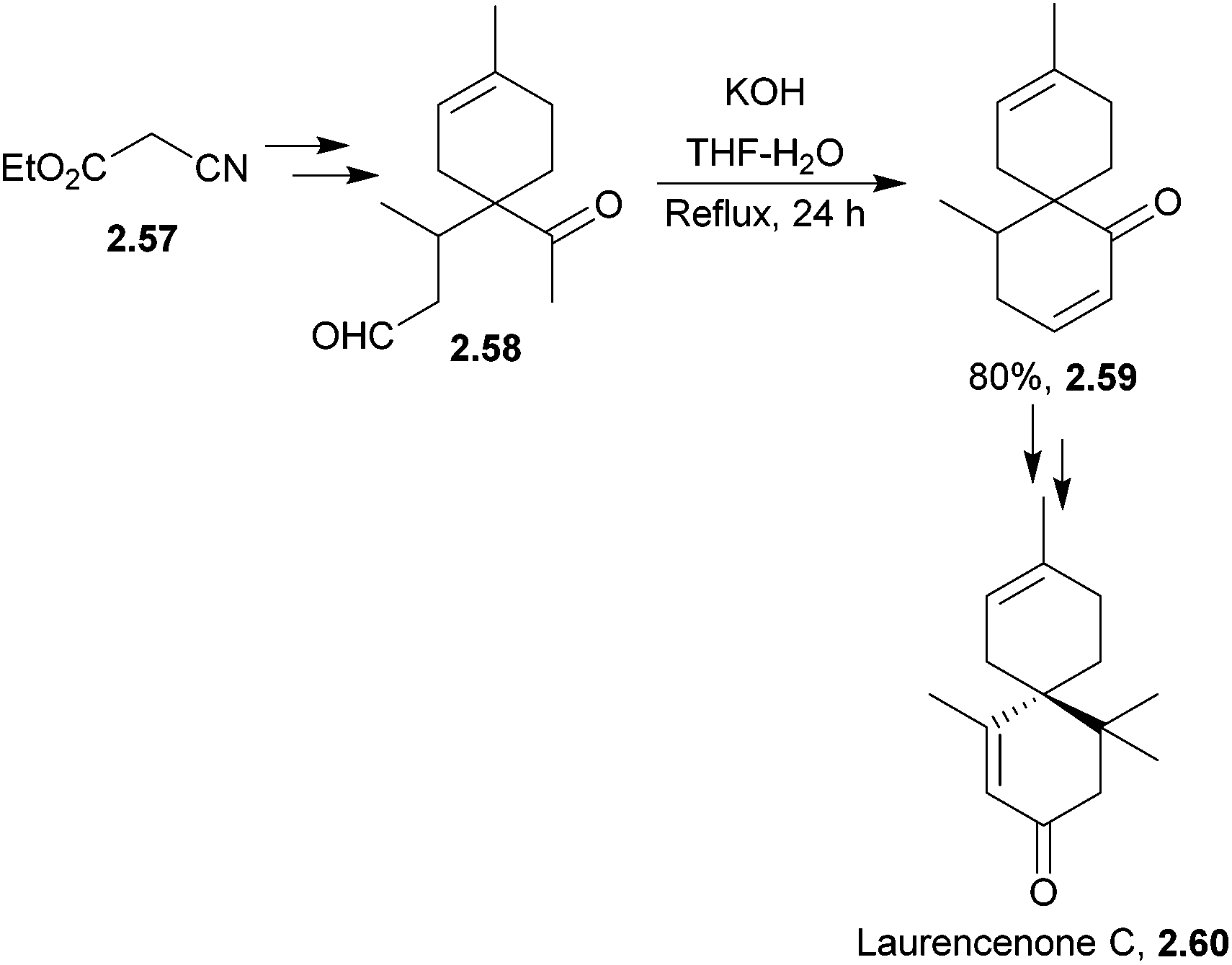 | ||
| Scheme 15 Aldol cyclisation in the synthesis of laurencenone C. | ||
Racemic majusculone also was synthesised from ethyl cyanoacetate in 15 steps71 (Scheme 16) by Zhu et al. in 2011. The key step in this particular synthesis was the tandem oxidation-aldol condensation of the diol to the keto aldehyde which underwent base-mediated cyclisation.
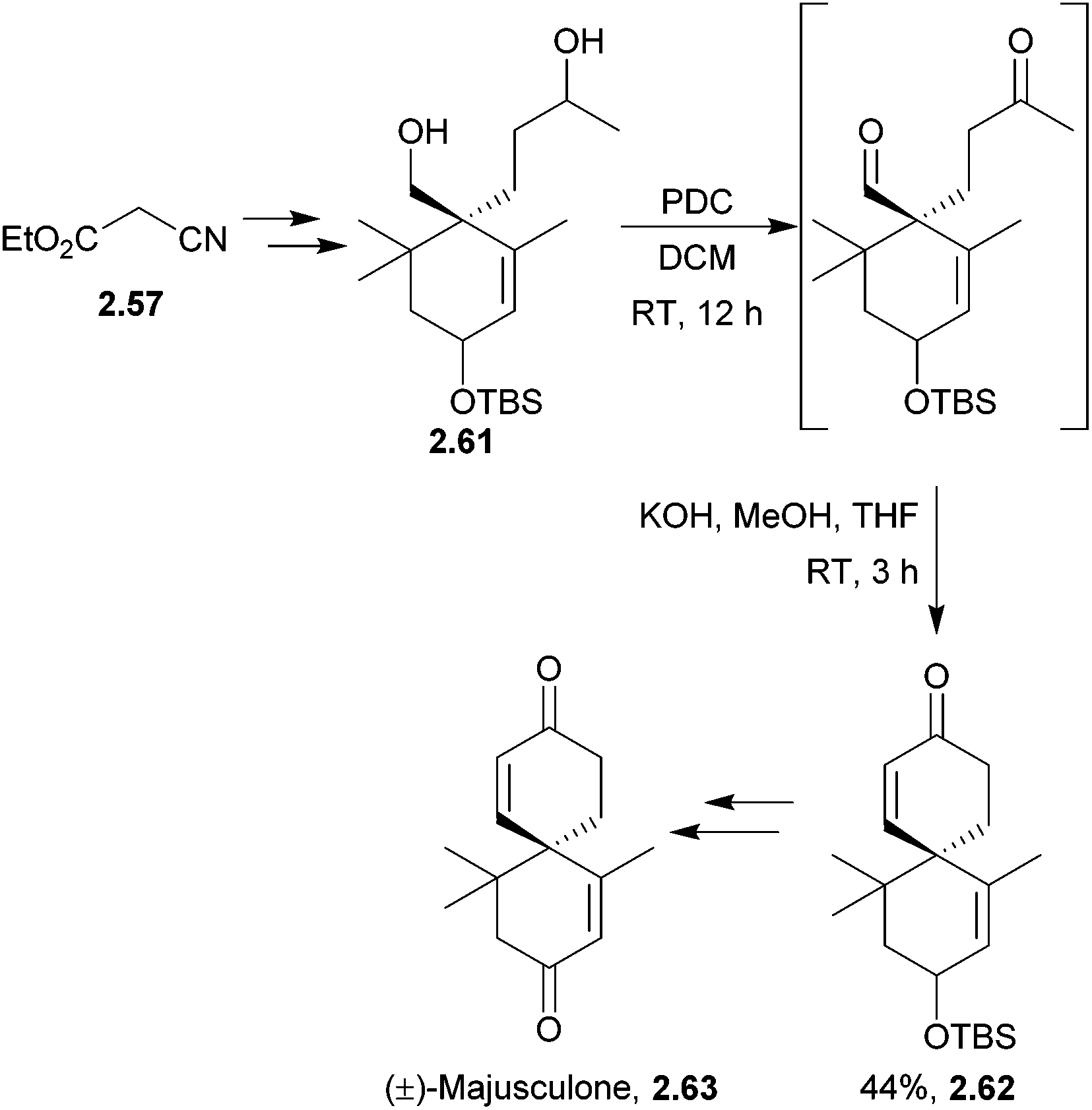 | ||
| Scheme 16 Oxidation followed by cyclisation in the synthesis of (±)-majusculone. | ||
3. Acid and base promoted spirocyclisation
Acids or bases can be used in a wider range of cyclisation reactions than just to promote the aldol reaction. For example, the related Robinson annulation and Prins cyclisation have been used to synthesise natural products containing spirocarbocycles as described below.3.1 Acoranes: acorenone, acorenone B and α-acorenol
(±)-Acorenone B has been synthesised from (±)-camporone.72 Formic acid was used to activate the allylic alcohol to attack by an alkene via an allylic cation to form the spiro centre (Scheme 17). | ||
| Scheme 17 Treatment of an intermediate with formic acid in the synthesis of acorenone B. The dr were not stated, but the product was isolated as a single diastereomer via preparative TLC. | ||
No mention was made of stereochemistry or diastereoselectivity in the original 1975 paper and thus this method may be less effective than the cyclisation in base which was described above.
In 1976, White et al. started from (R)-(+)-limonene to synthesise (−)-acorenone B in 11 steps.73 The key step was an acid-catalysed ring-opening of a cyclopropane to reveal the spiro[4.5]decanone (Scheme 18).
 | ||
| Scheme 18 Ring-opening of an intermediate 3.5 in the synthesis of (−)-acorenone B. | ||
If lithium in liquid ammonia was used to perform a reductive scission of the cyclopropane bond, inversion of the stereochemistry of the methyl group was observed.
Pesaro et al. used a Robinson annulation in 1978 to synthesise both (−)-acorenone and (−)-acorenone B, in 4 steps starting from (+)-p-menth-1-ene74 (Scheme 19).
 | ||
| Scheme 19 Treatment of an aldehyde with methyl vinyl ketone in base in the synthesis of (−)-acorenone and (−)-acorenone B. | ||
The reaction gave the key spiro intermediate in low yield, with the stereoselectivity of the reaction controlled by the isopropyl and methyl groups, which direct the electrophilic addition of the methyl vinyl ketone to the less hindered face.
More recently, Sakai et al. used a Lewis acid to effect a one-pot transformation to a spiro compound en route to racemic acorenone B (Scheme 20).75 The mechanism is thought to proceed via an aldol condensation, followed by hemiacetalisation and finally a Grob-type fragmentation to realise the intermediate.
 | ||
| Scheme 20 Three-step procedure in the synthesis of acorenone B. | ||
In 2014, Chen et al. used the reduction of a cycloheptatriene to trigger an acid-induced rearrangement in the formal synthesis of α-acorenol (Scheme 21).76
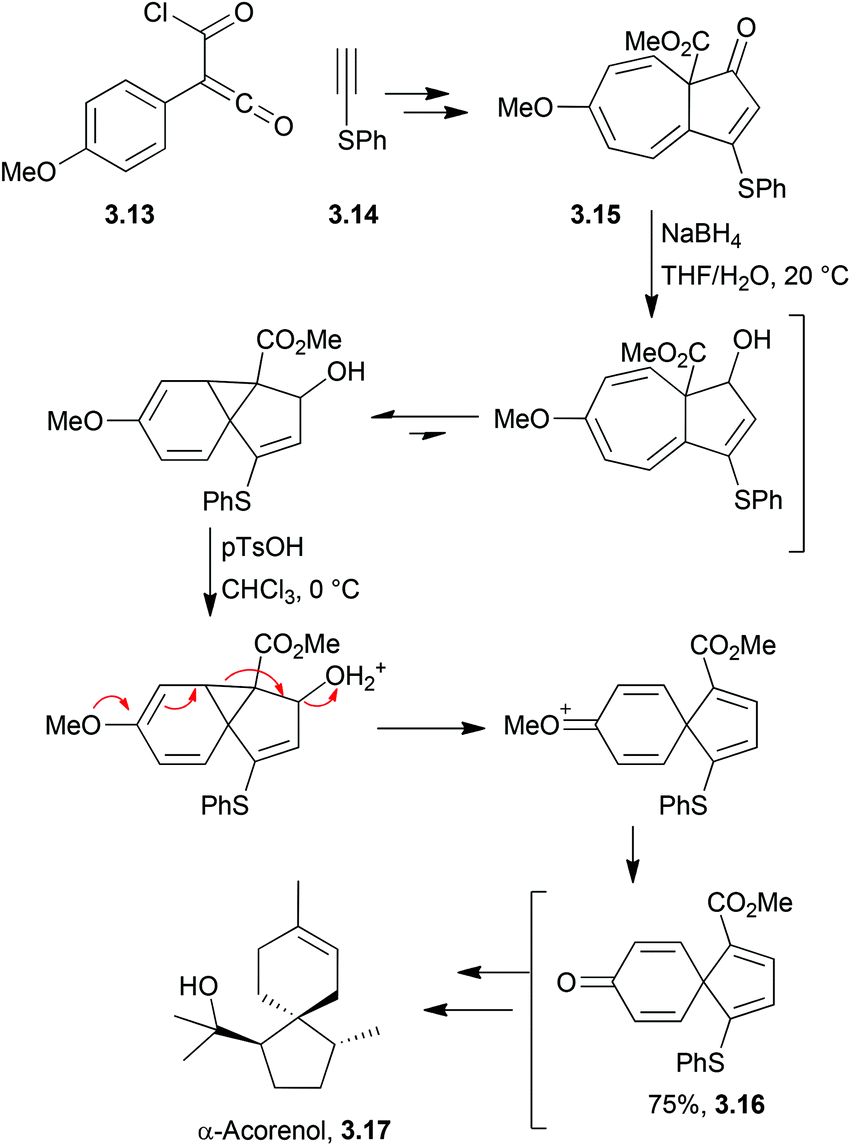 | ||
| Scheme 21 Reduction and acid-induced rearrangement in the synthesis of α-acorenol. | ||
3.2 Spirovetivanes: agarospirol, hinesol, β-vetivone, α-vetispirene and β-vetispirene
Acetal formation followed by reduction and treatment with acid was used to form the molecules hinesol, β-vetivone, and α- and β-vetispirenes77,78 (Scheme 22). These syntheses were published in a series of articles from Yamada et al. in 1973. | ||
| Scheme 22 Three-step sequence in the synthesis of β-vetispirene. | ||
A Lewis acid-catalysed Prins cyclisation was used by McCurry et al. in 1973 in the synthesis of racemic β-vetivone and β-vetispirene79 (Scheme 23).
 | ||
| Scheme 23 Prins cyclisation followed by chair-chair interconversion in the synthesis of β-vetivone. Diastereomers 3.25 and 3.26 were separable, but the dr was not stated. | ||
Of the four possible diastereomers, the two shown in Scheme 23 constituted 97% of the reaction mixture. The mechanism is thought to proceed via a 6-membered ring transition state.
Deslongchamps et al. have synthesised racemic agarospirol and hinesol from a keto ester.80,81 The spirocycle was formed by treatment of a cyclopropane intermediate with acid (Scheme 24).
 | ||
| Scheme 24 Treatment of an intermediate with acid in the synthesis of hinesol. | ||
The intermediate is thought to undergo facile cyclopropane ring-opening under acid conditions via the enol.
Spiroannulation of 5-methyl-1,3-cyclohexanedione with an alkyl halide in base was used to synthesise β-vetivone82 (Scheme 25) by Stork et al. in 1973.
 | ||
| Scheme 25 Spiroannulation in base during the synthesis of β-vetivone. | ||
A similar method was used by Markó et al. in 2004 to form racemic agarospirol, hinesol and α-vetispirene. A spiro diketone was formed by condensation of a silyl enol ether and an ortho ester to form a β-ketoketal followed by base-catalysed intramolecular cyclisation83 (Scheme 26).
 | ||
| Scheme 26 Base-catalysed intramolecular cyclisation during the synthesis of agarospirol. The de of intermediate 3.45 was >95%. | ||
In 1988, Posner et al. used a tandem oxidation-acid-promoted cyclisation in the third asymmetric total synthesis of (−)-β-vetivone (Scheme 27).84
 | ||
| Scheme 27 Oxidation then cyclisation in the synthesis of β-vetivone. | ||
Acidic or basic conditions are commonly employed in the key cyclisation step of (±)-hinesol.85,86 A base-catalysed reverse Prins reaction was used by Marshall et al. in 197087 (Scheme 28).
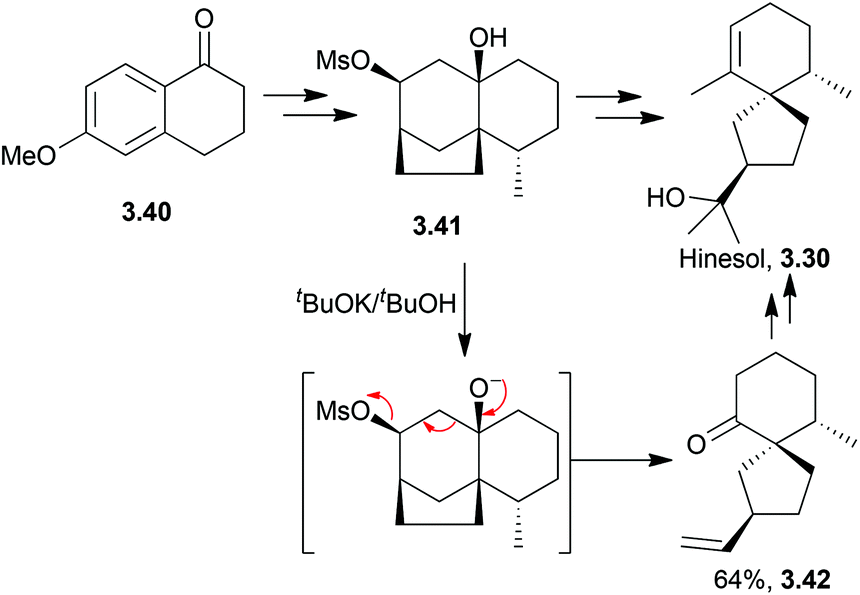 | ||
| Scheme 28 Basic conditions for the spirocyclisation step during the synthesis of hinesol. | ||
3.3 Spirovetivadienes: solavetivone, lubimin, oxylubimin, premnaspirodiene and hinesene
Solavetivone is a spirovetivadiene phytoalexin produced by tobacco plants infected with the tobacco mosaic virus.88–91 Lubimin and oxylubimin are two of several stress metabolites produced by fungally-infested white potato tubers.92,93 Premnaspirodiene and hinesene are diastereomers: (−)-premnaspirodiene is the (2R,5S,10R)-isomer whereas hinesene has the (2R,5S,10S) configuration.Racemic solavetivone was synthesised through a Diels–Alder reaction followed by π-cyclisation85 (Scheme 29) by Murai et al. in 1984. The starting material was 3,5-dimethylanisole and the procedure required 12 steps to give the product in 3% overall yield. The Murai group used this methodology to synthesise lubimin and oxylubimin in a series of follow-up articles.94,95
 | ||
| Scheme 29 Oxalic acid-induced cyclisation in the synthesis of (±)-solavetivone. | ||
Other conditions attempted for the π-cyclisation were unsuccessful, including using SnCl4 in DCM at −78 °C, acetic acid at 110 °C and formic acid at 50 °C.
Premnaspirodiene and hinesene have both been synthesised from α-santonin by Pedro et al. in 2004.96 The key step was an acid-promoted rearrangement of the carbon skeleton (Scheme 30) via a tertiary carbocation formed by ring-opening of the epoxide.
 | ||
| Scheme 30 Acid-promoted rearrangement of the carbon skeleton in the synthesis of premnaspirodiene. | ||
3.4 Vitrenal
Takahashi synthesised racemic vitrenal in 12 steps and 7% overall yield from the monoterpene piperitenone97 in 1982. The key step was an acid promoted acetal hydrolysis and enol ether trapping (Scheme 31). | ||
| Scheme 31 Acid-catalysed hydrolysis in the synthesis of (±)-vitrenal. | ||
3.5 Spirolaurenone
Spirolaurenone is a bromine-containing sesquiterpenoid98 based upon the spirolaurane skeleton.99Total synthesis of (±)-spirolaurenone proceeded from a bromohydrin of homogeranonitrile in 13 steps and 2% overall yield100 as described in 1982 by Murai et al. The key step was the cleavage of a cyclopropane ring under acidic conditions to give a mixture of endo and exo alkenes which were separated by preparative HPLC (Scheme 32). The endo isomer was used to reach the natural product.
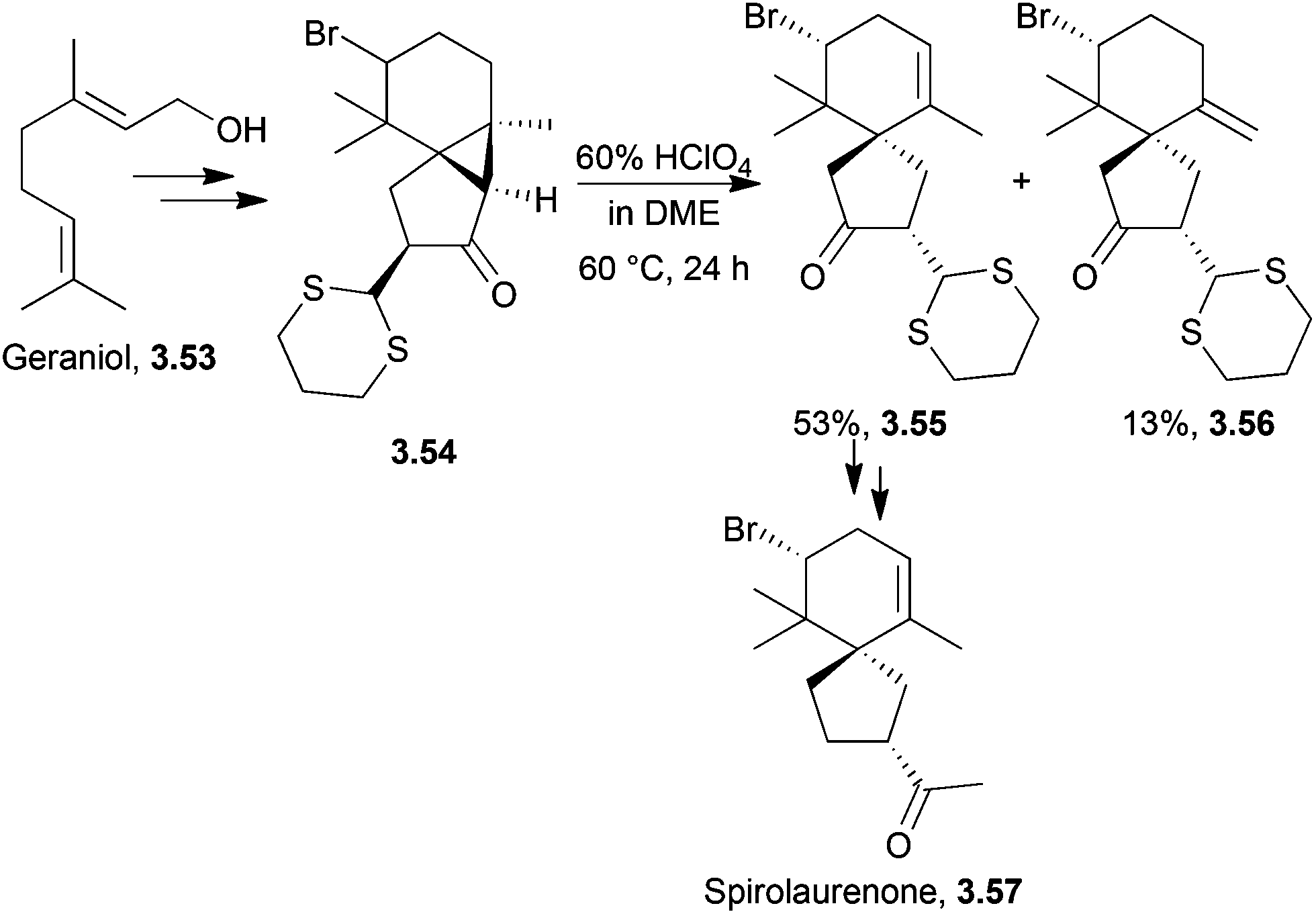 | ||
| Scheme 32 Spirocyclisation step in the synthesis of spirolaurenone. | ||
3.6 Chamigrenes: α-chamigrene and majusculone
Unlike its halogenated derivatives, α-chamigrene was isolated from the five-flavour berry, Schisandra chinensis, rather than a marine organism. Racemic α-chamigrene was synthesised by Canonne et al. in 1991 by intramolecular enolate alkylation promoted by KH101 (Scheme 33). | ||
| Scheme 33 Spirocyclisation step in the synthesis of α-chamigrene. | ||
The first enantioselective synthesis of (+)-majusculone was achieved by alkylidene carbene insertion promoted by KHMDS102 (Scheme 34) as described by Taber et al. in 2007.
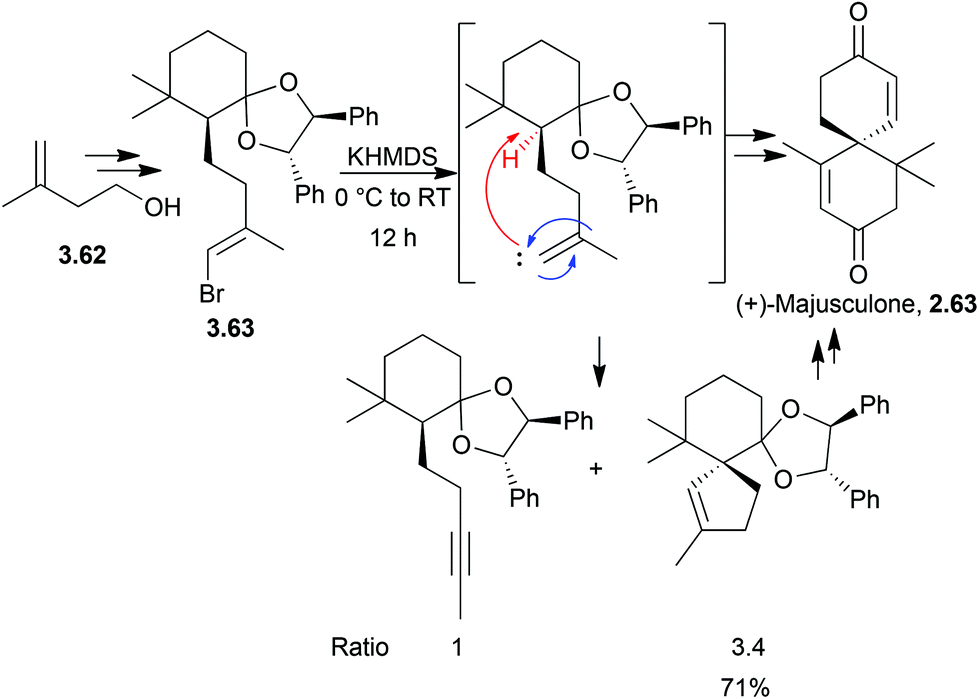 | ||
| Scheme 34 Spirocyclisation in the synthesis of (+)-majusculone. | ||
α-Elimination generated the carbene which inserted into a C–H bond shown in red to give the spirocycle. An alkyne by-product from the 1,2-rearrangement was also observed, shown in blue. Concerns over the ability of the carbene to insert into the deactivated, congested methine were not realised.
3.7 Spirocurcasone
Spirocurcasone is a diterpenoid with a novel carbon skeleton, isolated from Jatropha curcas.103 A high-yielding one-pot semisynthesis of (+)-spirocurcasone from the natural products curcasones A and B has been reported very recently by Qin et al. in 2014 (Scheme 35).104 | ||
| Scheme 35 Use of TMEDA in the synthesis of spirocurcasone. | ||
In the absence of a metal, the TMEDA is thought to act as a base. Various conditions were screened, and MeOH was found to give higher conversions of the starting material over toluene or DCM, and the relatively mild temperature of 50 °C was more effective than 90 °C or reflux.
4. Ring-closing metathesis
Spirocarbocycles can be synthesised using a variety of metal complexes.105 Ring-closing metathesis106,107 typically uses a ruthenium-based catalyst such as Grubbs’ I or Grubbs’ II to react two terminal alkenes intramolecularly to give an alkene and ethene. Kotha et al. have comprehensively reviewed the synthesis of spirocyclics by ring-closing metathesis in 2003.1084.1 Illudins
The illudins are sesquiterpenoids isolated from fungi, including the highly poisonous Jack-o′ lantern mushroom, Omphalotus illudens.109–111 The illudins themselves are extremely toxic. Illudins M and S show potent cytotoxic activity in vitro, but are much less effective in vivo.112,113 There has been considerable work towards developing anti-cancer derivatives, which are now in clinical trials. These include bicyclic and isomeric analogues of illudin M,114 acylfulvene and hydroxymethylacylfulvene115,116 (Fig. 5). The latter is now in Phase II clinical trials against ovarian, prostate, and gastrointestinal cancers.117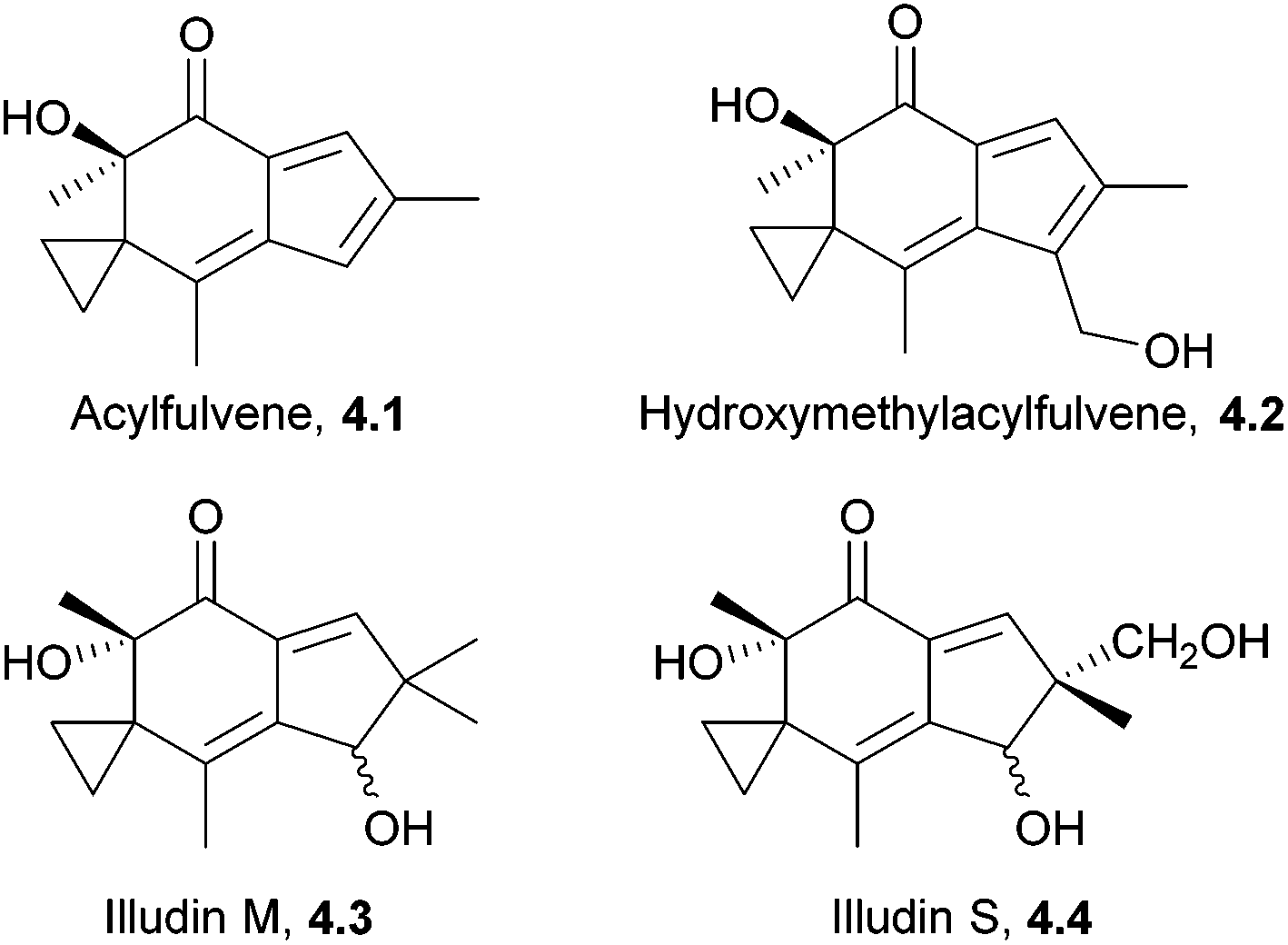 | ||
| Fig. 5 Acylfuvene, hydroxymethylacylfulvene and illudins M and S. | ||
The illudin core has been synthesised by Movassaghi et al. using an enyne ring-closing metathesis cascade118 (Scheme 36) in 2009.
 | ||
| Scheme 36 Enyne ring-closing metathesis in the synthesis of the illudin core. | ||
With Grubbs’ II catalyst, conversion to the desired product was 90% by 1H NMR spectroscopy, whereas with Grubbs’ I catalyst the conversion was only 25% to an unwanted side-product that contained a five-membered ring (Scheme 37).
 | ||
| Scheme 37 Enyne ring-closing metathesis in the synthesis of the illudin core to give an unwanted side-product. | ||
Illudins M and S have also been previously synthesised by Matsumoto et al., using a basic aldol ring-closing reaction.119,120
4.2 Acoranes: trichoacorenol, α-acorenol, β-acorenol, acorone, isoacorone, acorenone and α-acoradiene
Trichoacorenol is a sesquiterpenoid121 produced by the fungi Trichoderma koningii and T. harzianum.121,122 The unnatural enantiomers (−)-trichoacorenol and (−)-acorenone have both been synthesised from (+)-(R)-pulegone122 by Dickschat et al. in 2011. The key step was a ring-closing metathesis which occurred in moderate yield for such a transformation (Scheme 38).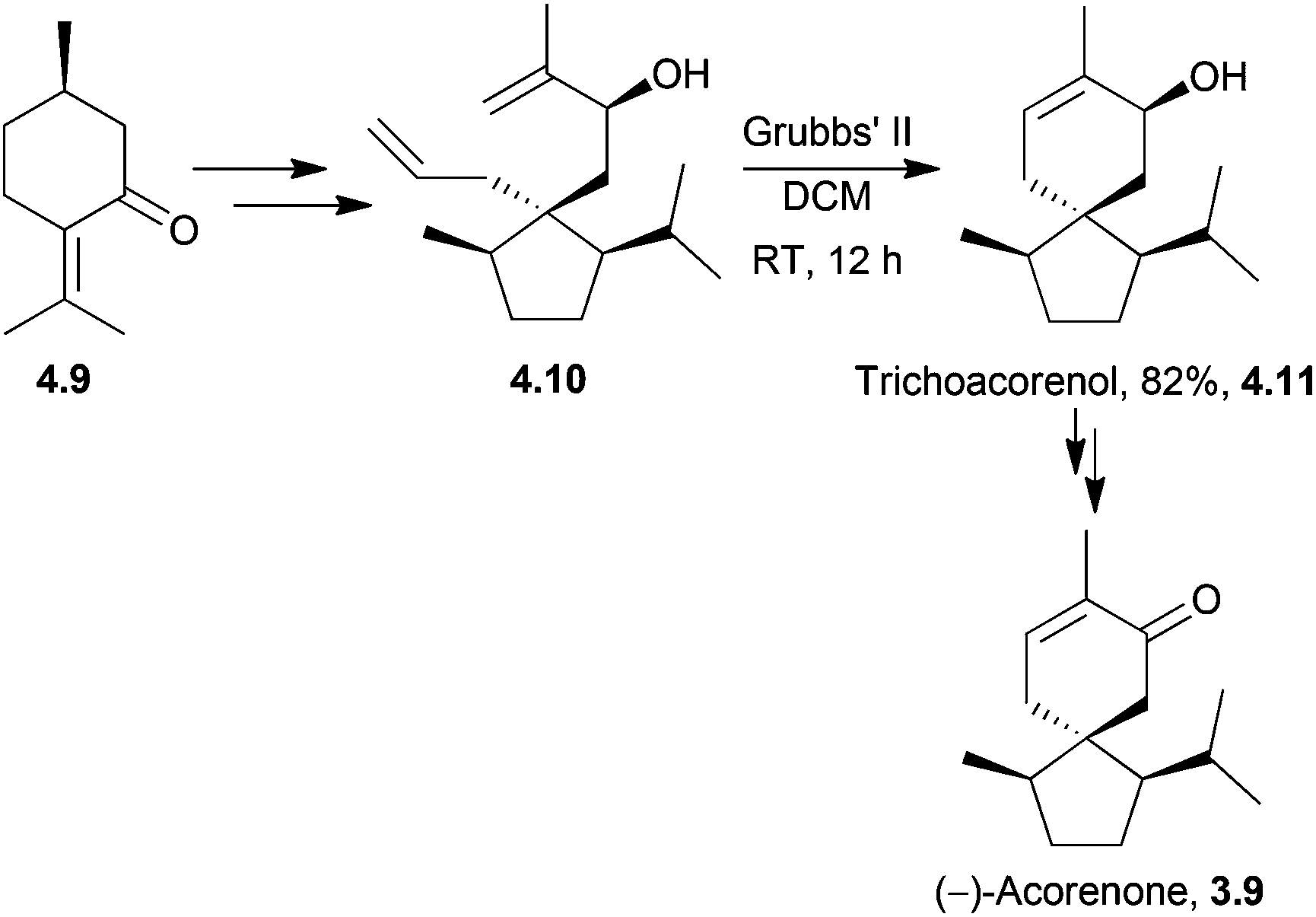 | ||
| Scheme 38 Ring-closing metathesis during the synthesis of (−)-trichoacorenol and (−)-acorenone. | ||
α-Acorenol and β-acorenol were synthesised starting from cyclohexan-1,4-dione in 7 steps and 67% overall yield33 by Srikrishna et al. in 2007. The key steps were an Ireland–Claisen rearrangement and ring-closing metathesis (Scheme 39). The same methodology had previously been applied to the formal syntheses of (±)-acorone and (±)-isoacorone.123
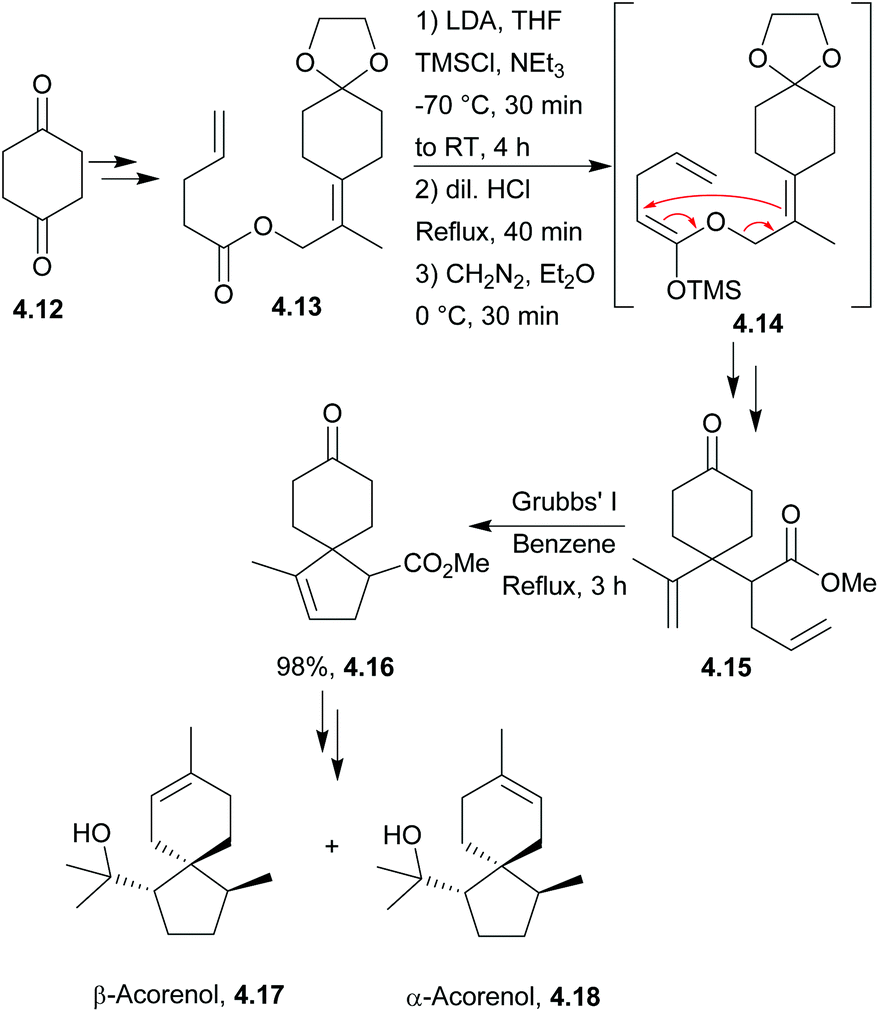 | ||
| Scheme 39 Ring-closing metathesis in the synthesis of (±)-α-acorenol and (±)-β-acorenol. | ||
The (1R,4R,5S) enantiomer of α-acoradiene has been synthesised by Mori et al. in a 14 step sequence, using Grubbs’ I catalyst in the key spirocyclisation (Scheme 40).124
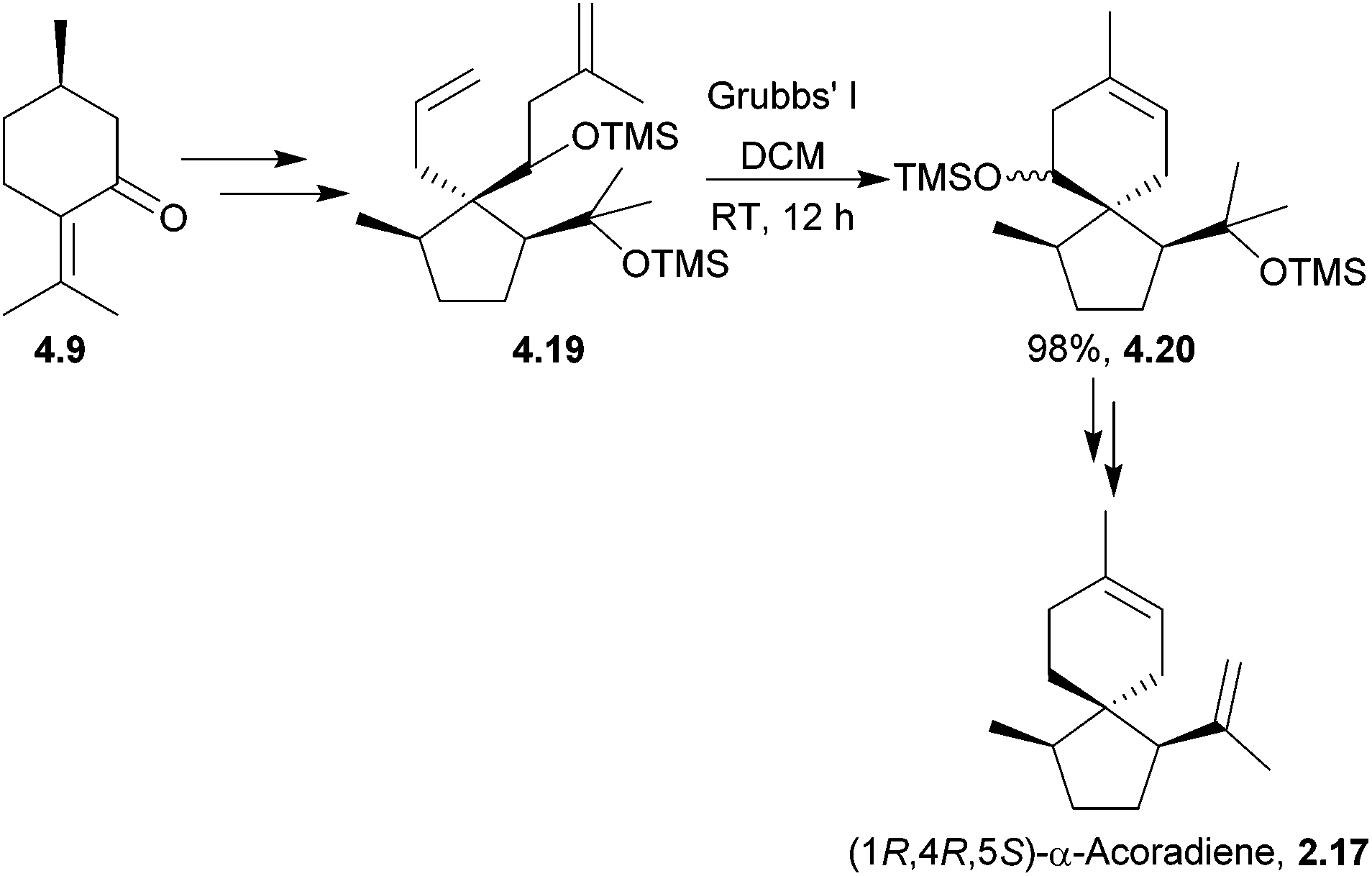 | ||
| Scheme 40 Ring-closing metathesis in the synthesis of α-acoradiene. | ||
The stereochemistry of the product formed did not match the stereochemistry of the broad-horned beetle pheromone, thus disproving the originally proposed structure and identifying the pheromone as (1S,4R,5R)-α-acoradiene.38
4.3 Spiroaxanes: axenol and gleenol
Axenol and gleenol are enantiomeric sesquiterpenes sharing the spiroaxane skeleton. They have both been isolated from the Eurypon marine sponges, but (−)-gleenol has also been found in a variety of coniferous trees, including Picea glehnii, P. koraiensis, Criptomeria japonica and Juniperus oxycedrus. Axenol has not been found to exhibit biological activity, but (+)-gleenol possesses termiticidal and anti-helmintic activities, as well as regulating the growth of plant seeds.(+)-Axenol and (−)-gleenol have been synthesised by Spitzner et al., using an organometallic domino reaction, with an unusual combination of methylenation followed by ring-closing metathesis125,126 (Scheme 41). The first step in the synthesis was a Michael addition with methyl vinyl ketone,127 followed by selective methylenation with the Kauffmann molybdenum complex which was generated in situ from MoOCl3(THF)2.128 A mixture of inseparable isomers was formed in the ratio 2:1, of which the major (R)-isomer (4.22) was the desired product. Grubbs’ I catalyst was added to the crude reaction mixture in a one-pot reaction to give epimers of the spiroketone 4.23.
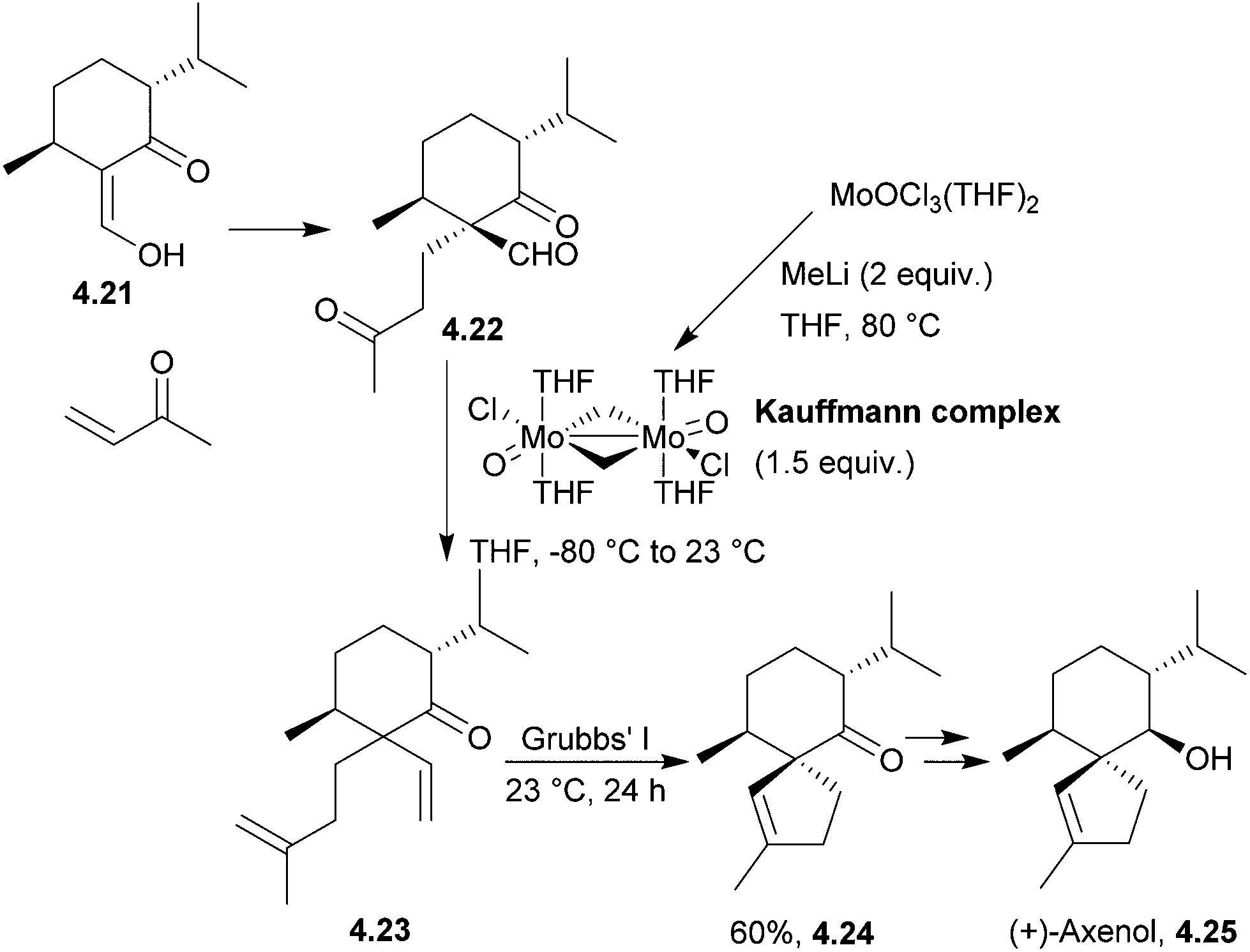 | ||
| Scheme 41 Ring-closing metathesis in the synthesis of (+)-axenol. | ||
A McMurry coupling was also directly attempted on the tricarbonyl, but the spiroketone product was not observed by GC-MS analysis.
Kobayashi et al. synthesised (−)-axenol in 2015 using ring-closing metathesis (Scheme 42).129 The key stereochemistry of the quaternary carbon in the intermediate was built using allylic substitution starting from a picolinate derived from menthol.
 | ||
| Scheme 42 Ring-closing metathesis in the synthesis of (−)-axenol. | ||
The TBS-protected alcohol was also considered, but gave lower conversions of substrate.
4.4 Spirocurcasone
The total synthesis of (−)-spirocurcasone was achieved by ring-closing metathesis of a pentaene130 by Ito et al. in 2013 (Scheme 43). No protecting groups were required in the 9-step procedure starting from (R)-perillaldehyde. | ||
| Scheme 43 Ring-closing metathesis in the synthesis of spirocurcasone. | ||
4.5 Sequosempervirin A
Sequosempervirin A is a novel spiro-norlignan compound isolated in 2004 from the branches and leaves of the California redwood Sequoia sempervirens.131 Three years later, the first total synthesis of sequosempervirin A used ring-closing metathesis as the spirocyclisation step leading to a cyclic allylic alcohol en route to the final product132 (Scheme 44).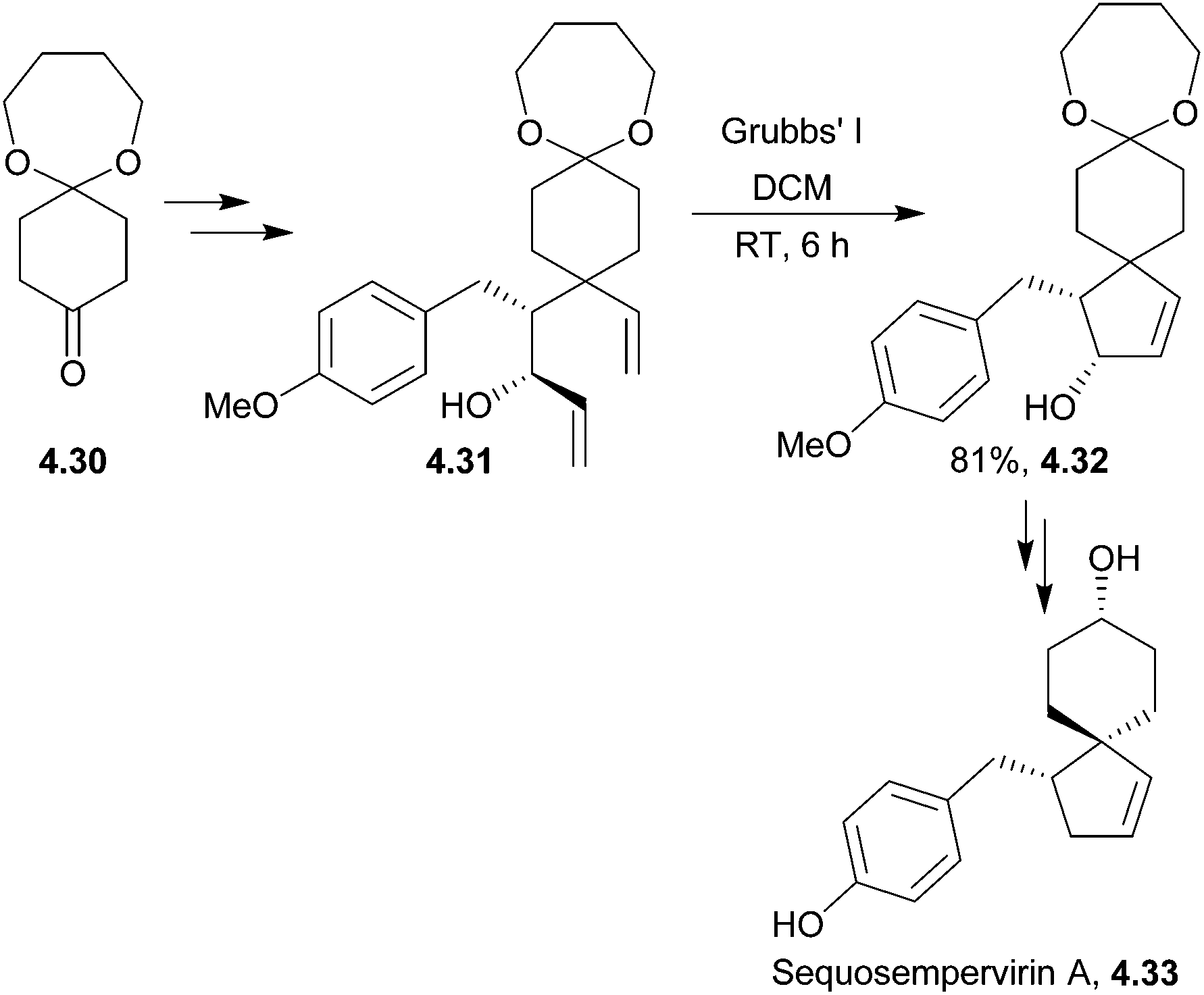 | ||
| Scheme 44 Ring-closing metathesis during the synthesis of sequosempervirin A. | ||
4.6 Chamigrenes: α-chamigrene, β-chamigrene, laurencenones B and C and elatol
Other members of the chamigrene natural product family not previously mentioned include β-chamigrene, laurencenone B and elatol. The potent and varied biological activity of elatol makes it a compound of significant interest. It has shown anti-biofouling activity,133 anti-bacterial activity,134–136 anti-fungal activity137 and cytotoxicity.138 It was isolated from red algae Laurencia sp., as were laurencenones B and C. Like α-chamigrene, β-chamigrene has been isolated from Schisandra chinensis, although it was first isolated from the leaf oil of the cypress Chamaecyparis taiwanensis.139(±)-α-Chamigrene was synthesised from cyclohexenecarboxylic acid140 by Srikrishna et al. in 2006. The procedure used an Ireland–Claisen rearrangement and ring-closing metathesis as previously demonstrated by the same group in the synthesis of (±)-isoacorone to give the product in 11 steps and 32% overall yield (Scheme 45).
 | ||
| Scheme 45 Ring-closing metathesis in the synthesis of racemic α-chamigrene. | ||
Racemic β-chamigrene and laurencenone C have been synthesised by an Ireland–Claisen rearrangement, followed by either ring-closing metathesis140 or an intramolecular type II carbonyl ene reaction141 by Srikrishna et al. The metathesis method required 11 steps to give the product in 32% overall yield starting from a cyclohexenecarboxylic acid. For laurencenone C, the initial method required 10 steps to give a 17% overall yield.
(+)-Laurencenone B, (+)-elatol142 and (−)-laurencenone C143 have all been synthesised by ring-closing metathesis by Stoltz et al. The starting material for both laurencenones was dimedone. The synthesis required 7 steps to give the products in 34% and 31% overall yield respectively (Scheme 46). (+)-Elatol required 9 steps to give the product in 11% overall yield.
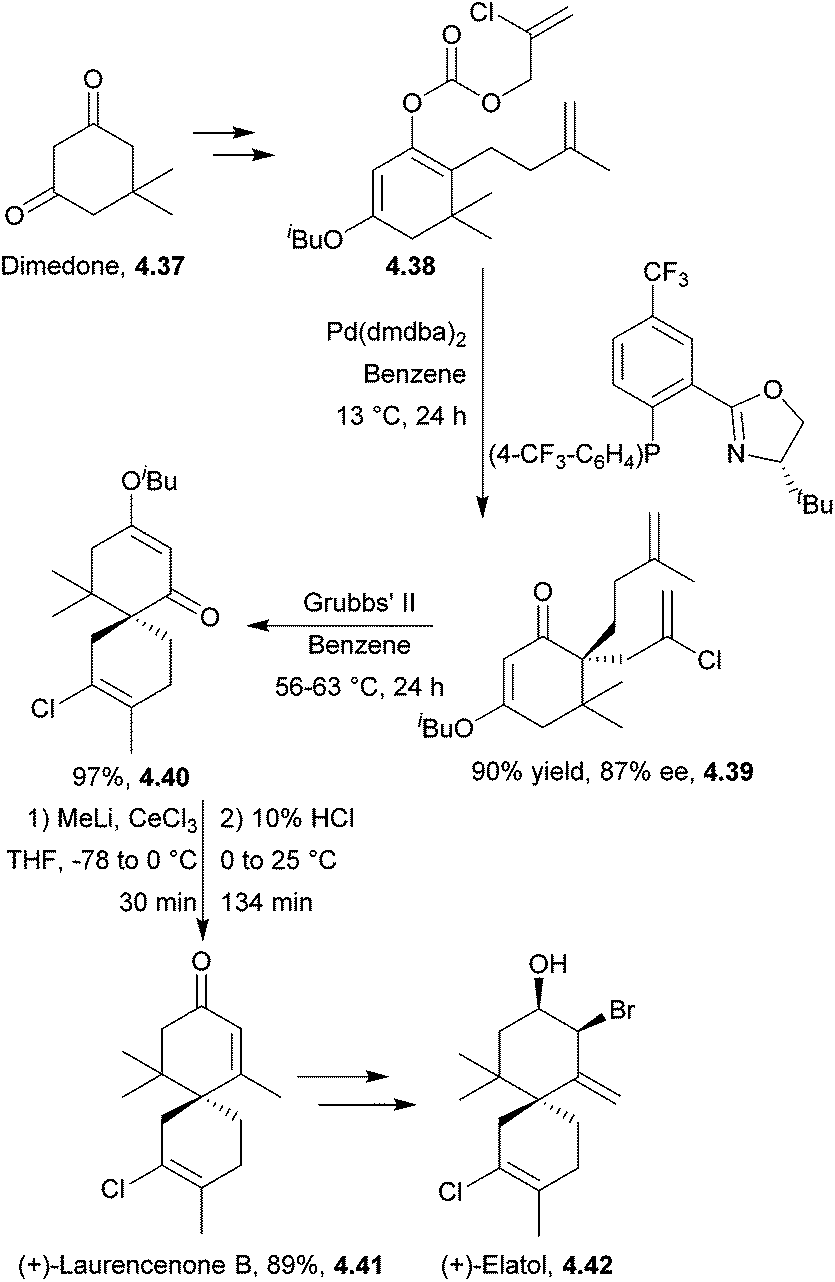 | ||
| Scheme 46 Ring-closing metathesis in the synthesis of (+)-laurencenone B and (+)-elatol. | ||
This catalytic asymmetric method was generalised to the chamigrene product family, using a palladium-based enantioselective decarboxylative allylation followed by ring-closing metathesis.143
5. Diels–Alder reaction
The Diels–Alder reaction is a valuable tool in total synthesis,144 since it forms two carbon–carbon bonds and up to four stereocentres. Its application to the synthesis of spirocarbocycle-containing natural products is described below.5.1 Acoranes: colletoic acid, β-, γ-, δ-acoradienes, acorone and isoacorone
For the avoidance of confusion, it should be noted that γ- and δ-acoradiene are also known as α- and β-alaskene respectively, due to their isolation from the Alaskan cedar, Chamaecyparis nootkatensis.145,146Retrosynthetic analysis of the acorane carbon skeleton reveals that the cyclohexene can be broken into two units, of which one is isoprene (Fig. 6).
 | ||
| Fig. 6 Retrosynthetic analysis of the carbon skeleton of colletoic acid. | ||
γ- and δ-Acoradiene were synthesised by Marx et al. as early as 1973147 using a method which was extended to β-acoradiene, acorone, and isoacorone two years later148 (Scheme 47). The starting material was (R)-pulegone, and a SnCl4-catalysed Diels–Alder reaction was used. The major products were those formed from the electronically favourable para orientation of the reagents, whilst the minor products were formed from the meta orientation. The Lewis acid increased selectivity for the products of para cycloaddition. The reaction had been performed without SnCl4, with heating to 100 °C and gave the same products, in the ratio 45:25:20:10. The transition state of the major isomer was formed by attack of the isoprene unit on the opposite face to the methyl group, reducing unfavourable steric interactions, and eventually leading to the formation of γ-acoradiene. The second isomer was formed by attack of the isoprene on the same face as the methyl group, which was then through further functionalisation converted to δ-acoradiene.
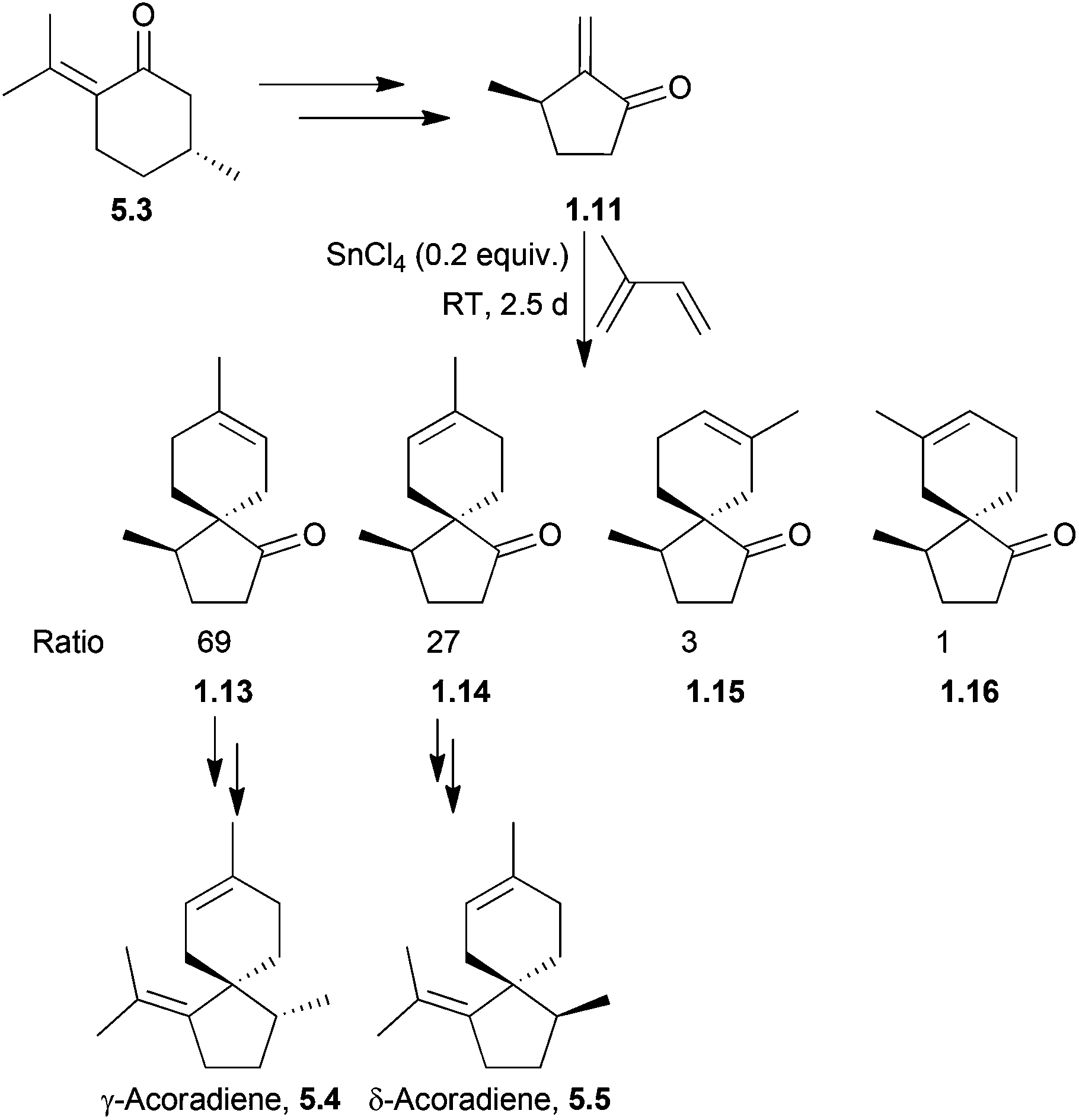 | ||
| Scheme 47 Lewis-acid catalysed Diels–Alder reaction with isoprene in the synthesis of γ- and δ-acoradienes. | ||
(+)-Colletoic acid is a sesquiterpenoid also from the acorane family and a potent inhibitor of the 11β-hydroxysteroid type 1 enzyme, making it a therapeutic target for metabolic syndrome.149 This compound was generated as a single enantiomer in 2013 by a diastereoselective intramolecular 5-exo-Heck reaction.150 At the same time, Nakada et al. used a stereoselective Lewis-acid catalysed Diels–Alder reaction with isoprene to create the spirocentre151,152 (Scheme 48). It was found that the starting material for the Diels–Alder reaction decomposed at high temperatures, but when run at −20 °C in the presence of the Lewis acid AlCl3, the reaction gave the desired product as a single isomer.
 | ||
| Scheme 48 Lewis-acid catalysed Diels–Alder reaction in the synthesis of (+)-colletoic acid. | ||
5.2 Ainsliadimers A and B and gochnatiolides A–C
The ainsliadimers and gochnatiolides are a related set of dimeric sesquiterpene lactones, biosynthesised from the guaianolide dehydrozaluzanin C.A biomimetic total synthesis of ainsliadimer A in 2010 by Lei et al. proceeded in 14 steps, starting from α-santonin153 (Scheme 49). The key step was a hydrogen bond-promoted [4 + 2]-hetero Diels–Alder dimerisation.
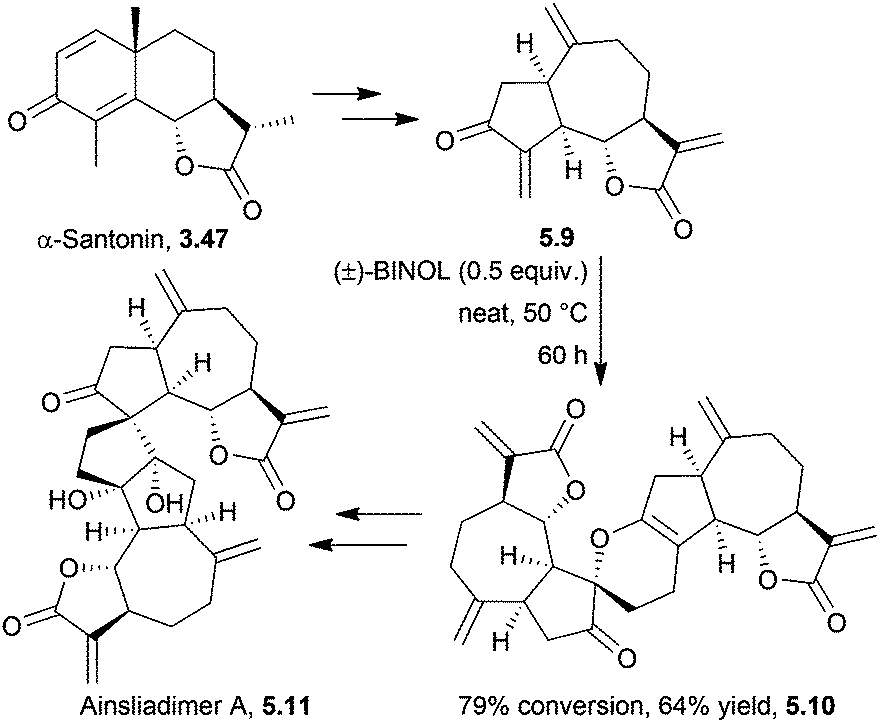 | ||
| Scheme 49 Hetero-[4 + 2] Diels–Alder reaction in the synthesis of ainsliadimer A. | ||
During an investigation into hydrogen bond donor catalysis, hydrogen-bonding solvents were used but only chloroform gave a trace amount of the dimer when the monomer was heated at 35 °C for 12 h. Additives including triethylamine or hydrochloric acid did not improve conversion, and longer reaction times and higher temperatures led to increased decomposition. Hydrogen bond donor catalysts were studied, and racemic BINOL was found to be particularly effective at catalysing the dimerisation.
More recently, Qin et al. reported the total syntheses of ainsliadimer B and gochnatiolides A and B which proceeded in 25 steps and 1% overall yield using a cross Diels–Alder cycloaddition154 (Scheme 50).
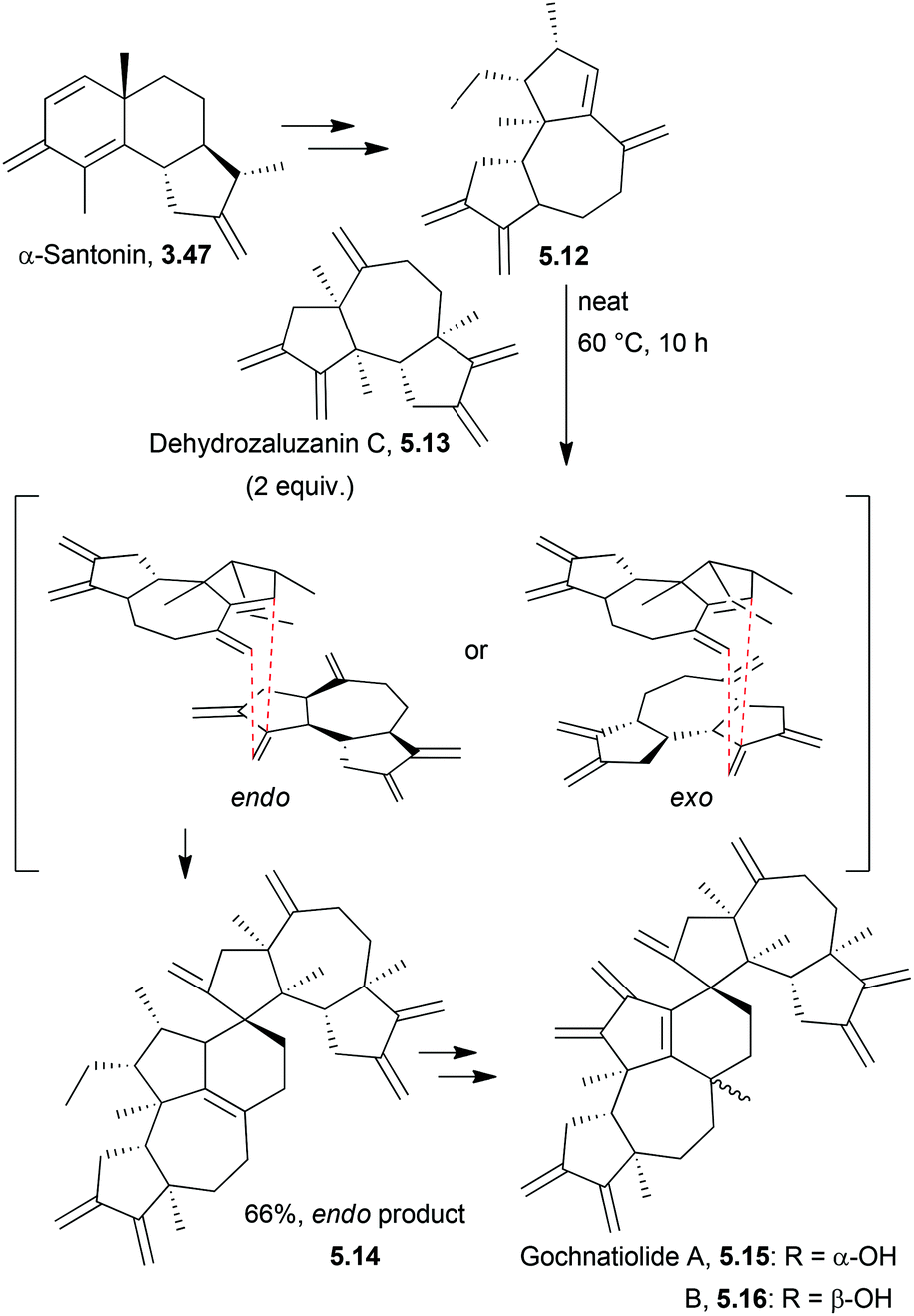 | ||
| Scheme 50 Cross Diels–Alder via an endo intermediate in the synthesis of ainsliadimer B and gochnatiolides A and B. | ||
The Diels–Alder reaction could proceed via either an endo or exo transition state, but only the product arising from the endo transition state was observed. The exo transition state would experience a high degree of steric interaction between the silyl groups on the bottom face of the diene and the seven-membered ring of the dehydrozazulin C. When attempted in a variety of solvents, the reaction proceeded slowly to give the Diels–Alder product in less than 15% yield and so the reaction was run neat. Addition of acids such as ceric ammonium sulfate, pyridinium p-toluenesulfonate, tosic acid and acetic acid failed to catalyse the reaction, instead causing the decomposition of the dehydrozaluzanin C product.
6. Claisen rearrangement
The Claisen rearrangement is a [3,3]-sigmatropic rearrangement where an allyl vinyl ether is converted to an unsaturated carbonyl.155,156 Its use in the synthesis of spiro sesquiterpenoids has been recently reviewed by Kobayashi et al.1576.1 Acoranes: acorone, isoacorone, α-acorenol and β-acorenol
As previously mentioned, (±)-acorone and (±)-isoacorone have been formally synthesised by the Claisen rearrangement followed by ring-closing metathesis. The Srikrishna group synthesised racemic α-acorenol and β-acorenol employing this strategy (see above).6.2 Spirovetivanes: agarospirol, hinesol, β-vetivone and α-vetispirene
(−)-Agarospirol,158,159 (+)-α-vetispirene, hinesol (an enantiomer of agarospirol) and β-vetivone have all been synthesised by the Kobayashi group using Claisen rearrangement methodologies160,161 (Scheme 51). | ||
| Scheme 51 Claisen rearrangement in the synthesis of agarospirol. The intermediate 6.3 was present in >95% dr. | ||
6.3 Spiroaxanes: axenol, axisonitrile-3 and gleenol
Axisonitrile-3 is a nitrogenous sesquiterpenoid of interest due to its significant biological activity, including anti-fouling activity against barnacle larvae,162 cytotoxicity163 and potent anti-malarial activity.164,165 It possesses an unusual isonitrile group. Axenol is a key intermediate in its synthesis.Racemic axenol and gleenol have been synthesised by the Kobayashi group in 2007,166,167 applying the same methodology as for the synthesis of other spiro[4.5]decanes described above.
Reduction and re-oxidation of the spiroketone gave a mixture of enantiomers in the ratio 77:23, which were epimerised by treatment with ethanolic KOH and then separated by column chromatography. Reduction with LiAlH4 gave a mixture of racemic gleenol and axenol (Scheme 52).
 | ||
| Scheme 52 Claisen rearrangement in the synthesis of (±)-gleenol and (±)-axenol. | ||
A Claisen rearrangement was used by the same group to synthesise (+)-axisonitrile-3 via (+)-gleenol and (−)-axamide-3168 in 2009 (Scheme 53).
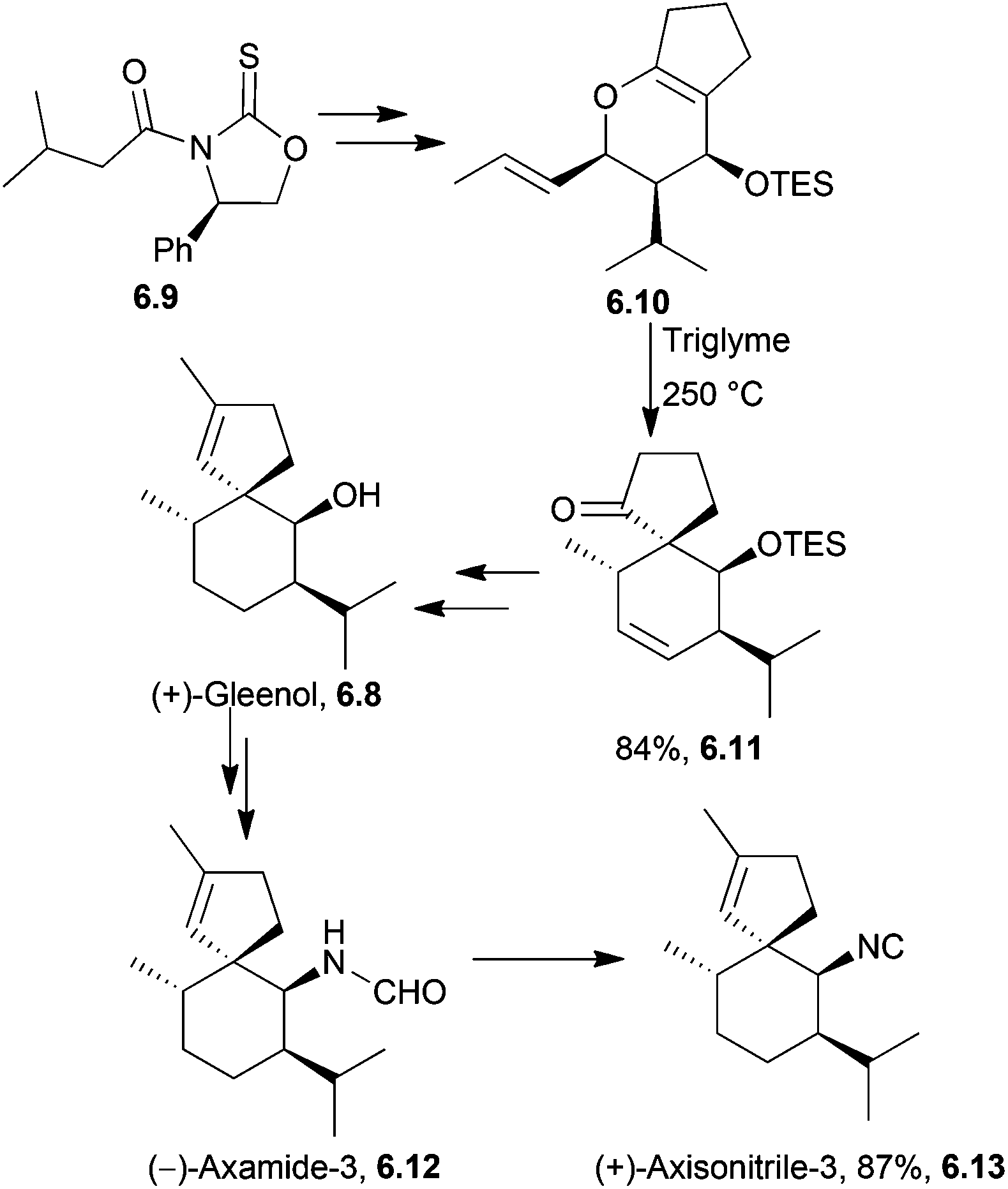 | ||
| Scheme 53 Claisen rearrangement in the synthesis of (+)-axisonitrile-3. The dr of the intermediate 6.11 was >95%. | ||
6.4 Erythrodiene and spirojatamol
Eilbracht et al. reported a formal synthesis of erythrodiene and spirojatamol which used a rhodium-based catalyst for a tandem Claisen rearrangement-hydroacylation of an allyl vinyl ether169 (Scheme 54). This method was also applied in 1996 for the synthesis of acoradienes and spirovetivanes as they share the spiro[4.5]decanone skeleton.170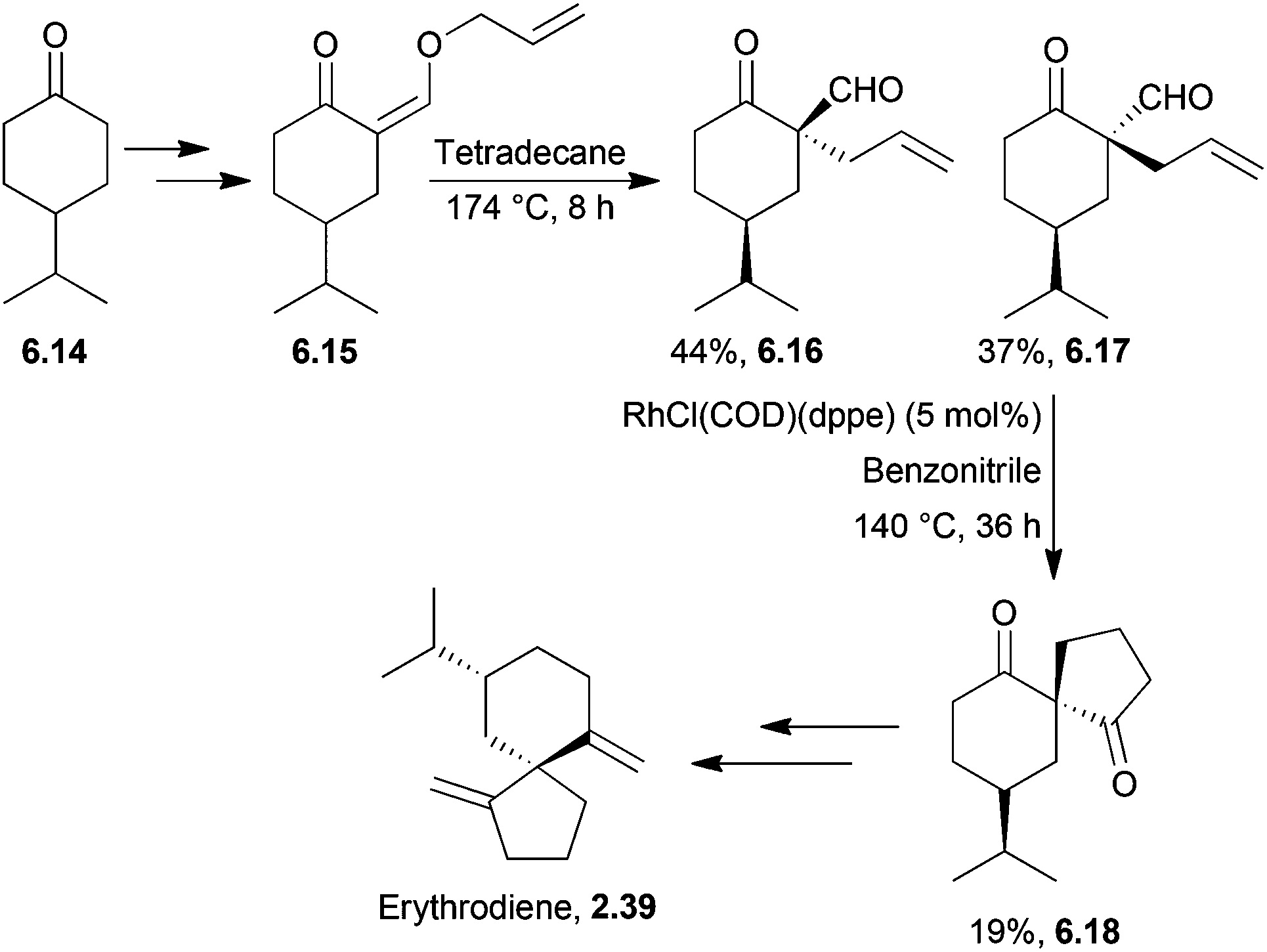 | ||
| Scheme 54 Rhodium-catalysed Claisen rearrangement in the synthesis of erythrodiene. | ||
The acoradiene and spirovetivane procedure was performed in one pot,171 and the particular rhodium catalyst chosen was selected because it did not decompose at the required higher temperatures and thus could catalyse the Claisen and hydroacylation reactions.172
7. Photochemical reaction
Photochemistry is a powerful tool used in total synthesis to access products that can be otherwise difficult to form. Its use in natural product synthesis has been extensively reviewed.173–1757.1 Acutumine
Acutumine is a tetracyclic alkaloid which shows selective T-cell cytotoxicity and anti-amnesic properties.176Castle et al. reported the first total synthesis of (−)-acutumine in 2009, which utilised a photochemical radical-polar crossover reaction to form the spirocycle177 (Scheme 55).
 | ||
| Scheme 55 Photochemical reaction in the synthesis of (−)-acutumine. No diastereomers were observed. | ||
The cascade process was observed at 0 °C with a sun-lamp. Optimisation experiments identified Et3Al as a more effective promoter than Et3B or Et2Zn, and 3-phenyl-2-(phenylsulfonyl)oxaziridine as a better hydroxylating agent than oxygen, DMDO or tBuOOH. Side-products were observed, with the alcohol group replaced by an iodide or hydrogen. Attempts were made to convert the iodide into the desired product by treatment with diethyl zinc, oxygen and the oxaziridine gave the desired product in 62% yield, thus increasing the overall yield to 66%. Samarium diiodide/HMPA was considered as a replacement for (Bu3Sn)2, but only gave a low yield of the desired product, and led to the formation of many side-products. This was attributed to the presence of functional groups in the starting material that could react with SmI2. The mechanism still requires further work to develop a better understanding.
Reisman et al. have attempted the synthesis of (−)-acutumine via a photochemical [2 + 2]-cycloaddition, but were only successful in constructing the aza-propellane core.178 (−)-Acutumine has also been synthesised through a Hosomi-Sakurai allylation in 17 steps179 by Herzon et al. in 2013.
7.2 Acoranes: α-acorenol, β-acorenol, (−)-acorenone and (±)-α-acoradiene
Photolysis was used in 1973 by Ramage et al. to stereospecifically synthesise (−)-α- and (+)-β-acorenol (Scheme 56).180 | ||
| Scheme 56 Photolysis in the synthesis of α-acorenol. | ||
An enone [2 + 2] photocycloaddition was used in 1982 by Baldwin et al. to synthesise (−)-acorenone181 (Scheme 57).
 | ||
| Scheme 57 Enone [2 + 2] photocycloaddition in the synthesis of (−)-acorenone. | ||
The key photochemical step was highly regio- and stereospecific, with the furanone reacting only in a head-to-tail manner. (−)-Acorenone was synthesised in a total of 13 steps in approximately 8% overall yield.
Irradiation of an enone was also used as the key step by Oppolzer et al. in 1983 in the synthesis of racemic α-acoradiene182 (Scheme 58).
 | ||
| Scheme 58 Photochemical cyclobutane formation followed by reductive fragmentation in the synthesis of racemic α-acoradiene. The configurations of 7.13 and 7.14 were not determined because this chirality was lost by reduction. | ||
The low yield was ascribed to the difficulty in separating the various isomers produced by the radical reaction. Formation of the diradical led to a loss of stereochemical information, and resulted in the formation of two enantiomers, of which reductive fragmentation of only one gave the desired product.
7.3 Spirovetivanes: lubiminol, agarospirol, hinesol, β-vetivone and α-vetispirene
Lubiminol is a spirovetivadiene phytoalexin isolated from infected potatoes183,184 and the red pepper Capsicum annuum.185A double diastereoselective intramolecular photocycloaddition has been used to synthesise the spiro centre of lubiminol (Scheme 59), followed by a radical fragmentation–rearrangement reaction, on which Crimmins et al. have published a series of articles.186–188
 | ||
| Scheme 59 Photocycloaddition during the synthesis of lubiminol. | ||
The reaction proceeded via a Dowd–Beckwith type ring expansion to give the spirocyclic intermediate.
Takeshita et al. used photocycloaddition followed by a base-catalysed benzilic acid rearrangement in 1995 to synthesise racemic hinesol and agarospirol189 (Scheme 60).
 | ||
| Scheme 60 Photochemical reaction in the synthesis of racemic hinesol and agarospirol. | ||
As the rearrangement was not triggered using triethylamine as a base, it was postulated that formation of a sodium chelated intermediate could assist in the rearrangement.
(−)-β-Vetivone has been synthesised by a dienone ring contraction of a spiro starting material with irradiation in acid to form a spiroketone.45 Photolysis of a cross-conjugated diene was used by Caine et al. to synthesise α-vetispirene190 (Scheme 61) in 1976.
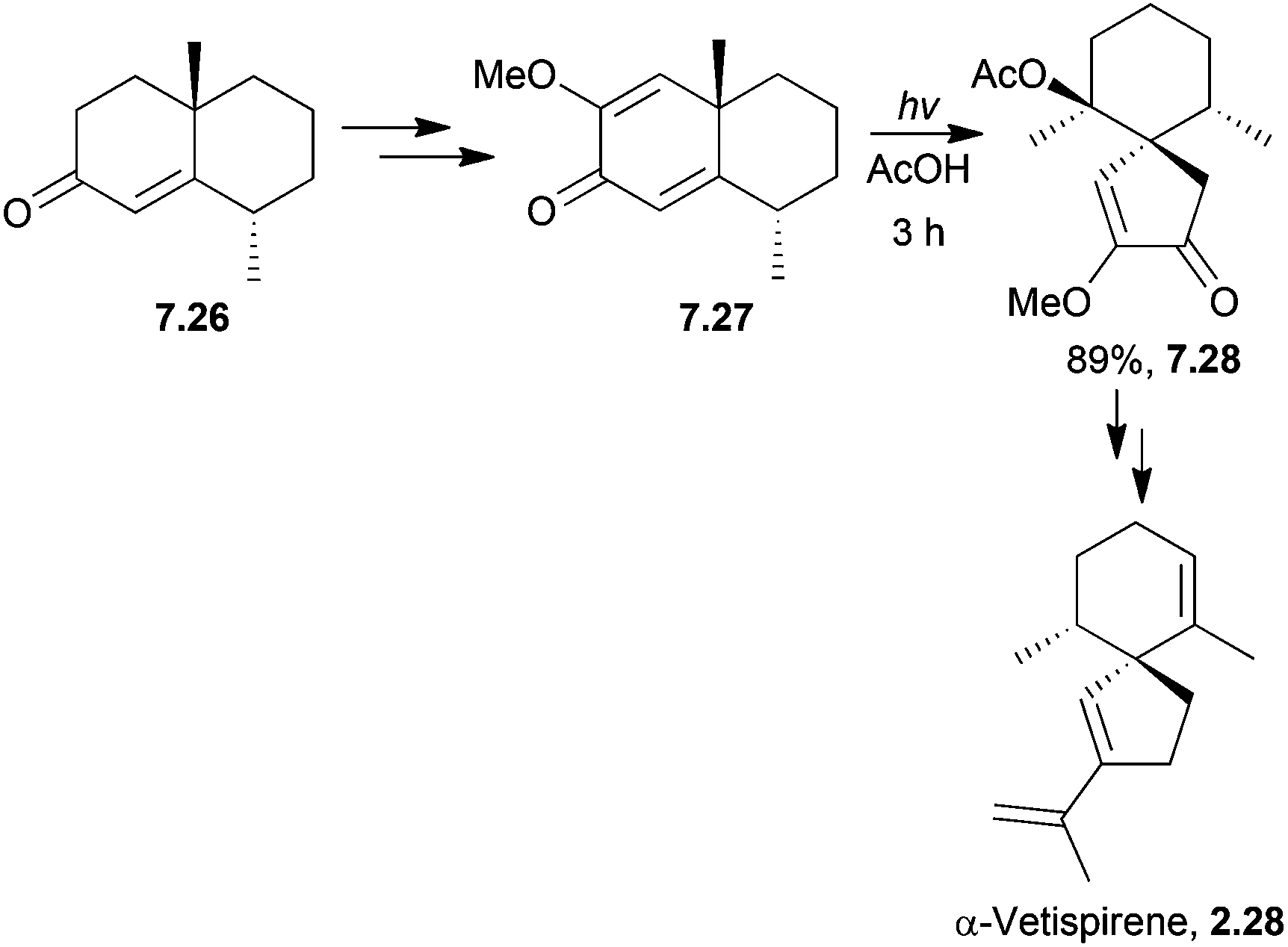 | ||
| Scheme 61 Photolysis in the synthesis of α-vetispirene. | ||
Glacial rather than aqueous acetic acid gave a dramatic improvement in yield (28% to 89%), and a reduction in side-product formation. The absence of water prevented hydrolysis of the enol ether.
7.4 Proaporphines
The proaporphines are a major group of isoquinoline alkaloids. They are biosynthetic precursors to aporphine alkaloids which exhibit a broad range of biological activity.Photolysis has been used by the Iwata and Fukumoto groups. to synthesise pronuciferine,191,192 orientalinone,193O-methyl-orientalinone,194 and O-methyl-kreysiginone195 (Scheme 62).
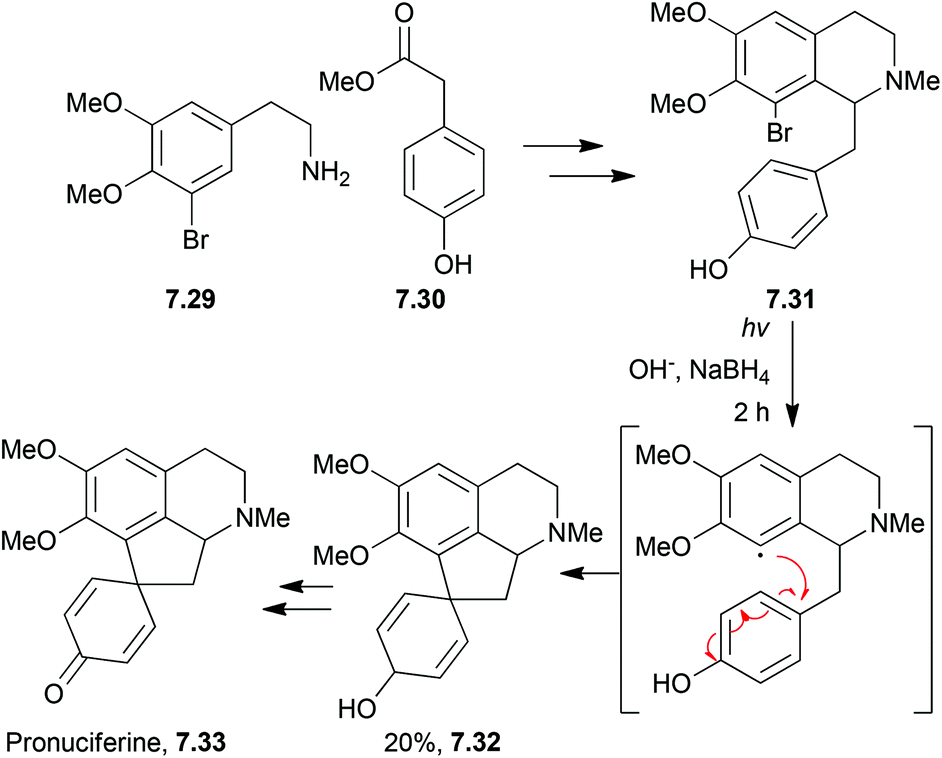 | ||
| Scheme 62 Photolysis of an intermediate in the synthesis of pronuciferine. | ||
Pronuciferine has been synthesised using methyl vinyl ketone in base,196 and more recently pronuciferine and stepharine have been synthesised by aromatic oxidation with a hypervalent iodine reagent.197 This led to the unprecedented C–C bond formation between the para position of a phenol and an enamide carbon.
7.5 Bilinderone and linderaspirone A
(±)-Bilinderone198 and (±)-linderaspirone A199 show promising activity in the improvement of insulin sensitivity. Linderaspirone A is a highly symmetrical compound with a [4.3.4.3]bispiro core, whereas bilinderone has a [4.5]spiro core.Linderones have been synthesised biomimetically by Wang et al. in 2011.200 The key step was a mild photochemical dimerisation. (±)-Linderaspirone A was synthesised by a [2 + 2]-cycloaddition-Cope rearrangement (Scheme 63), whilst (±)-bilinderone was synthesised by a [2 + 2]-cycloaddition-radical rearrangement.
 | ||
| Scheme 63 Photochemical dimerisation in the synthesis of linderaspirone A and bilinderone. | ||
Irradiation of the neat starting material as a solid state thin-film produced a mixture of products, including linderaspirone A, bilinderone, epi-bilinderone and an additional side product containing an unusual spiro[3.4] system. Photolysis in various solvents, including methanol, acetone, THF, acetonitrile, toluene and benzene all gave yields of linderaspirone A and bilinderone in less than 20%.
In 2013, Hu et al. synthesised linderaspirone A in 3 steps as well as bilinderone using a Darzens-type cyclopentadione reaction (Scheme 64).201 The starting material was the same as was used above.
 | ||
| Scheme 64 Photochemical reaction in the synthesis of linderaspirone A. | ||
When performed under oxygen rather than argon, the yield of linderaspirone A increased from 38% to 50%.
8. Conclusions
Spirocarbocycles are important structural motifs found in drugs, ligands and natural products, and so strategies towards their challenging synthesis are key. This review has examined several common reactions used to form these quaternary centres, including the use of acid and base, particularly in the aldol reaction. Ring-closing metathesis, the Diels–Alder reaction, Claisen rearrangements and photochemistry have also been used to synthesise these spirocarbocycles.As seen above, the aldol reaction has been successful on a wide range of carbon skeletons, although it lacks stereoselectivity unless the substrate already contains the desired chirality. High yields are achievable with suitable starting materials but if a mixture of products are obtained, the efficiency decreases. This is similarly true for those reactions effected using acid or base, but since these do not operate under one mechanism, it is difficult to generalise. Stereoselectivity depends on both the reagents and the reaction conditions: for example, the possibility of a 6-membered chair transition state can increase selectivity. It is well-known that ring-closing metathesis typically proceeds in excellent yields and, like the Claisen rearrangement, is highly atom efficient. This also applies to the Diels–Alder reaction, which can be chemo-, regio-, diastereo- and enantioselective. It is important to note, however, that a substrate's suitability for these methods depends on its carbon skeleton and structure. Finally, photochemical reactions are often not the obvious first choice for many organic chemists, and as shown above, their use is again highly substrate-dependent. It can be possible to get complex mixtures of products, but on occasion high yields are achievable, as shown by several examples above.
More work is required to develop new and improved routes to these compounds and others in the literature that are yet to be synthesised. As highlighted above, some natural products with biological applications still require syntheses. Examples of compounds discovered in the last ten years with biological activity which are yet to be synthesised include biyouyanagiol,202 spiromamakone A,203 and dibelamcandal A204 (Fig. 7).
 | ||
| Fig. 7 Unsynthesised natural products containing spirocarbocycles. | ||
Other natural products would benefit from more efficient routes, for example by the application of modern techniques to improve selectivity in older methods. Whilst progress over the last fifty years has been substantial, there is potential for further synthesis and exploitation of natural products containing spirocarbocycles.
Acknowledgements
The authors would like to gratefully acknowledge financial support from the Royal Society (IRB) and the EPSRC (LKS).Notes and references
- G. Moss, Pure Appl. Chem., 1999, 71, 531–558 CAS.
- L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023–1052 CrossRef CAS PubMed.
- G. Singh and Z. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- A. Pfau and P. Plattner, Helv. Chim. Acta, 1939, 22, 640–654 CrossRef PubMed.
- J. A. Marshall and P. Johnson, J. Am. Chem. Soc., 1967, 89, 2750–2751 CrossRef CAS.
- P. Johnson and J. Marshall, Chem. Commun., 1968, 391 Search PubMed.
- Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed.
- K. Ding, Chem. – Asian J., 2009, 4, 32–41 CrossRef CAS PubMed.
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- M. Sannigrahi, Tetrahedron, 1999, 55, 9007–9071 CrossRef CAS.
- E. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef.
- K. Fuji, Chem. Rev., 1993, 93, 2037–2066 CrossRef CAS.
- R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC.
- J. Peng, M. A. Avery and M. T. Hamann, Org. Lett., 2003, 5, 4575–4578 CrossRef CAS PubMed.
- J. Peng, K. Walsh, V. Weedman, J. D. Bergthold, J. Lynch, K. L. Lieu, I. A. Braude, M. Kelly and M. T. Hamann, Tetrahedron, 2002, 58, 7809–7819 CrossRef CAS.
- T. J. Reddy, G. Bordeau and L. Trimble, Org. Lett., 2006, 8, 5585–5588 CrossRef CAS PubMed.
- B. R. Moser, J. Nat. Prod., 2008, 71, 487–491 CrossRef CAS PubMed.
- D. F. Taber and K. V. Taluskie, J. Org. Chem., 2006, 71, 2797–2801 CrossRef CAS PubMed.
- D. F. Taber, J.-M. Joerger and K. V. Taluskie, Pure Appl. Chem., 2008, 80, 1141–1148 CrossRef CAS.
- D. F. Taber and J. Joerger, J. Org. Chem., 2008, 73, 4155–4159 CrossRef CAS PubMed.
- B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506–7525 CrossRef CAS PubMed.
- T. Machajewski and C. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352–1375 CrossRef CAS.
- G. Guella, F. Dini and F. Pietra, Angew. Chem., Int. Ed., 1999, 38, 1134–1136 CrossRef CAS.
- K. C. Nicolaou, H. Zhang and A. Ortiz, Angew. Chem., Int. Ed., 2009, 48, 5642–5647 CrossRef CAS PubMed.
- K. C. Nicolaou, M. P. Jennings and P. Dagneau, Chem. Commun., 2002, 2480–2481 RSC.
- K. C. Nicolaou, W. Tang, P. Dagneau and R. Faraoni, Angew. Chem., Int. Ed., 2005, 44, 3874–3879 CrossRef CAS PubMed.
- K. C. Nicolaou, H. Zhang, A. Ortiz and P. Dagneau, Angew. Chem., Int. Ed., 2008, 47, 8605–8610 CrossRef CAS PubMed.
- K. C. Nicolaou, A. Ortiz and H. Zhang, Angew. Chem., Int. Ed., 2009, 48, 5648–5652 CrossRef CAS PubMed.
- K. C. Nicolaou, A. Ortiz, H. Zhang, P. Dagneau, A. Lanver, M. P. Jennings, S. Arseniyadis, R. Faraoni and D. E. Lizos, J. Am. Chem. Soc., 2010, 132, 7138–7152 CrossRef CAS PubMed.
- G. Saielli, K. C. Nicolaou, A. Ortiz, H. Zhang and A. Bagno, J. Am. Chem. Soc., 2011, 133, 6072–6077 CrossRef CAS PubMed.
- J. Vrkoc, V. Herout and F. Sorm, Collect. Czech Chem. Commun., 1961, 26, 3183–3185 CrossRef CAS.
- B. Tomita, T. Isono and Y. Hirose, Tetrahedron Lett., 1970, 11, 1371–1372 CrossRef.
- A. Srikrishna and R. Ramesh Babu, Tetrahedron Lett., 2007, 48, 6916–6919 CrossRef CAS PubMed.
- D. McCrae and L. Dolby, J. Org. Chem., 1977, 42, 1607–1610 CrossRef CAS.
- S. Martin and T. Chou, J. Org. Chem., 1978, 43, 1027–1031 CrossRef CAS.
- A. Srikrishna, P. P. Kumar and R. Viswajanani, Tetrahedron Lett., 1996, 37, 1683–1686 CrossRef CAS.
- A. Srikrishna and P. P. Kumar, Tetrahedron, 2000, 56, 8189–8195 CrossRef CAS.
- T. Tashiro, S. Kurosawa and K. Mori, Biosci. Biotechnol. Biochem., 2004, 68, 663–670 CrossRef CAS.
- B. Trost, K. Hiroi and N. Holy, J. Am. Chem. Soc., 1975, 97, 5873–5877 CrossRef CAS.
- I. Ahuja, R. Kissen and A. M. Bones, Trends Plant Sci., 2012, 17, 73–90 CrossRef CAS PubMed.
- N. Andersen, M. Falcone and D. Syrdal, Tetrahedron Lett., 1970, 11, 1759–1762 CrossRef.
- E. Wenkert, B. L. Buckwalter, A. A. Craveiro, E. L. Sanchez and S. S. Sathe, J. Am. Chem. Soc., 1978, 100, 1267–1273 CrossRef CAS.
- R. Hutchins, N. Natele, I. Taffer and R. Zipkin, Synth. Commun., 1984, 14, 445–451 CrossRef CAS PubMed.
- M. L. Maheshwari, S. C. Bhattacharyya and K. Varma, Tetrahedron, 1965, 21, 115–138 CrossRef.
- C. Hughes, M. Deighton and R. Ramage, J. Chem. Soc., Chem. Commun., 1975, 662–663 Search PubMed.
- G. Balme, Tetrahedron Lett., 1985, 26, 2309–2310 CrossRef CAS.
- D. Coxon, K. Price and B. Howard, Tetrahedron Lett., 1974, 15, 2921–2924 CrossRef.
- A. Srikrishna and S. S. V. Ramasastry, Tetrahedron Lett., 2005, 46, 7373–7376 CrossRef CAS PubMed.
- Y. Tokunaga, M. Yagihashi, M. Ihara and K. Fukumoto, J. Chem. Soc., Perkin Trans. 1, 1997, 189–190 RSC.
- H. Huang and C. J. Forsyth, Tetrahedron Lett., 1993, 34, 7889–7890 CrossRef.
- H. Huang and C. J. Forsyth, J. Org. Chem., 1995, 60, 2773–2779 CrossRef CAS.
- W. Oppolzer and F. Flachsmann, Helv. Chim. Acta, 2001, 84, 416–430 CrossRef CAS.
- M. Lachia, F. Dénès, F. Beaufils and P. Renaud, Org. Lett., 2005, 7, 4103–4106 CrossRef CAS PubMed.
- A. Matsuo, S. Uto, H. Nozaki and M. Nakayama, J. Chem. Soc., Perkin Trans. 1, 1984, 215–221 RSC.
- M. Kodama, U. Tambunan, T. Tsunoda and S. Ito, Bull. Chem. Soc. Jpn., 1986, 59, 1897–1900 CrossRef CAS.
- E. Boeren, M. El-Sohly, C. Turner and A. Salemink, Experientia, 1976, 33, 848 CrossRef.
- C. Bercht, J. van Dongen, W. Heerma, R. Lousberg and F. Kueppers, Tetrahedron, 1976, 32, 2939–2943 CrossRef CAS.
- T. Ottersen, A. Aasen, F. S. El-Feraly and C. Turner, J. Chem. Soc., Chem. Commun., 1976, 580–581 RSC.
- F. S. El-Feraly, M. Elsohly, E. Boeren and C. Turner, Tetrahedron, 1977, 33, 2373–2378 CrossRef CAS.
- Y. Shoyama and I. Nishioka, Chem. Pharm. Bull., 1978, 26, 3641–3646 CrossRef CAS.
- L. Crombie, W. Crombie and S. Jamieson, Tetrahedron Lett., 1979, 20, 661–664 CrossRef.
- J. Kettenes-van den Bosch and C. Salemink, Recl.: J. R. Neth. Chem. Soc., 1978, 97, 221–222 CAS.
- L. Crombie, P. Tuchinda and M. J. Powell, J. Chem. Soc., Perkin Trans. 1, 1982, 1477–1484 RSC.
- L. Crombie, M. Powell and P. Tuchinda, Tetrahedron Lett., 1980, 21, 3603–3606 CrossRef CAS.
- F. El-Feraly and Y. Chan, J. Nat. Prod., 1981, 44, 557–561 CrossRef CAS.
- N. Natale, B. Marron, E. Evain and C. Dodson, Synth. Commun., 1984, 14, 599–603 CrossRef CAS PubMed.
- J. Novak and C. A. Salemink, J. Chem. Soc., Perkin Trans. 1, 1982, 2403–2405 RSC.
- M. Suzuki, E. Kurosawa and K. Kurata, Bull. Chem. Soc. Jpn., 1987, 60, 3795–3796 CrossRef CAS.
- D. Kennedy, I. Selby and R. Thomson, Phytochemistry, 1988, 27, 1761–1766 CrossRef CAS.
- K.-C. Chu, H.-J. Liu and J.-L. Zhu, Synlett, 2010, 3061–3064 CAS.
- J. Zhu, P. Huang, R. You, F. Lee, S. Taso and I. Chen, Synthesis, 2011, 715–722 CrossRef CAS.
- H. Wolf and M. Kolleck, Tetrahedron Lett., 1975, 16, 451–454 CrossRef.
- J. Ruppert, M. Avery and J. White, J. Chem. Soc., Chem. Commun., 1976, 978 RSC.
- M. Pesaro and J.-P. Bachmann, J. Chem. Soc., Chem. Commun., 1978, 203–204 RSC.
- S. Nagumo, H. Suemune and K. Sakai, J. Chem. Soc., Chem. Commun., 1990, 1778–1779 RSC.
- X. D. Yue, L. Chen and W. D. Z. Li, Tetrahedron, 2014, 70, 5505–5512 CrossRef CAS PubMed.
- K. Aoki, K. Yamada, H. Nagase, Y. Hayakawa and Y. Hirata, Tetrahedron Lett., 1973, 14, 4967–4970 CrossRef.
- K. Yamada, H. Nagase, Y. Hayakawa, K. Aoki and Y. Hirata, Tetrahedron Lett., 1973, 14, 4963–4966 CrossRef.
- P. McCurry and R. Singh, Tetrahedron Lett., 1973, 14, 3325–3328 CrossRef.
- M. Mongrain, J. Lafontaine, A. Bélanger and P. Deslongchamps, Can. J. Chem., 1970, 48, 3273–3274 CrossRef CAS.
- J. Lafontaine, M. Mongrain, M. Sergent-Guay, L. Ruest and P. Deslongchamps, Can. J. Chem., 1980, 58, 2460–2476 CrossRef CAS.
- G. Stork, R. Danheiser and B. Ganem, J. Am. Chem. Soc., 1973, 95, 3414–3415 CrossRef CAS.
- N. Maulide, J.-C. Vanherck and I. E. Markó, Eur. J. Org. Chem., 2004, 3962–3967 CrossRef CAS PubMed.
- G. Posner and T. Hamill, J. Org. Chem., 1988, 53, 6031–6035 CrossRef CAS.
- A. Murai, S. Sato and T. Masamune, Bull. Chem. Soc. Jpn., 1984, 57, 2276–2281 CrossRef CAS.
- S. N. Janaki and G. S. R. Subba Rao, J. Chem. Soc., Perkin Trans. 1, 1997, 195–200 RSC.
- J. A. Marshall and S. F. Brady, J. Org. Chem., 1970, 35, 4068–4077 CrossRef CAS.
- T. Ito and T. Takahashi, Agric. Biol. Chem., 1979, 43, 413–414 CrossRef CAS.
- T. Fujimori, R. Uegaki, Y. Takagi, S. Kubo and K. Kato, Phytochemistry, 1979, 18, 2032 CrossRef CAS.
- R. Uegaki, T. Fujimori, S. Kubo and K. Kato, Phytochemistry, 1981, 20, 1567–1568 CrossRef CAS.
- R. Uegaki, S. Kubo and T. Fujimori, Phytochemistry, 1988, 27, 365–368 CrossRef CAS.
- G. Birnbaum, C. Huber and M. Post, J. Chem. Soc., Chem. Commun., 1976, 330–331 RSC.
- A. Stoessl, J. Stothers and E. Ward, Can. J. Chem., 1978, 56, 645–653 CrossRef CAS.
- A. Murai, S. Sato and T. Masamune, Bull. Chem. Soc. Jpn., 1984, 57, 2286–2290 CrossRef CAS.
- A. Murai, S. Sato and T. Masamune, Bull. Chem. Soc. Jpn., 1984, 57, 2291–2294 CrossRef CAS.
- G. Blay, L. Cardona, A. M. Collado, B. García, V. Morcillo and J. R. Pedro, J. Org. Chem., 2004, 69, 7294–7302 CrossRef CAS PubMed.
- H. Magari, H. Hirota, T. Takahashi, A. Matsuo, S. Uto, H. Nozaki, M. Nakayama and S. Hayashi, Chem. Lett., 1982, 1143–1146 CrossRef CAS.
- M. Suzuki, N. Kowata and E. Kurosawa, Tetrahedron, 1980, 36, 1551–1556 CrossRef CAS.
- M. Suzuki, E. Kurosawa and T. Irie, Tetrahedron Lett., 1970, 11, 4995–4998 CrossRef.
- A. Murai, K. Kato and T. Masamune, Tetrahedron Lett., 1982, 23, 2887–2890 CrossRef CAS.
- J. Plamondon and P. Canonne, Tetrahedron Lett., 1991, 32, 589–592 CrossRef CAS.
- D. F. Taber, M. I. Sikkander and P. H. Storck, J. Org. Chem., 2007, 72, 4098–4101 CrossRef CAS PubMed.
- G. Chianese, E. Fattorusso, O. O. Aiyelaagbe, P. Luciano, H. C. Schröder, W. E. G. Müller and O. Taglialatela-Scafati, Org. Lett., 2011, 13, 316–319 CrossRef CAS PubMed.
- X. Li, Y. Yang, X. Peng, M. Li, L. Li, X. Deng, H. Qin, J. Liu and M. Qiu, Org. Lett., 2014, 16, 2196–2199 CrossRef CAS PubMed.
- V. A. D'yakonov, O. A. Trapeznikova, A. de Meijere and U. M. Dzhemilev, Chem. Rev., 2014, 114, 5775–5814 CrossRef PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS PubMed.
- A. Gradillas and J. Pérez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086–6101 CrossRef CAS PubMed.
- S. Kotha and E. Manivannan, ARKIVOC, 2003, 67–76 CAS.
- M. Anchel, A. Hervey and W. Robbins, Proc. Natl. Acad. Sci. U. S. A., 1950, 36, 300–305 CrossRef CAS.
- T. McMorris and M. Anchel, J. Am. Chem. Soc., 1963, 85, 831–832 CrossRef CAS.
- T. McMorris and M. Anchel, J. Am. Chem. Soc., 1965, 87, 1594–1600 CrossRef CAS.
- M. Kelner, T. McMorris, W. Beck, J. Zamora and R. Taetle, Cancer Res., 1987, 47, 3186–3189 CAS.
- T. McMorris, M. Kelner, W. Wang, L. Estes, M. Montoya and R. Taetle, J. Org. Chem., 1992, 57, 6876–6883 CrossRef CAS.
- F. Kinder, R. Wang, W. Bauta and K. Bair, Bioorg. Med. Chem. Lett., 1996, 6, 1029–1034 CrossRef CAS.
- M. Movassaghi, G. Piizzi, D. S. Siegel and G. Piersanti, Angew. Chem., Int. Ed., 2006, 45, 5859–5863 CrossRef CAS PubMed.
- D. S. Siegel, G. Piizzi, G. Piersanti and M. Movassaghi, J. Org. Chem., 2009, 74, 9292–9304 CrossRef CAS PubMed.
- T. C. McMorris, M. D. Staake and M. J. Kelner, J. Org. Chem., 2004, 69, 619–623 CrossRef CAS PubMed.
- M. Movassaghi, G. Piizzi, D. S. Siegel and G. Piersanti, Tetrahedron Lett., 2009, 50, 5489–5492 CrossRef CAS PubMed.
- T. Matsumoto, H. Shirahama, A. Ichihara, H. Shin, S. Kagaqa, F. Sakan, S. Matsumoto and S. Nishida, J. Am. Chem. Soc., 1968, 90, 3280–3281 CrossRef CAS.
- T. Matsumoto, H. Shirahama, A. Ichihara, H. Shin, S. Kagawa, F. Sakan and K. Miyano, Tetrahedron Lett., 1971, 2049–2052 CrossRef CAS.
- Q. Huang, Y. Tezuka, Y. Hatanaka, T. Kikuchi, A. Nishi and K. Tubaki, Chem. Pharm. Bull., 1995, 43, 1035–1038 CrossRef CAS.
- N. L. Brock and J. S. Dickschat, Eur. J. Org. Chem., 2011, 5167–5175 CrossRef CAS PubMed.
- A. Srikrishna and M. Rao, Indian J. Chem., Sect. B, 2008, 47, 1423–1429 Search PubMed.
- S. Kurosawa, M. Bando and K. Mori, Eur. J. Org. Chem., 2001, 4395–4399 CrossRef CAS.
- K. Oesterreich, I. Klein and D. Spitzner, Synlett, 2002, 1712–1714 CAS.
- K. Oesterreich and D. Spitzner, Tetrahedron, 2002, 58, 4331–4334 CrossRef CAS.
- E. J. Corey and S. Nozoe, J. Am. Chem. Soc., 1965, 87, 5728–5733 CrossRef CAS.
- T. Kauffmann, Angew. Chem., Int. Ed., 1997, 36, 1258–1275 CrossRef PubMed.
- T. Ozaki and Y. Kobayashi, Synlett, 2015, 1085–1088 CAS.
- H. Abe, A. Sato, T. Kobayashi and H. Ito, Org. Lett., 2013, 15, 1298–1301 CrossRef CAS PubMed.
- Y.-M. Zhang, N.-H. Tan, M. He, Y. Lu, S.-Q. Shang and Q.-T. Zheng, Tetrahedron Lett., 2004, 45, 4319–4321 CrossRef CAS PubMed.
- S. Maity and S. Ghosh, Tetrahedron Lett., 2007, 48, 3355–3358 CrossRef CAS PubMed.
- I. Granado and P. Caballero, Sci. Mar., 1995, 59, 21–39 Search PubMed.
- J. D. Martin, C. Perez and J. L. Ravelo, J. Am. Chem. Soc., 1986, 108, 7801–7811 CrossRef CAS PubMed.
- C. S. Vairappan, M. Daitoh, M. Suzuki, T. Abe and M. Masuda, Phytochemistry, 2001, 58, 291–297 CrossRef CAS.
- C. S. Vairappan, Biomol. Eng., 2003, 20, 255–259 CrossRef CAS.
- G. M. König and A. D. Wright, J. Nat. Prod., 1997, 60, 967–970 CrossRef PubMed.
- T. Dias, I. Brito, L. Moujir, N. Paiz, J. Darias and M. Cueto, J. Nat. Prod., 2005, 68, 1677–1679 CrossRef CAS PubMed.
- Y. Ohta and Y. Hirose, Tetrahedron Lett., 1968, 2483–2485 CrossRef CAS.
- A. Srikrishna, B. V. Lakshmi and M. Mathews, Tetrahedron Lett., 2006, 47, 2103–2106 CrossRef CAS PubMed.
- A. Srikrishna and R. Ramesh Babu, Tetrahedron, 2008, 64, 10501–10506 CrossRef CAS PubMed.
- D. E. White, I. C. Stewart, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 810–811 CrossRef CAS PubMed.
- D. E. White, I. C. Stewart, B. A. Seashore-Ludlow, R. H. Grubbs and B. M. Stoltz, Tetrahedron, 2010, 66, 4668–4686 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- N. H. Andersen and D. D. Syrdal, Tetrahedron Lett., 1970, 26, 2277–2280 CrossRef.
- N. Andersen and D. Syrdal, Tetrahedron Lett., 1972, 10, 899–902 CrossRef.
- J. N. Marx and L. R. Norman, Tetrahedron Lett., 1973, 4375–4378 CrossRef CAS.
- J. N. Marx and L. Norman, J. Org. Chem., 1975, 40, 1602–1606 CrossRef CAS.
- A. Aoyagi, M. Ito-kobayashi, Y. Ono, Y. Furukawa, M. Takahashi, Y. Muramatsu, M. Umetani and T. Takatsu, J. Antibiot., 2008, 61, 136–141 CrossRef CAS PubMed.
- T. Ling, E. Griffith, K. Mitachi and F. Rivas, Org. Lett., 2013, 15, 5790–5793 CrossRef CAS PubMed.
- T. Sawada and M. Nakada, Org. Lett., 2013, 15, 1004–1007 CrossRef CAS PubMed.
- T. Sawada and M. Nakada, Org. Lett., 2014, 16, 1537 CrossRef.
- C. Li, X. Yu and X. Lei, Org. Lett., 2010, 12, 4284–4287 CrossRef CAS PubMed.
- D. Xia, Y. Du, Z. Yi, H. Song and Y. Qin, Chem. – Eur. J., 2013, 19, 4423–4427 CrossRef CAS PubMed.
- F. E. Ziegler, Acc. Chem. Res., 1977, 10, 227–232 CrossRef CAS.
- A. M. M. Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef CAS PubMed.
- A. Nakazaki and S. Kobayashi, Synlett, 2012, 1427–1445 CAS.
- I. Yosioka, H. Hikino and Y. Sasaki, Chem. Pharm. Bull., 1961, 9, 84–85 CrossRef.
- I. Yosioka and T. Kimura, Chem. Pharm. Bull., 1965, 13, 1430–1434 CrossRef CAS.
- A. Nakazaki and S. Kobayashi, Chem. Lett., 2007, 36, 42–43 CrossRef CAS.
- A. Nakazaki, T. Era, Y. Numada and S. Kobayashi, Tetrahedron, 2006, 62, 6264–6271 CrossRef CAS PubMed.
- T. Okino, E. Yoshimura, H. Hirota and N. Fusetani, Tetrahedron, 1996, 52, 9447–9454 CrossRef CAS.
- H. Prawat, C. Mahidol, S. Wittayalai, P. Intachote, T. Kanchanapoom and S. Ruchirawat, Tetrahedron, 2011, 67, 5651–5655 CrossRef CAS PubMed.
- A. Wright, H. Wang, M. Gurrath, G. M. König, G. Kocak, G. Neumann, P. Loria, M. Foley and L. Tilley, J. Med. Chem., 2001, 44, 873–885 CrossRef CAS PubMed.
- C. Angerhoffer and J. Pezzuto, J. Nat. Prod., 1992, 55, 1787–1789 CrossRef.
- A. Nakazaki, H. Miyamoto, K. Henmi and S. Kobayashi, Synlett, 2005, 1417–1420 CAS.
- A. Nakazaki, T. Era and S. Kobayashi, Chem. Pharm. Bull., 2007, 55, 1606–1609 CrossRef CAS.
- K. Tamura, A. Nakazaki and S. Kobayashi, Synlett, 2009, 2449–2452 CAS.
- T. Sattelkau and P. Eilbracht, Tetrahedron Lett., 1998, 39, 9647–9648 CrossRef CAS.
- T. Sattelkau, C. Hollmann and P. Eilbracht, Synlett, 1996, 1221–1223 CrossRef CAS.
- T. Sattelkau and P. Eilbracht, Tetrahedron Lett., 1998, 39, 1905–1908 CrossRef CAS.
- P. Eilbracht, A. Gersmeier, D. Lennartz and T. Huber, Synthesis, 1995, 330–334 CrossRef CAS.
- Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, 2010 Search PubMed.
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- B.-W. Yu, J.-Y. Chen, Y.-P. Wang, K.-F. Cheng, X.-Y. Li and G.-W. Qin, Phytochemistry, 2002, 61, 439–442 CrossRef CAS.
- F. Li, S. S. Tartakoff and S. L. Castle, J. Org. Chem., 2009, 74, 9082–9093 CrossRef CAS PubMed.
- R. Navarro and S. E. Reisman, Org. Lett., 2012, 14, 4354–4357 CrossRef CAS PubMed.
- S. M. King, N. A. Calandra and S. B. Herzon, Angew. Chem., Int. Ed., 2013, 52, 3642–3645 CrossRef CAS PubMed.
- G. Guest, C. R. Hughes, A. Sattar and R. Ramage, J. Chem. Soc., Chem. Commun., 1973, 526–527 RSC.
- S. Baldwin and J. Fredericks, Tetrahedron Lett., 1982, 23, 1235–1238 CrossRef CAS.
- W. Oppolzer, F. Zutterman and K. Baettig, Helv. Chim. Acta, 1983, 66, 522–533 CrossRef CAS PubMed.
- A. Stoessl and E. Ward, Tetrahedron Lett., 1976, 17, 3271–3274 CrossRef.
- N. Katsui, A. Matsunaga, H. Kitahara, F. Yagihashi, A. Murai, T. Masamune and N. Sato, Bull. Chem. Soc. Jpn., 1977, 50, 1217–1225 CrossRef CAS.
- Y. Kawaguchi, T. Ochi, Y. Takaishi, K. Kawazoe and K.-H. Lee, J. Nat. Prod., 2004, 67, 1893–1896 CrossRef CAS PubMed.
- M. T. Crimmins, C. M. Dudek and A. W. Cheung, Tetrahedron Lett., 1992, 33, 181–184 CrossRef CAS.
- M. T. Crimmins, Z. Wang and L. A. McKerlie, Tetrahedron Lett., 1996, 37, 8703–8706 CrossRef CAS.
- M. T. Crimmins, Z. Wang and L. A. McKerlie, J. Am. Chem. Soc., 1998, 120, 1747–1756 CrossRef CAS.
- T. Hatsui, J. Wang and H. Takeshita, Bull. Chem. Soc. Jpn., 1995, 68, 2393–2399 CrossRef CAS.
- D. Caine, A. Boucugnani, S. Chao, J. Byron Dawson and P. Ingwalson, J. Org. Chem., 1976, 41, 1539–1544 CrossRef CAS.
- Z. Horii, C. Iwata and Y. Nakashita, Chem. Pharm. Bull., 1978, 26, 481–483 CrossRef CAS.
- Z. Horii, Y. Nakashita and C. Iwata, Tetrahedron Lett., 1971, 12, 1167–1168 CrossRef.
- T. Kametani, K. Fukumoto, S. Shibaya, H. Nemoto, T. Nakano, T. Sugahara, T. Takahashi, Y. Aizawa and M. Toriyama, J. Chem. Soc., Perkin Trans. 1, 1972, 1435–1441 RSC.
- T. Kametani, T. Sugahara, H. Sugi, S. Shibuya and K. Fukumoto, Tetrahedron, 1971, 27, 5993–5998 CrossRef CAS.
- T. Kametani, T. Sugahara, H. Sugi, S. Shibuya and K. Fukumoto, J. Chem. Soc., Chem. Commun., 1971, 724 RSC.
- K. Bernauer, Experientia, 1964, 15, 380–381 CrossRef.
- T. Honda and H. Shigehisa, Org. Lett., 2006, 8, 657–659 CrossRef CAS PubMed.
- F. Wang, Y. Gao, L. Zhang and J.-K. Liu, Org. Lett., 2010, 12, 2354–2357 CrossRef CAS PubMed.
- F. Wang, Y. Gao, L. Zhang, B. Bai, Y. Hu, Z. Donh, Q. Zhai, H. Zhu and J. Liu, Org. Lett., 2010, 12, 3196–3199 CrossRef CAS PubMed.
- H. Tan, C. Zheng, Z. Liu and D. Z. Wang, Org. Lett., 2011, 13, 2192–2195 CrossRef CAS PubMed.
- X. Hu, Y. Wang, F. Xiao, W. Liu, Q. Zhang and X. Li, Asian J. Org. Chem., 2013, 2, 216–219 CrossRef PubMed.
- N. Tanaka, Y. Kashiwada, S. Y. Kim, W. Hashida, M. Sekiya, Y. Ikeshiro and Y. Takaishi, J. Nat. Prod., 2009, 72, 1447–1452 CrossRef CAS PubMed.
- S. A. Van Der Sar, J. W. Blunt and M. H. G. Munro, Org. Lett., 2006, 8, 2059–2061 CrossRef CAS PubMed.
- Z. Song, X. Xu, W. Deng and S. Peng, Org. Lett., 2011, 13, 462–465 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |