
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Smaller, faster, better: modular synthesis of unsymmetrical ammonium salt-tagged NHC–gold(I) complexes and their application as recyclable catalysts in water†
Katrin
Belger
and
Norbert
Krause
*
Organic Chemistry, Dortmund University of Technology, Otto-Hahn-Str. 6, D-44227 Dortmund, Germany. E-mail: norbert.krause@tu-dortmund.de; Fax: (+)49 231 755 3884
First published on 8th July 2015
Abstract
Facile access towards a small library of unsymmetrical ammonium salt-tagged N-heterocyclic carbene gold(I) complexes is described, and their application as recyclable catalysts in cyclization reactions of acetylenic carboxylic acids and amides to lactones and lactams, respectively, in aqueous media is demonstrated. Catalyst 1ab was applied in the synthesis of 2-epi-clausemarine A (16).
Introduction
Sustainable chemistry is a frequently used term which indicates economic, ecological friendly and safe transformations. Accordingly, the development of new chemical reactions should combine waste prevention, use of nonhazardous solvents, and renewable raw materials.1 In organometallic chemistry, main objectives are reusable catalysts, easy separation of products, and prevention of side reactions.2 To achieve these goals, the use of water-soluble catalysts represents a desirable pathway. For this purpose, N-heterocyclic carbene (NHC) metal complexes were linked to water-soluble polymers3,4 or silica-based surfaces4 and carbohydrates.5 Moreover, they were functionalized with hydrophilic groups, such as carboxylates6,7 or sulfonates,7,8 resulting in improved water solubility and consequently an easier separation of the product and the catalyst, and also the opportunity for catalyst recycling.9,10Since the first synthesis of a sulfonated NHC–metal complex by Herrmann et al., the number of reports about water-soluble NHC ligands has been increasing steadily. However, ammonium salt-tagged NHC ligands still play a minor role in transition metal catalysis,8 and only a limited number of publications deal with a direct synthesis of the ammonium function.11,12 More often, an amino group of the final NHC–metal complex is quaternized with a Brønsted acid.13–15 Recently, we reported the direct synthesis of ammonium salt-tagged IMesAuCl complexes and demonstrated their catalytic activity in cycloisomerization reactions of allenic and acetylenic alcohols in aqueous medium.11 Moreover, we could successfully demonstrate their high stability and recyclability in water.
Here, we report an even more facile access towards ammonium salt-functionalized NHC–gold complexes. A shortened synthetic pathway is achieved by the application of a modular system which provides access to a small library of gold catalysts 1. These ligands consist of an arylimidazole and a benzylic linker with an attached ammonium group (Fig. 1). The ammonium salt is introduced by aminoalkylation with trimethyl-, triethyl-, or tributylamine.
 | ||
| Fig. 1 Modular system for the synthesis of ammonium salt-tagged gold catalysts 1. | ||
Results and discussion
The synthesis of the ammonium salt-tagged NHC–gold complexes 1 started with the formation of N-arylimidazoles 2a and 2b (Scheme 1). These were obtained from commercially available 2,4,6-trimethylaniline and 2,6-diisopropylaniline, respectively, according to a literature procedure with similar yields to those reported.16 The second building block 3 was formed by reduction of methyl 4-(chloromethyl)benzoate 5 with diisobutylaluminum hydride (DIBAL-H; 96% yield) and tetrahydropyranyl-(THP)-protection of alcohol 6 (63% yield). | ||
| Scheme 1 Synthesis of building blocks 2 and 3 (DIBAL-H = diisobutylaluminum hydride; DCM = dichloromethane; DHP = 3,4-dihydro-2H-pyran; PPTS = pyridinium p-toluenesulfonate). | ||
The coupling of these building blocks was performed by heating 2a or 2b in acetonitrile in the presence of 3 to form the imidazolium salts 7a and 7b with 78% and 76% yield, respectively (Scheme 2). Here, the THP ether was cleaved during the acidic work-up. The alcohols 7 were chlorinated with thionyl chloride (90/96% yield) and an aminoalkylation with trimethyl-, triethyl-, or tributylamine gave the desired carbene precursors 9 (66–86% yield). These imidazolium salts were transformed into the corresponding gold(I) complexes in the presence of (Me2S)AuCl and potassium tert-butoxide. The NHC gold complexes 1 were obtained with 71–86% yield.
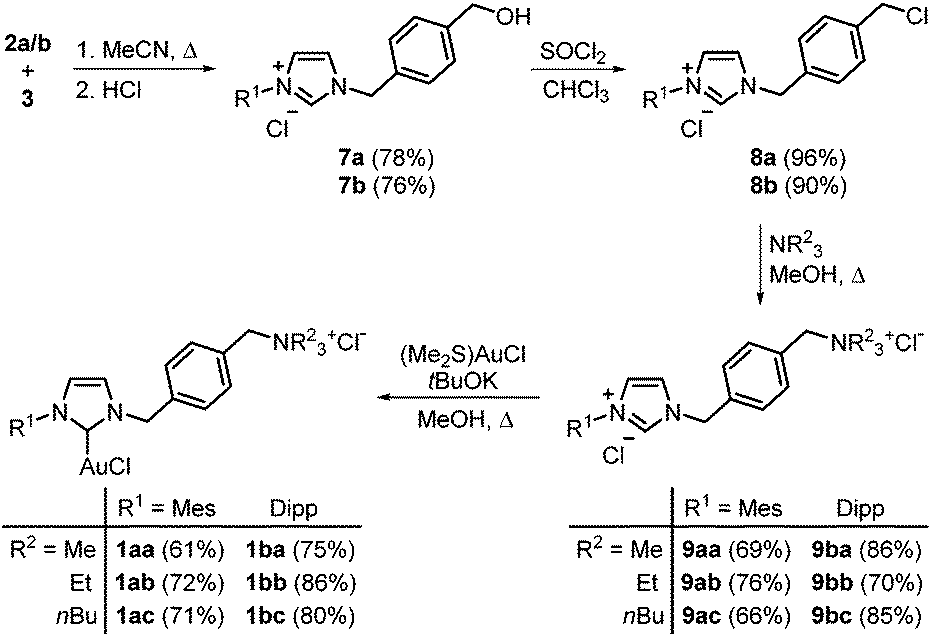 | ||
| Scheme 2 Synthesis of unsymmetrical ammonium salt-tagged gold(I) complexes 1. | ||
The catalytic activity of the new unsymmetrical ammonium salt-tagged gold catalysts 1 was investigated in cycloisomerization reactions of acetylenic carboxylic acids and amides. The gold-catalyzed lactonization of acetylenic carboxylic acids in water was previously examined by Cadierno et al.17 who used zwitterionic water-soluble gold complexes with sulfonate and pyridinium groups. Related studies were reported by Navarro Ranninger18 who applied platinum complexes and also investigated the hydrolysis of the lactone and its mechanism. Furthermore, copper and palladium complexes were also used in lactonization reactions of unsaturated carboxylic acids in aqueous medium.19,20 The corresponding cyclization of acetylenic amides to lactams was mainly performed in the presence of TBAF or bases in organic solvents.21,22 The only example of a metal-catalyzed cyclization was published by Nagasaka who used a lithium hexamethyldisilazide/AgOTf system.23 As far as we know, this reaction has not yet been carried out in water.
As a benchmark reaction, we first investigated the cyclization of 4-pentynoic acid (10a) to lactone 11a in the presence of gold catalyst 1ab at room temperature in water (Table 1). A recycling of catalyst 1ab after product extraction with diethyl ether showed decreasing conversions (99–79%) and yields (78–63%) over five cycles. As the measured pH value of pentynoic acid (pKa = 4.21 (ref. 24)) in water is 2.44, the gold complex slowly decomposed with the formation of a black precipitate in the acidic medium. The use of a triethylammonium acetate buffer solution with pH = 7 led to high conversions over five cycles and enhanced yields of 89–81%. Notably, an activation of the gold catalyst with a silver salt is not necessary. At present, it is not clear whether complex 1 or a cationic gold species formed by dissociation of chloride is the catalytically active species, even though the rather high concentration of the strongly coordinating chloride anion (from the ammonium chloride side chain) in the reaction mixture certainly disfavors the latter.25

Next, we applied the full library of ammonium salt-tagged gold catalysts 1 to the cycloisomerization of different acetylenic carboxylic acids in aqueous buffer solution (Table 2). It turned out that the mesityl-substituted catalysts 1aa–ac gave better results than their 2,6-diisopropylphenyl-substituted counterparts 1ba–1bc. In particular, catalyst 1ab gave high yields of lactones 11 (84–94%) in all cases. Even though the corresponding complex 1aa showed a similar reactivity, it gave lower yields (69–86%) than 1ab. These results were similar to 1bb (71–86%). The addition of THF as the cosolvent to carboxylic acids 10c–e was necessary in order to dissolve the (otherwise insoluble) substrates. Moreover, for carboxylic acid 10e the temperature had to be raised to 50 °C to decrease the reaction time to 0.5–4 h (otherwise, full conversion was obtained only after 24 h at room temperature).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Catalyst | 1aa | 1ab | 1ac | 1ba | 1bb | 1bc | |
| Substrate | R1, R2 | Yieldb (%) | |||||
| a Reaction times required for full conversion are given in the ESI. b Isolated yield. c 0.5 M solution of the carboxylic acid in THF was used. d At 50 °C. | |||||||
| 10a | H, H | 69 | 89 | 85 | 77 | 78 | 77 |
| 10b | Me, H | 77 | 85 | 83 | 67 | 71 | 65 |
| 10c | iPr, H | 86 | 94 | 91 | 72 | 78 | 71 |
| 10d | Ph, H | 72 | 84 | 77d | 77 | 86 | 74d |
| 10e , | Ph, Ph | 81 | 84 | 72 | 71 | 76 | 69 |

In order to render the transformation even more sustainable, we decreased the catalyst loading. As shown in Table 3, similar results were obtained for the cycloisomerization of carboxylic acid 10a to lactone 11a with 1 mol% of 1ab instead of 2.5 mol% (entry 2 vs. 1). Even lower catalyst loadings of 0.5 or 0.1 mol% afforded incomplete conversion within 1 h (entries 3 and 4). However, increasing the reaction time to 3 h (with 0.5 mol% of 1ab; 79% yield) or 7 h (with 0.1 mol% 1ab; 74% yield) was sufficient to give a full conversion of 10a to 11a.

A possible side reaction to the cyclization is the gold-catalyzed hydration of the triple bond of the acetylenic acid in the aqueous reaction medium. This has been observed previously by other groups,17,18 but not (to this point) with the ammonium salt-tagged gold catalysts 1. Cadierno et al.17 have detected the formation of ketoacid 12a from 10a with gold complexes within 1 h at rt, which led to moderate yields of lactone 11a (50%) in pure water (ratio of 11a/12a = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). With our catalyst 1ab, however, there were no traces of 12a or any other side product within this time (Table 1). Only after a prolonged reaction time of 24 h in pure water, we could isolate 21% of ketoacid 12a together with 50% of lactone 11a (Scheme 3). In the triethylammonium acetate buffer solution, similar yields were obtained after 24 h (12a: 18%; 11a: 54%). After 3 days in buffer solution, 43% of 12a and 23% of 11a could be isolated, whereas in the absence of a gold catalyst, there was no formation of 12a. If we assume that the cyclization of 10a to 11a is irreversible, the ketoacid 12a is not formed by hydration of 10a but rather by a slow gold-catalyzed hydrolytic ring-opening of the lactone 11a. Accordingly, we could not observe any formation of acetophenone when phenylacetylene was heated to 60 °C for 24 h with gold complex 1ab in water. Under these conditions, only the more electron-rich 1-ethynyl-4-methoxybenzene gave 2% of the corresponding ketone.
:1). With our catalyst 1ab, however, there were no traces of 12a or any other side product within this time (Table 1). Only after a prolonged reaction time of 24 h in pure water, we could isolate 21% of ketoacid 12a together with 50% of lactone 11a (Scheme 3). In the triethylammonium acetate buffer solution, similar yields were obtained after 24 h (12a: 18%; 11a: 54%). After 3 days in buffer solution, 43% of 12a and 23% of 11a could be isolated, whereas in the absence of a gold catalyst, there was no formation of 12a. If we assume that the cyclization of 10a to 11a is irreversible, the ketoacid 12a is not formed by hydration of 10a but rather by a slow gold-catalyzed hydrolytic ring-opening of the lactone 11a. Accordingly, we could not observe any formation of acetophenone when phenylacetylene was heated to 60 °C for 24 h with gold complex 1ab in water. Under these conditions, only the more electron-rich 1-ethynyl-4-methoxybenzene gave 2% of the corresponding ketone.
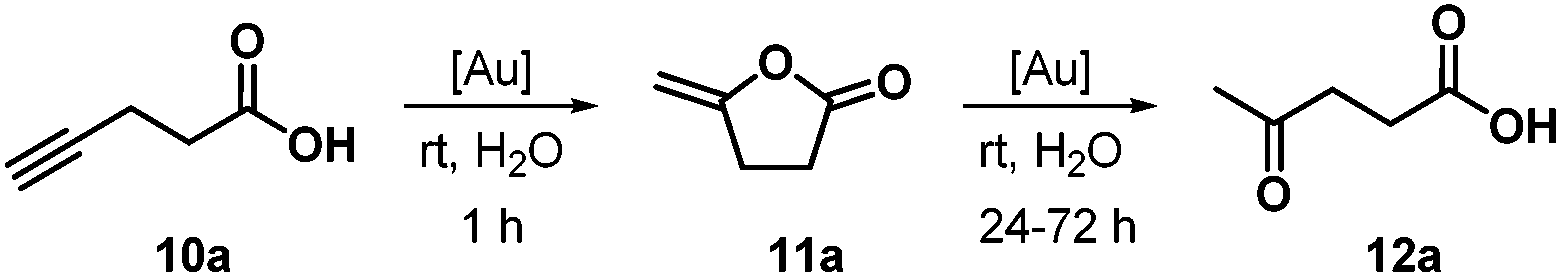 | ||
| Scheme 3 Gold-catalyzed cycloisomerization of 4-pentynoic acid (10a) and hydrolytic ring-opening of lactone 11a. | ||
In analogy to the acetylenic carboxylic acids 10, the corresponding tosylamides 1326 can be smoothly cyclized to the lactams 14 with the unsymmetrical ammonium salt-tagged gold catalysts 1 in aqueous medium (Table 4). As the measured pH value of amide 13a in water is 3.72 (calcd pKa = 6.31), we again used a buffer solution to avoid degradation of the catalyst. In general, all gold catalysts afforded high yields of lactams 13. Only complexes 1ac/1bc with tributylammonium groups needed extended reaction times of 2–4 h which has also been observed for the formation of lactones 11. In contrast to lactone 11a, treatment of lactam 13a with gold catalyst 1ab for 24 h at 50 °C did not afford any other product.

In order to apply the new gold catalysts 1 in target-oriented synthesis, we have chosen 2-epi-clausemarine A (16), an epimer of the furanocoumarin 15 which was isolated recently by Wu et al.27 from Clausena lansium, a grape-like fruit in Southeast Asia (Fig. 2). This contains an α-substituted lactone ring which can be formed by gold-catalyzed cyclization of a suitable acetylenic acid.
 | ||
| Fig. 2 Furanocoumarin clausemarine A (15) isolated from Clausena lansium, and its 2-epimer 16. | ||
The substrate required for the gold-catalyzed step, the hydroxycarboxylic acid 17 (Scheme 4), was synthesized by Evans alkylation (see the ESI† for details). With 1 mol% of our gold catalyst 1ab in aqueous triethylammonium acetate solution containing THF as the cosolvent, the desired lactone 18 was obtained with 77% yield. Hydrogenation of the double bond and subsequent oxidation of the hydroxy group with 2-iodoxybenzoic acid (IBX) gave the cis-(S,S)-diastereomer 19. Other hydrogenation catalysts such as PtO2 led to an opening of the lactone ring to the corresponding saturated hydroxycarboxylic acid. Treatment of 19 with vinylmagnesium bromide and PBr3 afforded the labile allyl bromide 20 as a mixture of E/Z-isomers. Finally, conversion of 20 according to a known procedure28 gave the target molecule 2-epi-clausemarine A (16).
 | ||
| Scheme 4 Synthesis of 2-epi-clausemarine A (16). | ||
Conclusions
We have developed new, rapid access to ammonium salt-tagged gold catalysts 1 with the possibility of further variation of their unsymmetrical structure. The modular approach allowed the synthesis of a small library of catalysts 1 with overall yields of 32–47% starting from building blocks 2 and 3. Gold complexes 1 catalyze the cycloisomerization of acetylenic carboxylic acids and amides to the corresponding lactones and lactams in aqueous medium with good to excellent yields. An activation of the gold catalyst with a silver salt is not necessary. The acid-promoted degradation of the catalysts in pure water can be prevented by adjustment of the pH value to 7. The recyclability of gold catalyst 1ab was demonstrated for the benchmark reaction of carboxylic acid 10a to lactone 11a. In contrast to previous gold catalysts operating in water, formation of ketoacids as a side product can be avoided with gold complexes 1. Moreover, we could successfully apply catalyst 1ab in the synthesis of 2-epi-clausemarine A (16). Further examples of sustainable gold catalysts will be reported in due course.Notes and references
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- (a) R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) K. Schröder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- M. T. Zarka, M. Bortenschlager, K. Wurst, O. Nuyken and R. Weberskirch, Organometallics, 2004, 23, 4817–4820 CrossRef CAS.
- W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860–873 CrossRef CAS PubMed.
- F. Tewes, A. Schlecker, K. Harms and F. J. Glorius, J. Organomet. Chem., 2007, 692, 4593–4602 CrossRef CAS PubMed.
- W. A. Herrmann, L. J. Goosen and M. J. Spiegler, J. Organomet. Chem., 1997, 547, 357–366 CrossRef CAS.
- L. R. Moore, S. M. Cooks, M. S. Anderson, H.-J. Schanz, S. T. Griffin, R. D. Rogers, M. C. Kirk and K. H. Shaughnessy, Organometallics, 2006, 25, 5151–5158 CrossRef CAS.
- L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751–2772 CrossRef PubMed.
- K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef CAS PubMed.
- K. Belger and N. Krause, Eur. J. Org. Chem., 2015, 220–225 CrossRef CAS PubMed.
- W. Wang, J. Wu, C. Xia and F. Li, Green Chem., 2011, 13, 3440–3445 RSC.
- İ. Özdemir, B. Yiğit, B. Çetinkaya, D. Ülkü, M. N. Tahir and C. J. Arıcı, J. Organomet. Chem., 2001, 633, 27–32 CrossRef.
- J. P. Jordan and R. H. Grubbs, Angew. Chem., Int. Ed., 2007, 46, 5152–5155 CrossRef CAS PubMed.
- S. L. Balof, S. J. P'Pool, N. J. Berger, E. J. Valente, A. M. Shiller and H.-J. Schanz, Dalton Trans., 2008, 5791–5799 RSC.
- J. Liu, J. Chen, J. Zhao, Y. Zhao, L. Li and H. Zhang, Synthesis, 2003, 2661–2666 CAS.
- (a) E. Thomás-Mendivil, P. Y. Toullec, J. Díez, S. Conejero, V. Michelet and V. Cadierno, Org. Lett., 2012, 14, 2520–2523 CrossRef PubMed; (b) E. Thomás-Mendivil, P. Y. Toullec, J. Borge, S. Conejero, V. Michelet and V. Cadierno, ACS Catal., 2013, 3, 3086–3098 CrossRef.
- (a) J. Alemán, V. Del Solar, L. Cubo, A. G. Quiroga and C. Navarro Ranninger, Dalton Trans., 2010, 39, 10601–10607 RSC; (b) J. Aleman, V. Del Solar and C. Navarro-Ranninger, Chem. Commun., 2010, 46, 454–456 RSC.
- T. L. Mindt and R. J. Schibli, Org. Chem., 2007, 72, 10247–10250 CrossRef CAS PubMed.
- (a) L. Zhou and H.-F. Jiang, Tetrahedron Lett., 2007, 48, 8449–8452 CrossRef CAS PubMed; (b) K. Ogata, D. Sasano, T. Yokoi, K. Isozaki, H. Seike, H. Takaya and M. Nakamura, Chem. Lett., 2012, 41, 498–500 CrossRef CAS; (c) J. García-Álvarez, J. Díez and C. Vidal, Green Chem., 2012, 14, 3190–3196 RSC.
- (a) M. C. De La Fuente and D. Dominguez, J. Org. Chem., 2007, 72, 8804–8810 CrossRef CAS PubMed; (b) P. A. Jacobi, S. C. Buddhu, D. Fry and S. Rajeswari, J. Org. Chem., 1997, 62, 2894–2906 CrossRef CAS PubMed; (c) P. A. Jacobi and S. Rajeswari, Tetrahedron Lett., 1992, 33, 6231–6234 CrossRef CAS.
- (a) M. M. Cid, D. Dominguez, L. Castedo and E. M. Vazquez-Lopez, Tetrahedron, 1999, 55, 5599–5610 CrossRef CAS; (b) J. Liu, Y. Zhang, G. Li, F. Roschangar, V. Farina, C. H. Senanayake and B. Z. Lu, Adv. Synth. Catal., 2010, 352, 2667–2671 CrossRef CAS PubMed.
- (a) Y. Koseki, S. Kusano and T. Nagasaka, Tetrahedron Lett., 1998, 39, 3517–3520 CrossRef CAS; (b) Y. Koseki, S. Kusano, D. Ichi, K. Yoshida and T. Nagasaka, Tetrahedron, 2000, 56, 8855–8865 CrossRef CAS.
- J. S. Lomas, J. Phys. Org. Chem., 2012, 25, 620–627 CrossRef CAS PubMed.
- B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015, 13, 7103–7118 CAS.
- Synthesized from the corresponding carboxylic acid according to: K. S. Feldman, M. M. Bruendl, K. Schildknegt and A. C. Bohnstedt, J. Org. Chem., 1996, 61, 5440–5452 CrossRef CAS.
- D.-Y. Shen, Y.-Y. Chan, T.-L. Hwang, S.-H. Juang, S.-C. Huang, P.-C. Kuo, T. D. Thang, E.-J. Lee, A. G. Damu and T.-S. Wu, J. Nat. Prod., 2014, 77, 1215–1223 CrossRef CAS PubMed.
- E. C. Row, S. A. Brown, A. V. Stachulski and M. S. Lennard, Bioorg. Med. Chem., 2006, 14, 3865–3871 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra. See DOI: 10.1039/c5ob01286d |
| This journal is © The Royal Society of Chemistry 2015 |