
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigins of observed reactivity and specificity in the addition of B2Cl4 and analogues to unsaturated compounds†
Cristina
Pubill-Ulldemolins
a,
Elena
Fernánez
b,
Carles
Bo
*a and
John M.
Brown
*c
aICIQ – Institute of Chemical Research of Catalonia, Avda. Països Catalans, 16. Tarragona 43007, Spain
bDept. Química Física i Inorgànica, Univ. Rovira i Virgili, C/Marcel-li Domingo s/n, Tarragona 43007, Spain
cChemistry Research Laboratory, Oxford University Mansfield Rd., Oxford OX1 3QY, UK. E-mail: john.brown@chem.ox.ac.uk
First published on 31st July 2015
Abstract
In 1954 Schlesinger and co-workers observed the direct reaction of diboron tetrachloride with simple organic compounds under mild conditions, the 1,2 addition product being formed with either ethylene or acetylene. In the following 25 years a series of addition reactions to simple alkenes, alkynes and dienes was demonstrated. B2F4 was shown to react in similar manner, albeit under more forcing conditions. Crucially, it was demonstrated that the addition to (E)- or (Z)-but-2-ene occurred with cis-stereospecificity. Only sporadic interest was shown in this field thereafter until catalysed addition reactions of diboron reagents were realized. Encouraged by this revival of interest through the discovery of transition-metal and nucleophilic catalysis of diboryl additions, DFT analysis of uncatalysed additions of B2X4 has been carried out and interpreted. This includes the relative reactivity of several B–B reagents with ethene, and that of B2Cl4vs. B2F4 additions, including benzene, naphthalene and C60 as reactants. This allows the analysis of relative reactivity vis-à-vis substitution on boron, and also direct comparison with hydroboration by HBCl2. [4 + 2] Addition of diboron reagents to dienes with B–B cleavage competes with direct [2 + 2] addition, favourably so for B2F4. The computational results demonstrate that the stereospecific addition to isomeric but-2-enes is a rare concerted [2σs + 2πs] process.
Introduction
The first addition of a boron reagent to an alkene was conducted by Schlesinger et al. in 1954,1a three years before H.C. Brown's seminal papers on hydroboration.2 In that first paper, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct formed at −80 °C between ethene and B2Cl41 was demonstrated, along with related products formed by addition of B2Cl4 to ethyne or cyclopropane, the latter occurring only at 0 °C. In their later full paper, addition of the boron reagent to a wide range of alkenes was carried out, including double addition to butadiene.1b The addition chemistry of B2F42 was similar but required much more forcing conditions. In other early work the stabilization of the reagent 1 by trichloroethene for addition reactions was noted, and reaction with both cis- and trans-but-2-ene observed although the stereochemical course could not be settled.3 For ethyne, cis-addition of 1 was defined, and later extended to higher alkynes.4 Slow reaction was observed with aromatic compounds, leading to a single electrophilic substitution product C6H5BCl2 from benzene, whilst a double addition product was obtained from naphthalene causing saturation of one ring.5 Progress was limited in these early papers by the instability and limited availability of B2Cl4, produced by an electric discharge through BCl3 at low temperatures.6 Later on, B2Cl4 became available on a 10 g. scale employing Cu and BCl3 in metal-vapour deposition.7 To this day more convenient syntheses of the reagent are lacking, although a simple route to its bis-dimethylamine adduct is known.8 Further work will be stimulated by the application of 1 in semiconductor doping.9 The analogous compound B2Br48 also adds easily across the double bond of alkenes,10 and is more readily accessible, encouraging a reappraisal of its reactivity.11 In summary, the concerted uncatalyzed addition of two heavy atoms from B2X4 to C–C unsaturation remains a rare and intriguing reaction type. The interest of these addition reactions for synthetic chemists was substantially enhanced when it was shown by oxidation and characterization of the ensuing chiral diols that the addition of B2Cl4 to isomeric butenes was cis-specific (Scheme 1).12 Addition of the reagent to cycloalkenes was likewise shown to be cis; with cyclohexa-1,3-diene, two sequential cis-specific additions occur on opposite faces.13 Selectivity in the addition of to the double bonds of methylenecyclopropane or vinylcyclopropane over competing C–C cleavage pathways was demonstrated.14 Further work showed that the addition of reagents 1 or 2 to buta-1,3-diene gave a 1:1 product that was assigned to 1,4-addition based on NMR spectra.15 After that, brief summaries appeared in a broader review of boron halide chemistry,16 otherwise the topic has been neglected, revived through the important discoveries of catalysed additions of diboryl compounds to alkenes and alkynes described below.
:1 adduct formed at −80 °C between ethene and B2Cl41 was demonstrated, along with related products formed by addition of B2Cl4 to ethyne or cyclopropane, the latter occurring only at 0 °C. In their later full paper, addition of the boron reagent to a wide range of alkenes was carried out, including double addition to butadiene.1b The addition chemistry of B2F42 was similar but required much more forcing conditions. In other early work the stabilization of the reagent 1 by trichloroethene for addition reactions was noted, and reaction with both cis- and trans-but-2-ene observed although the stereochemical course could not be settled.3 For ethyne, cis-addition of 1 was defined, and later extended to higher alkynes.4 Slow reaction was observed with aromatic compounds, leading to a single electrophilic substitution product C6H5BCl2 from benzene, whilst a double addition product was obtained from naphthalene causing saturation of one ring.5 Progress was limited in these early papers by the instability and limited availability of B2Cl4, produced by an electric discharge through BCl3 at low temperatures.6 Later on, B2Cl4 became available on a 10 g. scale employing Cu and BCl3 in metal-vapour deposition.7 To this day more convenient syntheses of the reagent are lacking, although a simple route to its bis-dimethylamine adduct is known.8 Further work will be stimulated by the application of 1 in semiconductor doping.9 The analogous compound B2Br48 also adds easily across the double bond of alkenes,10 and is more readily accessible, encouraging a reappraisal of its reactivity.11 In summary, the concerted uncatalyzed addition of two heavy atoms from B2X4 to C–C unsaturation remains a rare and intriguing reaction type. The interest of these addition reactions for synthetic chemists was substantially enhanced when it was shown by oxidation and characterization of the ensuing chiral diols that the addition of B2Cl4 to isomeric butenes was cis-specific (Scheme 1).12 Addition of the reagent to cycloalkenes was likewise shown to be cis; with cyclohexa-1,3-diene, two sequential cis-specific additions occur on opposite faces.13 Selectivity in the addition of to the double bonds of methylenecyclopropane or vinylcyclopropane over competing C–C cleavage pathways was demonstrated.14 Further work showed that the addition of reagents 1 or 2 to buta-1,3-diene gave a 1:1 product that was assigned to 1,4-addition based on NMR spectra.15 After that, brief summaries appeared in a broader review of boron halide chemistry,16 otherwise the topic has been neglected, revived through the important discoveries of catalysed additions of diboryl compounds to alkenes and alkynes described below.
 | ||
| Scheme 1 Stereochemical aspects of B2Cl4 addition chemistry with simple reactants. | ||
Compared to diboron dihalide species, boron derivatives which contain B–O or B–N bonds are more stable and less reactive as a consequence of the π donation from the lone pair of O and N substituents to the empty pz boron orbital.17 The lowered reactivity laid the foundations for catalytic addition; Miyaura and co-workers utilised a Pt[0] complex to activate tetraalkoxydiborons and thus catalysed cis-1,2-diboron addition to unsaturated substrates.18a This was subsequently extended to alkene additions,18b and further to catalytic asymmetric synthesis.18c Catalytic diboration has been extended to other metals and even demonstrated with Au nanoparticles,19 but not all such catalysts necessarily activate the diboron reagent by oxidative addition (Scheme 2a).20 Instead σ-bond metathesis between the metal entity and diboron reagent was suggested to play a key role (Scheme 2b).21 Although tetraalkoxydiborons are inactive under mild conditions for the diboration of unsaturated compounds, they can be catalytically activated by the addition of a Lewis base (A).22 A reactive Lewis acid–base adduct [A → B(OR)2–B(OR)2] can be formed under these conditions which facilitates the transfer of a boryl moiety with enhanced nucleophilic character (Scheme 2c). For reaction with electrophilic alkenes, the Lewis base may be a stable carbene,22b or a chiral phosphine capable of inducing product asymmetry.22c In the latter case a phosphonium cation formed by reaction of phosphine and electrophilic alkene forms an ion-pair with the reacting borate.22d Remarkably, addition to unactivated alkenes has been accomplished with catalytic methoxide/MeOH as Lewis base.23 Finally, we note that activation of modified dihalodiboranes Ar2X2B2 with a range of phosphine and N-heterocyclic carbenes reveals a rich chemistry that has just begun to be explored.24
 | ||
| Scheme 2 Activation pathways for bis(pinacolato)diboron that promote 1,2-diboration of alkenes (a) oxidative addition; (b) σ-bond metathesis; (c) through a Lewis base adduct. | ||
Significant questions remained unanswered with regard to the original addition reaction – e.g. why is such high reactivity observed with B2Cl4, and why is the reaction with 1,2-disubstituted alkenes stereospecific?
Results and discussion
Relative reactivity of various B–B compounds towards ethylene
The molecular structure of B2Cl4 is known both in the gas phase and in the solid state. IR/Raman and electron diffraction analyses confirm D2d symmetry,25 with a rotational barrier <2 kcal mol−1; in the crystalline state the molecule is planar, however.26 For B2F4, the planar form is marginally preferred over the orthogonal form.27 The CSD X-ray database contains ca. 100 B2X4 (X = N, O, S, Hal) structures, for which non-planar entities close to the orthogonal structure predominate over near-planar geometries. Notable exceptions lie in cases where X4 = O4, for which the planar form is generally preferred in the crystal state.28Our initial DFT calculations conformed well with these observations (Scheme 3), finding two stationary states with fairly closely matched energies in all cases except B2Cl41, one corresponding to the planar form, and the other closer to the orthogonal D2d form. In the exploratory phase of the work, three different functionals (B3LYP, M06-2x and ωB97X-D),29–31 were employed, with comparable results insofar as the ground-state structure and relative energies of the two conformers of the B2Cl4 reagent were concerned. The planar form was marginally the more stable one in the case of B2F42, and significantly so for the catechol derivative; only in the thiocatechol case did the X–B–B–X dihedral angle deviate far from 90° in the nonplanar form. The relevant observations are in accord with previous spectroscopic and computational work on the tetrahalodiboranes (ref. 25–27).
 | ||
| Scheme 3 DFT computed B2X4 initial states. Electronic energies relative to the planar conformer (ωB97X-D, 6-311G(d,p), gas phase, ZPE corrected) and X–B–B–X dihedral angles for the orthogonal isomer are shown. For 8, only the latter was found. The ESI† also shows results with other functionals. | ||
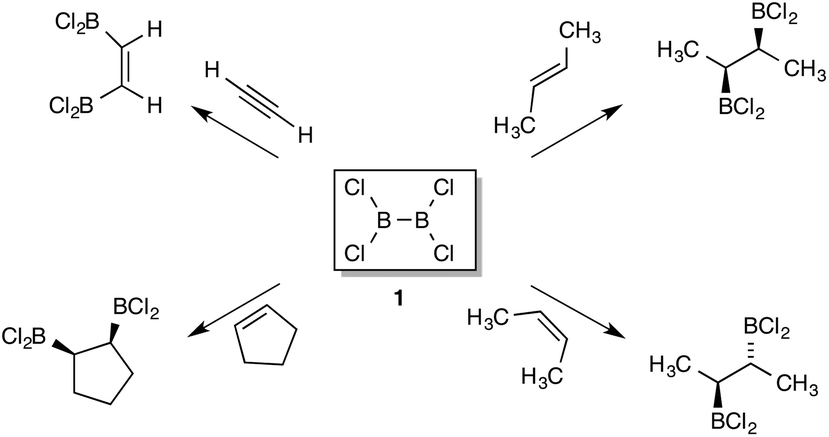
The choice of the DFT functional became more critical when considering the reactions of diboryls with alkenes. According to Urry and Schlesinger's original paper,1a the initial addition of B2Cl41 to C2H4 occurred to a very significant extent in 4 h. at −80 °C; the half-life at this temperature then translates to a Gibbs free energy value, ΔG‡ = 16.9 kcal mol−1. Comparable reaction with B2F42 only occurred at higher temperatures. Other addition reactions of B2Cl4 from different research groups were conducted in diverse ways; between −80 °C and 20 °C, neat, in solvent or in the vapour phase. For this reason all the computed results in the paper arise from the zero-point corrected electronic energy, in order to provide a basis for comparison that avoids the complications of varying TΔS and solvent effects. Given the lack of quantitative experimental data for comparison, comparisons of relative rather than absolute energy are needed. Reaction between C2H4 and 1 was examined by using several commonly used DFT functional: the ZPE-corrected transition state energy for 1,2-addition being 22.8 kcal mol−1 (ΔG0‡ = 35.3 kcal mol−1) above the isolated reactants using B3LYP as functional and the 6-311G(d,p) basis set,29a reduced to 15.6 kcal mol−1 (ΔG0‡ = 27.6 kcal mol−1) when D3-dispersion was included.29b Using the Truhlar functional M06-2X,30 a much lower value of 7.4 kcal mol−1 was obtained (ΔG0‡ = 19.6 kcal mol−1), whilst applying the ωB97X-D functional gave a value of 12.0 kcal mol−1 (ΔG0‡ = 23.7 kcal mol−1).31 It was decided that the last-named functional would be the most suitable for general analysis of B2X4 addition chemistry, and the body of results described here were obtained using this throughout, together with the 6-311G(d,p) basis set. At this stage we compared the reactivity of different B2X4 reagents towards C2H4 shown in Fig. 1; only the halo-compounds (X = F, Cl) had been applied in alkene additions before, and the fluoride is the less reactive of the two.1b When dispersion corrections were included in the functional an energetically favourable van der Waals (vdW) complex between the halodiborane or hydroxydiborane reactants and C2H4 was located as a separate stationary state for 1–3, although not for 4–7. Stabilization energies E for these vdW complexes varied between 3.8 and 5.5 kcal mol−1.
 | ||
| Fig. 1 (a)–(g) Transition-state structures for the addition of borane reagents to ethane with energies in red (kcal mol−1) relative to the isolated reactants and numbering as in Scheme 3; structures 4, 6 and 7 are drawn truncated. (h) shows superimposition of transition states for 1 and 3, DFT level as Scheme 3. | ||
For the diboron tetrahalides 1, 2 and 8 the four atoms involved in bond making and breaking are close to coplanar in the transition-state; the same is true for other examples. The structure of the transition states shows one near-tetrahedral boron atom B with advanced bonding to both carbons, the second one B′ less strongly involved and closer to its original trigonal geometry. The leading boron exhibits a BCC angle close to 70° at the transition state, conserved throughout the series. This resembles the analogous TS for simple hydroborations, derived in published computations,32 and this encouraged comparison of our results for B2Cl4 with those for hydroboration by HBCl2 (vide infra). The spread of transition-state energies is substantial, demonstrating high sensitivity of B–B bond activation to the substituents on boron. On this basis, the experimentally observed unreactivity of di-oxoboron reagents in addition to alkenes under non-catalytic conditions is corroborated, although a lower barrier for the sulfur analogue (Fig. 1f vs. 1e) is clearly predicted.
Analysis of the IRC's for the pathway with both 1 and 2 in their reaction with ethene (Fig. 2) demonstrates that the coplanar alignment of reactants (B′BCC′ = 0°) observed at the transition state is already present in the early phase of reaction, and persists right through to an initially B–C–C–B eclipsed product geometry. The coplanarity of the reacting atoms also suggests that vdW complexes are not directly involved on the reacting pathway, since these complexes possess C2 symmetry with pronounced twisting between the two components.
 | ||
| Fig. 2 IRC traces for 1 (blue) and 2 (red) reacting with ethene according to Fig. 1. | ||
The most obvious factor favouring addition to ethylene of B2Cl41 or B2Br48 over the related reactions of B2F4 and B2(OR)4 is the later transition states for 1 and 8. Compared to the other examples, they exhibit longer B–B and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and a shorter C–B bond. The energy differences involved are substantial although the geometric changes are very subtle, as witness the superimposed transition states from 1 and 4 shown in Fig. 1h. The observed variations in activation energy do not follow trends in BDE's,33 or B–B bond lengths, which do not vary much.34 Hence other possible reasons need to be considered. The energy differences observed for the simple reagents correlate with the value of the 11B–11B sym-stretching frequency, derived from computed Raman spectra of the D2h isomers using the same conditions as Fig. 1. For B2Cl4 this is observed at a frequency of 1111 cm−1 and for B2Br48 at 1102 cm−1 (D2d isomer) but for the less reactive B2F4 and B2(OH)4 at frequencies of 1395 cm−1 and 1423 cm−1 respectively. For diboryls 6 and 7 the B–B stretch is split through coupling to sym- and asymm-aromatic C–C stretching modes, and this complicates detailed analysis, although the same trend is observed, since the sulfur-substituted diboryl B2(Scat)27 possesses an activation energy for addition to ethene 5.2 kcal mol−1 lower than its oxo-analogue 6. In accord with this, the 11B–11B stretching frequencies in the computed Raman spectra of 7 are at 1131, 1177 cm−1 for B2Scat27 and 1377, 1449 cm−1 for B2Cat26. The softer symmetrical B–B stretching vibration in 1, 7 and 8 is also associated with later transition states compared with 2, 3, 4 and 6, as witness the shorter C–B and longer B–B′ bond lengths seen in Fig. 1. Both boron atoms are rehybridized towards sp3 at the transition states, less so for B′ (see ESI† pp. 83 and 84).
C bonds and a shorter C–B bond. The energy differences involved are substantial although the geometric changes are very subtle, as witness the superimposed transition states from 1 and 4 shown in Fig. 1h. The observed variations in activation energy do not follow trends in BDE's,33 or B–B bond lengths, which do not vary much.34 Hence other possible reasons need to be considered. The energy differences observed for the simple reagents correlate with the value of the 11B–11B sym-stretching frequency, derived from computed Raman spectra of the D2h isomers using the same conditions as Fig. 1. For B2Cl4 this is observed at a frequency of 1111 cm−1 and for B2Br48 at 1102 cm−1 (D2d isomer) but for the less reactive B2F4 and B2(OH)4 at frequencies of 1395 cm−1 and 1423 cm−1 respectively. For diboryls 6 and 7 the B–B stretch is split through coupling to sym- and asymm-aromatic C–C stretching modes, and this complicates detailed analysis, although the same trend is observed, since the sulfur-substituted diboryl B2(Scat)27 possesses an activation energy for addition to ethene 5.2 kcal mol−1 lower than its oxo-analogue 6. In accord with this, the 11B–11B stretching frequencies in the computed Raman spectra of 7 are at 1131, 1177 cm−1 for B2Scat27 and 1377, 1449 cm−1 for B2Cat26. The softer symmetrical B–B stretching vibration in 1, 7 and 8 is also associated with later transition states compared with 2, 3, 4 and 6, as witness the shorter C–B and longer B–B′ bond lengths seen in Fig. 1. Both boron atoms are rehybridized towards sp3 at the transition states, less so for B′ (see ESI† pp. 83 and 84).
Relative reactivity of substituted alkenes towards B2Cl4
These results encouraged analysis of addition reactions with B2Cl41. For ethyne, a vdW complex with the reagent was located with the partners orthogonal to one another; the cis-1,2-addition to ethyne was comparable in energy to that of ethene. Exploring the series of simple alkylethenes showed modest changes in TS energy with increasing methylation, and likewise to increasing stability of vdW complexes. For propene and 2-methylbut-2-ene, two isomeric pathways were located depending on which sp2-carbon had the shorter B–C bond at the TS. Tetramethylethylene did not permit location of either vdW or TS states. The energy of the gauche-product was also computed, and revealed the destabilizing effects of additional methylation. The comparison between 1 and 2 was extended to propene. B2F4 formed stronger vdW complexes and also showed transition state energy barriers ≥10 kcal mol−1 higher (Table 1, entry 5). The comparative reactivity of 1 and the hydroborating agent HBCl29 was also computed for the range of substrates.| Entry | Reactant | vdW (1)c | vdW (9)e | TS (1)c | TS(9)e |
|---|---|---|---|---|---|
| a Calculations were carried out using Gaussian09 rev D1, with the ωB97x-D functional and 6-311G(d,p) basis set; ZPE corrected. b prim, sec and tert-refer to the more strongly B-bonded carbon at the TS. c ZPE-corrected energies in kcal mol−1. d B2F42 as reagent. e HBCl29 as reagent. f The isomeric vdW complex was not found. | |||||
| 1 | C2H2 | −3.5 | — | 12.2 | — |
| 2 | C2H4 | −3.9 | −2.7 | 12.0 | 8.9 |
| 3b | C3H6(prim) | −5.5 | −3.2 | 10.4 | 5.4 |
| 4b | C3H6(sec) | −5.5 | −4.1 | 12.2 | 9.7 |
| 5d | C3H6(prim) | −7.2 | — | 10.1 | — |
| 6 | (E)-C4H8 | −5.6 | −3.0 | 10.7 | 6.4 |
| 7 | (Z)-C4H8 | −6.8 | −4.8 | 11.1 | 6.0 |
| 8b | C5H10(sec) | −7.8 | −5.2 | 13.5 | 3.0 |
| 9b | C5H10(tert) | −7.8 | —f | 12.5 | 8.5 |
| 10 | C10H8 | −7.8 | — | 26.7 | — |
| 11 | C6H6 | −5.7 | — | 36.9 | — |
Hydroboration of alkenes by BH3 decreases in reactivity with increasing alkyl substitution of the substrate,35 in contrast to conventional electrophilic additions.36 Results from the present calculations for B2Cl4 addition do not fit either pattern, the activation energy being comparable for propene and the but-2-ene isomers (entries 3, 6 and 7; Table 1), but all lower than for ethene or ethyne (entries 1 and 2). Only for the disfavoured pathway with propene and both pathways for trimethylethylene (TME) is there a significant increase in activation energy vs. ethene (entries 4, 8 and 9). For unsymmetrical alkenes, it is favourable to form the prim-C–B bond in propene first, but by contrast the tert-C–B bond in trimethylethylene (TME) first; compare entries 3,4 and entries 8,9.
The TS dipole moment in additions of 1 is 4.6 D for ethene and increases only marginally with increasing substitution to 5.2 D for 2-methylbutene, implying significant polar character throughout, as in electrophilic addition. (Z)-Butene shows higher affinity for vdW complexation with 1 than (E)-butene, but is the less reactive (entries 6 and 7). In entry 10, cis-addition of B2Cl4 to naphthalene occurs through an energetically accessible and exergonic pathway, in accord with the original experiments.5 This contrasts with the higher energy seen in addition to benzene (entry 11) where monosubstitution rather than addition had been observed experimentally. Addition to benzene is only very weakly exergonic, encouraging reversibility. In both cases a strong vdW interaction between the reactants was observed, with the B–B bond centrally aligned over the arene (Fig. 3); see ESI† for details.
 | ||
| Fig. 3 Comparison of TS structures for addition of B2Cl4 to benzene and naphthalene; distances in Å. | ||
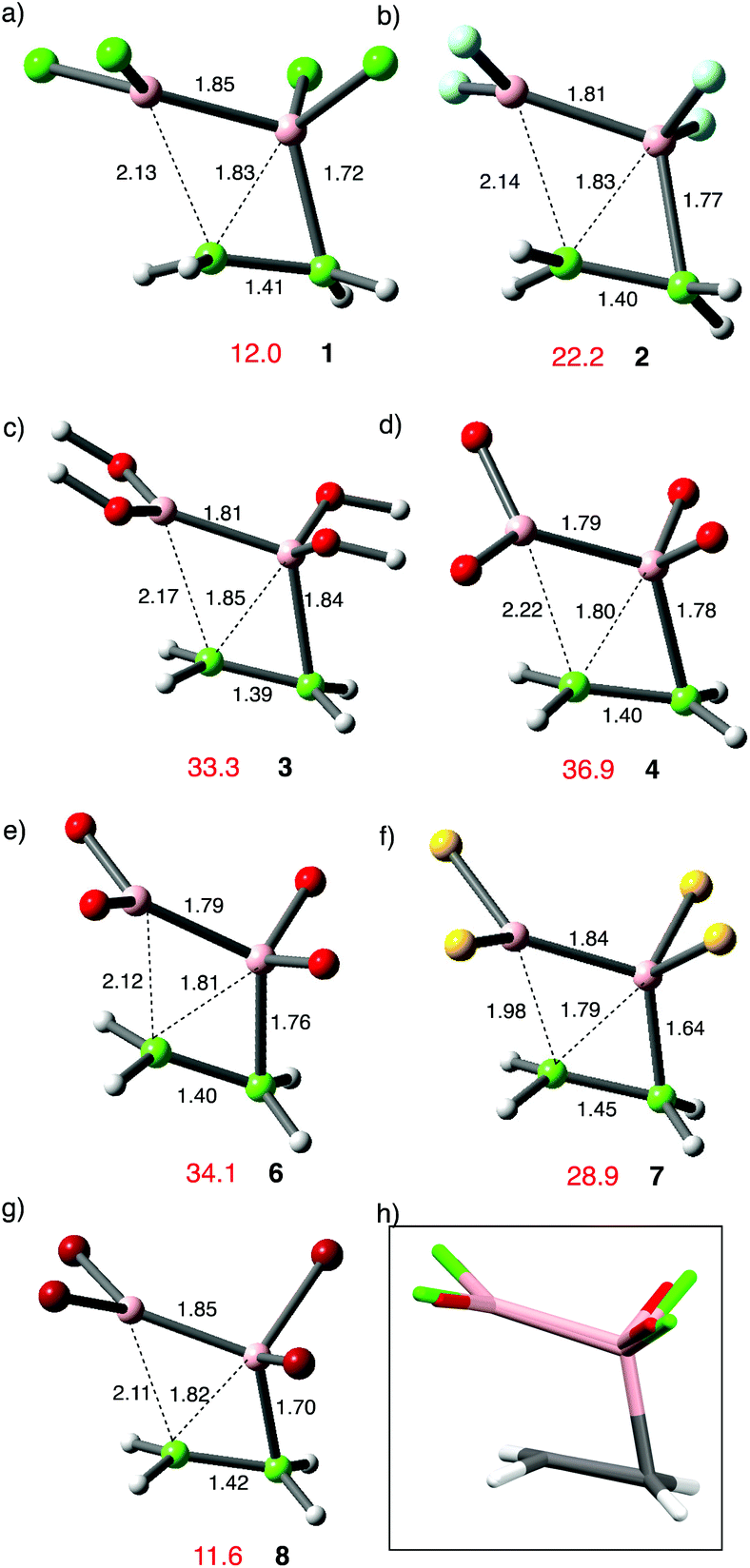
Dichloroborane has occasionally been employed as a hydroborating reagent. Aside from a series of observations in hexane solvent, mainly using fluoroalkene reactants, relatively slow rates have been reported for this addition reaction.37 This is a consequence of strong donor complexes to solvent when reaction is carried out in THF or ether; the reactions are faster in hydrocarbon media. Table 1 summarizes the results of a parallel set of calculations carried out on addition of HBCl29 to alkenes. For each case a vdW complex was located (two in the case of propene) rather less favourable than the corresponding complexes from 1. The transition states were readily defined, and the anti-Markovnikov pathway is strongly preferred (entries 3 vs. 4; 8 vs. 9).38 There is a broad correspondence between the two addition reactions, with the TS for hydroboration 3–5 kcal mol−1 lower in energy than the TS for diboration. Entry 8 is an exception, for which addition of 9 to trimethylethylene (TME) is particularly favourable relative to the addition of 1. The transition state structures for both pairs of regioisomeric pathways in addition to TME (entries 8 and 9) are shown in Fig. 4.
 | ||
| Fig. 4 Comparative geometries for trimethylethylene reacting with 1 (a,b) or 9 (c,d). The upper structures show advanced boron bonding to the secondary-carbon, and the lower structures to the tertiary-carbon. Distances are in Å, TS energies in kcal mol−1 relative to the reactants; ωB97x-D/6-311G(d,p). | ||
In these examples steric effects involving BCl2 and the tertiary CMe2 moiety are manifested at their transition states in different ways. For the sec-pathway with 1 shown in (a), there is a short B–C bond at 1.62 Å, but the BCC′ angle is widened to 78° from the ideal 70° found with ethene. For the tert-pathway shown in (b) a torsional twist of B′BCC′ to 20° reduces the steric clash between B′Cl2 and CMe2. This steric clash is absent in (c) but present again in (d) and contributes to their 5.5 kcal mol−1 difference in TS energy. The sec-pathway possesses the shorter leading B–C bond at 1.70 Å vs. 1.76 Å. The influence of steric effects on reactivity was reinforced by a failure to find a transition state structure for addition of B2Cl4 to tetramethylethylene.
Polarity effects on reactivity
The trends observed for reactivity in B2Cl4 addition vs. methyl substitution suggested that the alkene has nucleophilic character at the transition-state, consistent with the higher dipole moments observed for the transition states compared to either reactants or products. To test this, the reaction of (E)-butene was repeated in solvent of varying polarity using the polarized continuum model (PCM). The results showed a clear trend; the energy is lower in heptane (ε = 1.92) than in the gas phase, further lowered in THF (ε = 7.58) and further still in CH3CN (ε = 37.5). The overall range of energies is 3.02 kcal mol−1 so that the positive influence of increased solvent polarity is substantial (Fig. 5). | ||
| Fig. 5 Solvent effects on the ZPE-corrected electronic TS energy of addition of B2Cl4 to (E)-butene; ωB97x-D/6-311G(d,p); PCM with SCRF model; values in kcal mol−1. | ||
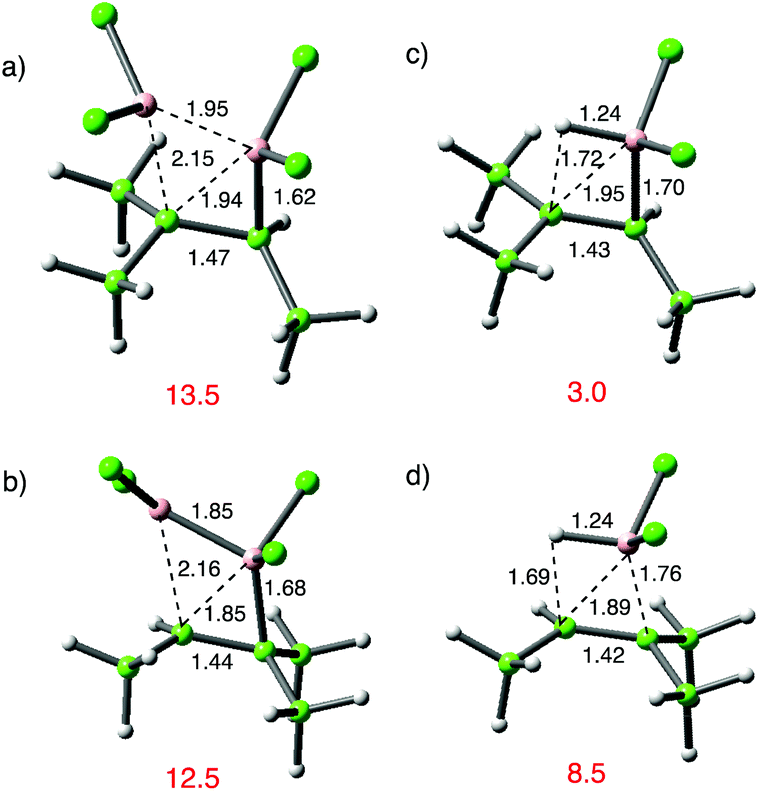
The results in Table 1 also make the prediction that an electrophilic alkene would show lower reactivity towards B2Cl4. Catalysed additions are well known in this sphere and generally lead to β-boration; with αβ-unsaturated carbonyl compounds this most likely arises through direct 1,4 addition. This is less likely, albeit still possible with unsaturated nitriles and hence the simple compounds acrylonitrile and (E)-dicyanoethene were selected.39 The computational results confirm that their reaction with B2Cl4 is less favourable than the typical examples in Table 1, as shown in Fig. 6. The much higher TS energies observed compared to typical B2Cl4 additions to alkenes in Table 1 are associated with distinct TS geometries. In the favoured case (a), the leading bond length from B is comparable to the alkene examples but the less developed bond from B′ is much longer. For the unfavourable addition in (b) it is the leading bond from B to the nitrile bound carbon that is anomalously long. For (E)-dicyanoethene in (c) both bonds are longer at the TS. When the less developed bond from B′ is to a cyano-substituted atom, that Cl2B′ group is twisted to minimize contact with the cyano-group. These results reaffirm preferred nucleophilic character in the alkene to facilitate reaction.
 | ||
| Fig. 6 Comparative geometries for the addition of B2Cl4 to electrophilic alkenes; (a) acrylonitrile, favoured regioisomer; (b) acrylonitrile, disfavoured regioisomer; (c) (E)-butene. ZPE-corrected TS electronic energies in kcal mol−1; ωB97x-D/6-311G(d,p). | ||
Reactivity of fullerene C60 towards B2Cl4
Covalent derivatives of C60 and other carbon allotropes are of interest in many applications and especially in the rapidly growing field of bioconjugates.40 This requires functionality in the fullerene, and at present there is a limited range of organic reactions that work well, and even fewer that involve controlled monoaddition.41 Indirect alkynyl group substitution is feasible, however and provides a route to ‘Click’ coupling chemistry.42 Hydroboration of fullerenes occurs, but leads only to isolation of di-and polyhydrides, or to products of further oxidation.43 An ability to form acid-stable C–B bonds to fullerenes in isolable intermediates is hence an attractive prospect since this opens new routes to their catalytic cross-coupling chemistry. Given the ease of addition of B2Cl41 to alkenes and to naphthalene, we wondered whether an addition product with C60 would be energetically accessible likewise.In any 1,2-addition to C60, there are two possible pathways depending on whether reaction occurs to the bond at a [6,6], or [6,5] ring junction. The latter involves a more drastic break in conjugation and is disfavoured.44 A stable vdW complex between C60 and B2Cl4 was found, 7.1 kcal mol−1 more stable than the isolated reactants. The transition state for the addition of B2Cl4 to the preferred [6,6] junction was readily located, with a calculated energy of 15.2 kcal mol−1 (Fig. 7). The alternative addition of B2Cl4 to the [6,5] ring junction involved a far higher energy transition state, and a product that is 19.5 kcal mol−1 less stable. Overall, these results endorse the established principle that reagents engaging by initial 1,2 addition exert a strong preference for the [6,6]-junction, observed through both reactivity and product stability.45 This accords with the relative energies of the related C60H2 isomers using the same DFT functional and basis set.46
 | ||
| Fig. 7 The favoured [6,6] and disfavoured [6,5] pathways for addition of B2Cl4 to C60. The quoted values of ZPE-corrected electronic energy are from DFT calculations with the ωB97-xD functional and 6-31G(d,p) basis set. | ||
1,2 vs. 1,4 addition possibilities; relative reactivity B2F4vs. B2Cl4
Alongside the several reports of 1,2-additions of B2X4 additions to alkenes there is a single report of 1,4-addition to buta-1,3-diene.15 This encouraged a broader analysis of the addition chemistry of dienes, and the results are shown in Fig. 8 below. Reactions of both B2Cl4 and B2F4 were analysed.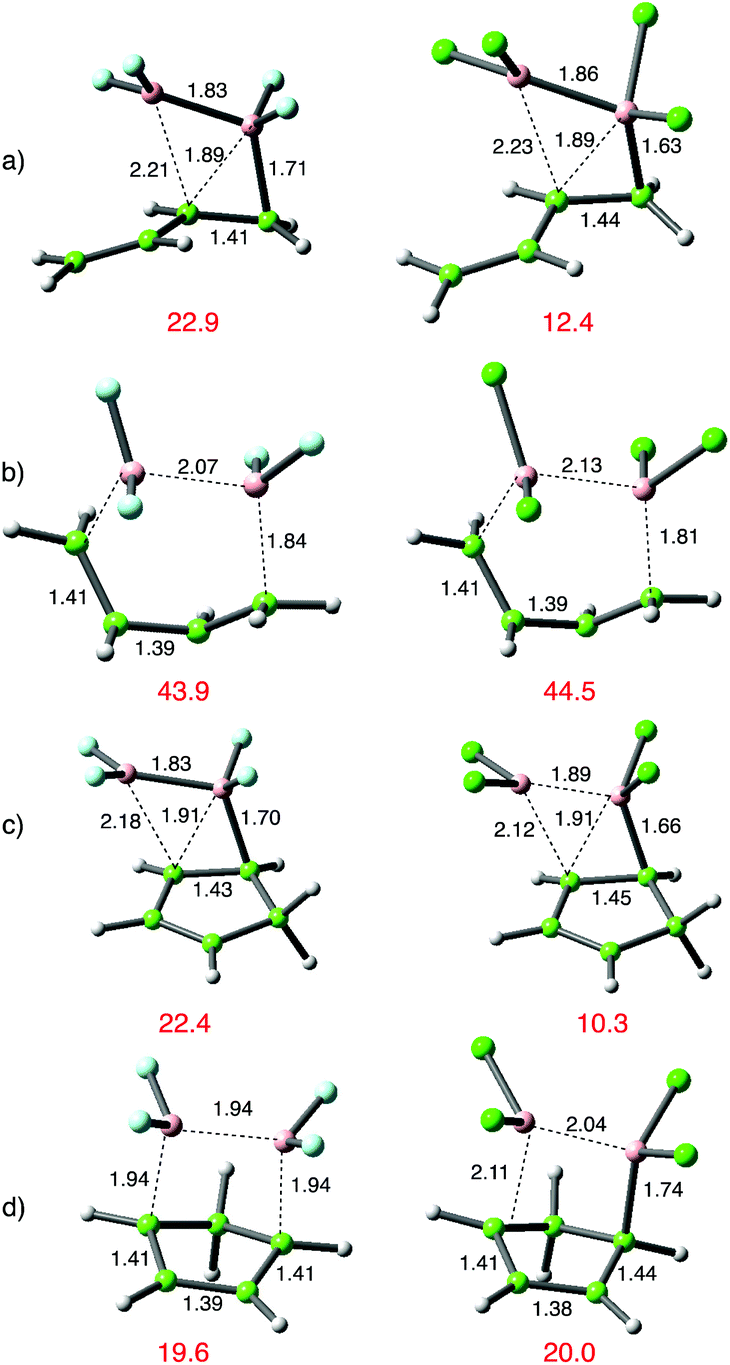 | ||
| Fig. 8 Transition-state structures for 1,2- or 1,4-addition to dienes, comparing B2F42 with B2Cl41; (a) 1,2-addition to s-trans-buta-1,3-diene; (b) the high energy 1,4-addition pathway; (c) 1,2-addition to cyclopentadiene and (d) 1,4 addition to cyclopentadiene. DFT as in Scheme 3. | ||
With buta-1,3-diene and B2Cl4, low energy 1,2-addition pathways were discovered for both the s-cis and s-trans conformations; this latter pathway was verified for B2F42. Attempts to find a normal 1,4-addition pathway from the s-cis isomers were not successful; the reaction course diverted to 1,2-addition. There is the intriguing suggestion in an early paper that the s-cis conformation might participate in a symmetry allowed [4πa + 2σa] cycloaddition,15 with orthogonal approach of the diboron reagent to the diene. We were unsuccessful in attempts to locate a transition state for this pathway, but the corresponding s-trans conformation proved more fruitful. A nicely symmetrical, albeit comparably high energy [4πs + 2σs] transition state was found for both B2Cl4 and B2F4 giving rise to the symmetrically 1,4-disubstituted (E)-but-2-ene.
With cyclopentadiene, both 1,2 and 1,4 addition pathways were located with B2Cl4, and the 1,2-addition shown in Fig. 5(c) was energetically favoured by ca. 10 kcal mol−1 over the same pathway for B2F4 and by a similar amount over the corresponding 1,4-addition shown in Fig. 5d). In contrast, the 1,4-addition pathway to cyclopentadiene with B2F4 was favoured over 1,2-addition, and was even lower in energy than the corresponding 1,4-addition with B2Cl4. Furthermore, the transition state (d) arising from B2F4 addition was symmetrical with both C–B bonds equal at 1.94 Å, whilst the TS from B2Cl4 lacked symmetry, with one C–B bond more advanced than the other, at 1.74 Å vs. 2.11 Å. The contrasting transition-state structures of the 1,2 and 1,4-addition routes for reaction of B2F4 with C4H6 and C5H6 are shown in Fig. 5(c) and (d).
Basis for the stereoselective pathway with but-2-enes
Experimental verification of the stereospecific addition of B2Cl4 to butenes by Rudolph and by Zeldin et al.,12 for a reaction that is (formally at least) a symmetry-forbidden 2πs + 2σs process requires further analysis. The frontier MO's were analysed at the B3LYP level as demonstrated in Fig. 9, and reveal the basis for a concerted, stereospecific reaction as is observed. This figure shows bonding orbitals of the reactants that are closely involved in generating the three highest energy orbitals of the TS, and the critical role of low-lying antibonding orbitals. Bonding between the proximal boron and its carbon is seen in TS(HOMO) and between the distal boron and its carbon in TS(HOMO–1). This requires a complex interplay between bonding and nonbonding orbitals of the reactants that is revealed by the TS fragment analysis shown. B2F4(LUMO) mixes strongly with the symmetry-matched C2H4(HOMO), and this makes the main contribution to TS(HOMO). The weaker involvement of B2F4(LUMO+1) leads to the less prominent distal bonding expressed in TS(HOMO−1). Overall, the combination of orbital interactions between B2F4 and the alkene permits a formally symmetry forbidden process to occur in a stereospecific manner. Orbital analysis of the TS for reaction between B2Cl4 and C2H4 is entirely comparable, save additional Cl-localised orbitals of comparable energy to (HOMO−1).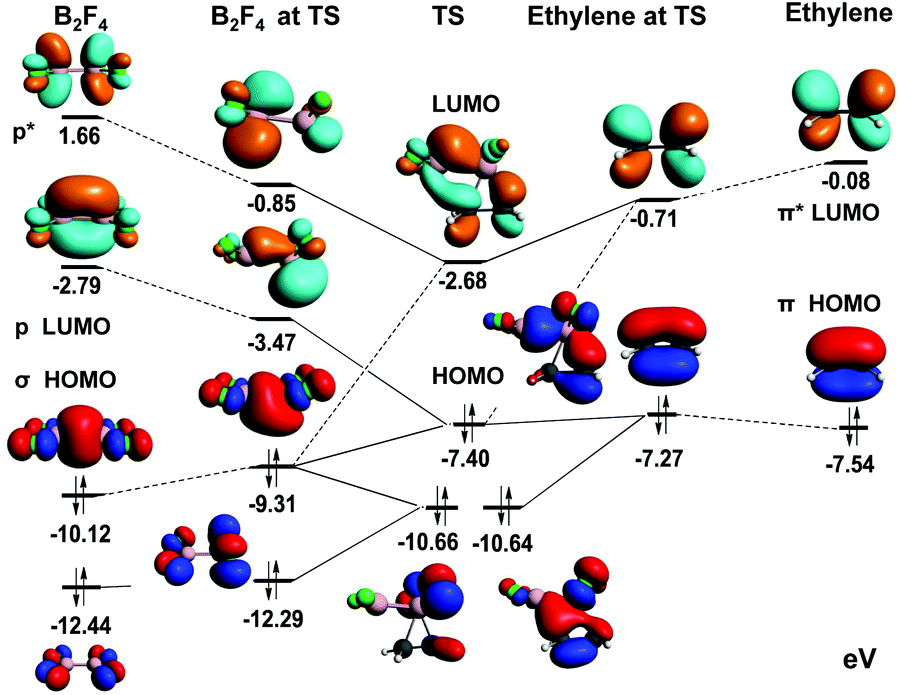 | ||
| Fig. 9 Orbital analysis of the reaction pathway for addition of B2F4 to C2H4. | ||
Computational details
In the early phase of the work geometries and analytical vibrational frequencies were computed at the B3LYP//6-311g(d,p) level in vacuo. The results obtained for addition of B2Cl4 to C2H4 were compared with calculations using functionals that included explicit treatment of dispersion energy, M06-2X, ωB97x-D and B3LYP-D3 (ref. 25–27). All of these were available within Gaussian09.47 (Rev. D-01). The choice for all further work was ωB97x-D, based on the reasonable values of activation energy obtained when compared to available experimental. ZPE-corrected electronic energies are normally reported; for the solvent effect data shown in Fig. 5, IEFPCM (Integral Equation Formalism of the Polarized Continuum Model) was employed. All transition states reported here possessed a single imaginary frequency of ≥300 cm−1.Summary and conclusions
This work was initially driven by the longstanding observation of stereoselectivity in the reaction between B2Cl4 and 1,2-disubstituted alkenes. The literature on B2Cl4 chemistry contains a number of interesting but scattered observations that provided the stimulus for a broader overview of its addition chemistry. Work in this area ceased thirty years ago, partly because of difficulties in accessibility of the reagent, but also because of its instability towards disproportionation to BCl3 and chloroboron clusters. Given the high level of current interest in catalytic additions of otherwise unreactive diboron compounds to unsaturated C–C bonds, this paper provides a set of benchmarks.The calculations herein confirm that the simple addition of B2Cl4 to alkenes uncatalysed by artefacts (e.g. HCl) is feasible, and of lower energy than all comparable B2X4 additions save B2Br4. Several novel observations arise from the current work. There is a robust transition state structure that operates across the B2X4 series studied and the reactivity in ethene addition correlates with the B–B sym-stretching vibrational frequency of the reagent. With increasing Me-substitution of the alkene, the polar character of B2Cl4 as electrophile initially reduces transition state energy, countered by increased steric effects with higher Me-substitution. Similar, but more pronounced trends are seen in a parallel analysis of HBCl2 addition. The ease of reaction extends to 1,4-addition of B2X4 to dienes, this being the preferred reaction with B2F4 and cyclopentadiene. Only transition states for 1,2-addition were located with s-cis-buta-1,3-diene although an unprecedented higher energy 1,4-trans-addition could be located for the s-trans-isomer using either B2Cl4 or B2F4. With cyclopentadiene, both 1,2 and 1,4 transition states were located. For B2Cl4, the 1,2-pathway is lower in energy. The reverse is true for B2F4, and in this case the 1,4 addition pathway is more favourable than for B2Cl4. The transition state for B2F4 addition retains σ-symmetry, whilst for B2Cl4 it is unsymmetrical. The asynchronous concerted pathway observed here for 1,4 addition of B2Cl4 contrasts with Diels–Alder addition of symmetrical dienophiles to cyclopentadiene, where symmetrical transition-states along a synchronous pathway are generally preferred.48
Given the range and ease of addition of B2Cl4 to unsaturated molecules found here, the topic merits experimental revival and extension. Easier access to diboron tetrahalides using modern synthetic methodologies would provide a crucial breakthrough. In particular, the easy addition reactions to naphthalene or C60 suggest a useful and mild method for the functionalization of aromatic carbon frameworks and assemblies in Materials Science. More generally, the reagents are shown to be electrophilic in their addition chemistry, permitting insight into their appropriate application. The concerted uncatalyzed addition of two heavy atoms to C–C unsaturation remains a rare and intriguing reaction type.
Acknowledgements
This research has been supported by the Spanish Ministerio de Economia y Competitividad MINECO under projects CTQ2013-43395-P and CTQ2014-52824-R, Consolider CTQ2014-52974-REDC, and Severo Ochoa Excellence Accreditation 2014-2018 EV-2013-0319, by the Generalitat de Catalunya (2014SGR409), and by the ICIQ Foundation. We thank NSCCS for access to the Slater Cluster at RAL under EPSRC support.Notes and references
- (a) G. Urry, J. Kerrigan, T. D. Parsons and H. I. Schlesinger, J. Am. Chem. Soc., 1954, 76, 5299–5301 CrossRef CAS; (b) P. Ceron, A. Finch, J. Frey, J. Kerrigan, T. Parsons, G. Urry and H. I. Schlesinger, J. Am. Chem. Soc., 1959, 81, 6368–6371 CrossRef CAS.
- (a) H. C. Brown and B. C. S. Rao, J. Org. Chem., 1957, 22, 1136–1137 CrossRef CAS; (b) H. C. Brown and B. C. S. Rao, J. Org. Chem., 1957, 22, 1137–1138 CrossRef CAS.
- J. Feeney, A. K. Holliday and F. J. Marsden, J. Chem. Soc., 1961, 356–360 RSC.
- (a) C. Chambers and A. K. Holliday, J. Chem. Soc., 1965, 3459–3462 RSC; (b) W. Siebert, M. Hildenbrand, P. Hornbach, G. Karger and H. Pritzkow, Z. Naturforsch., B: Chem. Sci., 1989, 44, 1179–1186 CrossRef CAS.
- W. B. Fox and T. Wartik, J. Am. Chem. Soc., 1961, 83, 498–499 CrossRef CAS.
- T. Wartik, R. Moore and H. I. Schlesinger, J. Am. Chem. Soc., 1949, 71, 3265–3266 CrossRef CAS; G. Urry, T. Wartik, R. E. Moore and H. I. Schlesinger, J. Am. Chem. Soc., 1954, 76, 5293–5298 CrossRef.
- (a) P. L. Timms, J. Chem. Soc., Dalton Trans., 1972, 830–832 RSC; (b) P. L. Timms, Inorg. Synth., 1979, 19, 74–78 CrossRef CAS PubMed.
- F. J. Lawlor, N. C. Norman, N. L. Pickett, E. G. Robins, P. Nguyen, G. Lesley, T. B. Marder, J. A. Ashmore and J. C. Green, Inorg. Chem., 1998, 37, 5282–5288 CrossRef CAS.
- P. Rothhardt, A. Wolf, D. Biro, U. Belledin and C. Wufka, Ger. Pat, DE102012025429A1, 2014 Search PubMed; World Pat, WO201409-6443A1, 2014 Search PubMed.
- L. Ahmed, J. Castillo, D. A. Saulys and J. A. Morrison, Inorg. Chem., 1992, 31, 706–710 CrossRef CAS.
- H. Noeth and H. Pommerening, Chem. Ber., 1981, 114, 398–399 CrossRef CAS PubMed.
- (a) R. W. Rudolph, J. Am. Chem. Soc., 1967, 89, 4216–4217 CrossRef CAS; (b) M. Zeldin, A. R. Gatti and T. Wartik, J. Am. Chem. Soc., 1967, 89, 4217–4218 CrossRef CAS.
- (a) M. Zeldin and A. Rosen, J. Organomet. Chem., 1971, 31, 319–328 CrossRef CAS; (b) M. Zeldin and T. Wartik, J. Am. Chem. Soc., 1966, 88, 1336–1338 CrossRef CAS.
- W. Haubold and K. Stanzl, Chem. Ber., 1978, 111, 2108–2112 CrossRef CAS PubMed.
- W. Haubold and K. Stanzl, J. Organomet. Chem., 1979, 174, 141–147 CrossRef CAS.
- J. A. Morrison, Chem. Rev., 1991, 91, 35–48 CrossRef CAS.
- Early papers: B2N4: (a) R. J. Brotherton, A. L. McClosky, L. L. Petterson and H. Steinberg, J. Am. Chem. Soc., 1960, 82, 6242–6245 CrossRef CAS; (b) H. Noth and W. Meister, Chem. Ber., 1961, 94, 509–514 CrossRef CAS PubMed;B2O4: (c) R. J. Brotherton, A. L. McCloskey, J. L. Boone and H. M. Manasevit, J. Am. Chem. Soc., 1960, 82, 6245–6248 CrossRef CAS.
- Inter alia: (a) T. Ishiyama, K. Nishijima, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 7219 Search PubMed; (b) R. T. Baker, P. Nguyen, T. B. Marder and S. A. Westcott, Angew. Chem., Int. Ed. Engl., 1995, 34, 1336–1338 Search PubMed; (c) J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702–8703 Search PubMed.
- (a) J. Ramírez, M. Sanaú and E. Fernández, Angew. Chem., Int. Ed., 2008, 47, 5194–5197 Search PubMed; (b) Q. Chen, J. Zhao, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2013, 15, 5766–5769 Search PubMed.
- (a) Q. Cui, D. G. Musaev and K. Morokuma, Organometallics, 1997, 16, 1355–1364 Search PubMed; (b) Q. Cui, D. G. Musaev and K. Morokuma, Organometallics, 1998, 17, 742–751 Search PubMed; (c) S. Sakaki and T. Kikuno, Inorg. Chem., 1997, 36, 226–229 Search PubMed.
- For pioneering works on Cu-catalysed β-boration through σ-bond metathesis see: (a) K. Takahashi, T. Isiyama and N. Miyaura, Chem. Lett., 2000, 982 Search PubMed; (b) H. Ito, H. Yamanaka, J. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821 Search PubMed; (c) K. Takahashi, T. Isiyama and N. Miyaura, J. Organomet. Chem., 2001, 625, 47 Search PubMed.
- (a) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem. – Eur. J., 2015, 21, 7082–7098 Search PubMed; (b) K. S. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253–7255 Search PubMed; (c) A. Bonet, H. Gulyas and E. Fernandez, Angew. Chem., Int. Ed., 2010, 49, 5130–5134 Search PubMed; (d) C. Pubill-Ulldemolins, A. Bonet, H. Gulyas, C. Bo and E. Fernandez, Org. Biomol. Chem., 2012, 10, 9677–9682 Search PubMed.
- A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyas and E. Fernandez, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 Search PubMed.
- (a) H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, T. Kupfer, K. Radacki, E. Siedler, A. Trumpp, K. Wagner and Ch. Werner, J. Am. Chem. Soc., 2013, 135, 8702 Search PubMed; (b) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kupfer and K. Radacki, Angew. Chem., Int. Ed., 2012, 51, 6267 Search PubMed; (c) H. Braunschweig, A. Damme and T. Kupfer, Chem. Commun., 2013, 49, 2774 Search PubMed.
- R. R. Ryan and K. Hedberg, J. Chem. Phys., 1969, 50, 4986–4995 Search PubMed; D. E. Mann and L. Fano, J. Chem. Phys., 1957, 26, 1665–1670 Search PubMed.
- M. Atoji, P. J. Wheatley and W. N. Lipscomb, J. Chem. Phys., 1957, 27, 196–199 Search PubMed.
- (a) L. Trefonas and W. N. Lipscomb, J. Chem. Phys., 1958, 28, 54–55 Search PubMed; (b) I. V. Kochikov and Y. I. Tarasov, Struct. Chem., 2003, 14, 227–238 Search PubMed; (c) D. Nori-Shargh, W. Zou and J. E. Boggs, Asian J. Spectrosc., 2010, 49–59 Search PubMed; (d) C. R. Watts and J. K. Badenhoop, J. Chem. Phys., 2008, 129, 104307 Search PubMed.
- CDS database: (a) F. H. Allen and W. D. S. Motherwell, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 407–422 Search PubMed; (b) F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 Search PubMed.
- (a) B3LYP: A. D. Becke, J. Chem. Phys., 1993, 98, 5648 Search PubMed; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 Search PubMed; (b) B3LYP-D3: S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 Search PubMed.
- M06-2x: (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- ωB97x-D: J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 Search PubMed.
- Examples of DFT calculations where heteroatom-substituted boron hydrides have been utilised: (a) S. W. Hadebe, H. G. Kruger and R. S. Robinson, Comput. Theor. Chem., 2011, 968, 26–30 Search PubMed; (b) S. W. Hadebe, R. S. Robinson and H. G. Kruger, S. Afr. J. Chem., 2009, 62, 84–87 Search PubMed.
- (a) M. Lein, A. Szabo, A. Kovacs and G. Frenking, Faraday Discuss., 2003, 124, 365–378 Search PubMed; (b) A. Szabo, A. Kovacs and G. Frenking, Z. Anorg. Allg. Chem., 2005, 631, 1803–1809 Search PubMed.
- (a) From electron diffraction: B2F4, 1.72 Å, B2Cl4 1.70 Å, B2Br4, 1.69 Å; D. D. Danielson and K. Hedberg, J. Am. Chem. Soc., 1979, 101, 3199–3203 Search PubMed ; from X-ray: (B2OR4) 1.696 ± 0.003 Å, 11 structures (ref. 28).
- H. C. Brown and A. W. Moerikofer, J. Am. Chem. Soc., 1963, 85, 2063–2068 Search PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, UK, 2nd edn, 2012, ch. 19, p. 429ff Search PubMed.
- (a) D. J. Pasto and P. Balasubramaniyan, J. Am. Chem. Soc., 1967, 89, 295–300 Search PubMed; (b) G. Zweifel, J. Organomet. Chem., 1967, 9, 215–221 Search PubMed; (c) H. C. Brown, G.-M. Chen, M. P. Jennings and P. V. Ramachandran, Angew. Chem., Int. Ed., 1999, 38, 2052–2054 Search PubMed; (d) P. V. Ramachandran and M. P. Jennings, Chem. Commun., 2002, 386–387 Search PubMed.
- The prototypical additon of BH3 to alkenes requires a molecular dynamics approach in order to correctly match the experimental regioselectivity, whilst QM-based transition-state theory favours the less substituted borane too strongly: (a) Y. Oyola and D. A. Singleton, J. Am. Chem. Soc., 2009, 131, 3130–3131 Search PubMed; (b) D. R. Glowacki, C. H. Liang, S. P. Marsden, J. N. Harvey and M. J. Pilling, J. Am. Chem. Soc., 2010, 132, 13621–13623 Search PubMed; (c) D. R. Glowacki, R. Lightfoot and J. N. Harvey, Mol. Phys., 2013, 111, 631–640 Search PubMed.
- (a) G. W. Kabalka, B. C. Das and S. Das, Tetrahedron Lett., 2002, 43, 2323–2325 Search PubMed; (b) M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. L. Santos, J. Org. Chem., 2011, 76, 3997–4007 Search PubMed.
- (a) N. F. Steinmetz, V. Hong, E. D. Spoerke, P. Lu, K. Breitenkamp, M. G. Finn and M. Manchester, J. Am. Chem. Soc., 2009, 131, 17093–17095 Search PubMed; (b) K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 Search PubMed.
- E.g.: (a) Y. Li, N. Lou and L. Gan, Org. Lett., 2015, 17, 524–527 Search PubMed; (b) J. Yang, L. B. Alemany, J. Driver, J. D. Hartgerink and A. R. Barron, Chem. – Eur. J., 2007, 13, 2530–2545 Search PubMed; (c) A. L. Balch, D. A. Costa, B. C. Noll and M. M. Olmstead, J. Am. Chem. Soc., 1995, 117, 8926–8932 Search PubMed.
- W.-B. Zhang, Y. Tu, R. Ranjan, R. M. Van Horn, S. Leng, J. Wang, M. J. Polce, C. Wesdemiotis, R. P. Quirk, G. R. Newkome and S. Z. D. Cheng, Macromolecules, 2008, 41, 515–517 Search PubMed , and references therein.
- (a) N. S. Schneider, A. D. Darwish, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1994, 463–464 Search PubMed; (b) C. C. Henderson, C. M. Rohlfing, K. T. Gillen and P. A. Cahill, Science, 1994, 264, 397–399 Search PubMed.
- See, for example: (a) N. Alegret, A. Rodriguez-Fortea and J. M. Poblet, Chem. – Eur. J., 2013, 19, 5061–5069 Search PubMed; (b) A. Rodriguez-Fortea, S. Irle and J. M. Poblet, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 350–367 Search PubMed; (c) I. Fernandez, M. Sola and F. M. Bickelhaupt, Chem. – Eur. J., 2013, 19, 7416–7422 Search PubMed; (d) S. Osuna, M. Swart and M. Sola, Phys. Chem. Chem. Phys., 2011, 13, 3585–3603 Search PubMed.
- Examples: (a) Y. Elemes, S. K. Silverman, C. Sheu, M. Kao, C. S. Foote, M. M. Alvarez and R. L. Whetten, Angew. Chem., Int. Ed., 1992, 31, 351–353 Search PubMed; (b) M. S. Meier and J. Kiegiel, Org. Lett., 2001, 3, 1717–1719 Search PubMed; (c) Y. Li, N. Lou and L. Gan, Org. Lett., 2015, 17, 524–527 Search PubMed; (d) M. Tsuda, T. Ishida, T. Nogami, S. Kurono and M. Ohashi, J. Chem. Soc., Chem. Commun., 1993, 1296–1298 Search PubMed; (e) N. Ikuma, Y. Susami and T. Oshima, Org. Biomol. Chem., 2010, 8, 1394–1398 Search PubMed.
- The favoured product from H2 addition at a (6,6) junction is 21.24 kcal mol−1 more stable than the reactants; from H2 addition at a (6,5) junction just 0.36 kcal mol−1 more stable than the reactants, on the basis of ZPE-corrected electronic energies (ex. DFT, see ESI†).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- E.g.: (a) A. M. Sarotti, Org. Biomol. Chem., 2014, 12, 187–199 Search PubMed; (b) L. Rulisek, P. Sebek, Z. Havlas, R. Hrabal, P. Capek and A. Svatos, J. Org. Chem., 2005, 70, 6295–6302 Search PubMed; (c) S. Yamabe, Y. Nishihara and T. Minato, J. Phys. Chem. A, 2002, 106, 4980–4987 Search PubMed; (d) cf. The experimental viewpoint; D. A. Singleton, B. E. Schulmeier, C. Hang, A. A. Thomas, S. W. Leung and S. R. Merrigan, Tetrahedron, 2001, 57, 5149–5160 Search PubMed; (e) D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9358 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DFT analyses described in the text. See DOI: 10.1039/c5ob01280e |
| This journal is © The Royal Society of Chemistry 2015 |