
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strand displacement and duplex invasion into double-stranded DNA by pyrrolidinyl peptide nucleic acids†
Peggy R.
Bohländer
a,
Tirayut
Vilaivan
*b and
Hans-Achim
Wagenknecht
*a
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: Wagenknecht@kit.edu; Fax: +49-721-608-44825; Tel: +49-721-608-47486
bOrganic Synthesis Research Unit, Department of Chemistry, Faculty of Science, Chulalongkorn University Phayathai Road, Patumwan, Bangkok 10330, Thailand. E-mail: vtirayut@chula.ac.th; Fax: +66-2-2187598; Tel: +66-2-2187627
First published on 22nd July 2015
Abstract
The so-called acpcPNA system bears a peptide backbone consisting of 4′-substituted proline units with (2′R,4′R) configuration in an alternating combination with (2S)-amino-cyclopentane-(1S)-carboxylic acids. acpcPNA forms exceptionally stable hybrids with complementary DNA. We demonstrate herein (i) strand displacements by single-stranded DNA from acpcPNA–DNA hybrids, and by acpcPNA strands from DNA duplexes, and (ii) strand invasions by acpcPNA into double-stranded DNA. These processes were studied in vitro using synthetic oligonucleotides and by means of our concept of wavelength-shifting fluorescent nucleic acid probes, including fluorescence lifetime measurements that allow quantifying energy transfer efficiencies. The strand displacements of preannealed 14mer acpcPNA–7mer DNA hybrids consecutively by 10mer and 14mer DNA strands occur with rather slow kinetics but yield high fluorescence color ratios (blue![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :yellow or blue:red), fluorescence intensity enhancements, and energy transfer efficiencies. Furthermore, 14mer acpcPNA strands are able to invade into 30mer double-stranded DNA, remarkably with quantitative efficiency in all studied cases. These processes can also be quantified by means of fluorescence. This remarkable behavior corroborates the extraordinary versatile properties of acpcPNA. In contrast to conventional PNA systems which require 3 or more equivalents PNA, only 1.5 equivalents acpcPNA are sufficient to get efficient double duplex invasion. Invasions also take place even in the presence of 250 mM NaCl which represents an ionic strength nearly twice as high as the physiological ion concentration. These remarkable results corroborate the extraordinary properties of acpcPNA, and thus acpcPNA represents an eligible tool for biological analytics and antigene applications.
:yellow or blue:red), fluorescence intensity enhancements, and energy transfer efficiencies. Furthermore, 14mer acpcPNA strands are able to invade into 30mer double-stranded DNA, remarkably with quantitative efficiency in all studied cases. These processes can also be quantified by means of fluorescence. This remarkable behavior corroborates the extraordinary versatile properties of acpcPNA. In contrast to conventional PNA systems which require 3 or more equivalents PNA, only 1.5 equivalents acpcPNA are sufficient to get efficient double duplex invasion. Invasions also take place even in the presence of 250 mM NaCl which represents an ionic strength nearly twice as high as the physiological ion concentration. These remarkable results corroborate the extraordinary properties of acpcPNA, and thus acpcPNA represents an eligible tool for biological analytics and antigene applications.
Introduction
Peptide nucleic acid (PNA) represents a very important nucleic acid analog and chemical-biological tool based on its important features like affinity, sequence selectivity and stability against degradation by nucleases and proteases.1 Especially the uncharged pseudopeptide backbone makes PNA an attractive scaffold for diagnostic applications. PNA with N-(2-aminoethyl)glycine backbone (aegPNA) is able to form complexes with double-stranded DNA either by binding from outside forming triplexes, or by duplex, double-duplex or triplex invasion.1–4 The attempt to increase affinity and, more importantly, sequence selectivity of PNA by incorporation of cyclic derivatives in the peptide backbone failed in many cases since the nucleic acid hybridization properties are deteriorated.5,6 Among several of those PNA architectures reported to date, and especially among those with cyclic structures as part of the backbone,7–9 the so-called acpcPNA system (Fig. 1) exhibits significant features. The peptide backbone consists of 4′-substituted proline units with (2′R,4′R) configuration in an alternating combination with (2S)-amino-cyclopentane-(1S)-carboxylic acids. As a result of this structural design, acpcPNA forms stable hybrids with complementary DNA but significantly less stable hybrids with complementary RNA.10–13 The recognition and binding to DNA shows high sequence selectivity and does not tolerate single base mismatches. The sequence selectivity can be visualized by fluorescent dyes such as pyrene,14,15 thiazole orange16 and nile red,17 that were synthetically incorporated into acpcPNA. Most importantly, self-paired hybrids are not formed by acpcPNA.11 This feature stands in contrast to many other PNA scaffolds that bind to themselves equally or even better than to naturally occurring nucleic acids.4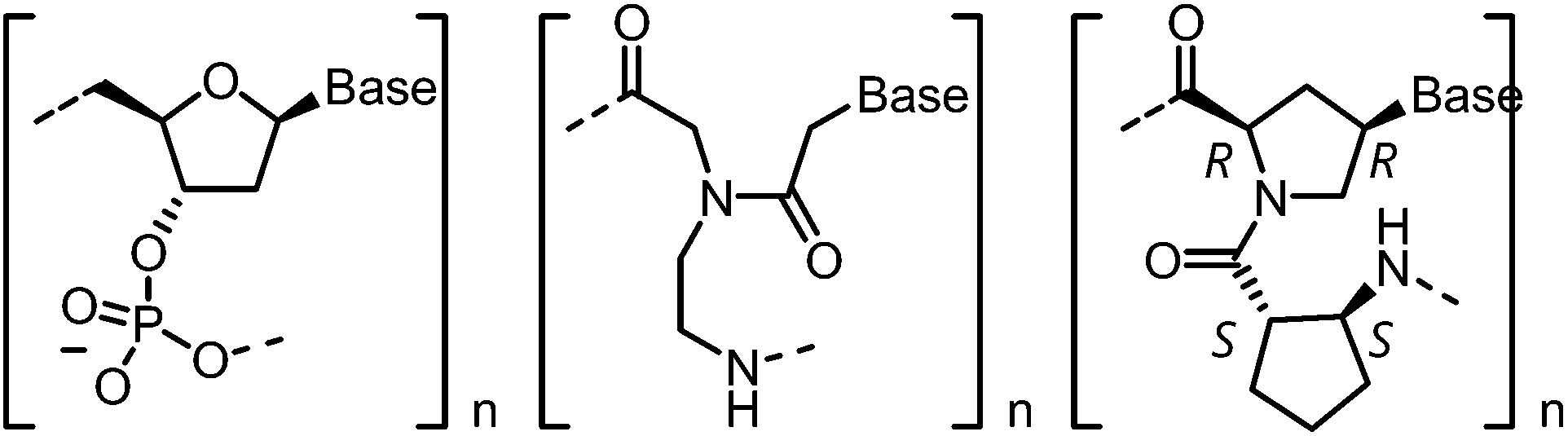 | ||
| Fig. 1 Monomeric unit of DNA (left), aegPNA (middle) and acpcPNA (right) in comparison. | ||
Base and/or PNA backbone modifications are traditionally required to destabilize PNA–PNA duplexes in order to allow efficient double duplex invasion of DNA.18,19 Alternatively, PNA modifications like carcinogenic acridine moieties or nucleobase substitutions of cytosine (G-clamp) also promote invasion into mixed sequence B-DNA.20–23 To prevent extensive or biologically harmful modifications, a chiral derivative of PNA called γPNA that exhibits even higher DNA affinity than normal PNA with extended strand length was employed. However, 20 equivalents of γPNA and incubating times of 8 hours are necessary to accomplish quantitative invasion and complex formation with double-stranded DNA.24 Moreover, limitations for applications in vivo still remain due to the high affinity of γPNA to both DNA and RNA. In order to demonstrate the advantages of acpcPNA in this regard we report herein (i) strand displacements by single-stranded DNA from acpcPNA–DNA hybrids, and by acpcPNA strands from DNA duplexes, and (ii) strand invasions by acpcPNA strands into double-stranded DNA with high efficiency. These processes have high biological significance; however, we studied them in vitro with model PNA strands by means of fluorescence color changes as well as fluorescent intensity enhancements using our concept of wavelength-shifting nucleic acid probes (“DNA traffic lights”).25
Results and discussion
Strand displacements
DNA strand displacements are biologically relevant and enzymatically driven processes inside living cells.26–29 Furthermore, these processes play a crucial role in DNA nanotechnology.30 Consecutive annealing and strand displacement steps represent the methodological basis to dynamically control the formation of highly ordered and self-assembled DNA nanoarchitectures, such as DNA switches, DNA tweezers,31–34 DNA-based logic gates,34–38 DNA walkers39–42 and others. In order to readout the strand displacement dynamics with acpcPNA not only by conventional fluorescence intensity changes at a single emission wavelength, we followed our concept of wavelength-shifting nucleic acid probes.25 Five years ago, we introduced this concept that is based on energy transfer between thiazole orange and thiazole red as fluorescent DNA base substitutions and applied it for molecular beacons.25,43 Moreover, we showed that this concept works also for pairs of fluorescent dyes attached to the 2′-positions in two different DNA strands and yields even better emission color contrasts as readout.44In order to apply such fluorescence color changes to directly report the dynamics of strand displacements in vitro we synthesized the 14mers PNA1 and PNA2 that carry the blue emitting dye 1 in the middle of the sequence (Fig. 2). These acpcPNA modifications were introduced by post-synthetic copper(I)-catalyzed cycloaddition between the precursor of dye 1 bearing an azide group and the synthesized precursors of PNA1 and PNA2 that carry an alkyne functionality. The only difference between the two PNA strands is the position of dye 1 as modification of the aminopyrrolidinecarboxylic acid spacers either on the carboxy or on the amino side of the central thymine unit in the sequence. All synthesized PNA strands bear five lysine residues at the C-terminus to enhance solubility. Five DNA counterparts of different lengths were synthesized. The shortest one, 7mer DNA1, was not labelled. The 10mers DNA2 and DNA4 were labelled by the yellow emitting dye 2 attached to the 2′-position of uridine next to the central adenosine unit (either on the 5′or 3′ side). The longest counterparts, 14mers DNA3 and DNA5, were modified at corresponding positions by the red emitting dye 3 (for the syntheses of dyes 1–3 and PNA/DNA labelling procedures see ESI†).44,45
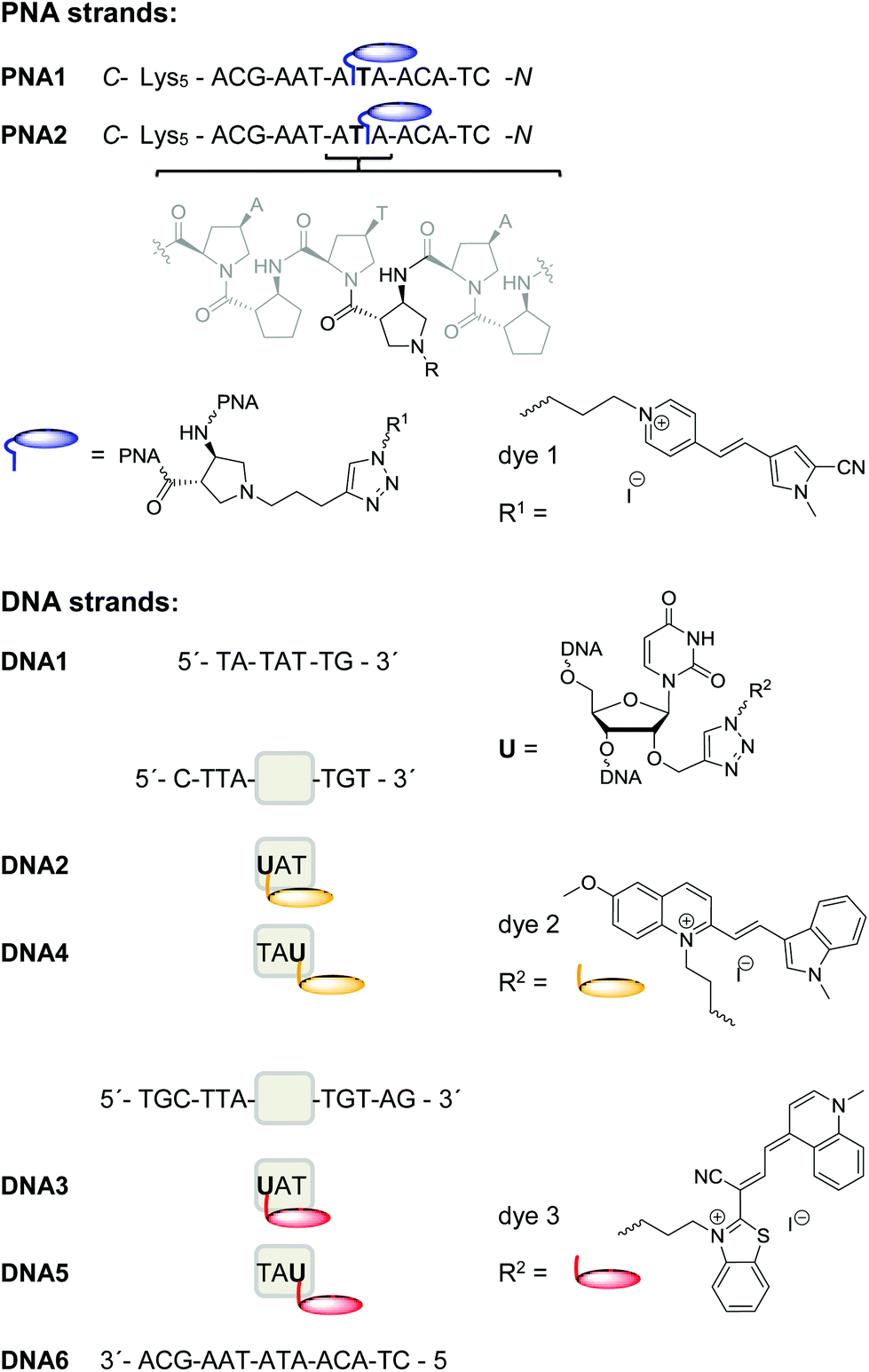 | ||
| Fig. 2 Sequences of PNA1, PNA2, DNA1–DNA6 and structures of acpcPNA and of dyes 1–3 as PNA or DNA labels, respectively. | ||
The dye 1-labeled PNA1 and PNA2 were separately annealed with the unmodified 7mer oligonucleotide counterstrand DNA1 to obtain the starting duplexes for strand displacement experiments. Both samples were consecutively titrated with 1.0 equivalent of DNA2 or DNA4, followed by DNA3 or DNA5, respectively (Fig. 3). The first titrations with DNA2 or DNA4 place the energy acceptor dye 2 into proximity to donor dye 1. If the excitation wavelength is kept constant at 389 nm, the efficient (vide infra) energy transfer between these two dyes causes significant fluorescence color changes from blue to yellow which were observed in all four cases (PNA1-DNA1 and PNA2-DNA1, each with DNA2 or DNA4). The second titrations with DNA3 or DNA5, respectively, remove dye 2 from the hybrids and place the energy acceptor dye 3 into close proximity to dye 1. As a result the fluorescence color changes from yellow to red; again at the same excitation wavelength (389 nm).
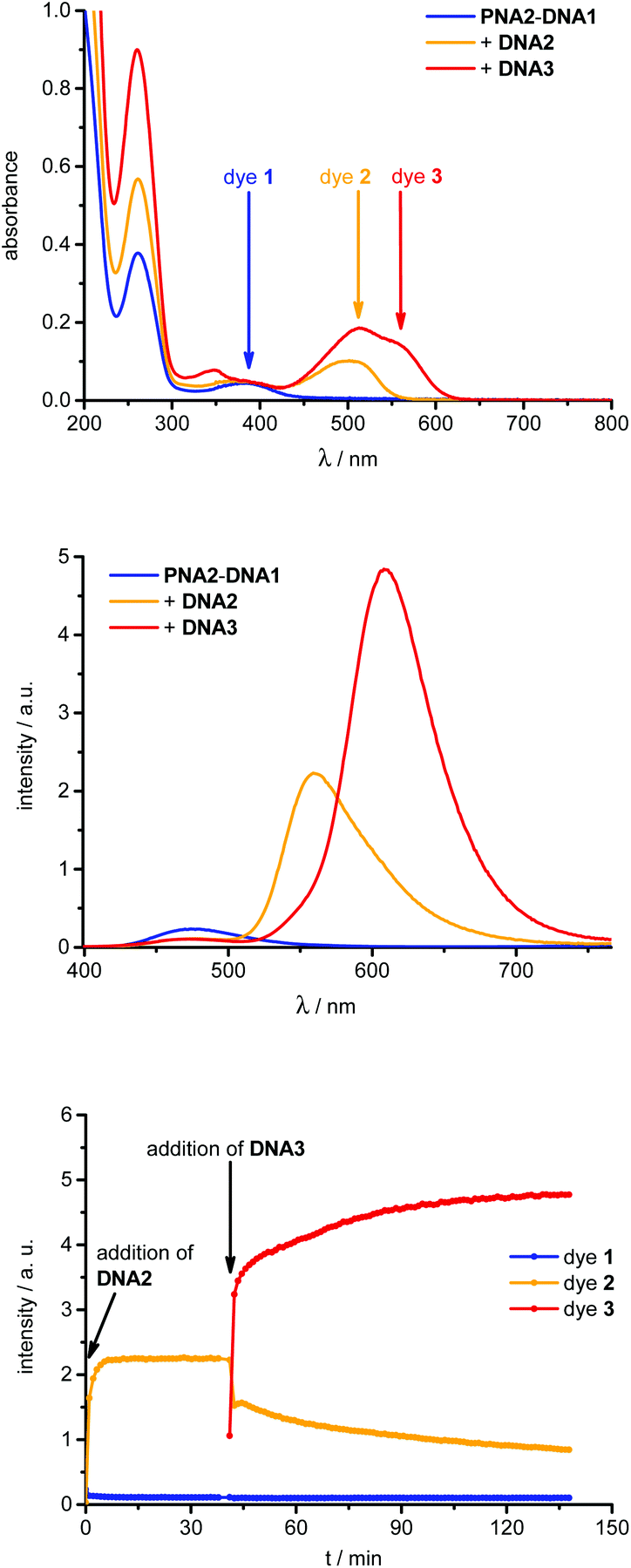 | ||
| Fig. 3 Optical spectra of the representative strand displacement experiment with PNA2–DNA1 (2.5 μM in 10 mM Na–Pi buffer, pH 7) by consecutive addition of DNA2 (1.0 equiv.) and DNA3 (1.0 equiv.): UV/Vis absorption (top), fluorescence (middle, λexc = 389 nm) and time-dependent fluorescence intensity changes (dye 1: λem = 478 nm, dye 2: λem = 560 nm, dye 3: λem = 606 nm) after addition of DNA2 (0–41 min, and DNA3 (41–138 min). | ||
From these titrations the following two observations have been made and further investigated. (i) The first strand displacements of 7mer DNA1 by 10mer DNA2 or DNA4 take place relatively fast (within 5 minutes). The second strand displacements of 10mer DNA2 or DNA4 as part of the intermediate acpcPNA hybrids by 14mer DNA3 or DNA5 occur significantly slower than the first displacement processes (approximately 1 h). This can be explained by the increasing affinity of acpcPNA to longer DNA pieces during the strand displacements and the exceptionally high stability of acpcPNA–DNA hybrids in general. Melting temperatures of the starting, intermediate and final acpcPNA–DNA hybrids were determined (Table 1). The representative titration of hybrid PNA2–DNA1 with DNA2 and DNA3 is shown in Fig. 3 and exhibits a Tm increase from 71.5 °C over 77.2 °C to >90 °C. As expected all PNA–DNA hybrids possess far higher Tm than comparable DNA–DNA duplexes (see ESI†). Both PNA strands, PNA1 and PNA2, form especially stable hybrids with unmodified DNA counterstrands as shown by Tm of >90 °C (see ESI†), whereas singly and doubly modified acpcPNA–DNA hybrids exhibit lower Tm values. The singly modified duplex DNA6–DNA3 exhibits a Tm which is around 50 °C lower in comparison to the corresponding acpcPNA–14mer DNA hybrid (see ESI†) and hence, affirms strong binding between acpcPNA and DNA even in the presence of an attached dye. In case of the majority of acpcPNA–10mer DNA hybrids, e.g.PNA1–DNA2, the Tm values show that the fluorescent dye labels are destabilizing and require carefully chosen placements in complementary hybrids.17 Especially the dye attachment in the hybrid PNA1–DNA4 causes less destabilizing effects and gives a Tm of >90 °C. (ii) Large fluorescence color ratios blue:yellow/blue:red and fluorescence intensity enhancements were accomplished by an efficient energy transfer (vide infra) from donor dye 1 to both acceptor dyes 2 and 3, respectively (Table 1). The highest blue:red contrast (I478 nm/I600 nm) of 1:223 is obtained during displacement of 10mer DNA4 by 14mer DNA5 yielding hybrid PNA2–DNA5. The enhancements of the yellow and red fluorescence signals were quantified by the fluorescence intensity increase factors (Table 1) that were determined for both acceptor dyes 2 or 3, respectively, at their corresponding emission maximum. The factors represent the ratios of the fluorescence intensities of the acceptor dyes after excitation at the absorption maximum of donor dye 1 to the fluorescence intensities of the acceptor dyes upon excitation at the same wavelength but in the absence of the donor dye 1. The latter fluorescence intensities were obtained with single-stranded DNA1 to DNA5. The largest fluorescence enhancement factor of 21.1 was obtained during the strand displacement of PNA1–DNA2 by DNA3.
| Duplex | Color contrast | Fluorescence intensity increase factora | Energy transfer efficiencyb | T m [°C] |
|---|---|---|---|---|
| a See definition in text. b Calculated by fluorescence lifetimes (see ESI). c λ = 260 nm, 5–95 °C, interval: 0.5 °C min–1, 2.5 μM duplex in 10 mM Na–Pi, pH 7. d I 478 nm/I560 nm. e I 478 nm/I606 nm. f I 478 nm/I579 nm. g I 478 nm/I600 nm. | ||||
| PNA1–DNA1 | — | — | — | 71.0 |
| PNA1–DNA2 | 1:34d |
16.6 | 0.82 ± 0.13 | 78.2 |
| PNA1–DNA3 | 1:141e |
21.1 | 0.99 ± 0.13 | > 90 |
| PNA1–DNA4 | 1:38f |
15.0 | 0.79 ± 0.12 | > 90 |
| PNA1–DNA5 | 1:173g |
8.9 | 0.98 ± 0.16 | > 90 |
| PNA2–DNA1 | — | — | — | 71.5 |
| PNA2–DNA2 | 1:20d |
10.8 | 0.78 ± 0.11 | 77.2 |
| PNA2–DNA3 | 1:47e |
20.1 | 0.98 ± 0.05 | > 90 |
| PNA2–DNA4 | 1:49f |
9.8 | 0.75 ± 0.10 | 82.7 |
| PNA2–DNA5 | 1:223g |
19.8 | 0.98 ± 0.07 | > 90 |
| DNA6–DNA3 | — | — | — | 32.7 |
In order to calculate energy transfer efficiencies, fluorescence lifetimes of dye 1 (λexc = 389 nm, λem = 478 nm) were determined for all modified PNA–DNA hybrids (see ESI†). In the absence of energy acceptor, dye 1 in hybrids of PNA1 and PNA2 with full length DNA counterstrands (see ESI†) exhibits lifetimes of approximately 1.6 ns and 1.5 ns, respectively. Due to the presence of acceptor dyes 2, or 3, respectively, these lifetimes are significantly shortened to the sub-ns range in all doubly labeled acpcPNA–DNA hybrids yielding excellent energy transfer efficiencies between 75% and 99%.
The following strand displacement experiment representatively shows the ability of acpcPNA to displace one DNA strand out of the 14mer duplex DNA6–DNA3. The recorded fluorescence of dye 3 shows a significant intensity increase due to the fact that the unmodified DNA6 is replaced by PNA1 bearing the energy donor which is dye 1 (Fig. 4). This clearly demonstrates that strand displacement of DNA6 by PNA1 occurs efficiently within several minutes due to the large increase in stability from 32.7 °C (DNA6–DNA3) to >90 °C (PNA1–DNA3). This kind of strand displacement is an important prerequisite for the in vitro strand invasion experiments described in the second part below.
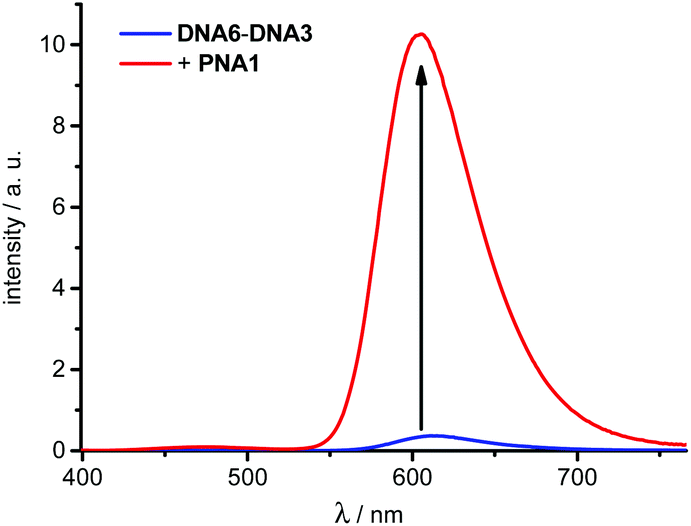 | ||
| Fig. 4 Fluorescence intensity change during the strand displacement of DNA6–DNA3 (2.5 μM in 10 mM Na–Pi buffer, pH 7) by PNA1 (1.0 equiv.), λexc = 389 nm, for UV/Vis absorption spectra and time-dependent fluorescence intensity changes see ESI.† | ||
Strand invasions
Antigene targeting of double stranded (ds) DNA by PNA is an important goal for biological applications inside living cells. It was shown10,11 that the conformationally restricted acpcPNA is not able to form triplex structures like conventional PNA that binds to dsDNA in several different binding modes, including triplex formation, triplex invasion, duplex invasion and double duplex invasion.4,46 In order to confirm that acpcPNA is able to undergo double duplex invasion and to follow this process by fluorescence spectroscopy, dsDNA7 to dsDNA10 were synthesized (Fig. 5). They were labeled by the blue emitting dye 1 or the red emitting dye 3, respectively, at the central A (dsDNA7, dsDNA9) or at the U on the 3′-side (dsDNA8, dsDNA10). PNA3 to PNA5 contain 14mer sequences that are complementary to the central part of the sequences of dsDNA7 to dsDNA10, and again 5 lysine residues to get enough solubility. Based on studies in other PNA systems47,19 it can be assumed that the lysine residues facilitate the strand invasion process by stabilizing the PNA–DNA duplex and destabilizing the PNA–PNA self duplex, although whether their presence is essential for the invasion is difficult to confirm due to the poor solubility of dye-labeled PNA without lysine. The stability of these double strands is only slightly diminished, if at all, by the attached dyes: the Tm difference is only between −1.7 °C and +0.1 °C, if compared to a 30mer, completely unmodified DNA double strand (DNA16–DNA15, Tm = 70.4 °C, see Table S5 in ESI†). PNA3 was modified with the yellow emitting dye 2 that serves as energy acceptor for dye 1 in dsDNA7 and dsDNA8, whereas PNA4 is modified with the green emitting dye 4 that represents the energy donor for dye 3 in dsDNA9 and dsDNA10 (for synthesis of dye 4, see ESI†). With respect to the biological context, PNA3 and PNA4 are complementary to the TIAM1 sequence that is regulated by miRNA-21 and miRNA-31 in colon carcinoma cells.46 The unmodified PNA5 serves as the fourth strand that was originally thought to be essential for the double duplex invasions. PNA6 bears a non-sense sequence for control experiments. Invasions were detected by blue:yellow or green:red fluorescence color contrasts (e.g. I475 nm/I585 nm) and by fluorescence intensity increase factors (Table 2) that were determined similarly as described above, for both acceptor dyes 2 or 3, respectively, at their corresponding emission maximum. The factors represent the ratios of the fluorescence intensities of the acceptor dyes after excitation at the absorption maxima of donor dyes 1 or 4, respectively, to the fluorescence intensities of the acceptor dyes upon excitation at the same wavelength but in the absence of the donor dyes 1 or 4. The latter fluorescence intensities were obtained with the non-invaded dsDNA7 to dsDNA10.
 | ||
| Fig. 5 Schematic drawing of double duplex invasion, sequences of PNA3–PNA6, structure of dye 4 and sequences of dsDNA7–dsDNA10 (from top to bottom). | ||
| Invasion complex | NaCla | Color contrast | Fluo-rescence intensity increase factorb | Energy transfer efficiencyc |
|---|---|---|---|---|
| a 250 mM. b I sample/IdsDNA (definition see text). c Calculated by fluorescence lifetimes (see ESI). d I 475 nm/I585nm. e I 477 nm/I580 nm. f I 472 nm/I583 nm. g I 475 nm/I574 nm. h I 528 nm/I604 nm. i I 528 nm/I600 nm. j I 477 nm/I613 nm. k I 529 nm/I611 nm. l I 528 nm/I608 nm. | ||||
| dsDNA7–PNA3–PNA5 | + | 1:2.1d |
18.1 | 0.67 ± 0.09 |
| − | 1:7.5e |
19.6 | 0.74 ± 0.12 | |
| dsDNA8–PNA3–PNA5 | + | 1:1.4f |
14.6 | 0.66 ± 0.15 |
| − | 1:4.8g |
20.8 | 0.76 ± 0.12 | |
| dsDNA9–PNA4–PNA5 | + | 1:43.0h |
16.6 | 0.97 ± 0.04 |
| − | 1:87.0i |
23.2 | 0.99 ± 0.01 | |
| dsDNA9–PNA4 | + | 1:23.7h |
14.4 | 0.94 ± 0.03 |
| − | 1:23.0i |
17.4 | 0.94 ± 0.13 | |
| dsDNA9–PNA6 | + | 1:2.2j |
1.8 | — |
| dsDNA10–PNA4–PNA5 | + | 1:28.6k |
10.0 | 0.93 ± 0.06 |
| − | 1:61.1l |
13.3 | 0.97 ± 0.02 | |
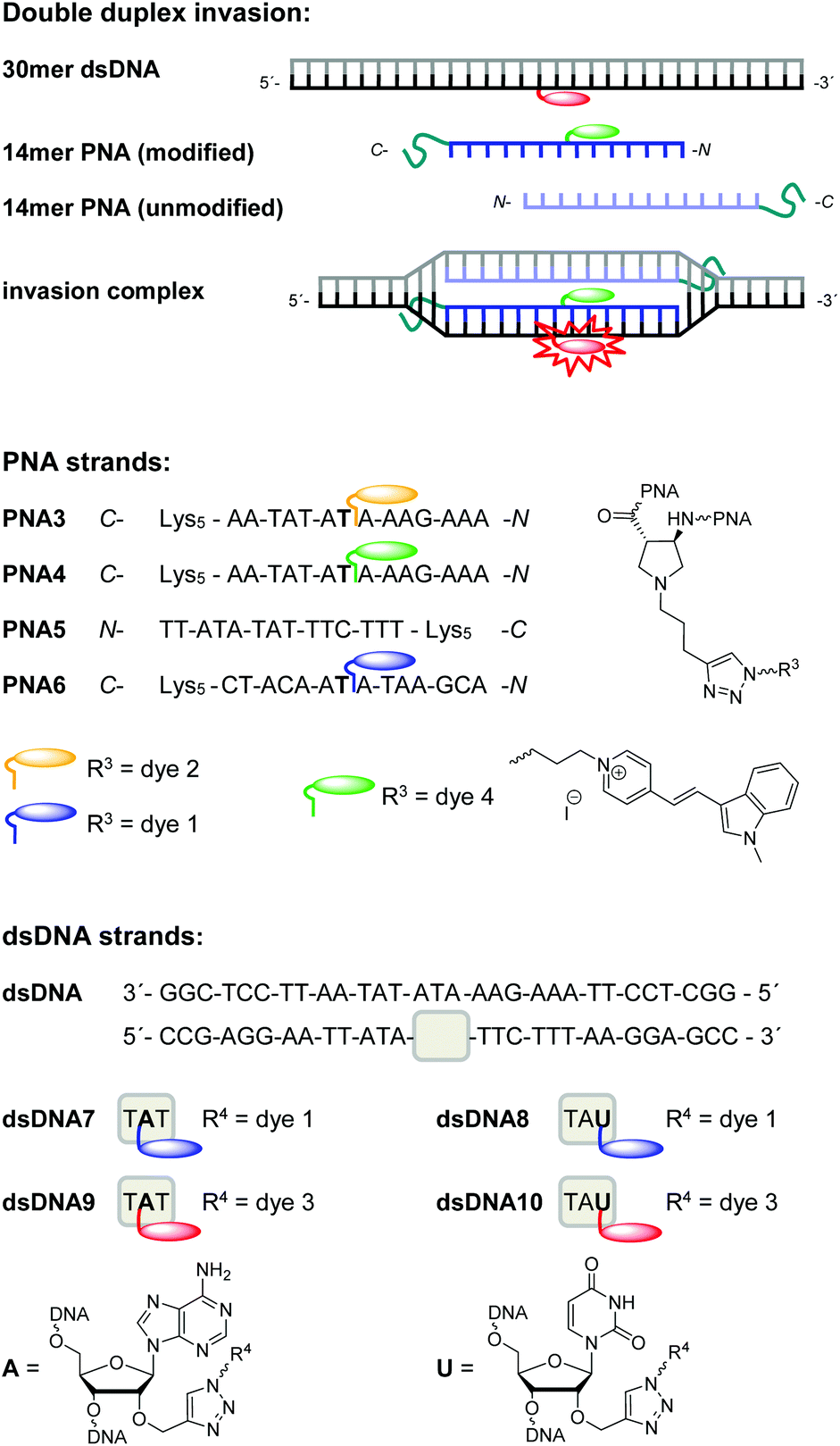
Energy transfer efficiencies (Table 2) of all invasion complexes were calculated from the corresponding fluorescence lifetimes of donor dyes 1 (λexc = 370 nm, λem = 475 nm) and 4 (λexc = 435 nm, λem = 528 nm). All strand invasion experiments were performed in presence and absence of 250 mM NaCl since we assumed that duplex invasions by acpcPNA might be salt-dependent processes. At first, we studied kinetics of strand invasion into dsDNA7 and dsDNA8 by 1.0 equiv. of PNA3 together with 1.0 equiv. of PNA5. Constant fluorescence intensities are reached within 60 minutes with NaCl and 90 minutes without NaCl (see ESI†). Although NaCl-free conditions show comparable kinetics, they yield stronger increase of emission intensities and considerably higher color contrasts blue:yellow. Obviously the dye interactions with DNA in the hybrids with DNA are salt dependent and alter the fluorescence intensities. Further titrations revealed that the fluorescence color change is maximized upon addition of 3.0 equiv. of acpcPNA (see ESI†). Hence, full double-duplex invasions into dsDNA7 and dsDNA8, respectively, were carried out by addition of 3.0 equiv. of PNA3 together with PNA5. In absence of acceptor dyes, the blue emitting dye 1 exhibit lifetimes of approximately 2.4–2.6 ns in dsDNA7 and dsDNA8. The lifetimes are shortened down to 0.6–0.9 ns in the invasion complexes with PNA3 and PNA5, which yields energy transfer efficiencies in the range from 66% to 76% (see ESI†). Moreover, strand invasion efficiencies could be determined based on these energy transfer efficiencies. As fluorescence reference for complete invasion, reference hybrids of PNA3 with full-length complementary DNA strands bearing dye 1 at the central A were annealed. The energy transfer efficiencies of these hybrids were measured and represent 100% reference values. Remarkably, invasion efficiencies of approximately 99% could be accomplished in all cases (see ESI†). Best results with this dye combination were accomplished e.g. with dsDNA7 in absence of NaCl (Fig. 6). The invasion by PNA3 together with PNA5 exhibits an intensity increase of the yellow emission by a factor of 19.6, a color contrast blue:yellow (I477 nm/I580 nm) of 1:7.5 and an energy transfer efficiency of 74%.
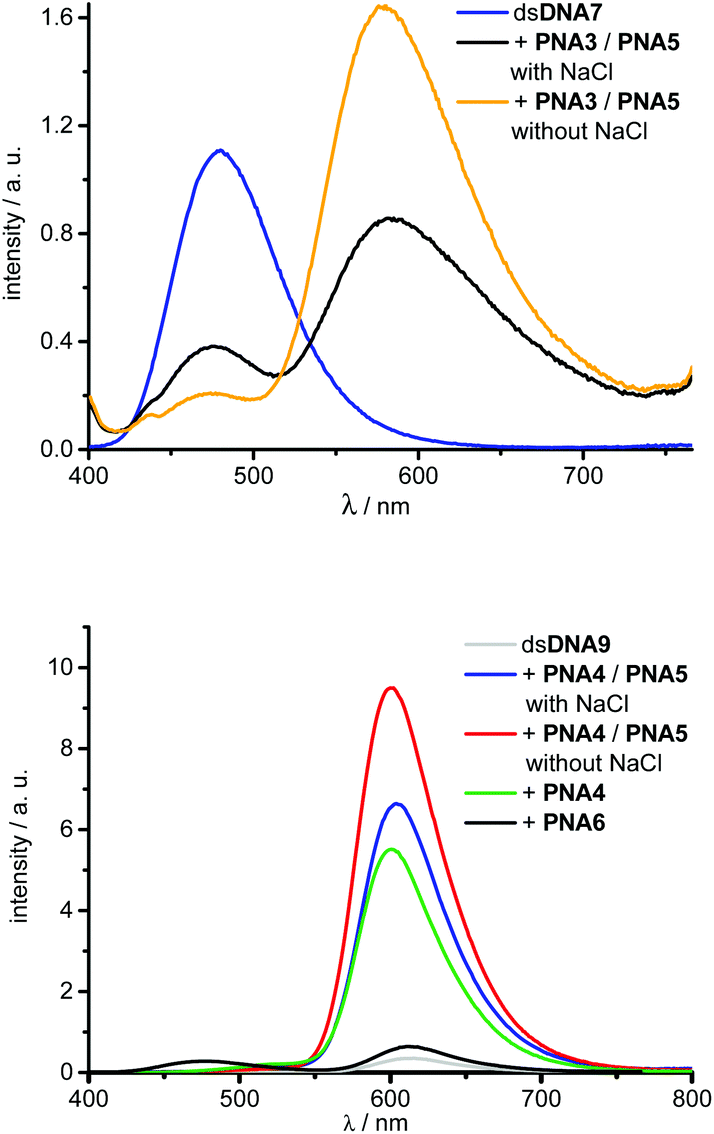 | ||
| Fig. 6 Representative fluorescence spectra before and after double duplex invasion: dsDNA7 (2.5 μM in 10 mM Na–Pi buffer, pH 7) invaded by 3.0 equiv. PNA3 together with PNA5, in absence and presence of NaCl (250 mM), λexc = 391 nm (top); dsDNA9 (2.5 μM in 10 mM Na–Pi buffer, pH 7) invaded by 1.5 equiv. PNA4 together with PNA5 in absence and presence of NaCl (250 mM), by 1.5 equiv. PNA4 without PNA5, and by 1.5 equiv. non-sense PNA6, λexc = 391 nm (bottom). | ||
Similarly, double-duplex invasion kinetics of dsDNA9 and dsDNA10 after adding 1.0 equivalent PNA4 together with PNA5 were recorded. It is important to note that titrations revealed that 1.5 equiv. PNA4 and PNA5 were sufficient for this type of strand invasions to obtain maximal fluorescence change (see ESI†). Hence, quantification of energy transfer efficiencies, fluorescence intensity increase factors and color contrasts green:red were carried out under these conditions, in absence and presence of NaCl, respectively (Table 2).
Remarkably, energy transfer from donor dye 4 to acceptor dye 3 occurs with 97–99% efficiency which yields very large increase factors and color contrasts. Here again, invasion efficiencies of 99% were achieved in all cases (see ESI†). Especially the modification at the central adenosine by dye 3 (dsDNA9) and invasion in the absence of NaCl turned out to be the optimal condition for double duplex invasion (Fig. 6). After addition of 1.5 equiv. of PNA3 together with PNA5 an increase factor of 23.3 and a color contrast green:red (I528 nm/I600 nm) of 1:87 could be accomplished. The observed variation of intensity enhancements and color contrasts could be assigned to the different anchor point of the post-synthetic DNA labeling by dye 3. Reference measurements with corresponding DNA duplexes bearing the dye 3 modification at an adenosine (dsDNA9) vs. uridine (dsDNA10) clearly show approximately 20% stronger fluorescence intensities (see ESI†). Importantly, the possibility of strand invasion of dsDNA9 by using only one PNA strand (PNA4) yields a D-loop and could be evidenced by energy transfer efficiency of 94% and invasion efficiency of 96% (see ESI†) which is only slightly lower compared to 99% invasion efficiencies obtained for double duplex invasions. Hence, the latter result demonstrates that single stranded acpcPNA may be sufficient to open double stranded DNA without requiring the other acpcPNA strand.
The duplex of the 14mer PNA with DNA containing a single mismatch base was still quite stable under the experimental conditions, therefore the invasion also proceeded with dsDNA target containing a mismatch at the invasion site (data not shown). In order to rule out that unspecific PNA aggregation to dsDNA occurs instead of the expected invasion, a random sequence and non-sense PNA6 was synthesized and modified with the blue donor dye 1 (Fig. 5). A rather small fluorescence enhancement after addition of 1.5 equiv. non-sense PNA6 to dsDNA9 was observed (Fig. 6, Table 2). As proven in strand displacement experiments above, an efficient energy transfer is only possible between dye 1 and dye 3 if they get in close proximity to each other. Thus, the very small intensity increase factor of 1.8 in case of PNA6 clearly excludes non-specific PNA–DNA aggregation.
In order to support the duplex invasions by PNA3 and PNA4, melting temperatures of dsDNAs, acpcPNA–DNA-hybrids, their DNA–DNA-analogs and of complexes invaded by 1.0 equiv. acpcPNA were determined, in the absence or presence of NaCl, respectively (see ESI†). Fig. 7 representatively displays the normalized absorption changes during Tm determination of the complex dsDNA8 invaded by only 1.0 equiv. PNA3 together with PNA5 without NaCl. It should be noted here that strand invasion with 1.0 equiv. of PNA3 is not complete. Accordingly, discrete melting temperature regions for each part of the invading complex are explicitly visible: (i) Comparison 8mer dsDNA13 (overhang on both ends of dsDNA8) that invasion takes place without dissociation of the remaining duplex regions on both sides of invasion, and with a locally displaced complementary strand. (ii) The melting transition of dsDNA8 which corresponds to the non-invaded duplex vanishes with more equivalents of PNA3/PNA5. (iii) The Tm of the DNA18–PNA3 hybrid is >90 °C represents the Tm and the exceptionally stability of the invaded duplex (for sequences of dsDNA13 and DNA18 see ESI†).
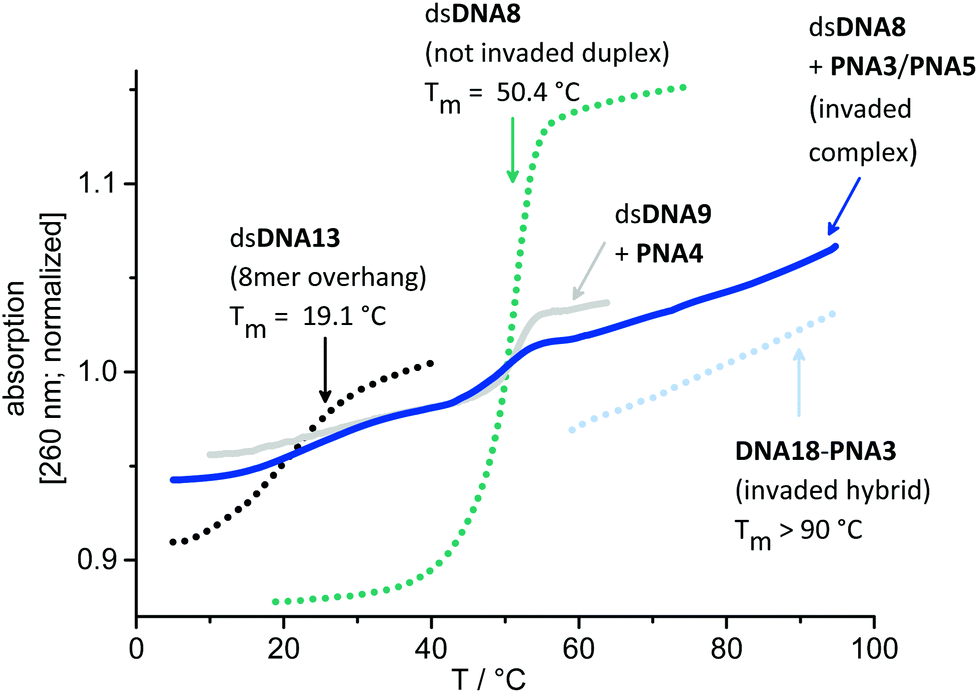 | ||
| Fig. 7 Representative, normalized melting temperature determination of dsDNA8 invaded by 1.0 equiv. PNA3 and PNA5 (blue line), and dsDNA9 invaded by PNA4 only (gray line), and those of references dsDNA8 (green dotted), 8mer dsDNA13 (black dotted) and DNA18–PNA3-hybrid (light blue dotted), in absence of NaCl. | ||
Experimental
All experimental details, syntheses and spectroscopic data are described in the ESI.†Conclusions
In conclusion to the first part of experiments, it became clear that displacements of DNA strands from acpcPNA–DNA hybrids can be followed by fluorescence color changes using our concept of wavelength probes (“DNA traffic lights”) and are possible with longer oligonucleotides than the preannealed ones. The kinetics of these strand displacements are rather slow due to the high stability of the acpcPNA–DNA hybrids. Moreover, acpcPNA is able to displace a DNA strand from double stranded DNA of the same length. There is important significance of these in vitro experiments for cell biological applications. The intracellular transport could potentially be facilitated by annealing acpcPNA to a short DNA counterstrand. In combination with attached positively charged lysine residues on the termini of acpcPNA strands, such acpcPNA/DNA hybrids may potentially be easier delivered into the cells.In the second part, we demonstrated that acpcPNA is able not only to displace one of the strands in double-stranded DNA of the same length, but also to invade into long double-stranded A–T rich DNA, remarkably with quantitative efficiency. Again, these processes can be followed by means of fluorescence color changes. Compared to conventional PNA systems which require 3 or more equiv. PNA, only 1.5 equiv. acpcPNA are sufficient to get efficient invasion. Although initial DNA duplex invasion experiments were performed with two non-self-pairing acpcPNA strands, single stranded acpcPNA was also able to invade into DNA duplex without requiring the other PNA strand, at least in the A–T rich sequence context being studied. Beyond that, our findings clearly confirm that the invasion takes place even at high ionic strength, a condition that is known to decrease the PNA duplex invasion efficiency. Therefore, acpcPNA exhibits sophisticated properties which are highly desirable in the field of biological analytics and antigene applications.
Acknowledgements
Financial support by the DFG (grant WA1387/17-1, GRK 2039), KIT and the Thailand Research Fund (DPG5780002) is gratefully acknowledged. P. Bohländer thanks the Karlsruhe House of Young Scientists (KHYS) for a fellowship to support a research stay in Bangkok.Notes and references
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 566–568 CrossRef CAS PubMed.
- P. E. Nielsen, Acc. Chem. Res., 1999, 32, 624–630 CrossRef CAS.
- P. E. Nielsen, Chem. Biodiversity, 2010, 7, 786–804 CAS.
- K. N. Ganesh and P. E. Nielsen, Curr. Org. Chem., 2000, 4, 931–943 CrossRef CAS.
- V. Kumar and K. N. Ganesh, Acc. Chem. Res., 2005, 38, 494–412 CrossRef PubMed.
- K. Matsumoto, E. Nakata, T. Tamura, I. Saito, Y. Aizawa and T. Morii, Chem. – Eur. J., 2013, 19, 5034–5040 CrossRef CAS PubMed.
- J. K. Pokorski, M. A. Witschi, B. L. Purnell and D. H. Appella, J. Am. Chem. Soc., 2004, 126, 15067–15073 CrossRef CAS PubMed.
- C. M. Micklitsch, B. Y. Oquare, C. Zhao and D. H. Appella, Anal. Chem., 2012, 85, 251–257 CrossRef PubMed.
- T. Vilaivan and C. Srisuwannaket, Org. Lett., 2006, 8, 1897–1900 CrossRef CAS PubMed.
- C. Vilaivan, C. Srisuwannaket, C. Ananthanawat, C. Suparpprom, J. Kawakami, Y. Yamaguchi, Y. Tanaka and T. Vilaivan, Artificial DNA: PNA & XNA, 2011, 2, 50–59 Search PubMed.
- J. Taechalertpaisarn, P. Sriwarom, C. Boonlua, N. Yotapan, C. Vilaivan and T. Vilaivan, Tetrahedron Lett., 2010, 51, 5822–5826 CrossRef CAS PubMed.
- T. Vilaivan, Acc. Chem. Res., 2015, 48, 1645–1656 CrossRef CAS PubMed.
- C. Boonlua, C. Vilaivan, H. A. Wagenknecht and T. Vilaivan, Chem. – Asian J., 2011, 6, 3251 CrossRef CAS PubMed.
- C. Boonlua, B. Ditmangklo, N. Reenabthue, C. Suparpprom, N. Poomsuk, K. Siriwong and T. Vilaivan, RSC Adv., 2014, 4, 8817–8827 RSC.
- B. Ditmangklo, C. Boonlua, C. Suparpprom and T. Vilaivan, Bioconjugate Chem., 2013, 24, 614–625 CrossRef CAS PubMed.
- N. Yotapan, C. Charoenpakdee, P. Wathanathavorn, B. Ditmangklo, H.-A. Wagenknecht and T. Vilaivan, Beilstein J. Org. Chem., 2014, 10, 2166–2174 CrossRef PubMed.
- J. Lohse, O. Dahl and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11804–11808 CrossRef CAS.
- T. Ishizuka, J. Yoshida, Y. Yamamoto, J. Sumaoka, T. Tedeschi, R. Corradini, S. Sforza and M. Komiyama, Nucleic Acids Res., 2008, 36, 1464–1471 CrossRef CAS PubMed.
- T. Bentin and P. E. Nielsen, J. Am. Chem. Soc., 2003, 125, 6378–6379 CrossRef CAS PubMed.
- S. Rapireddy, G. He, S. Roy, B. A. Armitage and D. H. Ly, J. Am. Chem. Soc., 2007, 129, 15596–15600 CrossRef CAS PubMed.
- V. Chenna, S. Rapireddy, B. Sahu, C. Ausin, E. Pedroso and D. H. Ly, ChemBioChem, 2008, 9, 2388–2391 CrossRef CAS PubMed.
- M. E. Hansen, T. Bentin and P. E. Nielsen, Nucleic Acids Res., 2009, 4498–4507 CrossRef CAS PubMed.
- G. He, S. Rapireddy, R. Bahal, B. Sahu and D. H. Ly, J. Am. Chem. Soc., 2009, 131, 12088–12090 CrossRef CAS PubMed.
- C. Holzhauser and H.-A. Wagenknecht, J. Org. Chem., 2013, 78, 7373–7379 CrossRef CAS PubMed.
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43, 1044–1056 RSC.
- P. V. Chang and C. R. Bertozzi, Chem. Commun., 2012, 48, 8864–8879 RSC.
- B. A. Armitage, Curr. Opin. Chem. Biol., 2011, 15, 806–812 CrossRef CAS PubMed.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- H. Yan, X. Zhang, Z. Shen and N. C. Seeman, Nature, 2002, 415, 62–65 CrossRef CAS PubMed.
- B. Chakraborty, R. Sha and N. C. Seeman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17245–17249 CrossRef CAS PubMed.
- K. Takahashi, S. Yaegashi, A. Kameda and M. Hagiya, in DNA computing, Springer, 2006, pp. 347–358 Search PubMed.
- G. Seelig, D. Soloveichik, D. Y. Zhang and E. Winfree, Science, 2006, 314, 1585–1588 CrossRef CAS PubMed.
- B. M. Frezza, S. L. Cockroft and M. R. Ghadiri, J. Am. Chem. Soc., 2007, 129, 14875–14879 CrossRef CAS PubMed.
- J. M. Picuri, B. M. Frezza and M. R. Ghadiri, J. Am. Chem. Soc., 2009, 131, 9368–9377 CrossRef CAS PubMed.
- L. Qian, D. Soloveichik and E. Winfree, in DNA computing and molecular programming, Springer, 2011, pp. 123–140 Search PubMed.
- W. B. Sherman and N. C. Seeman, Nano Lett., 2004, 4, 1203–1207 CrossRef CAS.
- J.-S. Shin and N. A. Pierce, J. Am. Chem. Soc., 2004, 126, 10834–10835 CrossRef CAS PubMed.
- H. Gu, J. Chao, S.-J. Xiao and N. C. Seeman, Nature, 2010, 465, 202–205 CrossRef CAS PubMed.
- I. K. Astakhova, K. Pasternak, M. A. Campbell, P. Gupta and J. Wengel, J. Am. Chem. Soc., 2013, 135, 2423–2426 CrossRef CAS PubMed.
- C. Holzhauser, R. Liebl, A. Goepferich, H.-A. Wagenknecht and M. Breunig, ACS Chem. Biol., 2013, 8, 890–894 CrossRef CAS PubMed.
- P. R. Bohländer and H. A. Wagenknecht, Eur. J. Org. Chem., 2014, 7547–7551 CrossRef PubMed.
- P. R. Bohländer and H.-A. Wagenknecht, Org. Biomol. Chem., 2013, 11, 7458–7462 Search PubMed.
- T. Bentin, H. J. Larsen and P. E. Nielsen, Biochemistry, 2003, 42, 13987–13995 CrossRef CAS PubMed.
- A. Abibi, E. Protozanove, V. V. Demidov and M. D. Frank-Kamenetskii, Biophys. J., 2004, 86, 3070–3078 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01273b |
| This journal is © The Royal Society of Chemistry 2015 |