
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced immunogenicity of multivalent MUC1 glycopeptide antitumour vaccines based on hyperbranched polymers†
M.
Glaffig
a,
B.
Palitzsch
a,
N.
Stergiou
b,
C.
Schüll
a,
D.
Straßburger
a,
E.
Schmitt
b,
H.
Frey
a and
H.
Kunz
*a
aJohannes Gutenberg-University Mainz, Institute of Organic Chemistry, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hokunz@uni-mainz.de
bUniversity Medical Center, Institute of Immunology, Johannes Gutenberg-University Mainz, Langenbeckstrasse 1, Geb. 708, 55101 Mainz, Germany
First published on 18th August 2015
Abstract
Enhancing the immunogenicity of an antitumour vaccine still poses a major challenge. It depends upon the selected antigen and the mode of its presentation. We here describe a fully synthetic antitumour vaccine, which addresses both aspects. For the antigen, a tumour-associated MUC1 glycopeptide as B-cell epitope was synthesised and linked to the immunostimulating T-cell epitope P2 derived from tetanus toxoid. The MUC1-P2 conjugate is presented multivalently on a hyperbranched polyglycerol to the immune system. In comparison to a related vaccine of lower multivalency, this vaccine exposing more antigen structures on the hyperbranched polymer induced significantly stronger immune responses in mice and elicited IgG antibodies of distinctly higher affinity to epithelial tumour cells.
Introduction
Cancer is still a major cause of mortality and morbidity in the world despite the immense progress made in the therapy of cancer and in the development of vaccines against cancer. The success of an antitumour vaccine very much depends upon the tumour-selectivity of the chosen antigen structure. Since antigens typically occurring on tumour cells are endogenous structures, they are of low immunogenicity. There are several possibilities to enhance the immunogenicity of these tumour-associated antigens to give efficient vaccines. One of them is to modify the architecture of the vaccine through coupling the antigen to a potent T-cell epitope peptide and by means of an optimal multiple antigen presentation. According to this approach, we here describe a fully synthetic antitumour vaccine with enhanced multivalent antigen presentation and compare its structure and immunological effect in mice with those of a related vaccine of lower valency.1As a promising antigen structure for an antitumour vaccine a glycopeptide of the tumour-associated epithelial mucin MUC1 was selected. MUC1 is expressed on the membrane surface of epithelial cells. In tumour cells it is over-expressed and structurally different from MUC1 on healthy cells.2,3 Compared to normal MUC1 which is highly glycosylated with long branched saccharide chains, tumour-associated MUC1 often carries truncated glycans.2,3 As a consequence, peptide epitopes within the protein backbone of the tumour-associated MUC1 are no longer shielded and become accessible to the immune system.4,5 Aberrant glycosylation patterns within the variable number of tandem repeat-domain (VNTR, one tandem repeat comprising 20 amino acid residues of the sequence PAHGVTSAPDTRPAPGSTAP) occur due to down-regulated glycosyl transferase activities in tumour cells. Therefore, tumour-associated carbohydrate antigens (TACAs), such as Tn-antigen, particularly occur on epithelial tumour cells.2,3
However, the immunogenicity of tumour-associated MUC1 as an endogenous structure is too low to elicit immune responses. To overcome this self-tolerance of the immune system, the synthetic MUC1 glycopeptide as a B-cell epitope needs to be linked to immune-stimulating components. These immunostimulants are or contain T-cell epitopes for activating T-helper cells which activate B-cells and induce antibody class switching. Especially the highly immunogenic protein tetanus toxoid (TTox), which contains several T-cell epitopes, effects strong immune responses in mice6,7 and provides, in addition, a nano-scaled platform for multiple antigen presentation. Because of disadvantages in tetanus toxoid-conjugated MUC1 vaccines, such as their insufficient characterisation and unwanted immune responses against the carrier, the construction of fully synthetic antitumour vaccines is of particular interest.
The potency of the immune response depends, among others, on the choice of the selected antigens, which should mimic the native structure of MUC1 glycopeptides on tumour cells, and on the mode of their presentation. To meet these requirements, a MUC1 antitumour vaccine based on hyperbranched polyglycerol (HPG), vaccine 2, was synthesised.1 This globular polymeric carrier contains flexible aliphatic polyether arms. In contrast to the use of linear polymeric carriers such as poly(N-(2-hydroxypropyl)meth-acrylamide) P(HPMA),8 hyperbranched polymer vaccines prevent entanglements of the bound antigens. Due to its dendrimer-like structure, there is plenty of space for coupling antigens which are exposed on the surface in a spherical way similar to the situation on a cell surface. Thus, an optimal multivalent presentation of the antigens to the immune system is ensured which is reflected in an enhanced immune response compared to that induced by P(HPMA)polymer-based vaccines.8 HPGs are biocompatible, non-immunogenic and soluble in water.9–11 In comparison to polyethylene glycol (PEG), HPG is more hydrophilic, exhibits a higher density of hydroxyl groups without a significant increase of viscosity.12 Therefore, hyperbranched polyglycerol as an inert carrier is considered attractive for biomedical applications.12,13
In order to prove the significance of multi antigen presentation in antitumour vaccines with respect to the immune response, the HPG-carrier was modified by adding more antigen binding sites. The numbers of linked antigens can readily be adjusted by the monomers glycidol (G)/glycidyl propargyl ether (GPE) ratio in the one-step synthesis of HPG.14 Therefore, a new multi-alkyne functionalised HPG 5 was synthesised which carries on average eight alkyne groups (see Experimental), instead of only five in the case of vaccine 2.1
For the (glyco)peptide antigen 3, the identical amino acid sequence as contained in vaccine 2 was chosen (PAHGVTSAPDTRPAPGSTAPPA). Since conformation of the glycopeptides, which is influenced by glycosylation, is crucial for tumour selectivitiy,15 the glycosylation pattern of 3 was modified in order to enhance the moderate and varying recognition of tumour cells observed for antibodies induced through vaccine 2 (see Immunological evaluation below). Instead of threonine-18, serine-17 was linked to Tn antigen and a second Tn antigen was coupled to threonine-11 in order to enhance the tumour-selectivity of the antigen. Hence, both immunodominant motifs PDTRP and GSTA considered the preferential binding sites for anti-MUC1 antibodies are glycosylated.16–18 As the immune-stimulating component the tetanus toxoid T-cell epitope P219,20 of the sequence QYIKANSKFIGITEL was used for the vaccine design. The components, MUC1 B-cell epitope and T-cell epitope P2, were separated by flexible immunologically silent oligoethylene glycol-spacers in order to prevent an influence of each other's conformation. The coupling of the MUC1-P2 conjugate 3 to the alkyne-functionalised HPG 5 was performed through an alkyne–azide click-reaction (Scheme 1).21,22
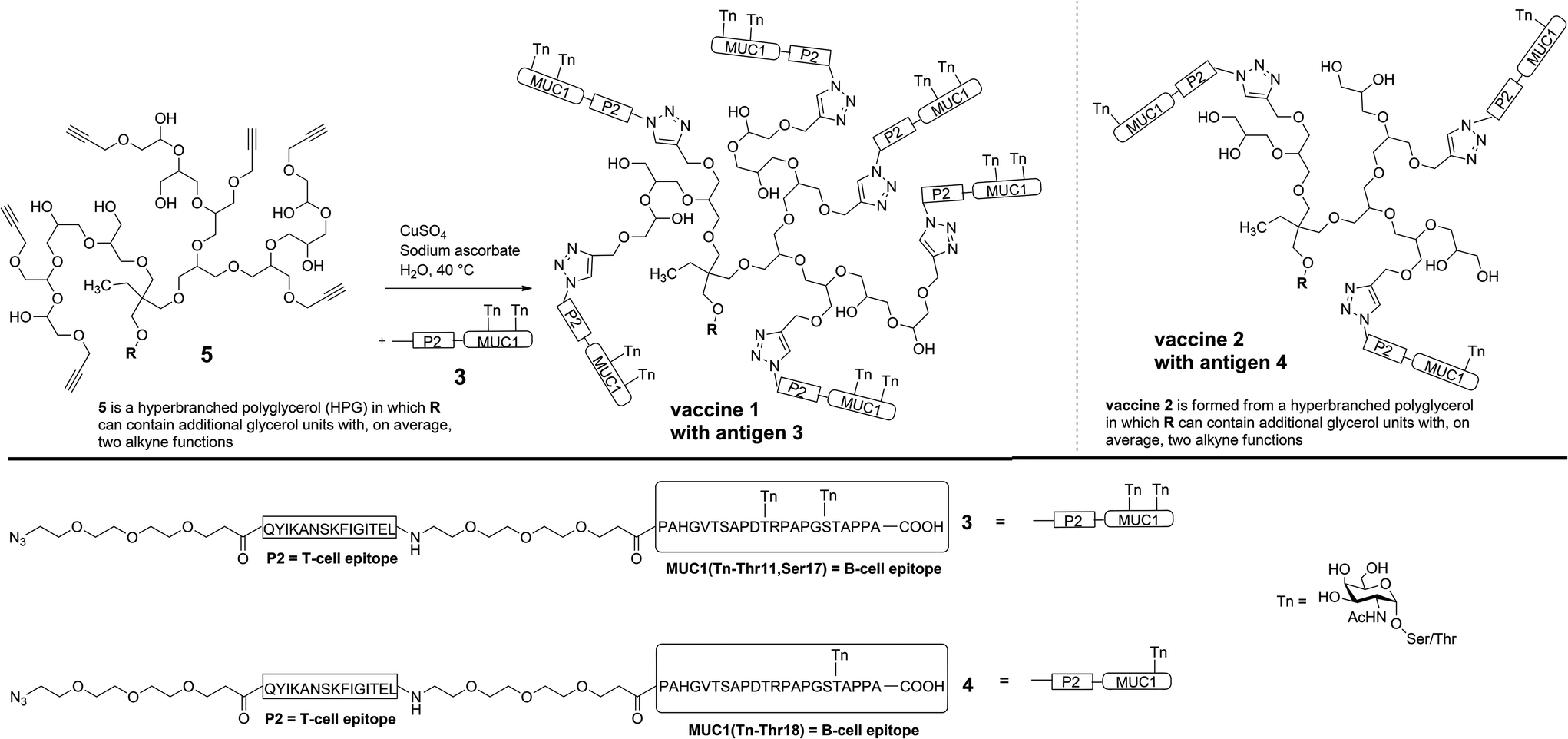 | ||
| Scheme 1 Synthesis of hyperbranched polyglycerol-based antitumour vaccine 1 by coupling antitumour antigen 3 with HPG 5via copper-catalysed azide–alkyne cycloaddition. For comparison on the right: structure of vaccine 2 (carrying antigen 4) which has less antigen binding sites on average than vaccine 1. | ||
Experimental
The (glyco)peptide 3 was synthesised via solid-phase peptide synthesis (SPPS) according to an established 9-flourenylmethoxycarbonyl (Fmoc) protocol.6 Starting from TentaGel resin preloaded with a trityl-anchored23 Fmoc-protected alanine, the MUC1 glycopeptide and the T-cell epitope P2 were assembled in a linear synthesis resulting in azido-terminated antigen 3 (Scheme 1). The coupling reactions of both glycosyl amino acids (two Tn antigens) and of the functional spacer molecules were performed under modified conditions (see ESI†). After coupling of the final building block (azido-functionalised spacer), release from resin and removal of all acid-labile protecting groups on the amino acid side chains were achieved using trifluoroacetic acid (TFA), triisopropylsilane (TIS) and water. The MUC1-P2 peptide still protected in the carbohydrates was purified by semi-preparative HPLC. The removal of the O-acetyl groups was performed using NaOMe in MeOH at pH below 10 to avoid elimination of the carbohydrate. After an additional purification by HPLC, the free glycopeptide 3 was obtained in an overall yield of 17%.Click ligation to the multi-alkyne functionalised hyperbranched polyglycerol 5 was achieved in degassed water at 40 °C within 4 d using copper sulphate and sodium ascorbate. The polymer as the coupling partner was synthesised with an average molecular weight of Mn = 2410 g mol−1 and contained, on average, eight alkyne groups (calculated from 1H NMR spectroscopy).14 For the click-type cycloaddition, the polymeric carrier was added to a slight excess of the glycopeptide 3 (1.1 eq. for each alkyne group) to guarantee that all accessible alkynes react. Purification was performed by ultrafiltration through a 30 kDa membrane in order to remove free peptides (4601.04 g mol−1) and copper salts. Subsequent lyophilisation gave the fully synthetic glycopeptide-hyperbranched polyglycerol vaccine 1.
The coupling was confirmed by analytic size exclusion chromatography (SEC) in hexafluoroisopropanol (HFIP) as the eluent. Compared to the elution volume of HPG 5 or the free (glyco)peptide antigen 3, respectively, the lower elution volume of 1 corresponds to the higher molecular weight indicating a successful coupling (Fig. 1). The size of the spherical vaccine 1 (>10 nm) reaches the dimension of small synthetic virus-like particles24 considered advantageous for the up-take by antigen-presenting dendritic cells.
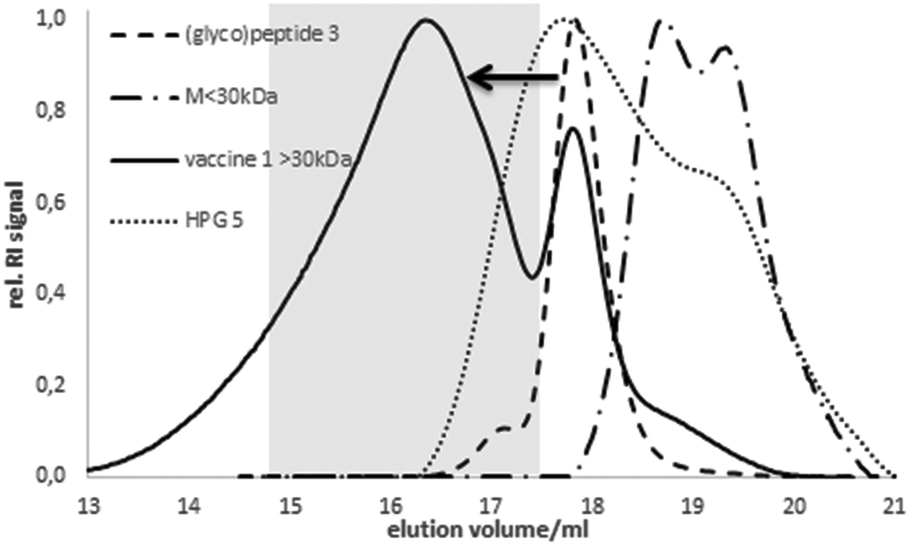 | ||
| Fig. 1 SEC elugrams of vaccine 1, MUC1-P2 antigen 3 and HPG 5 (eluent: HFIP). | ||
Immunological evaluation
In order to evaluate the immunological potential of the enhanced HPG-based vaccine 1 carrying more antigens than 2, three female Balb/c mice were immunised three times at intervals of two weeks.‡ The first administration was performed subcutaneously with complete Freund's adjuvant (CFA), whereas the second and third immunisations (two booster immunisations) were administrated intraperitoneally with incomplete Freund's adjuvant (IFA). Five days after the first and the second boost, blood was drawn from tail vein of each mouse in order to quantify the vaccine-induced antibodies against the tumour-associated MUC1. To this end, enzyme-linked immunosorbent assay (ELISA) experiments were performed using the corresponding MUC1 glycopeptide of 3 conjugated to bovine serum albumin (BSA) as the coating material for the microtitre plates. In contrast to the vaccine 2, the titres of 1 were significantly higher for all three mice showing the importance of the multivalency of the antigen presentation (Fig. 2). Addition of HPG 5 to the diluted serum did not reduce this binding. Considering the interval in which blood was drawn from mice for quantifying the induced antibodies (vaccine 1 after 33 days, vaccine 2 after 47 days, see Fig. 2) the superior immunological potency of vaccine 1 over vaccine 2 becomes even more evident. Comparing the antibody titres to those induced by other vaccines, the titres elicited by vaccine 1 do not reach the levels of those elicited through protein-based (like TTox) antitumour vaccines (endpoint titres of 500000–1 million)6,7 because TTox contains several potent T-helper cell epitopes and possible attachment sites (lysine residues) for antigen presentation. Nevertheless, the titres induced by vaccine 1 are among the highest of all fully synthetic vaccines.1,25–27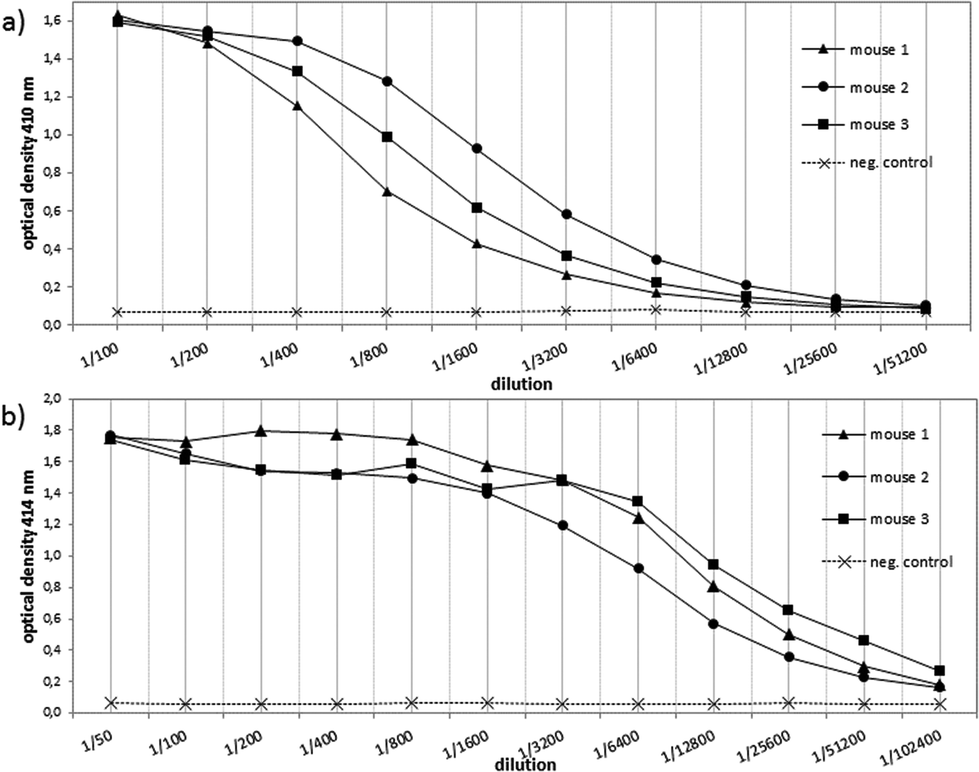 | ||
| Fig. 2 ELISA analysis of the antisera induced; (a) by vaccine 2 (after two boosts within 6 weeks on day 47) and (b) by vaccine 1 with significantly higher titers (after two boosts within 4 weeks on day 33). | ||
Isotype analysis of the induced antibodies showed prevailing IgG1 antibodies indicating MHC-II mediated immune responses and the installation of an immunological memory. IgM was also induced (Fig. 3b, right). IgM is the first immunoglobulin expressed by mature B-cells after initial exposure to an antigen. Its relatively high amounts can probably be traced back to the shorter immunisation intervals. In contrast to vaccine 2 with lower exposure of the antigen, vaccine 1 in all three mice also elicits high titres of the most potent IgG subclasses, IgG2a and IgG2b, capable of inducing antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).28,29
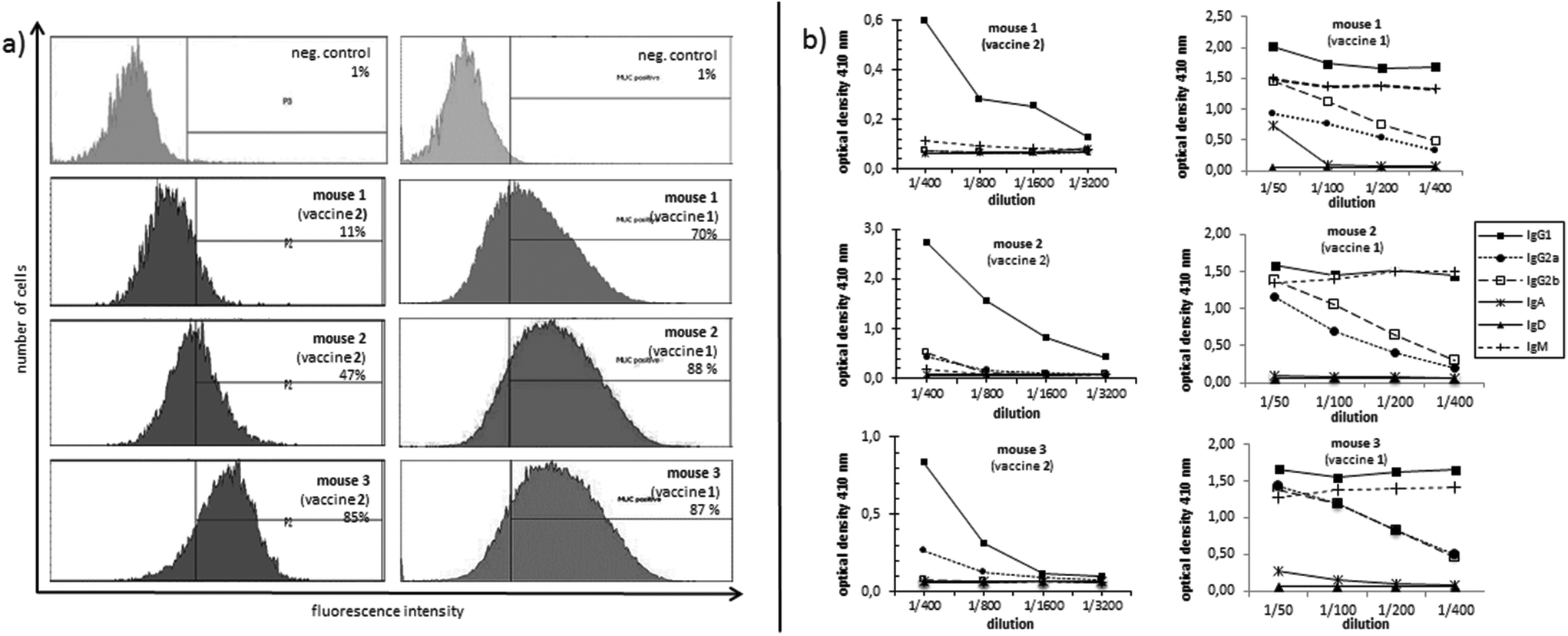 | ||
| Fig. 3 (a) Flow cytometric analysis of the binding of induced antibodies by vaccine 2 (left) and by vaccine 1 (right); neg. control: serum of a non-immunised mouse. (b) Analysis of the antibody subtype induced by vaccine 2 (left) and by vaccine 1 (right). | ||
In order to analyse the capability of the induced antibodies in terms of their binding to tumour cells, MUC1-expressing human breast tumour cells (cell line T47D) were incubated with the induced antisera. The recognition of tumour cells was recorded by flow cytometry (FACS). The antibodies induced through the more multivalent vaccine 1 invariably show distinctly stronger binding (70%, 87%, and 88%, see Fig. 3a, right) to tumour cells for the antisera of all three mice compared to the sera induced by vaccine 2 (11%, 47%, 85%, Fig. 3a, left). This result suggests that the selected MUC1 antigen 3 in vaccine 1 is more tumour-specific than the antigen sequence 4.
Conclusions
The fully synthetic MUC1-P2 antitumour vaccine 1 based on a hyperbranched polyglycerol as an inert polymer carrier with on average eight antigen binding sites induced stronger immune responses in mice than vaccine 2 which has only about five binding sites. This result gives evidence, that the concept of multivalent antigen presentation is valid. The enhanced antigen multivalency obviously enables a stronger avidity between the presented MUC1 antigens and B-cell receptors, which leads to a more efficient vaccine uptake.30 In contrast to the antisera elicited by vaccine 2, the binding of the antibodies induced by vaccine 1 to breast tumour cells is stronger throughout the antisera of all immunised animals. The modified glycosylation in the MUC1 B-cell epitope 3 obviously reflects more accurately the natural tumour-associated antigen on the surface of the tumour cells. In addition, the higher density of the bound antigen in vaccine 1 probably mimics the situation on a tumour cell more optimal than the structure of vaccine 2.Acknowledgements
The authors thank Lutz Nuhn for the SEC analysis and Kerstin Niederer for helpful discussions during this work. This work was supported by the Fonds der Chemischen Industrie and by the Deutsche Forschungsgemeinschaft (SFB 1066).Notes and references
- M. Glaffig, B. Palitzsch, S. Hartmann, C. Schüll, L. Nuhn, B. Gerlitzki, E. Schmitt, H. Frey and H. Kunz, Chem. – Eur. J., 2014, 20(15), 4232 CrossRef CAS PubMed.
- R. Singh and D. Bandyopadhyay, Cancer Biol. Ther., 2007, 6(4), 481 CrossRef CAS.
- J. Taylor-Papadimitriou, J. Burchell, D. Miles and M. Dalziel, Biochim. Biophys. Acta, 1999, 1455(2–3), 301 CrossRef CAS.
- S. Gendler, J. Taylor-Papadimitriou, T. Duhig, J. Rothbard and J. Burchell, J. Biol. Chem., 1988, 263(26), 12820 CAS.
- A. Girling, J. Bartkova, J. Burchell, S. Gendler, C. Gillett and J. Taylor-Papadimitriou, Int. J. Cancer, 1989, 43(6), 1072 CrossRef CAS PubMed.
- A. Kaiser, N. Gaidzik, U. Westerlind, D. Kowalczyk, A. Hobel, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2009, 48(41), 7551 CrossRef CAS PubMed.
- A. Hoffmann-Röder, A. Kaiser, S. Wagner, N. Gaidzik, D. Kowalczyk, U. Westerlind, B. Gerlitzki, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2010, 49(45), 8498 CrossRef PubMed.
- L. Nuhn, S. Hartmann, B. Palitzsch, B. Gerlitzki, E. Schmitt, R. Zentel and H. Kunz, Angew. Chem., Int. Ed., 2013, 52(40), 10652 CrossRef CAS PubMed.
- A. Zarrabi, M. A. Shokrgozar, M. Vossoughi and M. Farokhi, J. Mater. Sci. Mater. Med., 2014, 25(2), 499 CrossRef CAS PubMed.
- C. Du, A. A. Mendelson, Q. Guan, R. Chapanian, I. Chafeeva, G. da Roza and J. N. Kizhakkedathu, Biomaterials, 2014, 35(5), 1378 CrossRef CAS PubMed.
- R. K. Kainthan and D. E. Brooks, Bioconjugate Chem., 2008, 19(11), 2231 CrossRef CAS PubMed.
- E. Mohammadifar, A. Nemati Kharat and M. Adeli, J. Mater. Chem. B, 2015, 3(19), 3896 RSC.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22(2), 190 CrossRef PubMed.
- C. Schüll, T. Gieshoff and H. Frey, Polym. Chem., 2013, 4(17), 4730 RSC.
- M. J. Scanlon, S. D. Morley, D. E. Jackson, M. R. Price and S. J. B. Tendler, Biochem. J., 1992, 284, 137 CrossRef CAS.
- M. A. Tarp, A. L. Sørensen, U. Mandel, H. Paulsen, J. Burchell, J. Taylor-Papadimitriou and H. Clausen, Glycobiology, 2007, 17(2), 197 CrossRef CAS PubMed.
- U. Westerlind, H. Schröder, A. Hobel, N. Gaidzik, A. Kaiser, C. M. Niemeyer, E. Schmitt, H. Waldmann and H. Kunz, Angew. Chem., Int. Ed., 2009, 48(44), 8263 CrossRef CAS PubMed.
- O. Blixt, E. Cló, A. S. Nudelman, K. K. Sørensen, T. Clausen, H. H. Wandall, P. O. Livingston, H. Clausen and K. J. Jensen, J. Proteome Res., 2010, 9(10), 5250 CrossRef CAS PubMed.
- D. Valmori, A. Pessi, E. Bianchi and G. Corradin, J. Immunol., 1992, 149(2), 717 CAS.
- P. Panina-Bordignon, A. Tan, A. Termijtelen, S. Demotz, G. Corradin and A. Lanzavecchia, Eur. J. Immunol., 1989, 19(12), 2237 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67(9), 3057 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 114(14), 2708 CrossRef.
- J. M. J. Fréchet and K. E. Haque, Macromolecules, 1975, 8(2), 130 CrossRef.
- A. Ghasparian, T. Riedel, J. Koomullil, K. Moehle, C. Gorba, D. I. Svergun, A. W. Periman, S. Mann, M. Tamborrini, G. Pluschke and J. A. Robinson, ChemBioChem, 2011, 12, 100–109 CrossRef CAS PubMed.
- B. Palitzsch, S. Hartmann, N. Stergiou, M. Glaffig, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2014, 53(51), 14245 CrossRef CAS PubMed.
- S. Hartmann, L. Nuhn, B. Palitzsch, M. Glaffig, N. Stergiou, B. Gerlitzki, E. Schmitt, H. Kunz and R. Zentel, Adv. Healthcare Mater., 2015, 4(4), 522 CrossRef CAS PubMed.
- (a) H. Cai, M.-S. Chen, Z.-Y. Sun, Y.-F. Zhao, H. Kunz and Y.-M. Li, Angew. Chem., Int. Ed., 2013, 52(23), 6106 CrossRef CAS PubMed; (b) H. Cai, Z.-Y. Sun, M.-S. Chen, Y.-F. Zhao, H. Kunz and Y.-M. Li, Angew. Chem., Int. Ed., 2014, 53(6), 1699 CrossRef CAS PubMed.
- P. Bruhns, Blood, 2012, 119(24), 5640 CrossRef CAS PubMed.
- F. Nimmerjahn, A. Lux, H. Albert, M. Woigk, C. Lehmann, D. Dudziak, P. Smith and J. V. Ravetch, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(45), 19396 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37(20), 2754 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01255d |
| ‡ The mice used were 6–10 weeks old. All mice used for this study were bred and housed in a specific pathogen-free colony at the animal facility of Johannes Gutenberg University following institutionally approved protocols (permission was obtained from the Landesuntersuchungsamt Koblenz, reference number: 23 177-07/G 08-1-019). |
| This journal is © The Royal Society of Chemistry 2015 |