
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective functionalisation of dibenzothiophenes through gold-catalysed intermolecular alkyne oxyarylation†
Matthew J.
Barrett
,
Paul W.
Davies
* and
Richard S.
Grainger
*
School of Chemistry, University of Birmingham, Haworth Building, Edgbaston, Birmingham, B15 2TT, UK. E-mail: p.w.davies@bham.ac.uk; r.s.grainger@bham.ac.uk
First published on 7th July 2015
Abstract
A protocol has been developed for direct Csp3–Csp2 bond formation at the 4- and 6-positions of dibenzothiophenes using a gold(I) catalyst with terminal alkynes and dibenzothiophene-S-oxides. The sulfoxide acts as a traceless directing group to avoid the need to prefunctionalise at carbon. The iterative use of this protocol is possible and has been employed in the preparation of novel macrocyclic structures. In addition, a cascade process shows how oxyarylations can be combined with other processes resulting in complex, highly efficient transformations.
Introduction
Dibenzothiophenes are aromatic sulfur-containing heterocycles of broad utility. The optical, redox and conducting properties of dibenzothiophenes and their corresponding S,S-dioxides have led to applications in materials science.1S-Substituted dibenzothiophenes are used as precursors to triphenylenes2 and as a platform for the transfer of reactive species such as F3C+ (Umemoto's reagent),3 atomic oxygen (O(3P)),4 nitrenes5 and carbenes.6 Biological and medicinal chemistry applications of dibenzothiophenes and their S-oxides have also been reported.7Functionalised dibenzothiophenes are generally prepared through one of two main approaches. Late stage formation of the dibenzothiophene core has been achieved through intramolecular C–S8 or C–C (biaryl)9 bond formation and benzannulation of thiophenes or benzothiophenes.10 Alternatively, dibenzothiophene undergoes regioselective bromination at the 2,8-positions11 or the 3,7-positions of the corresponding S,S-dioxide.12 Substitution at the 4- and 6-positions however requires stoichiometric metallation using organolithium or organoaluminium reagents.13,14 Here we report a catalysis-based approach for direct carbon–carbon bond formation at the unfunctionalised 4- and 6-positions of dibenzothiophenes under mild and functional group tolerant conditions.
Our interests in aromatic S-oxide chemistry15 and π-acid catalysis16 led us to investigate whether 4-substituted dibenzothiophenes could be accessed in an expedient fashion from dibenzothiophene S-oxides by a gold-catalysed alkyne oxyarylation.17–19 This approach should be regiospecific, installing a Csp3–Csp2 bond with transfer of the oxygen atom to generate the synthetically versatile α-arylcarbonyl motif (Scheme 1).20
 | ||
| Scheme 1 Proposed regiospecific functionalisation of dibenzothiophenes using the S-oxide as a traceless directing group. | ||
Following the introduction of alkyne oxyarylation with sulfoxides in gold-catalysed intramolecular cycloisomerisations by the groups of Toste17a and Zhang,17b the viability of an intermolecular process was shown by Ujaque, Asensio and co-workers (Scheme 2).17c
 | ||
| Scheme 2 General schematic for gold-catalysed oxyarylation reaction of alkynes with sulfoxides. | ||
This and subsequent17d,e studies established that such processes are regiospecific by virtue of proceeding via a [3,3]-sigmatropic rearrangement of the vinyl gold carbenoid B formed on attack of the sulfoxide to the gold–alkyne complex (Scheme 2, A → C).21
Despite sulfoxide-based alkyne oxyarylation offering substantial potential for atom-economic, functional group tolerant and direct intermolecular aryl C–H functionalisation routes into challenging aromatic substitution patterns, they have been rarely employed in synthesis. In large part this can be assigned to the challenges of ensuring that the key aromaticity-disrupting [3,3]-sigmatropic rearrangement (B → C) is favoured over elimination of a sulfide nucleofuge (B → D), or competing inter- or intra-molecular attack of a nucleophile (B → E).17,22,23 In addition, structural elaboration of the sulfoxide must not prevent it from being sufficiently nucleophilic to intercept the alkyne–gold complex A, yet not force further reaction at B to afford the biscarbonyl G alongside two equivalents of sulfide.24,25
Results and discussion
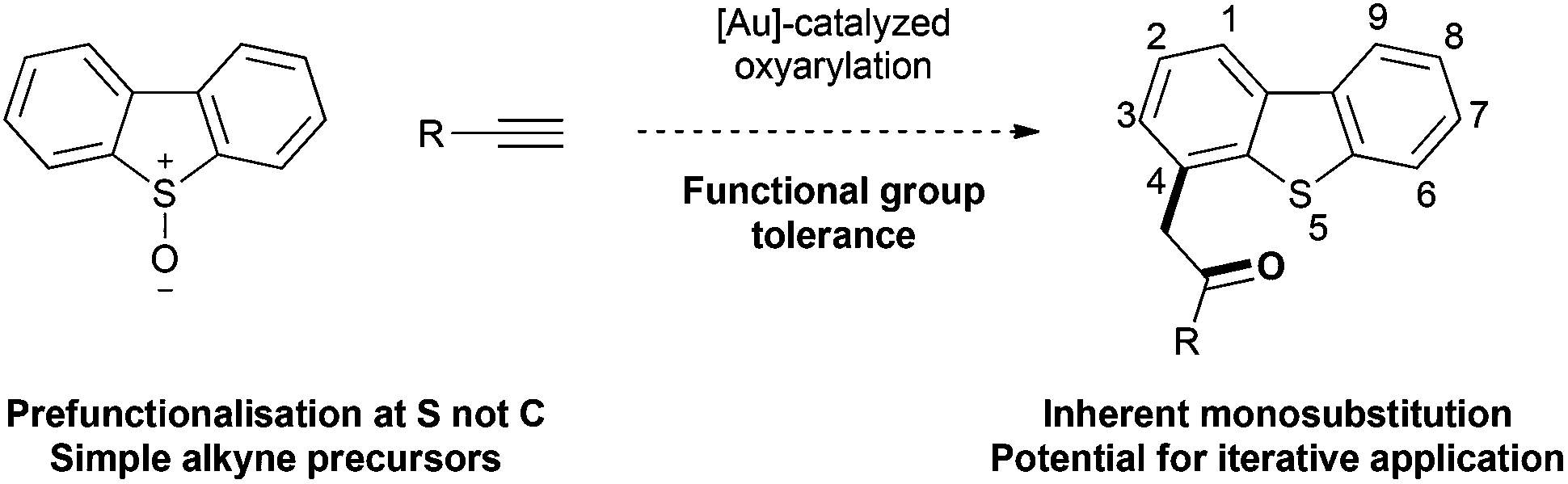
The viability of selective elaboration of a dibenzothiophene through an alkyne oxyarylation approach was investigated using dibenzothiophene-S-oxide 1 and hex-1-yne 2a. Applying the combination of Ph3PAuCl/AgSbF6 in superheated CH2Cl2 from Asensio's work17c generated a mixture of dibenzothiophenes 3a and 4 in high yield with the desired oxyarylation product 3a as the minor component (Table 1, entry 1).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Gold catalyst | Solvent | Time/h | Temp/°C | Conc. M | Yield 1b/% | Yield 3ab/% | Yield 4b/% | Ratio 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 :4 |
| a 1 (0.10 mmol), 2a (0.20 mmol). b Yields calculated by 1H-NMR spectroscopy against a known quantity of internal standard (1,2,4,5-tetramethylbenzene). c Catalyst prepared by in situ combination of equimolar quantity of the (Ligand)AuCl with the appropriate Ag(counterion) salt. d Due to overlap with unidentified resonances estimated yields were determined. XPhos = 2-dicyclohexylphosphino-2,4,6-triisopropylbiphenyl; JohnPhos = (2-biphenyl)di-tert-butylphosphine; Ar = (2,4-di-tert-butylphenyl). | |||||||||
| 1b | Ph3PAuCl/AgSbF6c | CH2Cl2 | 16 | 70 | 1.0 | 0 | 37 | 61 | 1:1.6 |
| 2 | Ph3PAuCl/AgSbF6c | ClCH2CH2Cl | 16 | 70 | 1.0 | 10d | 20 | 30d | — |
| 3 | Ph3PAuCl/AgSbF6c | CH3NO2 | 16 | 70 | 1.0 | 0 | 39 | 36 | 1.1:1 |
| 4 | Ph3PAuCl/AgOTsc | CH3NO2 | 16 | 70 | 1.0 | 12 | 41 | 31 | 1.3:1 |
| 5 | AuCl | CH3NO2 | 16 | 70 | 1.0 | 52 | 4 | 22 | 1:5.5 |
| 6 | AuPicolinateCl2 | CH3NO2 | 16 | 70 | 1.0 | 36 | 4 | 24 | 1:6.0 |
| 7 | (p-F3CC6H4)3PAuCl/AgOTsc | CH3NO2 | 16 | 70 | 1.0 | 5 | 42 | 24 | 1.8:1 |
| 8 | XPhosAuCl/AgOTsc | CH3NO2 | 16 | 70 | 1.0 | 51 | 10 | 24 | 1:2.4 |
| 9 | JohnPhosAuCl/AgOTsc | CH3NO2 | 16 | 70 | 1.0 | 52 | 8 | 24 | 1:3.0 |
| 10 | (ArO)3PAuCl/AgOTsc | CH3NO2 | 16 | 70 | 1.0 | <5 | 47 | 17 | 2.8:1 |
| 11 | (ArO)3PAu(NCCH3)SbF6 | CH3NO2 | 16 | 70 | 1.0 | <5 | 48 | 16 | 3.0:1 |
| 12 | (ArO)3PAu(NCCH3)SbF6 | CH3NO2 | 3 | 70 | 0.1 | <5 | 44 | 20 | 2.2:1 |
| 13 | (ArO)3PAu(NCCH3)SbF6 | CH3NO2 | 3 | RT | 0.1 | 8 | 67 | 10 | 6.7:1 |
| 14 | (ArO)3PAu(NCCH3)SbF6 | CH2Cl2 | 3 | RT | 0.1 | <5 | 54 | 14 | 3.9:1 |
| 15 | (ArO)3PAu(NCCH3)SbF6 | CH3CN | 3 | RT | 0.1 | 29 | 17 | 5 | 3.4:1 |
| 16 | (ArO)3PAu(NCCH3)SbF6 | Toluene | 3 | RT | 0.1 | 0 | 84 | 8 | 10.5:1 |
| 17 | (ArO)3PAu(NCCH3)SbF6 | Toluene | 3 | 0 | 0.1 | 0 | 91 | 8 | 11.4:1 |
An investigation of the reaction conditions was undertaken to explore the factors favouring the rearrangement pathway over those leading to S–O bond cleavage and formation of 4 (Table 1). The most significant factors identified in this study proved to be the use of electron-deficient rather than electron-rich ligands on gold (compare entries 7 and 10 vs. 4, 8 and 9) and the use of lower reaction temperatures (compare entries 13 and 14 vs. 11 and 1), which differ substantially from those conditions previously reported for the intermolecular oxyarylation reaction with sulfoxides.17c,d These observations are in keeping with higher temperature and electron-density at the gold centre being likely to increase the rate of elimination of the sulfide nucleofuge (Scheme 2, B → D).26,27
Little counterion effect was observed and similar results were obtained with the single component catalyst system (entries 10 and 11). Re-evaluating the solvent showed CH2Cl2 to be poor and that excellent selectivity was ultimately obtained in toluene at 0 °C using (2,4-di-tert-BuC6H3O)3PAu(NCCH3)SbF6,16d,28 affording 3a in high yield (entry 17).
The use of dibenzothiophene-S-oxide 1 with different terminal alkynes 2b–k was then studied in the oxyarylation reaction: chloro, aryl, vinyl and phthalimide substituents were well-tolerated as were the methyl and silyl-ethers, affording products 3b–k in generally good yields (Table 2, entries 2–7).29 The α-hydroxyketone oxyarylation product 3g was also formed in high yield (entry 8) despite the potential for oxetan-3-one formation by intramolecular capture of the vinylgold intermediate by the propargylic alcohol, as reported using cationic gold(I) catalysts and pyridine-N-oxides.30 This protocol proved to be robust: a very similar yield was obtained even when the reaction was run open to the air and using non-dried toluene with only 1 mol% catalyst loading on larger scale (entries 4 and 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Cond. | Mmol | Cat./mol% | Time/h | Yielda/% |
| a Yields of isolated material after flash chromatography. b Reactions stirred for 4 h at 0 °C then warmed to rt over 16 hours. c Yield calculated by 1H-NMR spectroscopy against a known quantity of internal standard (1,2,4,5-tetramethylbenzene). | ||||||
| 1 | n Bu | A | 0.2 | 5 | 0.75 | 87 3a |
| 2 | (CH2)3Cl | A | 0.2 | 5 | 0.75 | 79 3b |
| 3 | (CH2)2Ph | A | 0.2 | 5 | 0.75 | 65 3c |
| 4 | CH2OMe | A | 0.2 | 5 | 0.75 | 87 3d |
| 5 | CH2OMe | A | 2.0 | 1 | 2 | 84 3d |
| 6 | (CH2)4OTBDPS | A | 0.5 | 1 | 2 | 82 3e |
| 7 | CH2NPhth | A | 2.0 | 5 | 20b | 52 3f |
| 8 | CH(OH)nC7H15 | A | 0.2 | 5 | 20b | 76 3g |
| 9 | Ph | A | 0.1 | 5 | 1.5 | 48c3h |
| 10 | Ph | B | 0.3 | 5 | 20 | 58 3h |
| 11 | 2-BrC6H4 | B | 0.2 | 5 | 20 | 40 3i |
| 12 | 4-MeOC6H4 | B | 0.2 | 5 | 20 | 42 3j |
| 13 | 2-Thienyl | B | 0.2 | 5 | 20 | 62 3k |

The use of phenyl acetylene gave lower yields and led to formation of significant quantities of dibenzothiophene 4 under the standard conditions (Table 2, entry 9). Further reducing the temperature, which in-turn necessitated a higher dilution to maintain solubility of 1, gave improved yields which were also seen with other aryl alkynes, including thiophene and o-bromobenzene (entries 10–13).
The 2,8-dibromo substitution pattern, which is useful for further transformations in materials science applications,1 was readily accommodated with S-oxide 5 reacting to afford the oxyarylation product 6 in good yield (Scheme 3).
 | ||
| Scheme 3 Use of substituted dibenzothiophene S-oxide. | ||
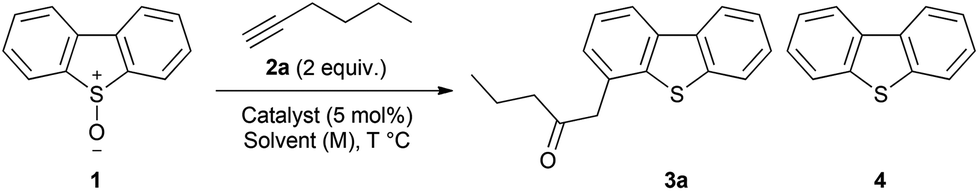
The use of an ynamide under these reaction conditions did not lead cleanly to the oxyarylation products, though the complex mixture formed did indicate that 1 was functioning as an effective oxidant. In order to benchmark the potential suitability of dibenzothiophene-S-oxide as an oxidant in gold catalysis it was applied under the conditions previously reported by Davies and co-workers for the oxidative transformation of ynamides using pyridine N-oxides (Scheme 4).31 Under those conditions 1 proved to be as, or more-, effective than the unsubstituted pyridine-N-oxide and substantially more effective than the diphenylsulfoxide in both the oxidative formation of α,β-unsaturated imide 8 and α-oxoimide 10. Hence 1 may be considered as an alternative reagent to diphenylsulfoxide in gold-catalysed oxidative processes.32,33
 | ||
| Scheme 4 Comparison of reactivity in oxidative transformation of ynamides. Yield of known compounds 8 and 10 determined by 1H NMR against an internal standard. | ||
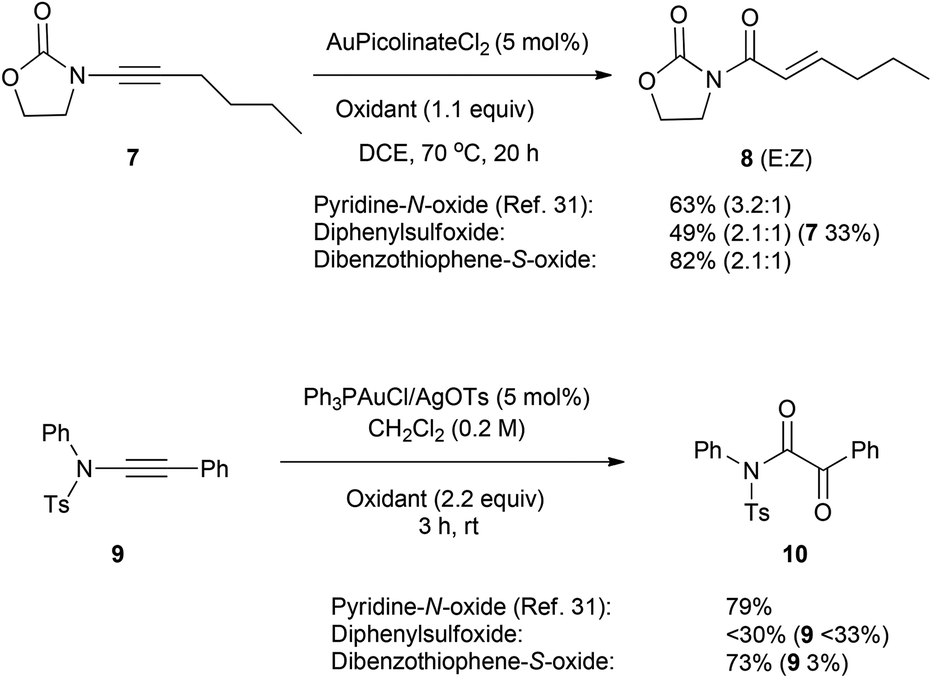
Toste and co-workers had previously reported that the gold-catalysed reaction of 1,6-enynes in the presence of excess diphenylsulfoxide led to the formation of aldehydes by intramolecular cyclisation and capture of the intermediate cyclopropyl gold carbene with sulfoxide.25 Given the higher reactivity observed of 1 compared to diphenylsulfoxide (Scheme 4), the reaction of enyne substrates 11 was studied to see whether 1 would be sufficiently nucleophilic to allow the intermolecular reaction of the sulfoxide at the gold–alkyne complex to compete with intramolecular cycloisomerisation. Under our standard conditions the 1,6-enynes 11a and 11c reacted cleanly to give the oxyarylation products 12a/c in high yield (Scheme 5). In contrast, the cinnamyl derivative 11b and the malonate-derived enyne 11d led to the aldehydes 13b/d with low conversion. On this basis, the relatively high efficacy of dibenzothiophene S-oxide 1 as a nucleophile towards gold alkyne complexes allows it to compete with an intramolecular enyne cycloisomerisation so long as the latter pathway is not strongly biased toward cyclisation by reactive rotamer effects or use of more electron-rich alkenes. Products arising from capture of the vinyl gold carbenoid by the tethered alkene were not observed.34
 | ||
| Scheme 5 The use of enynes in the oxyarylation process and application in an iterative approach to access 4,6-disubstituted dibenzothiophenes and subsequently macrocycles. a 2 mol% for 11a and 5 mol% for 11b–d. | ||
Iterative application of the oxyarylation reaction was then tested to selectively functionalise both the 4- and 6-positions of dibenzothiophene (Scheme 5). The gold-catalysed reactions of 14, from selective oxidation of 12a using mCPBA,35 with 1,6- and 1,7-enynes 11a and 15 afforded high yields of the 4,6-disubstituted dibenzothiophenes 16a/b respectively. A similar iterative process was also successfully applied to 3d (see ESI† for details). While a higher catalyst loading and dilution were required for the second iteration, the compatibility of this approach with the flanking alkene and keto-functionality highlights the potential of using intermolecular oxyarylation approaches with substantially more-functionalised sulfoxides. Ring-closing metathesis of dienes 16a/b furnished the new symmetrical and unsymmetrical macrocyclic products 17a/b in good yield, isolated as trans double bond isomers. The double bond geometry in 17a was determined to be trans through X-ray crystallography (Fig. 1).‡
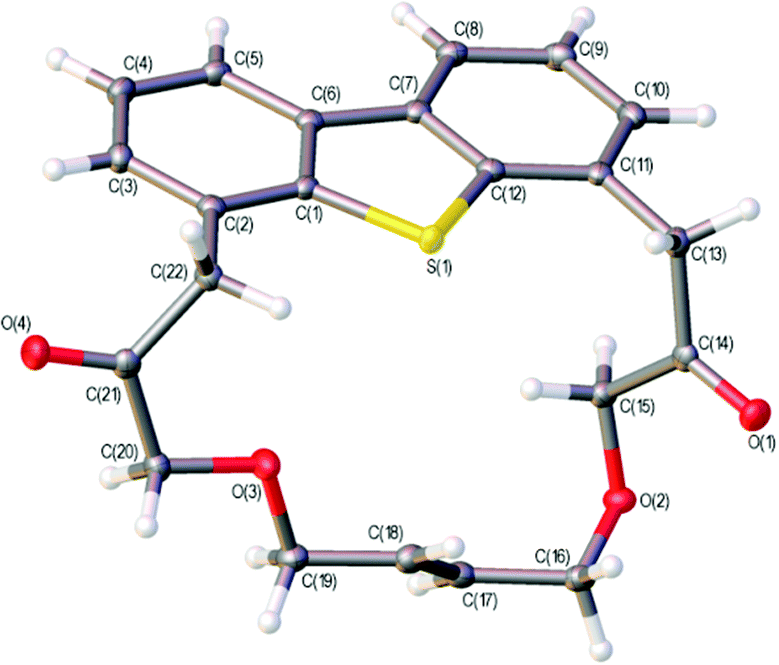 | ||
| Fig. 1 Crystal structure of macocycle 17a with ellipsoids drawn at the 50% probability level. | ||
In addition to regiospecific formation of the Csp2–Csp3 bond the simultaneous installation of a methylenecarbonyl moiety introduces a potentially useful handle for elaboration. We explored this in two ways: first, a classical Fischer-indole synthesis from 3h (yield unoptimised, Scheme 6) affords the 3-dibenzothiophene indole motif 18.7i Thus an alternative is proffered to the standard cross-coupling strategies requiring prefunctionalisation of substrates for the formation of biaryl-linkages at the 4-position of dibenzothiophene. Second, a cascade process using 1,6-diyne 19 provides direct access into the α-arylated cyclohexenone 20 in a single step (Scheme 6). Gold-catalysed cycloisomerisation of the 1,5-ketoalkyne generated from intermolecular oxyarylation results in formation of five new bonds across the alkyne including three carbon–carbon bonds at one carbon. The formation of bisketone 21 as a side-product alongside the major product 20 is consistent with the hydration/aldol dehydration pathway Davies and Detty-Mambo previously reported in cycloisomerisation of alkynes tethered to unactivated, enolisable ketones in the presence of cationic gold(I) species.36
 | ||
| Scheme 6 Utilising the introduced ketomethylene group in (a) formation of a dibenzothienylindole as alternative to cross coupling, (b) cascade catalysis. | ||
Conclusions
Conditions have been developed for regioselective formation of Csp2–Csp3 bonds at the 4- and 6-positions of dibenzothiophenes without prior C-functionalisation. Selectivity for the oxyarylation pathway is favoured by lower temperature and electron-poor ligands on gold. The reactions allow for the introduction of a variety of functionality under robust, scalable conditions. Substantially more elaborate aryl sulfoxides can be used in the oxyarylation approach as demonstrated in an iterative application, which in conjunction with enyne substrates was used to access new macrocyclic structures. In addition, the use of the oxyarylation reaction as the basis for further cascade process development has been demonstrated.Experimental†
General oxyarylation procedure 1 (GP1), Table 2, conditions A
The dibenzothiophene-S-oxide (1 eq.) and alkyne (2 eq.) were stirred in toluene (0.1 M) until dissolved. The mixture was then cooled in an ice bath at 0 °C and the catalyst, (2,4-di-tert-butylC6H3O)3PAu(NCCH3)SbF6 (1–5 mol%), was added. The reaction mixture was stirred until TLC showed consumption of dibenzothiophene-S-oxide, filtered through a pad of silica, washing with CH2Cl2 before being concentrated and the residue purified by column chromatography.General oxyarylation procedure 2 (GP2), Table 2, conditions B
The dibenzothiophene-S-oxide (1 eq.) and alkyne (2 eq.) were stirred in toluene (0.01 M) until dissolved. The mixture was then cooled in a (NaCl/ice) bath to −10 °C and the catalyst, (2,4-di-tert-butylC6H3O)3PAu(NCCH3)SbF6 (5 mol%) was added. The reaction mixture was stirred for 6 hours at this temperature and then allowed to warm to stir at rt for 14 hours, filtered through a pad of silica washing with CH2Cl2 before being concentrated and the residue purified by column chromatography.Alkynes 11a, 3-(prop-2-yn-1-yloxy)prop-1-ene (54 wt% in Et2O), and 15, 4-(prop-2-yn-1-yloxy)but-1-ene (77 wt% in Et2O), were both used with a diethyl ether impurity.
1-(Dibenzo[b,d]thiophen-4-yl)hexan-2-one (3a)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40.0 mg, 0.2 mmol), 1-hexyne (23 μl, 0.4 mmol), toluene (2 mL) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). The reaction was stirred for 45 minutes at 0 °C. Column chromatography (1:19 EtOAc:hexane) afforded 3a (49 mg, 87%) as a white solid; Rf 0.28 (1:19 EtOAc:hexane); mp: 43–45 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.20–8.13 (m, 1H), 8.10 (d, J 7.2, 1H), 7.92–7.82 (m, 1H), 7.51–7.43 (m, 3H), 7.32 (d, J 7.2, 1H), 3.96 (s, 2H), 2.51 (t, J 7.4, 2H), 1.63–1.50 (m, 2H), 1.33–1.19 (m, 2H), 0.85 (t, J 7.3, 3H); 13C-NMR (101 MHz, CDCl3): δ = 207.5 (C), 140.0 (C), 139.1 (C), 136.1 (C), 136.0 (C), 129.2 (C), 128.0 (CH), 127.0 (CH), 125.2 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.6 (CH), 49.4 (CH2), 42.0 (CH2), 26.0 (CH2), 22.3 (CH2), 14.0 (CH3); IR (neat): ν = 3057, 2957, 2930, 2872, 1708, 1584, 1404, 749; HR-MS (ES-TOF): m/z: calcd for C18H18ONaS: 305.0976, found 305.0978 [M + Na]+.
6-Chloro-1-(dibenzo[b,d]thiophen-4-yl)hexan-2-one (3b)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40.0 mg, 0.2 mmol), 6-chloro-1-hexyne (48.5 μL, 0.4 mmol), toluene (2 mL) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). The reaction was stirred for 45 minutes at 0 °C. Column chromatography (9:11 CH2Cl2:hexane) afforded 3b as a yellow oil (50 mg, 79%); Rf 0.44 (9:11 CH2Cl2:hexane); 1H-NMR (300 MHz, CDCl3): δ = 8.18–8.12 (m, 1H), 8.09 (dd, J 7.9 and 0.9, 1H), 7.91–7.83 (m, 1H), 7.54–7.42 (m, 3H), 7.32 (d, J 7.2, 1H), 3.95 (s, 2H), 3.50–3.42 (m, 2H), 2.59–2.50 (m, 2H), 1.76–1.66 (m, 4H); 13C-NMR (101 MHz, CDCl3): δ = 206.6 (C), 139.9 (C), 139.0 (C), 136.2 (C), 136.1 (C), 129.0 (C), 128.0 (CH), 127.0 (CH), 125.2 (CH), 124.8 (CH), 123.0 (CH), 122.0 (CH), 120.7 (CH), 49.4 (CH2), 44.7 (CH2), 41.1 (CH2), 31.8 (CH2), 21.1 (CH2); IR (neat): ν = 3060, 2953, 1711, 1584, 1443, 1401, 749; HR-MS (ES-TOF): m/z: calcd for C18H17ONaS35Cl: 339.0586, found 339.0574 [M + Na]+.
1-(Dibenzo[b,d]thiophen-4-yl)-4-phenylbutan-2-one (3c)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40.0 mg, 0.2 mmol), 4-phenyl-1-butyne (56 μl, 0.4 mmol), toluene (2 mL) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). The reaction was stirred for 45 minutes at 0 °C. Column chromatography (1:1 hexane:CH2Cl2) afforded 3c (43 mg, 65%) as a white solid; Rf 0.78 (3:7 EtOAc:hexane); mp: 102–104 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.04–7.96 (m, 1H), 7.93 (d, J 7.8, 1H), 7.74–7.66 (m, 1H), 7.38–7.26 (m, 3H), 7.15–6.93 (m, 6H), 3.78 (s, 2H), 2.79–2.63 (m, 4H); 13C-NMR (101 MHz, CDCl3): δ = 206.3 (C), 140.9 (C), 140.0 (C), 139.0 (C), 136.2 (C), 136.1 (C), 128.9 (C), 128.6 (2CH), 128.5 (2CH), 128.0 (CH), 127.0 (CH), 126.2 (CH), 125.2 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.7 (CH), 49.6 (CH2), 43.7 (CH2), 29.9 (CH2); IR (neat): ν = 3058, 3027, 2877, 1706, 1601, 1583, 1403, 1046, 746; HR-MS (ES-TOF): m/z: calcd for C22H18ONaS: 353.0976, found 353.0991 [M + Na]+.
1-(Dibenzo[b,d]thiophen-4-yl)-3-methoxypropan-2-one (3d)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40.0 mg, 0.2 mmol), methyl propargyl ether (33.8 μl, 0.4 mmol), toluene (2 mL) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). The reaction was stirred for 45 minutes at 0 °C. Column chromatography (CH2Cl2) afforded 3d (47 mg, 87%) as a yellow solid; Rf 0.31 (3:7 EtOAc:hexane); mp: 51–53 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.19–8.13 (m, 1H), 8.10 (d, J 7.7, 1H), 7.91–7.83 (m, 1H), 7.52–7.43 (m, 3H), 7.34 (d, J 7.2, 1H), 4.13 (s, 2H), 4.03 (s, 2H), 3.41 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 204.7 (C), 140.0 (C), 138.9 (C), 136.2 (C), 136.1 (C), 128.2 (C), 128.1 (CH), 127.1 (CH), 125.2 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 122.0 (CH), 77.3 (CH2), 59.5 (CH3), 45.5 (CH2); IR (neat): ν = 2903, 1712, 1590, 1427, 1394, 1316, 1102, 759; HR-MS (ES-TOF): m/z: calcd for C16H14O2NaS: 293.0612, found 293.0610 [M + Na]+. Open-flask protocol: To a 25 mL RBF under an atmosphere of air was added dibenzothiophene-S-oxide 1 (401 mg, 2.0 mmol), methyl propargyl ether (338 μl, 4.0 mmol) and toluene (technical grade) (20 mL). The flask was placed in an ice bath and (2,4-di-tert-butylC6H3O)3PAu(NCCH3)SbF6 (22.4 mg, 0.002 mmol, 1 mol%) was added. The reaction was stirred at this temperature for 2 hours until TLC indicated reaction completion. Column chromatography (CH2Cl2) afforded 3d (456 mg, 84%).
6-((tert-Butyldiphenylsilyl)oxy)-1-(dibenzo[b,d]thiophen-4-yl)hexan-2-one (3e)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (100 mg, 0.5 mmol), tert-butyl(hex-5-yn-1-yloxy)diphenylsilane (336 mg, 1.0 mmol), toluene (5 mL) and catalyst (11.2 mg, 2 mol%). The reaction was stirred for 2 hours at 0 °C. Column chromatography (3:2 hexane:CH2Cl2) afforded 3e (222 mg, 82%) as a viscous oil; Rf 0.31 (3:2 hexane:CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ = 8.20–8.13 (m, 1H), 8.09 (dd, J 7.9 and 0.9, 1H), 7.88–7.82 (m, 1H), 7.67–7.59 (m, 4H), 7.51–7.32 (m, 9H), 7.30 (d, J 7.3, 1H), 3.93 (s, 2H), 3.60 (t, J 6.2, 2H), 2.51 (t, J 7.3, 2H), 1.77–1.60 (m, 2H), 1.53–1.40 (m, 2H), 1.01 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ = 207.2 (C), 140.0 (C), 139.1 (C), 136.2 (C), 136.1 (C), 135.7 (4CH), 134.1 (2C), 129.7 (2CH), 129.2 (C), 128.0 (CH), 127.7 (4CH), 127.0 (CH), 125.2 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.6 (CH), 63.6 (CH2), 49.4 (CH2), 42.0 (CH2), 32.0 (CH2), 27.0 (3CH3), 20.4 (CH2), 19.4 (C); IR (neat): ν = 2930, 2856, 1713, 1588, 1427, 1105; HR-MS (ES-TOF): m/z: calcd for C34H36O2NaSiS: 559.2103, found 559.2102 [M + Na]+.
2-(3-(Dibenzo[b,d]thiophen-4-yl)-2-oxopropyl)isoindoline-1,3-dione (3f)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (200 mg, 1.0 mmol), N-propargyl phthalimide (370 mg, 2.0 mmol) and catalyst (22.4 mg, 0.04 mmol, 2 mol%) for 4 hours at 0 °C and stirring for a further 16 hours at rt. The precipitate formed was washed with toluene and then recrystallized from hot EtOH affording 3f as yellow crystals (201 mg, 52%); Rf 0.65 (3:7 EtOAc:hexane); mp: 190–192 °C (EtOH); 1H-NMR (300 MHz, CDCl3): δ = 8.20–8.15 (m, 1H), 8.13 (dd, J 7.9 and 1.0, 1H), 7.90–7.85 (m, 1H), 7.85–7.78 (m, 2H), 7.75–7.67 (m, 2H), 7.54–7.44 (m, 3H), 7.40 (d, J 7.3, 1H), 4.56 (s, 2H), 4.11 (s, 2H); 13C-NMR (101 MHz, CDCl3): δ = 199.0 (C), 167.7 (2C), 140.0 (C), 139.0 (C), 136.5 (C), 136.1 (C), 134.2 (2CH), 132.1 (2C), 128.0 (CH), 127.5 (C), 127.2 (CH), 125.4 (CH), 124.8 (CH), 123.6 (2CH), 123.1 (CH), 122.0 (CH), 121.1 (CH), 46.7 (CH2), 46.3 (CH2); IR (neat): ν = 2970, 1769, 1735, 1698, 1470, 1409, 1067; HR-MS (ES-TOF): m/z: calcd for C23H15NO3NaS: 408.0670, found 408.0667 [M + Na]+.
1-(Dibenzo[b,d]thiophen-4-yl)-3-hydroxydecan-2-one (3g)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40 mg, 0.2 mmol), dec-1-yn-3-ol (64 μl, 0.4 mmol), toluene (2 mL) and catalyst (11.2 mg, 5 mol%) for 4 hours at 0 °C and stirring for a further 16 hours at rt. Purification of the reaction mixture with column chromatography (9:1 hexane:EtOAc), followed by recrystallization from hot MeOH afforded 3g (54 mg, 76%); Rf 0.25 (9:1 hexane:EtOAc); mp: 52–54 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.21–8.14 (m, 1H), 8.11 (d, J 7.9, 1H), 7.91–7.82 (m, 1H), 7.54–7.43 (m, 3H), 7.33 (d, J 7.2, 1H), 4.38 (dd, J 7.4 and 3.6, 1H), 4.07 (s, 2H), 3.33 (s, 1H), 2.02–1.88 (m, 1H), 1.75–1.62 (m, 1H), 1.59–1.16 (m, 10H), 0.88 (t, J 6.6, 3H); 13C-NMR (101 MHz, CDCl3): δ = 208.9 (C), 139.9 (C), 138.9 (C), 136.3 (C), 136.1 (C), 128.1 (CH), 127.9 (C), 127.1 (CH), 125.2 (CH), 124.8 (CH), 123.0 (CH), 122.0 (CH), 121.0 (CH), 76.4 (CH), 44.3 (CH2), 34.0 (CH2), 31.9 (CH2), 29.5 (CH2), 29.2 (CH2), 24.9 (CH2), 22.8 (CH2), 14.2(CH3); IR (neat): ν = 3446, 2924, 2854, 1714, 1585, 1443, 1402, 1047, 749; HR-MS (ES-TOF): m/z: calcd for C22H26O2NaS: 377.1551, found 377.1565 [M + Na]+.
2-(Dibenzo[b,d]thiophen-4-yl)-1-phenylethanone (3h)
Prepared according to GP2 using dibenzothiophene-S-oxide 1 (60.0 mg, 0.3 mmol), phenylacetylene (65 μl, 0.6 mmol), toluene (0.01 M, 30 mL) and catalyst (16.8 mg, 0.03 mmol, 5 mol%). Column chromatography (19:1 hexane:EtOAc), followed by recrystallization from hot EtOAc afforded 3h (53 mg, 58%) as a white solid; Rf 0.33 (19:1 hexane:EtOAc); mp: 127–129 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.19–8.12 (m, 1H), 8.12–8.05 (m, 3H), 7.89–7.82 (m, 1H), 7.63–7.54 (m, 1H), 7.52–7.40 (m, 5H), 7.34 (d, J 6.9, 1H), 4.55 (s, 2H); 13C-NMR (101 MHz, CDCl3): δ = 196.4 (C), 139.9 (C), 139.1 (C), 136.7 (C), 136.2 (2C), 133.5 (CH), 129.5 (C), 128.9 (2CH), 128.7 (2CH), 127.9 (CH), 127.0 (CH), 125.1 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.6 (CH), 44.7 (CH2); IR (neat): ν = 3056, 2924, 2856, 1685, 1580, 1440, 1206, 908; HR-MS (ES-TOF): m/z: calcd for C20H14ONaS: 325.0663, found 325.0660 [M + Na]+.
1-(2-Bromophenyl)-2-(dibenzo[b,d]thiophen-4-yl)ethanone (3i)
Prepared according to GP2 using dibenzothiophene-S-oxide 1 (40.0 mg, 0.2 mmol), 1-bromo-2-ethynylbenzene (50 μL, 0.4 mmol), toluene (20 mL, 0.01 mmol) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). Column chromatography (19:1 hexane:EtOAc) followed by recrystallization from hot EtOH afforded 3i (30.5 mg, 40%) as white needles; Rf 0.20 (19:1 hexane:EtOAc); mp: 93–95 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.18–8.12 (m, 1H), 8.09 (dd, J 7.7 and 1.1, 1H), 7.89–7.82 (m, 1H), 7.64–7.59 (m, 1H), 7.51–7.42 (m, 3H), 7.41–7.35 (m, 2H), 7.35–7.24 (m, 2H), 4.53 (s, 2H); 13C-NMR (101 MHz, CDCl3): δ = 200.2 (C), 141.4 (C), 140.2 (C), 139.1 (C), 136.2 (C), 136.1 (2C), 133.7 (CH), 131.8 (CH), 128.8 (CH), 128.3 (CH), 127.6 (CH), 127.0 (CH), 125.1 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.8 (CH), 118.8(C), 48.7 (CH2); IR (neat): ν = 3054, 2940, 1703, 1591, 1441, 1332, 989, 742; HR-MS (ES-TOF): m/z: calcd for C20H14OS79Br: 380.9949, found 380.9948 [M + H]+.
2-(Dibenzo[b,d]thiophen-4-yl)-1-(4-methoxyphenyl)ethanone (3j)
Prepared according to GP2 using dibenzothiophene-S-oxide 1 (40.0 mg, 0.2 mmol), 1-ethynyl-4-methoxybenzene (52 μl, 0.4 mmol), toluene (20 mL) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). Column chromatography (19:1 hexane:EtOAc) afforded 3j (28 mg, 42%) as a white solid; Rf 0.18 (19:1 hexane:EtOAc); mp: 113–115 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.19–8.12 (m, 1H), 8.11–8.01 (m, 3H), 7.90–7.82 (m, 1H), 7.51–7.39 (m, 3H), 7.34 (d, J 7.2, 1H), 6.98–6.89 (m, 2H), 4.49 (s, 2H), 3.86 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 195.0 (C), 163.8 (C), 139.8 (C), 139.1 (C), 136.3 (C), 136.2 (C), 131.0 (2CH), 129.9 (C), 129.7 (C), 127.8 (CH), 126.9 (CH), 125.1 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.4 (CH), 114.0 (2CH), 55.6 (CH3), 44.4 (CH2); IR (neat): ν = 2910, 1717, 1593, 1508, 1400, 1167, 751; HR-MS (ES-TOF): m/z: calcd for C21H17O2NS: 333.0949, found 333.0950 [M + H]+.
2-(Dibenzo[b,d]thiophen-4-yl)-1-(thiophen-2-yl)ethanone (3k)
Prepared according to GP2 using dibenzothiophene-S-oxide 1 (40 mg, 0.2 mmol), 2-ethynylthiophene (44 μl, 0.4 mmol), toluene (20 mL, 0.01 mmol) and catalyst (11.2 mg, 0.02 mmol, 5 mol%). Purification of the reaction mixture by column chromatography (9:1 hexane:EtOAc) afforded 3k (38 mg, 62%) as an orange oil; Rf 0.65 (9:1 hexane:EtOAc); 1H-NMR (300 MHz, CDCl3): δ = 8.19–8.11 (m, 1H), 8.09 (dd, J 7.5 and 1.2, 1H), 7.90–7.82 (m, 2H), 7.65 (dd, J 4.9 and 0.7, 1H), 7.51–7.38 (m, 4H), 7.11 (dd, J 4.9 and 4.0, 1H), 4.46 (s, 2H); 13C-NMR (101 MHz, CDCl3): δ = 189.2 (C), 143.9 (C), 139.9 (C), 138.9 (C), 136.2 (C), 136.2 (C), 134.4 (CH), 132.9 (CH), 129.2 (C), 128.4 (CH), 127.8 (CH), 127.0 (CH), 125.1 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.7 (CH), 45.5 (CH2); IR (neat): ν = 3092, 3074, 1641, 1410, 1276, 1057, 750; HR-MS (EI-TOF): m/z: calcd for C18H13OS2: 308.0330, found 308.0329 [M + H]+.
1-(2,8-Dibromodibenzo[b,d]thiophen-4-yl)-3-methoxypropan-2-one (6)
Sulfoxide 5 (71.6 mg, 0.2 mmol) was added to a 50 mL RBF with methyl propargyl ether (34 μl, 0.4 mmol) and CHCl3 (30 mL). catalyst (11.2 mg, 0.01 mmol, 5 mol%) was added and the mixture was stirred at rt for 17 hours. Purification by column chromatography (CH2Cl2) afforded 6 (58 mg, 70%) as a white solid; Rf 0.07 (19:1 hexane:EtOAc); mp: 139–141 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.18 (s, 1H), 8.12 (s, 1H), 7.68 (d, J 8.5, 1H), 7.56 (dd, J 8.5 and 1.3, 1H), 7.46 (s, 1H), 4.12 (s, 2H), 4.00 (s, 2H), 3.45 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 204.1 (C), 139.4 (C), 138.0 (C), 136.6 (C), 136.4 (C), 131.5 (CH), 130.6 (CH), 130.0 (C), 125.0 (CH), 124.3 (CH), 123.8 (CH), 119.1 (C), 119.0 (C), 77.6 (CH2), 59.6 (CH3), 44.8 (CH2); IR (neat): ν = 3067, 2901, 1723, 1567, 1410, 1319, 1072, 1042, 746; HR-MS (ES-TOF): m/z: calcd for C16H12O2NaS79Br81Br: 450.8802, found 450.8801 [M + Na]+.
1-(Allyloxy)-3-(dibenzo[b,d]thiophen-4-yl)propan-2-one (12a)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (100 mg, 0.5 mmol), 3-(prop-2-yn-1-yloxy)prop-1-ene 11a (54 wt% in Et2O, 177 mg, 1.0 mmol), and catalyst (11.2 mg, 0.02 mmol, 2 mol%). The reaction mixture was stirred for 4 hours at 0 °C and left to warm to rt for a further 16 hours. Column chromatography (CH2Cl2) afforded 12a (119 mg, 80%) as a yellow oil; Rf 0.34 (CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ = 8.19–8.14 (m, 1H), 8.10 (dd, J 7.9 and 1.0, 1H), 7.90–7.83 (m, 1H), 7.52–7.43 (m, 3H), 7.35 (d, J 7.2, 1H), 5.89 (ddt, J 17.2, 10.4 and 5.8, 1H), 5.27 (dd, J 17.2 and 1.5, 1H), 5.21 (dd, J 10.4 and 1.5, 1H), 4.18 (s, 2H), 4.06 (s, 2H), 4.06–4.02 (m, 2H); 13C-NMR (101 MHz, CDCl3): δ = 205.0 (C), 140.0 (C), 139.0 (C), 136.2 (C), 136.1 (C), 133.8 (CH), 128.3 (C), 128.1 (CH), 127.0 (CH), 125.2 (CH), 124.7 (CH), 123.0 (CH), 122.0 (CH), 120.8 (CH), 118.3 (CH2), 74.8 (CH2), 72.6 (CH2), 45.6 (CH2); IR (neat): ν = 2901, 1726, 1554, 1443, 1402, 1096, 912; HR-MS (ES-TOF): m/z: calcd for C18H16O2SNa: 319.0769, found 319.0775 [M + Na]+.N-Allyl-N-(3-(dibenzo[b,d]thiophen-4-yl)-2-oxopropyl)-4-methylbenzenesulfonamide (12c)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40 mg, 0.2 mmol), N-allyl-4-methyl-N-(prop-2-yn-1-yl)benzenesulfonamide 11c (96 mg, 0.4 mmol), and catalyst (11.2 mg, 0.02 mmol, 5 mol%). The reaction mixture was stirred for 2.5 hours at 0 °C. Column chromatography (9:1 hexane:EtOAc) followed by recrystallization from hot MeOH afforded 12c (66 mg, 74%) as a white solid; Rf 0.31 (9:1 hexane:EtOAc); mp: 104–106 °C; 1H NMR (300 MHz, CDCl3): δ = 8.21–8.14 (m, 1H), 8.11 (dd, J 8.1, 0.9, 1H), 7.91–7.82 (m, 1H), 7.65 (d, J 8.3, 2H), 7.53–7.43 (m, 3H), 7.32 (d, J 7.2, 1H), 7.21 (d, J 8.1, 2H), 5.58 (ddt, J 16.9, 10.1 and 6.8, 1H), 5.03 (dd, J 10.1, 1.1, 1H), 4.96 (dd, J 16.9, 1.1, 1H) 4.10 (s, 2H), 4.01 (s, 2H), 3.78 (d, J 6.7, 2H), 2.36 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 201.9 (C), 143.7 (C), 140.0 (C), 139.0 (C), 136.3 (C), 136.2 (C), 136.0 (C), 132.1 (CH), 129.8 (2CH), 128.2 (CH), 128.0 (C), 127.6 (2CH), 127.1 (CH), 125.3 (CH), 124.8 (CH), 123.0 (CH), 122.0 (CH), 120.9 (CH), 120.5 (CH2) 54.4 (CH2), 51.4 (CH2), 46.4 (CH2), 21.6 (CH3); IR (neat): ν = 1731, 1443, 1397, 1153, 1045, 924, 752; HR-MS (ES-TOF): m/z: calcd for C25H24NO3S2: 450.1198, found 450.1180 [M + H]+.
1-(Allyloxy)-3-(5-oxidodibenzo[b,d]thiophen-4-yl)propan-2-one (14)
mCPBA (72.3 mg, 0.42 mmol, 1.1 equiv.) was added in 5 portions over 10 minutes to a solution of 12a (113 mg, 0.38 mmol) in CH2Cl2 (10 mL) at 0 °C. The reaction was allowed to warm to rt over 2 hours, washed with NaHCO3 (4 × 10 mL), extracted with CH2Cl2 (4 × 10 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by column chromatography (4:1 hexane:EtOAc to EtOAc) afforded firstly 12a (25 mg, 22%) and then 14 (74 mg, 62%) as a white solid; Rf 0.37 (3:7 hexane:EtOAc); mp: 98–100 °C; 1H-NMR (300 MHz, CDCl3): δ = 7.96 (d, J 7.5, 1H), 7.80 (d, J 7.6, 1H), 7.74 (d, J 7.5, 1H), 7.64–7.53 (m, 2H), 7.50 (td, J 7.5 and 0.9, 1H), 7.29 (d, J 7.6, 1H), 5.94 (ddt, J 17.2, 10.5 and 5.7, 1H), 5.32 (dd, J 17.2 and 1.5, 1H), 5.24 (dd, J 10.5 and 1.2, 1H), 4.38–4.17 (m, 4H), 4.11 (d, J 5.8, 2H); 13C-NMR (101 MHz, CDCl3): δ = 204.6 (C), 144.7 (C), 144.0 (C), 137.7 (C), 137.3 (C), 135.0 (C), 133.9 (CH), 132.7 (CH), 132.7 (CH), 131.7 (CH), 129.7 (CH), 127.5 (CH), 122.2 (CH), 121.0 (CH), 118.3 (CH2), 75.1 (CH2), 72.7 (CH2), 42.5 (CH2); IR (neat): ν = 3050, 2857, 1725, 1551, 1485, 1424, 1321, 1161, 1145, 1070, 1045, 1012, 762; HR-MS (ES-TOF): m/z: calcd for C18H17O3S: 313.0898, found 313.0906 [M + H]+.
3,3′-(Dibenzo[b,d]thiophene-4,6-diyl)bis(1-(allyloxy)propan-2-one) (16a)
Sulfoxide 14 (68 mg, 0.22 mmol) and enyne 11a (54 wt% in Et2O, 78.3 mg, 0.44 mmol) were dissolved in toluene (8.8 mL, 0.025 M). After stirring for 20 minutes the reaction mixture was transferred to an ice bath at 0 °C. (2,4-Di-tert-butylC6H3O)3PAu(NCCH3)SbF6 (12.3 mg, 0.011 mmol, 5 mol%) was added with a further portion (6.1 mg, 5.5 μmol, 2.5 mol%) added after 3 hours with the reaction mixture then stirred for a further 1 hour at 0 °C. The reaction mixture was filtered through a plug of silica and washed with CH2Cl2 (10 mL). The reaction mixture was concentrated under reduced pressure and purified by column chromatography (4:1 hexane:EtOAc) providing 16a (65 mg, 73%) as a white solid; Rf 0.58 (1:1 hexane:EtOAc); mp: 77–80 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.08 (d, J 7.7, 2H), 7.47 (app t, J 7.6, 2H), 7.34 (d, J 7.2, 2H), 5.90 (ddt, J 17.2, 10.4 and 5.7, 2H), 5.27 (app d, J 17.2, 2H), 5.21 (app d, J 10.4, 2H) 4.17 (s, 4H), 4.11–4.01 (m, 8H); 13C-NMR (101 MHz, CDCl3): δ = 204.8 (2C), 139.5 (2C), 136.6 (2C), 133.8 (2CH), 128.3 (2CH), 128.3 (2C), 125.4 (2CH), 121.0 (2CH), 118.3 (2CH2), 74.8 (2CH2), 72.6 (2CH2), 45.6 (2CH2); IR (neat): ν = 2855, 1722, 1574, 1426, 1390, 1331, 1164, 1060, 1045; HR-MS (ES-TOF): m/z: calcd for C24H24O4NaS: 431.1293, found 431.1288 [M + Na]+.
1-(Allyloxy)-3-(6-(3-(but-3-en-1-yloxy)-2-oxopropyl)dibenzo[b,d]thiophen-4-yl)propan-2-one (16b)
Sulfoxide 14 (60 mg, 0.192 mmol) and enyne 15 (77 wt% in Et2O, 50 mg, 0.348 mmol) were dissolved in toluene (0.025 M, 10 mL). After stirring for 20 minutes at rt the reaction was transferred to an ice bath at 0 °C and (2,4-di-tert-butylC6H3O)3PAu(NCCH3)SbF6 (7.5 mol%) was added. The reaction was stirred for 4 hours filtered through a pad of silica washing with CH2Cl2, concentrated and purified by column chromatography (4:1 hexane:EtOAc) to afford 16b (68 mg, 83%) as an off white solid; Rf 0.81 (1:1 hexane:EtOAc); mp: 57–59 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.02 (dd, J 7.9 and 0.8, 2H), 7.40 (app t, J 7.6, 2H), 7.28 (d, J 7.3, 2H), 5.91–5.68 (m, 2H), 5.21 (dd, J 17.2 and 1.6, 1H), 5.15 (dd, J 10.2 and 1.6, 1H), 5.04 (dd, J 17.2 and 1.6, 1H), 4.97 (dd, J 10.2 and 1.6, 1H), 4.10 (s, 4H), 4.02–3.96 (m, 6H), 3.48 (t, J 6.7, 2H), 2.37–2.27 (m, 2H); 13C-NMR (101 MHz, CDCl3): δ = 205.1 (C), 204.9 (C), 139.6 (2C), 136.7 (2C), 134.9 (CH), 133.8 (CH), 128.3 (2CH), 128.2 (2C), 125.4 (2CH), 121.0 (2CH), 118.3 (CH2), 117.0 (CH2), 75.9 (CH2), 74.8 (CH2), 72.6 (CH2), 71.3 (CH2), 45.6 (2CH2), 34.2 (CH2); IR (neat): ν = 2860, 1721, 1644, 1575, 1476, 143, 1061, 913, 776; HR-MS (ES-TOF): m/z: calcd for C25H26O4NaS: 445.1450, found 445.1429 [M + Na]+.
Macrocycle 17a
[1,3-Bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)(tricyclohexylphosphine)ruthenium (10.4 mg, 0.012 mmol) was added to a solution of 16a (100 mg, 0.245 mmol) in CH2Cl2 (25 mL). The reaction mixture was heated to reflux for 1 hour, allowed to cool, concentrated and purified by column chromatography (3:7 hexane:EtOAc) to afford 17a (80 mg, 86%) as a white solid; Rf 0.58 (1:1 hexane:EtOAc); mp: 165–167 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.08 (d, J 7.2, 2H), 7.48 (app t, J 7.6, 2H), 7.39 (d, J 7.0, 2H), 5.74–5.60 (m, 2H), 4.19 (s, 4H), 4.02 (s, 4H), 3.96 (dd, J 3.0 and 1.3, 4H); 13C-NMR (101 MHz, CDCl3): δ = 205.0 (2C), 139.2 (2C), 136.7 (2C), 130.2 (2CH), 128.1 (2CH), 128.1 (2C), 125.7 (2CH), 121.1 (2CH), 74.3 (2CH2), 71.1 (2CH2), 45.6 (2CH2); IR (neat): ν = 2855, 1722, 1574, 1426, 1390, 1331, 1144, 1060, 1045, 919, 776, 731; HR-MS (ES-TOF): m/z: calcd for C22H20O4NaS: 403.0980, found 403.0996 [M + Na]+.
Macrocycle 17b
[1,3-Bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)(tricyclohexylphosphine)ruthenium (5.0 mg, 0.006 mmol) was added to a solution of 16b (50 mg, 0.118 mmol) in CH2Cl2 (25 mL). The reaction mixture was heated to reflux for 2 hours, concentrated and purified by column chromatography (3:7 hexane:EtOAc) to afford 17b (35 mg, 70%) as a white solid; Rf 0.48 (1:1 hexane:EtOAc); mp: 138–139 °C; 1H-NMR (300 MHz, CDCl3): δ = 8.07 (d, J 7.8, 2H), 7.48 (td, J 7.6 and 3.1, 2H), 7.37 (d, J 5.1, 2H), 6.11–5.95 (m, 1H), 5.69 (dt, J 15.0 and 5.6, 1H), 4.18 (s, 4H), 4.15–4.05 (m, 6H), 3.64 (t, J 5.8, 2H), 2.42 (dd, J 11.6 and 5.6, 2H); 13C-NMR (101 MHz, CDCl3): δ = 206.6 (C), 205.8 (C), 139.2 (C), 139.1 (C), 136.7 (C), 136.6 (C), 132.0 (CH), 128.7 (CH), 128.7 (CH), 128.4 (C), 128.4 (C) 127.4 (CH), 125.4 (2CH), 121.0 (CH), 120.9 (CH), 76.3 (CH2), 74.4 (CH2), 71.7 (CH2), 71.2 (CH2), 44.7 (CH2), 44.2 (CH2), 32.8 (CH2); IR (neat): ν = 2861, 1720, 1644, 1575, 1426, 1143, 1062, 914, 775; HR-MS (ES-TOF): m/z: calcd for C23H22O4NaS: 417.1137, found 417.1136 [M + Na]+.
3-(Dibenzo[b,d]thiophen-4-yl)-2-phenyl-1H-indole (18)
To 3h (55 mg, 0.183 mmol, 1.0 eq.) was added AcOH (0.80 ml), TFA (0.28 ml) and phenylhydrazine (45 μL, 0.46 mmol, 2.5 eq.) in a sealed (Ace) tube. The reaction was stirred at 100 °C for 28 hours at which point reaction completion was observed by TLC. The mixture was added to ice/water (10 mL), the mixture was extracted with CH2Cl2 (10 mL × 3) and the organic portions were washed with HCl (1 M, 5 mL), water (5 mL), dried over Na2SO4, concentrated and purified by column chromatography (9:1 hexane:EtOAc) to afford 18 (45.6 mg, 66%) as a viscous orange oil; Rf 0.31 (9:1 hexane:EtOAc); 1H-NMR (300 MHz, CDCl3): δ = 8.46 (s, 1H), 8.21 (ddd, J 7.6, 5.6 and 1.4, 2H), 7.77–7.70 (m, 1H), 7.59–7.48 (m, 3H), 7.48–7.41 (m, 3H), 7.40–7.34 (m, 2H), 7.33–7.20 (m, 4H), 7.13 (td, J 7.6 and 0.9, 1H); 13C-NMR (101 MHz, CDCl3): δ = 141.4 (C), 140.1 (C), 136.1 (C), 136.0 (2C), 134.8 (C), 132.5 (C), 130.5 (C), 129.3 (CH), 128.9 (2CH), 128.8 (C), 127.9 (CH), 127.3 (2CH), 126.7 (CH), 125.0 (CH), 124.3 (CH), 123.0 (CH), 122.9 (CH), 121.8 (CH), 120.4 (CH), 120.4 (CH), 120.3 (CH), 113.5 (C), 111.1 (CH); IR (neat): ν = 3408, 3057, 1578, 1487, 1442, 1384, 1253, 905, 742, 693; HR-MS (ES-TOF): m/z: calcd for C26H18NS: 376.1160, found 376.1170 [M + H]+.
Diethyl 4-(dibenzo[b,d]thiophen-4-yl)-3-methyl-5-oxocyclohex-3-ene-1,1-dicarboxylate (20) and diethyl 2-(3-(dibenzo[b,d]thiophen-4-yl)-2-oxopropyl)-2-(2-oxopropyl)malonate (21)
Prepared according to GP1 using dibenzothiophene-S-oxide 1 (40 mg, 0.2 mmol), diyne 19 (48.6 mg, 0.4 mmol), toluene (2 mL) and catalyst (11.2 mg, 5 mol%). The reaction was stirred for 2 hours at 0 °C before allowing to warm to rt for 17 hours. Column chromatography (3:7 hexane:CH2Cl2 to CH2Cl2) afforded 20 (49 mg, 57%) as a colourless oil and 21 (15 mg, 17%) as a colourless oil.
20
R
f 0.48 (3:7 hexane:EtOAc); 1H NMR (300 MHz, CDCl3): δ = 8.13 (ddd, J 12.9, 5.7 and 2.4, 2H), 7.83–7.75 (m, 1H), 7.55–7.37 (m, 3H), 7.11 (dd, J 7.2 and 1.0, 1H), 4.44–4.21 (m, 4H), 3.32–2.94 (m, 4H), 1.85 (s, 3H), 1.32 (t, J 7.1, 3H), 1.31 (t, J 7.1, 3H),; 13C NMR (101 MHz, CDCl3): δ = 192.4 (C), 169.9 (C), 169.8 (C) 156.3 (C), 156.3 (C), 140.1 (C), 139.4 (C), 136.1 (C), 136.1 (C) 135.8 (C), 128.2 (CH), 126.8 (CH), 124.7 (CH), 124.5 (CH), 122.8 (CH), 121.8 (CH), 121.0 (CH), 62.6 (CH2), 62.4 (CH2), 55.0 (C), 42.6 (CH2), 37.5 (CH2), 22.7 (CH3), 14.2 (2CH3); IR (neat): ν = 2982, 1729, 1673, 1302, 1250, 1167, 752; HR-MS (ES-TOF): m/z: calcd for C25H24O5NaS: 459.1242, found 459.1229 [M + Na]+.
21
R
f 0.42 (7:3 hexane:EtOAc); 1H NMR (300 MHz, CDCl3): δ = 8.18–8.12 (m, 1H), 8.09 (dd, J 7.9 and 0.9, 1H), 7.87–7.81 (m, 1H), 7.50–7.42 (m, 3H), 7.31 (d, J 7.0, 1H), 4.13 (q, J 7.1, 2H), 4.12 (q, J 7.1, 2H), 3.97 (s, 2H), 3.50 (s, 2H), 3.31 (s, 2H), 1.99 (s, 3H), 1.17 (t, J 7.0, 6H); 13C NMR (101 MHz, CDCl3): δ = 206.0 (C), 204.6 (C), 169.5 (2C), 139.9 (C), 139.1 (C), 136.3 (C), 136.0 (C), 128.4 (CH), 128.2 (C), 127.1 (CH), 125.3 (CH), 124.8 (C), 122.9 (C), 122.0 (CH), 120.8 (CH), 62.1 (2CH2), 53.2 (C), 49.4 (CH2), 45.8 (CH2), 44.9 (CH2), 30.2 (CH3), 14.0 (2CH3); IR (neat): ν = 2982, 2930, 1719, 1444, 1403, 1364, 1201, 1096, 754; HR-MS (ES-TOF): m/z: calcd for C25H26O6SNa: 477.1348, found 477.1339 [M + Na]+.
Acknowledgements
The authors thank Dr Louise Male (University of Birmingham) for X-ray crystallography and the EPSRC/University of Birmingham for financial support (studentship to MJB). The facilities used in this research were part supported through Birmingham Science City AM2 by Advantage West Midlands and the European Regional Development Fund.Notes and references
- For recent examples see: (a) S.-C. Dong, L. Zhang, J. Liang, L.-S. Cui, Q. Li, Z.-Q. Jiang and L.-S. Liao, J. Phys. Chem. C, 2014, 118, 2375–2384 CrossRef CAS; (b) L. Yao, S. Sun, S. Xue, S. Zhang, X. Wu, H. Zhang, Y. Pan, C. Gu, F. Li and Y. Ma, J. Phys. Chem. C, 2013, 117, 14189–14196 CrossRef CAS; (c) L. Yu, J. Liu, S. Hu, R. He, W. Yang, H. Wu, J. Peng, R. Xia and D. D. C. Bradley, Adv. Funct. Mater., 2013, 23, 4366–4376 CrossRef CAS PubMed; (d) L. Ying, Y.-H. Li, C.-H. Wei, M.-Q. Wang, W. Yang, H.-B. Wu and Y. Cao, Chin. J. Polym. Sci., 2013, 31, 88–97 CrossRef; (e) S. Cai, X. Hu, J. Han, Z. Zhang, X. Li, C. Wang and J. Su, Tetrahedron, 2013, 69, 1970–1977 CrossRef CAS PubMed.
- D. Vasu, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 7162–7166 CrossRef CAS PubMed.
- (a) T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156–2164 CrossRef CAS; (b) C. Zhang, Org. Biomol. Chem., 2014, 12, 6580–6589 RSC.
- (a) J. Korang, W. R. Grither and R. D. McCulla, J. Am. Chem. Soc., 2010, 132, 4466–4476 CrossRef CAS PubMed; (b) M. Nag and W. S. Jenks, J. Org. Chem., 2005, 70, 3458–3463 CrossRef CAS PubMed; (c) M. Nag and W. S. Jenks, J. Org. Chem., 2004, 69, 8177–8182 CrossRef CAS PubMed; (d) A. B. Thomas and A. Greer, J. Org. Chem., 2003, 68, 1886–1891 CrossRef PubMed; (e) D. D. Gregory, Z. Wan and W. S. Jenks, J. Am. Chem. Soc., 1997, 119, 94–102 CrossRef CAS; (f) Z. Wan and W. S. Jenks, J. Am. Chem. Soc., 1995, 117, 2667–2668 CrossRef CAS.
- (a) V. Desikan, Y. Liu, J. P. Toscano and W. S. Jenks, J. Org. Chem., 2007, 72, 6848–6859 CrossRef CAS PubMed; (b) V. Desikan, Y. Liu, J. P. Toscano and W. S. Jenks, J. Org. Chem., 2008, 73, 4398–4414 CrossRef CAS PubMed; (c) T. Nakahodo, M. Okuda, H. Morita, T. Yoshimura, M. O. Ishitsuka, T. Tuchiya, Y. Maeda, H. Fujihara, T. Akasaka, X. Gao and S. Nagase, Angew. Chem., Int. Ed., 2008, 47, 1298–1300 CrossRef CAS PubMed; (d) H. Morita, A. Tatami, T. Maeda, B. J. Kim, W. Kawashima, T. Yoshimura, H. Abe and T. Akasaka, J. Org. Chem., 2008, 73, 7159–7163 CrossRef CAS PubMed.
- (a) W. S. Jenks, M. J. Heying, S. A. Stoffregen and E. M. Rockafellow, J. Org. Chem., 2009, 74, 2765–2770 CrossRef CAS PubMed; (b) S. A. Stoffregen, M. J. Heying and W. S. Jenks, J. Am. Chem. Soc., 2007, 129, 15746–15747 CrossRef CAS PubMed.
- (a) Y. Gao, K. J. Kellar, R. P. Yasuda, T. Tran, Y. Xiao, R. F. Dannals and A. G. Horti, J. Med. Chem., 2013, 56, 7574–7589 CrossRef CAS PubMed; (b) C. Cano, K. Saravanan, C. Bailey, J. Bardos, N. J. Curtin, M. Frigerio, B. T. Golding, I. R. Hardcastle, M. G. Hummersone, K. A. Menear, D. R. Newell, C. J. Richardson, K. Shea, G. C. M. Smith, P. Thommes, A. Ting and R. J. Griffin, J. Med. Chem., 2013, 56, 6386–6401 CrossRef CAS PubMed; (c) J. Korang, I. Emahi, W. R. Grither, S. M. Baumann, D. A. Baum and R. D. McCulla, RSC Adv., 2013, 3, 12390–12397 RSC; (d) M. R. Schrimpf, K. B. Sippy, C. A. Briggs, D. J. Anderson, T. Li, J. Ji, J. M. Frost, C. S. Surowy, W. H. Bunnelle, M. Gopalakrishnan and M. D. Meyer, Bioorg. Med. Chem. Lett., 2012, 22, 1633–1638 CrossRef CAS PubMed; (e) M. Zhang, G. E. Ravilious, L. M. Hicks, J. M. Jez and R. D. McCulla, J. Am. Chem. Soc., 2012, 134, 16979–16982 CrossRef CAS PubMed; (f) S. R. Patpi, L. Pulipati, P. Yogeeswari, D. Sriram, N. Jain, B. Sridhar, R. Murthy, T. A. Devi, S. V. Kalivendi and S. J. Kantevari, J. Med. Chem., 2012, 55, 3911–3922 CrossRef CAS PubMed; (g) C. Cano, O. R. Barbeau, C. Bailey, X.-L. Cockcroft, N. J. Curtin, H. Duggan, M. Frigerio, B. T. Golding, I. R. Hardcastle, M. G. Hummersone, C. Knights, K. A. Menear, D. R. Newell, C. J. Richardson, G. C. M. Smith, B. Spittle and R. J. Griffin, J. Med. Chem., 2010, 53, 8498–8507 CrossRef CAS PubMed; (h) W. Kemnitzer, N. Sirisoma, S. Jiang, S. Kasibhatla, C. Crogan-Grundy, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2010, 20, 1288–1292 CrossRef CAS PubMed; (i) M.-J. R. P. Queiroz, A. S. Abreu, M. S. D. Carvalho, P. M. T. Ferreira, N. Nazareth and M. S.-J. Nascimento, Bioorg. Med. Chem., 2008, 16, 5584–5589 CrossRef CAS PubMed; (j) J. J. J. Leahy, B. T. Golding, R. J. Griffin, I. R. Hardcastle, C. Richardson, L. Rigoreau and G. C. M. Smith, Bioorg. Med. Chem. Lett., 2004, 14, 6083–6087 CrossRef CAS PubMed; (k) Y. Mori, S. Taneda, H. Hayashi, A. Sakushima, K. Kamata, A. K. Suzuki, S. Yoshino, M. Sakata, M. Sagai and K.-I. Seki, Biol. Pharm. Bull., 2002, 25, 145–146 CrossRef CAS; (l) D. A. Patrick, J. E. Hall, B. C. Bender, D. R. McCurdy, W. D. Wilson, F. A. Tanious, S. Saha and R. R. Tidwell, Eur. J. Med. Chem., 1999, 34, 575–583 CAS; (m) A. M. El-Naggar, F. S. M. Ahmed and S. G. Donia, J. Indian Chem. Soc., 1983, 60, 479–482 CAS.
- (a) X.-D. Xiong, C.-L. Deng, X.-S. Peng, Q. Miao and H. N. C. Wong, Org. Lett., 2014, 16, 3252–3255 CrossRef CAS PubMed; (b) P. Zhao, H. Yin, H. Gao and C. Xi, J. Org. Chem., 2013, 78, 5001–5006 CrossRef CAS PubMed; (c) T. H. Jepsen, M. Larsen, M. Jørgensen and M. B. Nielsen, Synthesis, 2013, 1115–1120 CAS; (d) X. Shang, W. Chen and Y. Yao, Synlett, 2013, 851–854 CAS; V. B. Pandya, M. R. Jain, B. V. Chaugule, J. Patel, B. M. Parmar, J. K. Joshi and P. R. Patel, Synth. Commun., 2012, 42, 497–505 Search PubMed; (e) S. Rodriguez-Aristegui, K. M. Clapham, L. Barrett, C. Cano, M. D.-E. Murr, R. J. Griffin, I. R. Hardcastle, S. L. Payne, T. Rennison, C. Richardson and B. T. Golding, Org. Biomol. Chem., 2011, 9, 6066–6074 RSC; (f) R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217–5220 CrossRef CAS PubMed; (g) T. H. Jepsen, M. Larsen, M. Jørgensen, K. A. Solanko, A. Bond, A. Kadziola and M. B. Nielsen, Eur. J. Org. Chem., 2011, 53–57 CrossRef CAS PubMed; (h) M. Kienle, A. Unsinn and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 4751–4754 CrossRef CAS PubMed.
- (a) R. Che, Z. Wu, Z. Li, H. Xiang and X. Zhou, Chem. – Eur. J., 2014, 20, 7258–7261 CrossRef CAS PubMed; (b) S. Trosien, P. Böttger and S. R. Waldvogel, Org. Lett., 2014, 16, 402–405 CrossRef CAS PubMed; (c) P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848–851 CrossRef CAS PubMed; (d) T. Wesch, A. Berthelot-Bréhier, F. R. Leroux and F. R. Colobert, Org. Lett., 2013, 15, 2490–2493 CrossRef CAS PubMed; (e) J. Chen and T. Murafuji, Organometallics, 2011, 30, 4532–4538 CrossRef CAS; (f) X. Xu, X. Li, A. Wang, Y. Sun, W. B. Schweizer and R. Prins, Helv. Chim. Acta, 2011, 94, 1754–1763 CrossRef CAS PubMed; (g) M. Black, J. I. G. Cadogan and H. McNab, Org. Biomol. Chem., 2010, 8, 2961–2967 RSC; (h) R. Sanz, Y. Fernández, M. P. Castroviejo, A. Pérez and F. J. Fañanás, J. Org. Chem., 2006, 71, 6291–6294 CrossRef CAS PubMed.
- (a) M. Nandakumar, J. Karunakaran and A. K. Mohanakrishnan, Org. Lett., 2014, 16, 3068–3071 CrossRef CAS PubMed; (b) S.-M. T. Toguem, I. Malik, M. Hussain, J. Iqbal, A. Villinger and P. Langer, Tetrahedron, 2013, 69, 160–173 CrossRef PubMed; (c) A. S. K. Hashmi, W. Yang and F. Rominger, Chem. – Eur. J., 2012, 18, 6576–6580 CrossRef CAS PubMed; (d) S.-M. T. Toguem, I. Knepper, P. Ehlers, T. T. Dang, T. Patonay and P. Langer, Adv. Synth. Catal., 2012, 354, 1819–1826 CrossRef CAS PubMed.
- W. Yang, Q. Hou, C. Liu, Y. Niu, J. Huang, R. Yang and Y. Cao, J. Mater. Chem., 2003, 13, 1351–1355 RSC.
- H. Sirringhaus, R. H. Friend, C. Wang, J. Leuningerb and K. J. Müllen, J. Mater. Chem., 1999, 9, 2095–2101 RSC.
- A. R. Katritzky and S. J. Perumal, J. Heterocycl. Chem., 1990, 27, 1737–1740 CrossRef CAS PubMed.
- K. Groll, T. D. Blake, A. Unsinn, D. Haas and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 11157–11161 CrossRef CAS PubMed.
- (a) C. Figliola, L. Male, S. L. Horswell and R. S. Grainger, Eur. J. Inorg. Chem., 2015, 3146–3156 CrossRef CAS PubMed; (b) C. Figliola, L. Male, P. N. Horton, M. B. Pitak, S. J. Coles, S. L. Horswell and R. S. Grainger, Organometallics, 2014, 33, 4449–4460 CrossRef CAS; (c) S. Allenmark, R. S. Grainger, S. Olsson and B. Patel, Eur. J. Org. Chem., 2011, 4089–4092 CrossRef CAS PubMed; (d) B. Patel, J. Carlisle, S. E. Bottle, G. R. Hanson, B. M. Kariuki, L. Male, J. C. McMurtrie, N. Spencer and R. S. Grainger, Org. Biomol. Chem., 2011, 9, 2336–2344 RSC; (e) R. S. Grainger, B. Patel, B. M. Kariuki, L. Male and N. Spencer, J. Am. Chem. Soc., 2011, 133, 5843–5852 CrossRef CAS PubMed; (f) R. S. Grainger, B. Patel and B. M. Kariuki, Angew. Chem., Int. Ed., 2009, 48, 4832–4835 CrossRef CAS PubMed; (g) R. S. Grainger, A. Procopio and J. W. Steed, Org. Lett., 2001, 3, 3565–3568 CrossRef CAS PubMed.
- Representative examples: (a) M. Garzón and P. W. Davies, Org. Lett., 2014, 16, 4850–4853 CrossRef PubMed; (b) H. V. Adcock, T. Langer and P. W. Davies, Chem. – Eur. J., 2014, 20, 7262–7266 CrossRef CAS PubMed; (c) M. Dos Santos and P. W. Davies, Chem. Commun., 2014, 50, 6001–6004 RSC; (d) E. Chatzopoulou and P. W. Davies, Chem. Commun., 2013, 49, 8617–8619 RSC; (e) P. W. Davies and S. J.-C. Albrecht, Synlett, 2012, 70–73 CrossRef CAS; (f) P. W. Davies, A. Cremonesi and L. Dumitrescu, Angew. Chem., Int. Ed., 2011, 50, 8931–8934 CrossRef CAS PubMed; (g) P. W. Davies and S. J.-C. Albrecht, Chem. Commun., 2008, 44, 238–240 RSC.
- Alkyne oxyarylation using S-oxides: (a) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160–4161 CrossRef CAS PubMed; (b) G. Li and L. Zhang, Angew. Chem., Int. Ed., 2007, 46, 5156–5159 CrossRef CAS PubMed; (c) A. B. Cuenca, S. Montserrat, K. M. Hossain, G. Mancha, A. Lledós, M. Medió-Simon, G. Ujaque and G. Asensio, Org. Lett., 2009, 11, 4906–4909 CrossRef CAS PubMed; (d) C. Li, K. Pati, G. Lin, S. Md, A. Sohel, H.-H. Hung and R.-S. Lui, Angew. Chem., Int. Ed., 2010, 49, 9891–9894 CrossRef CAS PubMed; (e) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512–8524 CrossRef CAS PubMed; (f) R. Fang and L. Yang, Organometallics, 2012, 31, 3043–3055 CrossRef CAS.
- For recent related transformations with a different mechanistic rationale: functionalisation of quinolones: (a) X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794–10798 CrossRef CAS PubMed; (b) U. Sharma, Y. Park and S. Chang, J. Org. Chem., 2014, 79, 9899–9906 CrossRef CAS PubMed. Formation of indolines: R. B. Dateer and S. Chang, J. Am. Chem. Soc., 2015, 137, 4908–4911 CrossRef CAS PubMed.
- For similar overall transformations by hydrofunctionalisation and then [3,3]-rearrangement see: (a) S. Ngwerume and J. E. Camp, Chem. Commun., 2011, 47, 1857–1849 RSC; (b) S. Ngwerume, W. Lewis and J. E. Camp, J. Org. Chem., 2013, 78, 920–934 CrossRef CAS PubMed. For a stepwise variation using N-hydroxy heterocycles see: (c) M. Kumar, M. Scobie, M. S. Mashuta, G. B. Hammond and B. Xu, Org. Lett., 2013, 15, 724–727 CrossRef CAS PubMed; (d) Y. Wang, L. Liu and L. Zhang, Chem. Sci., 2013, 4, 739–746 RSC; (e) Y. Wang, L. Ye and L. Zhang, Chem. Commun., 2011, 47, 7815–7817 RSC; (f) M. Kumar, M. Scobie, M. S. Mashuta, G. B. Hammond and B. Xu, Org. Lett., 2013, 15, 724–727 CrossRef CAS PubMed.
- In the course of preparing our work for publication, a report appeared which includes one example of an acid catalysed reaction of dibenzothiophene S-oxide with an ynamide to provide a C-4 substituted dibenzothiophene: B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718–8721 CrossRef CAS PubMed.
- (a) General reviews: A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3439 CrossRef PubMed; (b) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed; (c) J. Xiao and X. Li, Angew. Chem., Int. Ed., 2011, 50, 7226–7236 CrossRef CAS PubMed; (d) M. Rudolph, A. Stephen and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC; (e) L.-P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129–3139 RSC; (f) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864–876 CrossRef CAS PubMed; (g) D. Qian and J. Zhang, Chem. Rec., 2014, 14, 280–302 CrossRef CAS PubMed; (h) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245–5256 RSC; (i) R. Dorel and A. M. Echavarren, Chem. Rev., 2015 DOI:10.1021/cr500691k.
- (a) P. W. Davies and S. J. C. Albrecht, Angew. Chem., Int. Ed., 2009, 48, 8372–8375 CrossRef CAS PubMed; (b) P. W. Davies, Pure Appl. Chem., 2010, 82, 1537–1544 CrossRef CAS.
- For a platinum-catalysed oxyarylation with nitrones, see: S. Bhunia, C.-J. Chang and R.-S. Liu, Org. Lett., 2012, 14, 5522–5525 CrossRef CAS PubMed.
- C.-F. Xu, M. Xu, Y.-X. Jia and C. Li, Org. Lett., 2011, 13, 1556–1559 CrossRef CAS PubMed.
- For gold-catalysed reactions where diphenylsulfoxide acts as an oxidant following cyclisation of a gold-alkyne complex see: (a) C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS PubMed; (b) H.-S. Yeom and S. Shin, Org. Biomol. Chem., 2013, 11, 1089–1092 RSC.
- For a rationalisation of decreasing temperature to slow down elimination of the nucleofuge in related nitrenoid chemistry see: B. Lu, Y. Luo, L. Liu, L. Ye, Y. Wang and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 8358–8362 CrossRef CAS PubMed.
- The role of the phosphite ligand to allow reaction at a vinyl gold carbenoid centre by disfavouring elimination of a nucleofuge to form the gold carbene centre was employed to rationalise chemoselectivity for intermolecular trapping pathways in gold-catalysed ynamide oxidation reaction. See ref. 16c.
- C. H. M. Amijis, V. López-Carillo, M. Raducan, P. Pérez-Galán, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef PubMed.
- The use of hex-1-yn-1-ylbenzene gave the α–β unsaturated ketone in a 58% yield (based on the dibenzothiophene-S-oxide).
- L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550–8551 CrossRef CAS PubMed.
- P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379–381 RSC.
- See ref. 24 for gold-catalysed double oxidation of triple bonds using diphenylsulfoxide (3 equiv.) with AuCl/AgSbF6 (4 mol%), under reflux in 1,2-DCE.
- For recent representative examples and overviews of gold catalysed reactions of alkynes with other nucleophilic oxidants see: (a) L. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed; (b) S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4229 CrossRef PubMed; (c) S. Shi, T. Wang, W. Yang, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2013, 19, 6576–6580 CrossRef CAS PubMed; (d) J. Fu, H. Shang, Z. Wang, L. Chang, W. Shao, Z. Yang and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 4198 CrossRef CAS PubMed; (e) F. Pan, S. Liu, C. Shu, R.-K. Lin, Y.-F. Yu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2014, 50, 10726–10729 RSC; (f) G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277–6282 CrossRef CAS PubMed.
- Oxidative cyclopropanation of enynes with pyridine-N-oxides: (a) D. Qian, H. Hu, F. Liu, B. Tang, W. Ye, Y. Wang and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 13751–13755 CrossRef CAS PubMed; (b) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 50, 6911–6914 CrossRef CAS PubMed; (c) K.-B. Wang, R.-Q. Ran, S.-D. Xiu and C.-Y. Li, Org. Lett., 2013, 15, 2374–2377 CrossRef CAS PubMed; (d) K. Ji and L. Zhang, Org. Chem. Front., 2014, 1, 34–38 RSC; (e) D. Qian and J. Zhang, Chem. Commun., 2011, 47, 11152–11154 RSC.
- The use of urea hydrogen peroxide afforded the sulfone preferentially and sodium metaperiodate gave no reaction.
- P. W. Davies and C. Detty-Mambo, Org. Biomol. Chem., 2010, 8, 2918–2922 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures, additional example of iterative process and NMR spectra for new compounds. CCDC 1405198. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01241d |
‡ Crystal structure determination of 17a. Crystal data for C22H20O4S, M = 380.44, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.0122(4) Å, b = 10.2941(6) Å, c = 10.6266(6) Å, α = 75.079(5)°, β = 73.655(4)°, γ = 75.040(5)°, V = 895.60(9) Å3, Z = 2, T = 100.00(10) K, μ(CuKα) = 1.826 mm−1, Dcalc = 1.411 g cm−3, 4979 reflections measured (8.85° ≤ 2Θ ≤ 136.478°), 3193 unique (Rint = 0.0141, Rsigma = 0.0201) which were used in all calculations. The final R1 was 0.0290 (I > 2σ(I)) and wR2 was 0.0733 (all data). CCDC 1405198. (no. 2), a = 9.0122(4) Å, b = 10.2941(6) Å, c = 10.6266(6) Å, α = 75.079(5)°, β = 73.655(4)°, γ = 75.040(5)°, V = 895.60(9) Å3, Z = 2, T = 100.00(10) K, μ(CuKα) = 1.826 mm−1, Dcalc = 1.411 g cm−3, 4979 reflections measured (8.85° ≤ 2Θ ≤ 136.478°), 3193 unique (Rint = 0.0141, Rsigma = 0.0201) which were used in all calculations. The final R1 was 0.0290 (I > 2σ(I)) and wR2 was 0.0733 (all data). CCDC 1405198. |
| This journal is © The Royal Society of Chemistry 2015 |