
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient synthesis and application of versatile nucleic acid lipid membrane anchors in the assembly and fusion of liposomes†
Oliver
Ries
,
Philipp M. G.
Löffler
and
Stefan
Vogel
*
University of Southern Denmark, Department of Physics, Chemistry and Pharmacy, Campusvej 55, 5230 Odense M, Denmark. E-mail: snv@sdu.dk
First published on 31st July 2015
Abstract
Hydrophobic moieties like lipid membrane anchors are highly demanded modifications for nucleic acid oligomers. Membrane-anchor modified oligonucleotides are applicable in biomedicine leading to new delivery strategies as well as in biophysical investigations towards the assembly and fusion of liposomes or the construction of DNA origami structures. We present herein the synthesis and applications of versatile lipid membrane anchor building blocks suitable for solid-supported oligonucleotide synthesis. These are readily synthesized in bulk in five to seven steps from commercially available precursors and can be incorporated at any position within an oligonucleotide without significantly altering the duplex stability and structure as was proven by thermal denaturation experiments and circular dichroism. Furthermore, their applicability could be demonstrated by the assembly and fusion of liposomes mediated by lipid-modified oligonucleotides.
Introduction
Lipid–oligonucleotide conjugates have been used in biomedical applications to develop new gene therapy strategies using lipidated oligonucleotides as synthetic vectors for the transport of oligonucleotides into cells, for enhancing gene delivery and gene silencing, in the development of new drug delivery systems (e.g. utilizing aptamers), as well as for the detection of RNA in living cells.1–3 Biophysical and nanotechnological studies encompass the controlled aggregation and fusion of liposomes and their tethering to supported bilayers, the detection of single base mismatches of captured probes by end-point affinity measurements or mass spectrometry, and the construction of artificial devices and DNA origami structures.1–5The assembly of DNA-functionalized liposomes has been reported earlier by us6–8 and others.9–11 Hybridization assays based on the thermal denaturation of oligonucleotides and the experimental data gained are crucial for the development and improvement of diagnostic assays targeting, e.g., single nucleotide polymorphism, deletions or insertions.12–15
The fusion of lipid bilayers is a ubiquitous mechanism in nature for the uptake and release of biomolecules, e.g. in signal transduction.16 Furthermore, liposomes can act as nanocontainers for chemical reagents and catalysts, leading to versatile nanoreactors upon fusion.17 First attempts made by Boxer18,19 and Höök20–22 have shown earlier the feasibility of liposome fusion mediated by lipid–oligonucleotide conjugates by mimicking the geometry of the natural SNARE complex.23
Results and discussion
Lipidated nucleic acid oligomers are often functionalized either on the 5′-end or the 3′-end.1 Here we introduce lipid membrane anchors derived from enantiomerically pure (R)-3-amino-1,2-propanediol (2) as building blocks in lipid–DNA conjugates. These are easily accessible in a small number of chemical steps from commercially available compounds and can be incorporated both terminally and internally into a nucleic acid oligomer in good coupling yield (70–90% based on the absorption of cleaved DMTr during synthesis).Membrane anchor synthesis
Building blocks 1a–c suitable for solid-supported DNA synthesis using standard phosphoramidite chemistry24 have been prepared from aminodiol 2 in five steps and overall yields of 16–33% (Scheme 1). Aminodiol 2 was chemoselectively acylated with palmitic acid, cholesterylic acid (S1) and phytanic acid (S3), respectively, utilizing acid chloride chemistry leading to amides 3a–c in good yield. The presence of magnesium oxide was crucial to prevent side reactions of the free hydroxyl groups.25 For the synthesis of cholesterylic acid26–28 (S1) and phytanic acid (S3) see ESI.† Direct coupling of phytenic acid (S2), an intermediate in the synthesis of S3, followed by reduction led to an undesired mixture of the 1,2- and 1,4-reduction products due to the present Michael acceptor (data not shown). Reduction of the amides 3a–c with lithium aluminium hydride yielded the corresponding secondary amines 4a–c in excellent yield without the need for further purification. A second acylation step, carried out under the same conditions as those mentioned above, gave di-lipidated amidodiols 5a–c in moderate to good yield. DMTr-protection of the primary alcohol under standard conditions and good yield (6a–c), followed by phosphoramidite formation resulted in lipid anchor building blocks 1a–c ready for solid-supported automated DNA synthesis. | ||
| Scheme 1 (a) For 3a: C15H31COCl, MgO, H2O, THF, 0 °C, 4 h; for 3b,c: S1 (for 3b) or S3 (for 3c), SOCl2, toluene, reflux, 2 h, then MgO, H2O, THF, 0 °C, 4–5 h; (b) LiAlH4, THF, reflux, 4 h; (c) for 5a: C15H31COCl, MgO, H2O, THF, 0 °C, 7 h; for 5b,c: S1 (for 5b) or S3 (for 5c), SOCl2, toluene, reflux, 2 h, then MgO, H2O, THF, 0 °C, 5–6 h; (d) DMTrCl, DMAP, TEA, DCE, 80 °C, 5 h; (e) iPr2NPCl(OCH2CH2CN), DIPEA, DCE, 0 °C, 3–6 h; f) oligonucleotide synthesis. | ||
Influence on thermal DNA stability
Lipid membrane anchor building blocks X–Z have been introduced into DNA oligonucleotides at terminal and internal positions. When incorporated terminally, the modification was followed by a single deoxythymidine to suppress foam-formation of the detergent-like amphiphiles. No evidence could be found that single strands or duplexes were less soluble in aqueous solutions compared to their unlipidated congeners in the concentrations used for the experiments. However, it is recommended to store stock solutions of lipid–DNA conjugates in a ddH2O/acetonitrile mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as reported by others.18
:1 v/v) as reported by others.18
The results of thermal denaturation experiments (Tm) are shown in Table 1. It was reported that DNA-amphiphiles might show broad transitions and hysteresis in Tm-experiments.8,29–31 All systems presented here gave sharp and clear transitions in UV absorption at 260 nm. Hysteresis was only observed when oligonucleotides bearing two X modifications, on both the ends of the same strand, were present (Table 1, entries 10 and 11) and in the case of a single modification Y (entry 4). Incorporation of modifications X and Z as overhang in a 17mer DNA sequence led to a slight stabilization of the corresponding dsDNA by ΔTm = 2.0 °C (entries 2 and 5), whereas modification Y at the same position resulted in a decrease of the transition temperature by ΔTm = −2.6 °C and a hysteresis of approx. 3.9 °C (entry 4). The same DNA strand bearing modification X significantly destabilized a DNA/RNA duplex (ΔTm = −7.0 °C, entry 3). When both DNA strands were modified with the same membrane anchor as overhang on the same side in the duplex, a strong stabilization of the resulting dsDNA could be observed. Stabilization increased thereby from modification X (ΔTm = +19.4 °C, entry 6), over Z (ΔTm = +25.8 °C, entry 8) to Y (ΔTm = +28.8 °C, entry 7). This observation can be explained by the increasing non-polar surface of the different modifications (X < Z < Y), exposed to the aqueous medium upon denaturation of the duplex, and is in accordance with the trend of free energy release for transferring a lipophilic membrane anchor from the aqueous phase to the lipid membrane.1,32,33 As the membrane anchors in both strands can interact with each other in the duplex, due to the hydrophobic effect, these attractive forces have to be overcome by thermal energy in the denaturation experiment.34 This argument could be supported by thermal denaturation of dsDNA formed by two 5′-X-modified strands, leading to a duplex with membrane anchor modifications on different ends. In this case no interaction of the hydrocarbon chains could be observed (ΔTm = 1.9 °C, entry 9). In fact, the slight stabilization corresponds well to that found for the single-modified duplex (entry 2).
| Sequencea |
T
mb [°C] |
ΔTmc [°C] |
|
|---|---|---|---|
|
a
X, Y, Z: lipid anchor monomers, Φ: abasic site.
b Conditions: [HEPES] 10 mM, [Na+] 110 mM, [Cl−] 108 mM adjusted to pH 7.0, [DNA] 1 μM each, error ±0.5 °C.
c Change in Tm value calculated relative to the DNA:DNA reference duplex.
|
|||
| 1 | 5′-TGT GGA AGA AGT TGG TG | 57.7 | — |
| 3′-ACA CCT TCT TCA ACC AC | |||
| 2 | 5′-TX TGT GGA AGA AGT TGG TG | 59.7 | 2.0 |
| 3′-ACA CCT TCT TCA ACC AC | |||
| 3 | 5′-TX TGT GGA AGA AGT TGG TG | 50.7 | −7.0 |
| 3′-r(ACA CCU UCU UCA ACC AC) | |||
| 4 | 5′-TY TGT GGA AGA AGT TGG TG | 55.1 | −2.6 |
| 3′-ACA CCT TCT TCA ACC AC | |||
| 5 | 5′-TZ TGT GGA AGA AGT TGG TG | 59.7 | 2.0 |
| 3′-ACA CCT TCT TCA ACC AC | |||
| 6 | 5′-TX TGT GGA AGA AGT TGG TG | 77.1 | 19.4 |
| 3′-TX ACA CCT TCT TCA ACC AC | |||
| 7 | 5′-TY TGT GGA AGA AGT TGG TG | 86.5 | 28.8 |
| 3′-TY ACA CCT TCT TCA ACC AC | |||
| 8 | 5′-TZ TGT GGA AGA AGT TGG TG | 83.5 | 25.8 |
| 3′-TZ ACA CCT TCT TCA ACC AC | |||
| 9 | 5′-TX TGT GGA AGA AGT TGG TG | 59.6 | 1.9 |
| 3′-ACA CCT TCT TCA ACC AC XT | |||
| 10 | 5′-TX TGT GGA AGA AGT TGG TG XT | 48.9 | −8.8 |
| 3′-ACA CCT TCT TCA ACC AC | |||
| 11 | 5′-TX TGT GGA AGA AGT TGG TG XT | 85.4 | 27.7 |
| 3′-TX ACA CCT TCT TCA ACC AC XT | |||
| 12 | 5′-TGA AGA AGG TGT TTG GGA AGA AGT | 63.1 | — |
| 3′-ACT TCT TCC ACA AAC CCT TCT TCA | |||
| 13 | 5′-TGA AGA AGG TGT XTG GGA AGA AGT | 61.6 | −1.5 |
| 3′-ACT TCT TCC ACA AAC CCT TCT TCA | |||
| 14 | 5′-TGA AGA AGG TGT XTG GGA AGA AGT | 61.9 | −1.2 |
| 3′-ACT TCT TCC ACA ΦAC CCT TCT TCA | |||
| 15 | 5′-TGA AGA AGG TGT XTG GGA AGA AGT | 72.5 | 9.4 |
| 3′-ACT TCT TCC ACA XAC CCT TCT TCA | |||
When modification X was introduced as overhang on both sides of a 17mer oligonucleotide, the corresponding dsDNA was significantly destabilized (ΔTm = −8.8 °C, entry 10) as seen earlier in similar systems.34 Furthermore, a strong hysteresis of 6.9 °C could be observed. This might be explained by the interaction of the hydrocarbon chains forming a hairpin-like structure. Duplex formation has to overcome these hydrophobic interactions. This is in contrast to nucleoside overhangs, which are known to stabilize dsDNA.35,36 When both single strands of a duplex bear modification X on both the ends, the stabilizing effect could be recovered (ΔTm = +27.7 °C, entry 11) even though the hysteresis increased to 11.0 °C further indicating intramolecular interactions of the hydrocarbon chains.
Incorporation of modification X into the middle of a DNA 24mer resulted in a slight decrease in thermal stability opposite deoxyadenosine (ΔTm = −1.5 °C, entry 13) as well as an abasic site (ΔTm = −1.2 °C, entry 14). In contrast, two X-units across from each other could stabilize dsDNA (ΔTm = +9.4 °C, entry 15), even though the effect is not as high as that for terminally modified duplexes.
Circular dichroism
Selected duplexes have been studied by circular dichroism (CD). Fig. 1A shows the CD spectra of selected, terminally modified duplexes as well as the unmodified dsDNA as the reference. CD spectra of duplexes terminally modified with lipid membrane anchors Y (olive, dotted) and Z (red, solid) on both DNA strands (Table 1, entries 7 and 8) are in good agreement with the reference spectrum (Table 1, entry 1) with a minimum at 245 nm and a broad maximum around 275 nm indicating a B-type duplex. The spectrum for the X-modified system (orange, dashed, Table 1, entry 6) resembles that of the aforementioned with a broad maximum at 275 nm, but shows a significantly reduced minimum at 245 nm, indicating a distortion of the duplex due to the interactions of the lipid moieties. The duplex consisting of one terminally X-modified DNA strand and the corresponding unmodified RNA counter strand (blue, Table 1, entry 3) shows an intense maximum at 266 nm and a weak minimum at 238 nm. This observation agrees with a structure closer to the RNA-A-type conformation than to the DNA-B-type, which is expected for a DNA/RNA hybrid duplex.37Fig. 1B shows the CD-spectra of 24mers bearing modification X in the middle of the strand across an abasic site (orange, dashed, Table 1, entry 14) or another X-unit (blue, dotted, Table 1, entry 15) as well as the unmodified DNA/DNA reference (black, solid, Table 1, entry 12). All spectra show an intense maximum around 277 nm and a weak minimum at 245 nm. Incorporation of modification X seems not to affect the B-type structure when incorporated in the middle of the strand even when two membrane anchor units are placed across from each other. | ||
| Fig. 1 CD spectra of unmodified DNA and examples of terminally (A) and internally (B) modified DNA duplexes; conditions: [DNA] 1 μM each, [HEPES] 10 mM, [Na+] 110 mM, [Cl−] 108 mM, adjusted to pH 7.0. | ||
Assembly of liposomes
In order to prove the usability of the lipid membrane anchors described here, assembly of liposomes has been performed applying the X-modified system from Table 1, entry 9. In this case, the membrane anchors are situated on different ends of the duplex leading to a DNA-tether between the liposomes after incorporation into the lipid bilayer and hybridisation. DNA-functionalized liposomes were prepared by the postinsertion technique.7,30,38,39 Successful incorporation of DNA–lipid conjugates into liposomes could be confirmed by a change of zeta-potential upon the addition of different amounts of the conjugates (see ESI† for further details). The data showed no indication of a saturation of the lipophilic reservoir of the liposomes towards the self-inserting amphiphilic oligonucleotides within the range of concentrations used in this study. Assembly and disassembly can be monitored by changes in optical density seen as apparent absorption due to increased light scattering by larger assemblies of liposomes. Fig. 2 shows the normalized apparent absorption at 260 nm for three heating–cooling cycles. Characteristic of this type of experiment is a strong and sharp decrease in apparent absorption upon heating (red curve) at the DNA thermal denaturation temperature. For DNA strands, lipid-modified on both the ends and hybridized with an unmodified DNA counter strand, differences in Tm values of 4–5 °C in the presence and absence of liposomes have been observed.7 Here, the difference in Tm values was only 0.5 °C, 60.1 °C in the presence and 59.6 °C in the absence of liposomes, indicating that attachment to liposomes did not alter the hybridisation of the DNA duplex.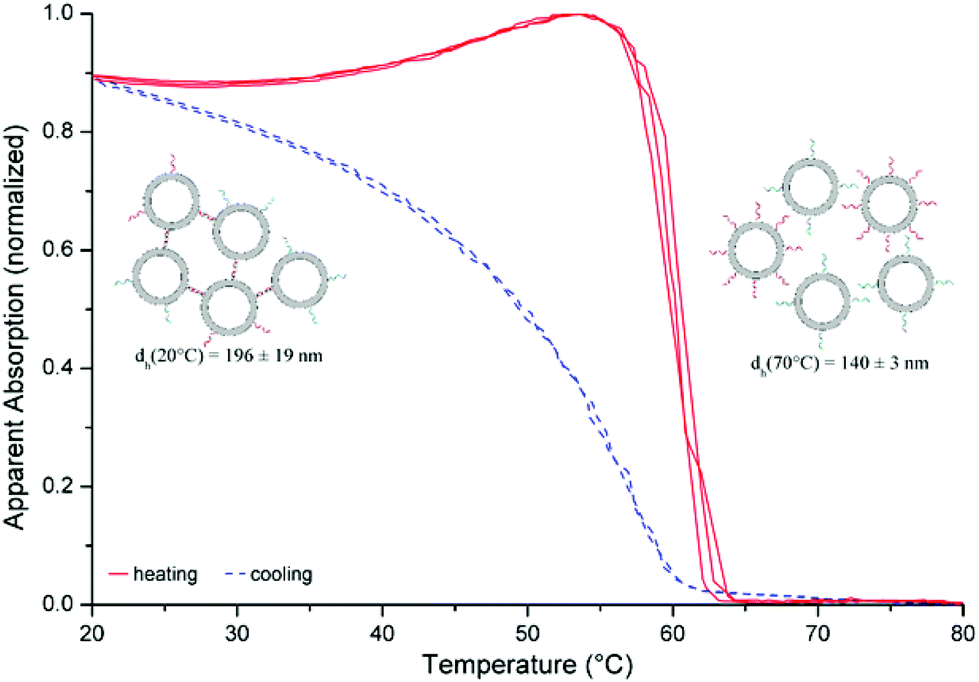 | ||
| Fig. 2 Assembly of liposomes: liposome–DNA conjugates (100 nM): [POPC] 50 μM, [DNA] 100 nM (strands: Table 1, entry 9), buffer: [HEPES] 10 mM, [Na+] 110 mM, [Cl−] 108 mM, adjusted to pH 7.0; dh: hydrodynamic diameter as determined by DLS (liposomes and DNA strands are not drawn to scale). | ||
Fusion of liposomes
DNA–lipid conjugates with the same setup have been used to mediate fusion of liposomes.18,19,21,22 We tested our system bearing two Z-modifications on the same side in the duplex (“DNA zipper”, Table 1, entry 8) for its fusogenicity with liposomes consisting of DOPC/DOPE/Chol (2:1:1, molar ratio) applying a frequently used FRET assay.40 In brief, one population of liposomes is membrane-labelled with NBD- and Rhodamine-functionalized lipids presenting a FRET pair, whereas a second population remains unlabelled. Upon fusion and mixing of lipids from different liposome populations, the FRET pair gets diluted within the lipid bilayer. Recovery of the NBD-fluorescence can be detected leading to a value for total lipid mixing, the sum of the mixing of the inner and outer lipid layers. For detection of the inner leaflet only, i.e. the detection of full fusion only, NBD-fluorophores on the outside can be selectively destroyed, yielding asymmetrically labelled liposomes.41 A 100% value can be obtained by lysis of the liposomes at the end of the experiment. Fig. 3 shows the normalized fluorescence time scan of NBD emission for a total lipid mixing ( ) and a fusion experiment (
) and a fusion experiment ( ) performed with a lipid–DNA conjugate, fitted by a double-exponential curve, and a corresponding control experiment without DNA (total lipid mixing: △, fusion:
) performed with a lipid–DNA conjugate, fitted by a double-exponential curve, and a corresponding control experiment without DNA (total lipid mixing: △, fusion:  ).22 It can be seen that most of the total lipid mixing, mainly mixing of the outer leaflet, proceeds within the first 5 min of the experiment, but still increases thereafter, while the actual fusion process reaches a maximum after approx. 15 min. From the fitting curves a value of 2.3% for total lipid mixing and 0.9% for fusion can be estimated. The corresponding control experiments showed only traces of total lipid mixing and no fusion. Following the fusion process by DLS led to an increase in the hydrodynamic diameter from 117 nm to 125 nm within the first minute. Thereafter the value stayed constant over time (for the graph see ESI†). Even though the system needs further improvement, these first results show that our new membrane anchor incorporated into DNA strands (Table 1, entry 8) is able to mediate fusion between liposomes.
).22 It can be seen that most of the total lipid mixing, mainly mixing of the outer leaflet, proceeds within the first 5 min of the experiment, but still increases thereafter, while the actual fusion process reaches a maximum after approx. 15 min. From the fitting curves a value of 2.3% for total lipid mixing and 0.9% for fusion can be estimated. The corresponding control experiments showed only traces of total lipid mixing and no fusion. Following the fusion process by DLS led to an increase in the hydrodynamic diameter from 117 nm to 125 nm within the first minute. Thereafter the value stayed constant over time (for the graph see ESI†). Even though the system needs further improvement, these first results show that our new membrane anchor incorporated into DNA strands (Table 1, entry 8) is able to mediate fusion between liposomes.
 | ||
Fig. 3 Amount of Total Lipid Mixing (TLM) and Fusion (F) mediated by lipid–DNA conjugates (LiDNA, TLM:  , F: , F:  ) and a control without LiDNA (TLM: △, F: ) and a control without LiDNA (TLM: △, F:  ) monitored by fluorescence spectroscopy (solid lines: double-exponential fit).22 DNA strands used: Table 1, entry 8. ) monitored by fluorescence spectroscopy (solid lines: double-exponential fit).22 DNA strands used: Table 1, entry 8. | ||
Conclusions
A short and facile synthesis of lipid membrane anchors bearing different lipophilic units suitable for solid-supported DNA synthesis has been achieved. The synthesis is not limited to the lipid moieties shown here, but can be easily extended to other lipophilic units and building blocks bearing two different anchor units. The lipid membrane anchors could be introduced at terminal and internal positions within DNA strands. Thermal denaturation experiments showed that a single lipid membrane anchor in a duplex leads to a weak stabilization or destabilisation (ΔTm = ±2 °C), whereas two modifications across from each other in different strands can stabilize a duplex by up to ΔTm = 29 °C. The duplex structure of modified dsDNA is not significantly disturbed as shown by CD spectroscopy. First applications towards the assembly and fusion of liposomes have demonstrated the versatility of the lipid membrane anchors described. Depending on the site of attachment of the lipid modification, a DNA strand can either be used for the assembly or for the fusion of liposomes.Experimental procedures
Materials and methods
400 MHz-1H, 101 MHz-13C, and 126 MHz-31P NMR spectra were recorded on a Bruker Avance III spectrometer. All 13C and 31P spectra are 1H-decoupled. All spectra were recorded at 25 °C and were referenced internally to solvent reference frequencies wherever possible. Chemical shifts (δ) are quoted in ppm, and coupling constants (J) are reported in Hz. Indexes a and b indicate diastereotope protons. Assignment of signals was carried out using 1H,1H-COSY, HSQC and HMBC spectra obtained on the spectrometer mentioned above. ESI mass spectrometry was performed on a Bruker micrOTOF-Q II system. MALDI mass spectrometry was performed on a Bruker Daltonics microflex LT or a Bruker Daltonics autoflex III smartbeam spectrometer. UV spectroscopy was carried out on a Varian Cary 100 or Cary 300 spectrometer and CD spectroscopy on a Jasco J-815 CD spectrometer. Fluorescence spectroscopy was performed on a Varian Cary Eclipse Fluorescence Spectrophotometer. Hydrodynamic diameters were determined on a Beckman Coulter DelsaMax Pro or a Malvern Nanosight LM14 instrument.:1) and purified on an HPLC (Dionex Ultimate 3000, Dionex Acclaim 120 C18 5 μm 120 Å 4.6 × 150 mm, 0.05 M TEAA–ACN/water (3:1) 10:90 → 100:0, 1 mL min−1) or UPLC system (Thermo Scientific Ultimate 3000, Merck Millipore Chromolith Performance RP18-e 4.6 × 100 mm, 0.05 M TEAA–ACN/water (3:1) 10:90 → 100:0, 1 mL min−1). Fractions were concentrated to dryness, dissolved in acetonitrile/water (1:1, v/v) and analysed by MALDI-TOF-MS.
Unlabelled DOPC/DOPE/Chol liposomes (2:1:1, molar ratio) or labelled liposomes (containing 1.5 mol% NBD–DPPE, 1.5 mol% Rhodamine Lissamine–DPPE) were obtained by mixing lipid stock solutions in CHCl3 and methanol, evaporation of the organic solvent and drying under vacuum. The lipid film was rehydrated in HEPES buffer, vortexed and extruded through a 100 nm polycarbonate filter (11×, Avanti Polar Lipids Handextruder). The mean hydrodynamic diameter was determined to be 131 nm by nanoparticle tracking analysis (NanoSight®) and liposomes were used on the same day.
For asymmetrically labelled liposomes, liposomes containing NBD/Rhodamine were treated with an aliquot of sodium dithionite solution ([sodium dithionite] 55 mM, [HEPES] 10 mM) for 4 min and purified by size exclusion chromatography using a prepacked NAP-10 column (elution buffer: [HEPES] 10 mM, [Na+] 110 mM, [Cl−] 108 mM, adjusted to pH 7.0). The liposome concentration was determined by measuring the Rhodamine fluorescence against a standard curve obtained from unreduced liposomes. More reliable results were obtained when the samples were treated with Triton X-100 prior to measurements.

The experimental data points were fitted with a double-exponential fit to calculate the average amount of total lipid mixing/fusion using the following formula:22
| y(t) = A0 + A1(1 − e−k1t) + A2(1 − e−k2t) |
Synthetic procedures
2-Cyanoethyl (1-(R)-(N-hexadecyl-N-palmitoyl-1-(dimethoxytriphenylmethyloxy)-3-amino-2-yl)diisopropylphosphoramidite (1a). 1.48 g (1.73 mmol) 6a, 1.20 mL (7.06 mmol) DIPEA in 15 mL DCE and 0.45 mL (2.02 mmol) CDP were used. Reaction time: 3 h. Column chromatography: petroleum ether/ethyl acetate 4
:1, 0.1% TEA. Yield: 1.46 g (1.38 mmol, 80%) as a clear oil. Rf 0.62 (petroleum ether/ethyl acetate 2:1); 1H NMR (400 MHz, CDCl3): δ 0.88 (t, 12H, J = 6.6, 4× palm-16-H3), 1.04 (d, 3H, J = 6.7, NiPr-CH3), 1.11 (d, 3H, J = 6.8, NiPr-CH3), 1.13–1.37 (m, 106H, 2× palm-3-H2, 4× palm-4-H2-15-H2, 2× NiPr-CH3), 1.39–1.67 (m, 8H, 2× palm-3-H2, 2× palm-2-H2), 2.14–2.28 (m, 2H, 1× palm-2-H2), 2.30–2.46 (m, 4H, 2× CHaCN, 1× palm-2-H2), 2.55–2.72 (m, 2H, 2× CHbCN), 2.99–3.34 (m, 8H, 2× 3-H2, 2× palm-1-H2), 3.38–3.97 (m, 24H, 2× 1-H2, 4× NiPr-CH, 2× POCH2, 4× DMTr-OCH3), 4.00–4.13 (m, 1H, 1× 2-H), 4.21–4.37 (m, 1H, 1× 2-H), 6.70–6.91 (m, 4H, DMTr-CH), 7.13–7.38 (m, 7H, DMTr-CH), 7.40–7.53 (m, 2H, DMTr-CH); 13C NMR (101 MHz, CDCl3): δ 14.24 (4× palm-C16), 20.13–20.65 (2× CH2CN), 22.81, 24.50–24.92, 26.90–27.22, 29.40–29.98, 32.04 (1× palm-C3, 2× palm-C4-C15, 8× NiPr-CH3), 25.48–25.72, 27.49, 29.04 (2× palm-C3, 2× palm-C2), 33.20–33.40 (1× palm-C2), 33.69–33.83 (1× palm-C2), 43.04–43.48 (4× NiPr-CH), 48.49 (1× C1), 49.40 (2× palm-C1), 49.79–50.09 (1× C1), 55.27, 55.30, 55.33 (4× DMTr-OCH3), 58.15–58.62 (2× POCH2), 64.54–65.03 (2× C3), 70.84–71.12 (1× C2), 71.75–72.12 (1× C2), 85.95, 86.36 (DMTr-CAr3), 113.10, 113.20, 113.24 (DMTr-CH), 117.47–117.99 (2× CN), 126.69–126.83, 126.97, 127.14, 127.75–127.98, 128.12–128.45, 129.24, 130.03–130.32 (DMTr-CH), 135.83–136.29, 139.62, 144.74–145.05, 158.44–158.75 (DMTr-C), 173.20–173.52 (2× palm-CO); 31P NMR (162 MHz, CDCl3): δ 149.27 (d, J = 125.16), 149.47 (d, J = 51.04); HRMS (ESI): calcd for C65H106N3NaO6P m/z 1078.7711 [M + Na]+, found 1078.7763.
2-Cyanoethyl (1-(2R)-(N-(cholest-5-en-3-carbonyl)-N-(chol-est-5-en-3-methyl)-1-(dimethoxytriphenylmethyloxy)-3-amino-2-yl)diisopropylphosphoramidite (1b). 1.03 g (0.88 mmol) 6b, 0.60 mL (3.53 mmol) DIPEA in 10 mL DCE and 0.25 mL (1.12 mmol) CDP were used. Reaction time: 4 h. Column chromatography: petroleum ether/ethyl acetate 4
:1, 0.1% TEA. Yield: 781 mg (0.57 mmol, 65%) as a clear foam. Rf 0.52 (petroleum ether/ethyl acetate 3:1); 1H NMR (400 MHz, CDCl3): δ 0.68 (s, 3H, chol-18-H3), 0.68 (s, 3H, chol-18-H3), 0.86 (d, 12H, J = 6.6, 2× chol-26-H3, 2× chol-27-H3), 0.92 (d, 6H, J = 6.2, 2× chol-21-H3), 0.94–1.72 (56H, 2× chol-19-H3, 4× NiPr-CH3, chol-CH2, chol-CH), 1.76–2.09 (m, 12H, chol-CH2), 2.25–2.77 (m, 4H, CH2CN, chol-CH2, chol-CH), 2.90–3.30 (m, 4H, 3-H2, chol-CH2), 3.32–3.86 (m, 6H, 1-H2, POCH2, 2× NiPr-CH), 3.74–3.85 (m, 6H, 2× DMTr-OCH3), 3.88–4.06 (m, 0.5H, 2-H), 4.19–4.36 (m, 0.5H, 2-H), 5.19–5.36 (m, 2H, 2× chol-6-H), 6.78–6.88 (m, 4H, DMTr-CH), 7.15–7.38 (m, 7H, DMTr-CH), 7.39–7.51 (DMTr-CH); 13C NMR (101 MHz, CDCl3): δ 11.98, 12.01 (chol-C18), 18.87 (chol-C21), 19.58, 19.61, 19.63 (chol-C19), 20.20–20.62 (CH2CN), 20.99, 21.06, 21.17 (chol-CH2), 22.96 (chol-C26, chol-C27), 23.97, 24.43 (chol-CH2), 24.65–24.88 (NiPr-CH3), 25.68, 26.81 (chol-CH2), 28.15 (chol-CH), 28.38, 29.84 (chol-CH2), 31.92, 31.95, 31.99 (chol-CH), 32.02, 32.06 (chol-CH2), 35.93 (chol-CH), 36.11, 36.34, 37.14, 37.21, 37.23, 37.26, 37.37, 37.40, 37.43 (chol-CH2), 38.06 (chol-CH), 38.90, 39.18, 39.33, 39.66, 39.94, 39.98 (chol-CH2), 42.03, 42.17 (chol-CH), 42.44 (chol-C), 43.20–43.47 (NiPr-CH), 48.95–49.78 (C1), 50.43, 50.49, 50.53, 50.63 (chol-CH), 54.64 (chol-CH2), 55.27, 55.29, 55.33 (DMTr-OCH3), 56.29, 56.33 (chol-CH), 56.94, 56.99 (chol-CH), 58.08–58.57 (POCH2), 60.51 (chol-C), 85.86, 86.48 (DMTr-CAr3), 113.12, 113.28 (DMTr-CH), 117.51–117.98 (CN), 119.99, 120.52, 120.65 (chol-C6), 127.99, 130.13, 130.14 (DMTr-CH), 135.97, 136.05, 142.00, 142.03, 142.10, 142.20, 142.53, 144.82, 145.02 (DMTr-C, chol-C), 158.50, 158.54, 158.66 (DMTr-C), 176.02, 176.05, 176.14 (chol-CO); 31P NMR (162 MHz, CDCl3): δ 149.30 (d, J = 118.2), 149.50 (d, J = 35.2); HRMS (ESI): calcd for C89H134N3NaO6P m/z 1394.9902 [M + Na]+, found 1394.9962.
2-Cyanoethyl (1-(2R)-(N-phytanyl-N-phytanoyl)-1-(dimethoxytriphenylmethyloxy)-3-amino-2-yl)diisopropylphosphoramidite (1c). 500 mg (0.52 mmol) 6c, 0.35 mL (2.06 mmol) DIPEA in 7 mL DCE and 0.15 mL (0.67 mmol) CDP were used. Reaction time: 6 h. Column chromatography: petroleum ether/ethyl acetate 6
:1, 0.1% TEA. Yield: 313 mg (0.27 mmol, 52%) as a clear oil. Rf 0.48 (petroleum ether/ethyl acetate 3:1); 1H NMR (400 MHz, CDCl3): δ 0.77–0.94 (m, 30H, phyt-3-CH3 phyt-7-CH3, phyt-11-CH3, phyt-16-H3), 0.98–1.60 (m, 55H, 1× phyt-2-H2, 1× phyt-3-H, 2× phyt-4-H2-6-H2, 2× phyt-7-H, 2× phyt-8-H2-10-H2, 2× phyt-11-H, 2× phyt-12-H2-14-H2, NiPr-CH3), 1.64–2.75 (m, 5H, 1× phyt-2-H2, 1× phyt-3-H, CH2CN), 1.52 (qqt, 2H, J = 6.6, 6.6, 6.6, 2× phyt-15-H), 3.02–3.98 (m, 16H, 1-H2, 3-H2, phyt-1-H2, POCH2, NiPr-CH, DMTr-OCH3), 4.00–4.14 (m, 0.5H, 2-H), 4.23–4.37 (m, 0.5H, 2-H), 6.73–6.91 (m, 4H, DMTr-CH), 7.13–7.38 (m, 7H, DMTr-CH), 7.40–7.52 (m, 2H, DMTr-CH); 13C NMR (101 MHz, CDCl3): δ 19.27–20.67 (phyt-3-CH3, phyt-7-CH3, phyt-11-CH3, CH2CN), 22.63, 22.73 (phyt-C16), 22.31–24.96, 30.18–31.16, 32.62–32.99, 37.12–37.76, 39.38 (phyt-C2, phyt-C3-C14, NiPr-CH3), 27.98 (phyt-C15), 40.23–41.18 (phyt-C2), 42.91–43.42 (NiPr-CH), 47.58–50.34 (C3, phyt-C1), 55.19 (DMTr-OCH3), 57.60–58.58 (POCH2), 64.14–64.86 (C1), 70.74–72.33 (C2), 85.70–86.39 (DMTr-CAr3), 113.00, 113.15, 126.65, 126.87, 127.70, 127.84, 128.07, 128.14, 128.27, 128.32, 129.89–130.29 (DMTr-CH), 117.29–117.96 (CN), 135.65–136.25, 144.58–145.00, 158.41, 158.57 (DMTr-C), 172.39–172.83 (phyt-CO); 31P NMR (162 MHz, CDCl3): δ 148.82–148.99 (m), 149.30–149.75 (m); HRMS (ESI): calcd for C73H122N3NaO6P m/z 1190.8963 [M + Na]+, found 1190.8945.
Acknowledgements
This work has been supported by BioNEC, a Centre of Excellence funded by the Villum Foundation for studies on biomolecular nanoscale engineering.Notes and references
- M. Schade, D. Berti, D. Huster, A. Herrmann and A. Arbuzova, Adv. Colloid Interface Sci., 2014, 208, 235 CrossRef CAS PubMed.
- A. Gissot, M. Camplo, M. W. Grinstaff and P. Barthélémy, Org. Biomol. Chem., 2008, 6, 1324 CAS.
- A. Patwa, A. Gissot, I. Bestel and P. Barthélémy, Chem. Soc. Rev., 2011, 40, 5844 RSC.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932 CrossRef CAS PubMed.
- K. Göpfrich, T. Zettl, A. E. C. Meijering, S. Hernández-Ainsa, S. Kocabey, T. Liedl and U. F. Keyser, Nano Lett., 2015, 15, 3134 CrossRef PubMed.
- U. Jakobsen and S. Vogel, in Methods in Enzymology, ed. N. Düzgüneş, Academic Press, Burlington, 2009, vol. 464, pp. 233–248 Search PubMed.
- U. Jakobsen, A. C. Simonsen and S. Vogel, J. Am. Chem. Soc., 2008, 130, 10462 CrossRef CAS PubMed.
- U. Jakobsen and S. Vogel, Bioconjugate Chem., 2013, 24, 1485 CrossRef CAS PubMed.
- C. A. Merkin, R. I. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef PubMed.
- S. R. Tabaei, P. Jönsson, M. Brändén and F. Höök, J. Struct. Biol., 2009, 168, 200 CrossRef CAS PubMed.
- T. Maruyama, H. Yamamura, M. Hiraki, Y. Kemori, H. Takata and M. Goto, Colloids Surf., B, 2008, 66, 119 CrossRef CAS PubMed.
- R. H. A. M. Vossen, E. Aten, A. Roos and J. T. den Dunnen, Hum. Mutat., 2009, 30, 860 CrossRef CAS PubMed.
- J. S. R. Farrar, H. Gudrun and C. T. Wittwer, in Molecular Diagnostics, ed. G. P. Patrinos and W. J. Ansorge, Academic Press, Burlington, 2009, pp. 229–245 Search PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed.
- J. C. Keer, in Essentials of Nucleic Acid Analysis: A Robust Approach, ed. J. T. Keer and L. Birch, RSC Publishing, Cambridge, 2008, ch. 7, pp. 132–166 Search PubMed.
- R. Jahn, T. Lang and T. C. Südhof, Cell, 2003, 112, 519 CrossRef CAS.
- P. A. Beales and T. K. Vanderlick, Adv. Colloid Interface Sci., 2014, 207, 290 CrossRef CAS PubMed.
- Y. M. Chan, B. van Lengerich and S. Boxer, Biointerphases, 2008, 3, FA17 CrossRef CAS PubMed.
- Y. M. Chan, B. van Lengerich and S. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979 CrossRef CAS PubMed.
- F. Höök, G. Stengel, A. B. Dahlin, A. Gunnarsson, M. P. Jonsson, P. Jönsson, E. Reimhult, L. Simonsson and S. Svedhem, Biointerphases, 2008, 3, FA108 Search PubMed.
- G. Stengel, R. Zahn and F. Höök, J. Am. Chem. Soc., 2007, 129, 9584 CrossRef CAS PubMed.
- G. Stengel, L. Simonsson, R. A. Campbell and F. Höök, J. Phys. Chem. B, 2008, 112, 8264 CrossRef CAS PubMed.
- H. R. Marsden, I. Tomatsu and A. Kros, Chem. Soc. Rev., 2011, 40, 1572 RSC.
- M. H. Caruthers, Acc. Chem. Res., 1991, 24, 278 CrossRef CAS.
- D. Kim, H. Rho, J. W. You and J. C. Lee, Tetrahedron Lett., 2002, 43, 277 CrossRef CAS.
- E. J. Corey and R. A. Sneen, J. Am. Chem. Soc., 1953, 75, 6234 CrossRef CAS.
- W. J. Gensler and G. M. Sherman, J. Org. Chem., 1958, 23, 1227 CrossRef CAS.
- S. M. Harwood, K. J. Toyne, J. W. Goodby, M. Parsley and G. W. Gray, Liq. Cryst., 2000, 27, 443 CrossRef CAS PubMed.
- R. G. Shea, J. C. Marsters and N. Bischofberger, Nucleic Acids Res., 1990, 18, 3777 CrossRef CAS PubMed.
- B. Waybrant, T. R. Pearce and E. Kokkoli, Langmuir, 2014, 30, 7465 CrossRef CAS PubMed.
- C. Gosse, A. Boutorine, I. Aujard, M. Chami, A. Kononov, E. Cogné-Laage, J. Allemand, J. Li and L. Jullien, J. Phys. Chem. B, 2004, 108, 6485 CrossRef CAS PubMed.
- C. T. Pool and T. E. Thompson, Biochemistry, 1998, 37, 10246 CrossRef CAS PubMed.
- J. R. Silvius, in Peptide-lipid interactions, ed. S. A. Simon and T. J. McIntosh, Elsevier, San Diego, 1st edn, 2002, pp. 371–395 Search PubMed.
- K. Rohr and S. Vogel, ChemBioChem, 2006, 7, 463 CrossRef CAS PubMed.
- H. Rosemeyer and F. Seela, J. Chem. Soc., Perkin Trans. 2, 2002, 746 RSC.
- F. Seela and R. Kröschel, Nucleic Acids Res., 2003, 31, 7150 CrossRef CAS PubMed.
- N. Sugimoto, M. Katoh, S. Nakano, T. Ohmichi and M. Sasaki, FEBS Lett., 1994, 354, 74 CrossRef CAS.
- T. Ishida, D. L. Iden and T. M. Allen, FEBS Lett., 1999, 460, 129 CrossRef CAS.
- J. N. Moreira, T. Ishida, R. Gaspar and T. M. Allen, Pharm. Res., 2002, 19, 265 CrossRef CAS.
- D. K. Struck, D. Hoekstra and R. E. Pagano, Biochemistry, 1981, 14, 4093 CrossRef.
- J. C. McIntyre and R. G. Sleight, Biochemistry, 1991, 30, 11819 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of compounds 3–6a–c and S1–S3, 1H-, 13C- and 31P-NMR-spectra of compounds 1–6 and S1–S3, characterization of oligonucleotides, DLS and zeta-potential experiments. See DOI: 10.1039/c5ob01207d |
| This journal is © The Royal Society of Chemistry 2015 |