
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ω-Transaminases for the amination of functionalised cyclic ketones
N.
Richter
ab,
R. C.
Simon
c,
H.
Lechner
c,
W.
Kroutil
bc,
J. M.
Ward
d and
H. C.
Hailes
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk; Tel: +44 (0)20 7679 7463
bACIB GmbH, c/o Department of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
cDepartment of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria
dThe Advanced Centre for Biochemical Engineering, Department of Biochemical Engineering, University College London, Bernard Katz Building, Gordon Street, London, WC1H 0AH, UK
First published on 14th July 2015
Abstract
The potential of a number of enantiocomplementary ω-transaminases (ω-TAms) in the amination of cyclic ketones has been investigated. After a preliminary screening of several compounds with increasing complexity, different approaches to shift the equilibrium of the reaction to the amine products were studied, and reaction conditions (temperature and pH) optimised. Interestingly, 2-propylamine as an amine donor was tolerated by all five selected ω-TAms, and therefore used in further experiments. Due to the higher conversions observed and interest in chiral amines studies then focused on the amination of α-tetralone and 2-methylcyclohexanone. Both ketones were aminated to give the corresponding amine with at least one of the employed enzymes. Moreover, the amination of 2-methylcyclohexanone was investigated in more detail due to the different stereoselectivities observed with TAms used. The highest yields and stereoselectivities were obtained using the ω-TAm from Chromobacterium violaceum (CV-TAm), producing 2-methylcyclohexylamine with complete stereoselectivity at the (1S)-amine position and up to 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity for the cis:trans [(1S,2R):(1S,2S)] isomer.
:1 selectivity for the cis:trans [(1S,2R):(1S,2S)] isomer.
Introduction
Single isomer chiral amines are one of the most common functional groups found in natural products and pharmaceutical compounds including, sitagliptin the antihyperglycemic drug for the treatment of diabetes,1 the antihypertensive dilevalol,2 and (S)-rivastigmine used for the treatment of Alzheimer's disease.3,4 Enantiopure amines are therefore highly desirable building blocks for the development of new pharmaceuticals. The frequent occurrence of such chiral amines has also highlighted interest in their preparation as synthons for use in the pharmaceutical sector. For instance, the aminotetraline motif is present in a variety of pharmaceuticals, such as the antidepressants sertraline and norsertraline with an α-aminoteraline core,5–7 and rotigotine a treatment for Parkinson's disease containing a β-aminotetraline core unit.8,9Traditionally single isomer chiral amines are generated from racemic mixtures using crystallisation methods, or they can be synthesised using chiral auxiliaries.7,10–12 In addition more recently a variety of organocatalytic, metal-dependent as well as chemo-enzymatic dynamic kinetic resolution methods have been developed to produce enantiopure amines.13–15 The requirement for metals in some of these systems such as lipase-catalysed dynamic kinetic resolutions is however a major backdraw when considering the sustainability of the process.14 An alternative method to generate enantiopure amines that is currently attracting significant interest is the use of ω-transaminases (ω-TAms).16–20 Despite the improved sustainability with this biocatalytic approach, one problem has been the issue of shifting the reaction equilibrium towards the amine product. However, in recent years efforts have been focussed on the development of methods to overcome this unfavourable equilibrium, via the chemical or enzymatic removal of the co-product or use of an excess of amine donor.1,21–30 The incorporation of enzymatic cascades has been particularly successful, including reuse of the co-product in a multi-enzymatic cascade with a carboligation step.31 Several of these studies used ω-TAms for the preparation of pharmaceutical intermediates or bioactive compounds,24,25,27–36 and have also lead to an industrial process.1
Here we describe the use of several ω-TAms in the asymmetric amination of several cyclic substrates. Moreover, different methods to shift the equilibrium towards the desired amine product were compared and reaction parameters optimised with the model compound cyclohexanone 1. The amination of two selected substrates was then investigated in further detail, to establish the different stereoselectivities of the ω-TAms used.
Results and discussion
Our aim was to test a panel of promising and recently reported ω-TAms against a range of functionalised cyclic ketones, including sterically challenging substrates, to determine substrate type acceptance by the selected ω-TAms. These were (S)-ω-TAms from Chromobacterium violaceum DSM30191 (CV-TAm),37Vibrio fluvialis (Vf-TAm),38Klebsiella pneumoniae KPN_0799 (Kp-TAm),39 and Pseudomonas putida PP_3718 (Pp-TAm).36 Also the (R)-ω-TAms from Mycobacterium vanbaalenii (Mv-TAm),40 and a variant from Arthrobacter sp. (ArRMut11).1 CV-TAm and Vf-TAm were selected as they have been used with a range of acyclic substrates and cyclic compounds such as cyclohexanone,19,20,31,37,38,41,42 while Pp-TAm has recently been reported to accept dopamine36 and Kp-TAm39 was selected as a promising TAm from screening our UCL TAm library. Mv-TAm has been used almost exclusively with ketones/amines as acyclic moietes,40,43,44 and ArRMut11 with 1,3-ketoamides to generate the (R)-functionality in sitagliptin as well as for example bicyclic tetralone and chromone substrates and a carbazolamine.1,28,34,45Ten ketone substrates 1–10 were selected, which would generate the corresponding amines 1a–10a, including cyclohexanone 1 and cyclopentanone 5 to determine the influence of ring size, as well as diketones (2,6), α,β-unsaturated ketones (4,7,9), α-tetralone 8 and ketones with α-methyl groups (3, and camphor 10). Initial assays used the ω-TAms (crude cell lysates) and either (R)- or (S)-α-methylbenzylamine (MBA) 11 as the amine donor, depending on the selectivity of the transaminase, with substrates 1–10: the product acetophenone was detected by HPLC at 254 nm (Fig. 1).37 This preliminary assay method highlighted substrates for further investigation. Control reactions were performed in the absence of amine acceptor and low levels of acetophenone were detected which were subtracted from assay results with amine acceptor present. The results indicated that several ketones showed good levels of conversion with the ω-TAms selected, particularly CV-TAm, Pp-TAm and ArRMut11. Cyclohexanone 1 was the best cyclic substrate for most ω-TAms with conversions of up to 40%. Interestingly, while the substitution at the α-position on the six-membered ring in 2-methylcyclohexanone 3 was particularly well tolerated with only slightly lower conversions, the presence of a conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond led to significantly less activity with 4. A similar reactivity pattern was observed with the five-membered rings: while cyclopentanone 5 was readily accepted by several ω-TAms, the corresponding enone 7 had negligible reactivity with all the ω-TAms used. This presumably reflects the modified steric demands and reactivity in the α,β-enones and less electrophilic carbonyl moiety. The diketones 2 and 6, and bicyclic compound 10 had negligible levels of acceptance. The bicyclic systems α-tetralone 8 (Kp-TAm, ArRMut11) and 8a-methyl-3,4,8,8a-tetrahydro-1,6(1H,7H)-naphthalenedione 9 were accepted with conversions at levels of 5–10% (ArRMut11), with even lower conversions for several of the other TAms. Tetralone 8 has previously been used as a substrate with ArRMut11 together with co-product removal to shift the equilibrium toward the desired amine, so these results using two TAms and no co-product removal were promising.28,45 The reaction of ArRMut11 with 9 has also recently been reported, and although products were observed by LC-MS analysis no products could be isolated.30 With the exception of the Vf-TAm which showed in general poor activity with cyclic substrates, the five other ω-TAms were studied in more detail, initially with ketone 1.
C double bond led to significantly less activity with 4. A similar reactivity pattern was observed with the five-membered rings: while cyclopentanone 5 was readily accepted by several ω-TAms, the corresponding enone 7 had negligible reactivity with all the ω-TAms used. This presumably reflects the modified steric demands and reactivity in the α,β-enones and less electrophilic carbonyl moiety. The diketones 2 and 6, and bicyclic compound 10 had negligible levels of acceptance. The bicyclic systems α-tetralone 8 (Kp-TAm, ArRMut11) and 8a-methyl-3,4,8,8a-tetrahydro-1,6(1H,7H)-naphthalenedione 9 were accepted with conversions at levels of 5–10% (ArRMut11), with even lower conversions for several of the other TAms. Tetralone 8 has previously been used as a substrate with ArRMut11 together with co-product removal to shift the equilibrium toward the desired amine, so these results using two TAms and no co-product removal were promising.28,45 The reaction of ArRMut11 with 9 has also recently been reported, and although products were observed by LC-MS analysis no products could be isolated.30 With the exception of the Vf-TAm which showed in general poor activity with cyclic substrates, the five other ω-TAms were studied in more detail, initially with ketone 1.
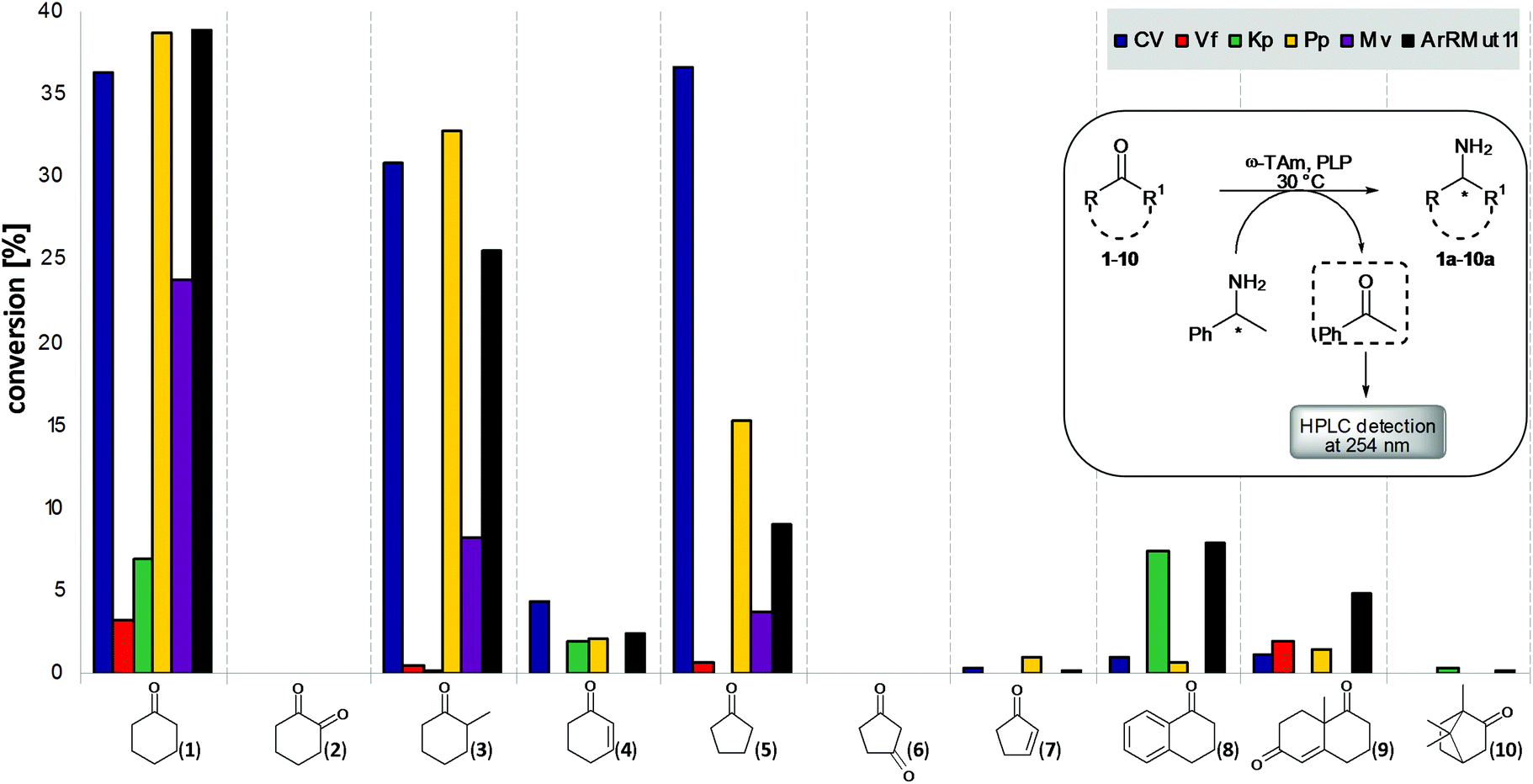 | ||
| Fig. 1 Initial screening results giving conversions for a range of cyclic ketone substrates employing the selected ω-TAms. (R)- or (S)-MBA 11 were used as amine donors and the product acetophenone was detected by HPLC analysis (see Scheme). All reactions were performed in triplicate and standard deviations were less than 10%. | ||
To facilitate a better comparability of the different TAm enzymes, the specific activity of crude cell extracts in the amination of 1 was investigated. In addition, freeze dried samples of clarified lysate were prepared as convenient preparations for storage and usage. The highest activity was obtained using crude cell extracts of CV-TAm and ArRMut11, with 0.58 U mg−1 and 0.39 U mg−1 of total protein, respectively (see Experimental). After lyophilisation the activity was decreased in most cases, though not significantly, with residual activities in the range of 55–100%. For this reason and ease of usage lyophilised cell extracts were used in all of the following studies.
To identify optimal reaction conditions, as well as investigating the reaction temperature and pH, a study comparing the use of MBA 11 to both enzyme-coupled and excess amine donor methods to shift the equilibrium towards the product amine was conducted using cyclohexanone 1 (Scheme 1). Two enzyme-coupled systems were used (Scheme 1a), where L- or D-alanine was used as the amine donor and the co-product pyruvate was removed by either a L-lactate dehydrogenase (LDH) (Scheme 1aA) or recycled by an alanine dehydrogenase (AlaDH) for (S)-selective ω-TAms (Scheme 1aB).18,28,46 The nicotinamide cofactor (NADH) required was recycled by employing standard techniques using formate dehydrogenase (FDH) or glucose dehydrogenase (GDH).18,28,46 An alternative enzyme-independent method was investigated using 2-propylamine (isopropylamine, IPA) 12 as the amine donor which generates acetone as a co-product (Scheme 1b).1,37,42
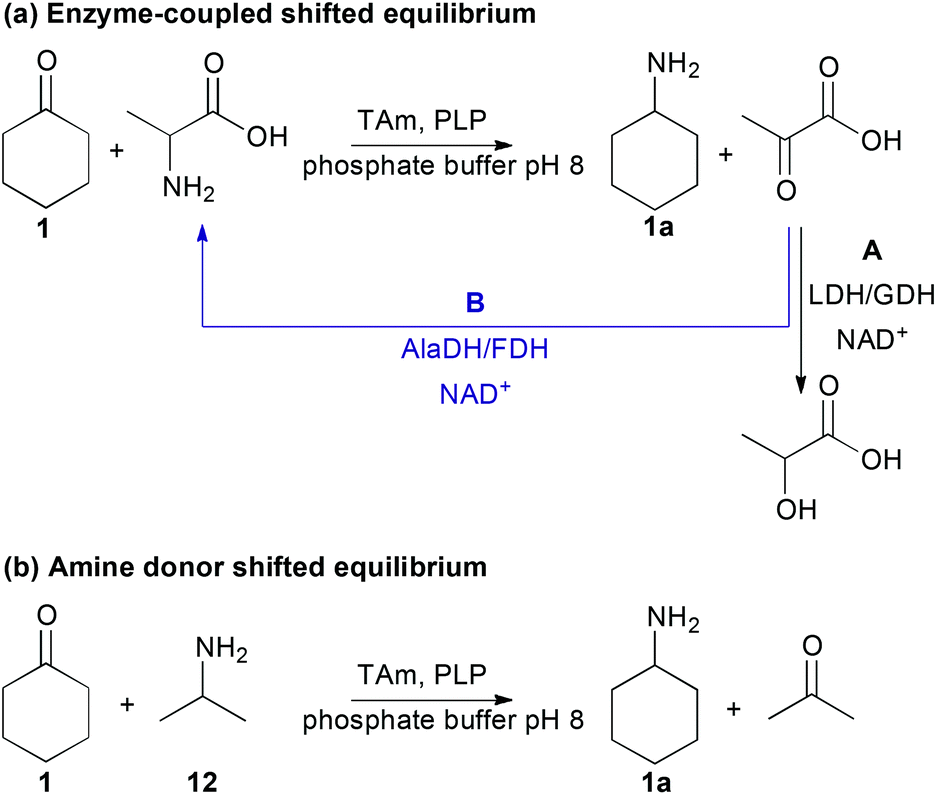 | ||
| Scheme 1 Methods used to shift the TAm reaction equilibrium toward the desired amine product, using 1 as the ketone acceptor. (a) Enzyme-coupled shifted equilibrium; (b) amine donor shifted equilibrium. | ||
Most enzymatic reactions proceeded with good conversions of up to 95% (Scheme 1, Table 1), especially compared to the conversions observed in the initial screening experiments using MBA 11 (7–39%), confirming the benefits of using shifting systems in ω-TAm reactions. Interestingly, four of the ω-TAm reactions showed similar or higher conversions using the IPA (12)-amine donor reaction systems compared to the use of enzyme coupled-systems. Only the Mv-TAm showed a slightly lower conversion (39%) compared to the AlaDH/FDH-system (50%). In general, this broad acceptance of IPA 12 as amine donor was unexpected since IPA does not appear to be an amine donor for many ω-TAms.47 To date, only a few have demonstrated high tolerance towards IPA 12 such as the engineered variant ArRMut11, and also CV-TAm which was used in the synthesis of (2S,3S)-2-aminopentane-1,3-diol, facilitating the use of this low cost amine donor to shift the equilibrium towards the product.1,42 The observation that all five of the ω-TAms investigated can be used with IPA 12 is notable, especially with respect to the applicability of these enzymes, due its low cost and more facile optimisation of reaction condition since only one enzyme is required. Moreover, the highly volatile co-product acetone can be readily removed, as recently demonstrated in the synthesis of sitagliptin.1
| TAm | MBA 11 amine donor (Fig. 1) conv. (%) | A LDH/GDH (Scheme 1a) conv. (%) | B AlaDH/FDH (Scheme 1a) conv. (%) | IPA 12 amine donor (Scheme 1b) conv. (%) |
|---|---|---|---|---|
| a 48 h reaction time. Reactions were performed in triplicate with standard deviations of less than 10%. Product 1a was detected by GC analysis for methods A, B and IPA 12, and acetophenone was detected by HPLC as previously using MBA 11 as the amine donor (MBA 11 was not used in a large excess as high numbers of equivalents have been found to have a detrimental effect on the transaminase reaction).42 | ||||
| CV-TAm | 36 | 92 | 88 | 94 |
| Kp-TAm | 7 | 0 | 0 | 22a |
| Pp-TAm | 39 | 91 | 88 | 95 |
| Mv-TAm | 24 | 31a | 50a | 39a |
| ArRMut11 | 39 | 0 | 0 | 93 |
Another interesting observation was that for the Kp-TAm and ArRMut11 no conversions were observed employing the enzyme-coupled systems, indicating that alanine was not accepted as an amine donor. While in engineering the Arthrobacter sp. ω-TAm to accept high IPA (12) and co-solvent concentrations the ability to use alanine has been lost, for the native Kp-TAm enzyme not to accept alanine as an amine donor is unexpected. However, since Kp is a member of the Class III transaminases and there is variable use of α-amino acids amongst the Class III transaminases, this finding is not too unusual.16
All further experiments were conducted using the IPA 12 shifting system because of the advantages outlined above. The amination of 1 was then performed at two different pHs and temperatures typically used in transamination reactions (Table 2). Optimal conditions which were used for further experiments were pH 8 and 30 °C for CV-TAm and Pp-TAm, pH 8 and 45 °C for Kp-TAm and Mv-TAm, while the best conditions for ArRMut11 were at pH 10 and 30 °C.
| TAm | pH 8, 30 °C conv. (%) | pH 8, 45 °C conv. (%) | pH 10, 30 °C conv. (%) | pH 10, 45 °C conv. (%) |
|---|---|---|---|---|
| n.d. – not determined. Conversions were determined in triplicate with errors below 10%. Product 1a detected by GC analysis. | ||||
| CV-TAm | 94 | 91 | 0 | n.d. |
| Kp-TAm | 5 | 18 | 8 | 14 |
| Pp-TAm | 94 | 10 | 0 | n.d. |
| Mv-TAm | 17 | 41 | 7 | 27 |
| ArRMut11 | 93 | n.d. | 94 | 90 |
The amination of 1, 3 and 8, to give 1a, 3a and 8a was then studied in more detail as initial experiments indicated reasonable levels of conversion (Table 3): compound 8a is an important chiral product and the potential to establish two stereogenic centres in 3a in a single step is particularly interesting. Ketones 1 and 3 were well accepted by the TAm enzymes, however ketone 8 was only accepted by ArRMut11 to give exclusively the α-aminotetraline (R)-8a, as determined by chiral GC analysis. This was consistent with previous reports using this ω-TAm with substrate 8.28,45 Only traces of 8a were observed with Kp-TAm and Mv-TAm (Table 3).
| TAm |
|
|
|
|||
|---|---|---|---|---|---|---|
| Conv. (%) | Conv. (%) | Amine config. |
cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :trans ratio :trans ratio |
Conv. (%) | Amine config. | |
| n.d. – not determined. The reactions were performed under the optimised conditions in triplicate with a standard deviation of under 10%. Products detected by GC and chiral GC analysis. Conversions were determined after 24 h (1a), 48 h (3a) and 144 h (8a). | ||||||
| CV-TAm | 94 | 58 | (1S) | 88:12 |
0 | — |
| Kp-TAm | 18 | 8 | (1S) | 63:37 |
1 | n.d. |
| Pp-TAm | 94 | 90 | (1S) | 43:57 |
0 | — |
| Mv-TAm | 41 | 10 | (1R) | 53:47 |
2 | n.d. |
| ArRMut11 | 94 | 91 | (1R) | 36:64 |
19 | (R) |
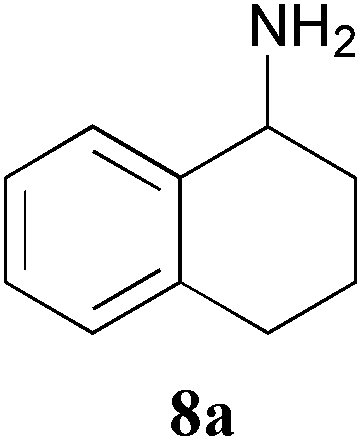
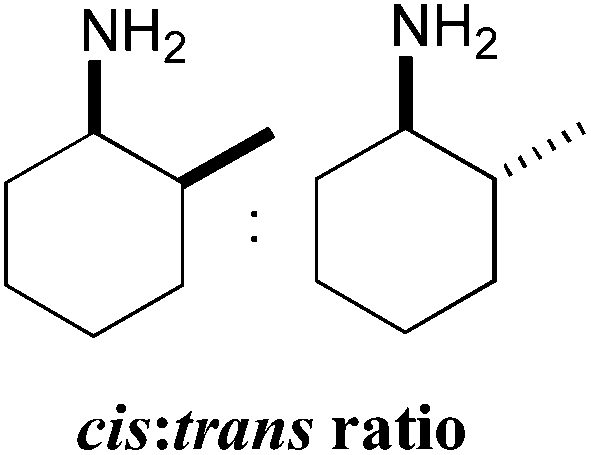
Amination of the α-substituted ketone 3 involves a dynamic kinetic resolution due to the chiral α-methyl group. Ketone 3 was accepted by all the selected ω-TAms, but conversion yields varied from 8% to 91%. Analysis of the products by GC indicated that CV-TAm preferentially gave cis-3a, while the trans-3a isomer was formed preferentially by Pp-TAm and ArRMut11-TAm.
A more detailed study of the amination of 3 was therefore performed, in order to evaluate the full product stereochemistry with the three most productive ω-TAms (Table 4). Samples were taken after 2 h, 4 h, 24 h and 48 h and conversions, cis:trans ratios and enantioselectivities were determined by GC analysis (Table 4). The data confirmed the stereoselectivities observed before (Table 3), but additionally by monitoring the amination over a period of time it became apparent that some selectivities decreased with increasing conversions/time. While CV-TAm was very selective for generating the cis-isomer the other enzymes seem to show lower selectivities. For example the ArRMut11 only showed a strong preference towards the formation of the trans-isomer at very low conversions (6% conversion and a cis:trans ratio of 20:80) while at a conversion of 20% the cis:trans ratio had increased to 40:60.
:trans ratios and ees over a reaction time of 48 h
| TAm | Time (h) | Conv. (%) |
|
|
|
|---|---|---|---|---|---|
| Conversions and cis:trans ratios were detected by achiral GC, while ee-values were determined using chiral GC; results were determined in triplicate with a standard deviation of less than 10%. |
|||||
| CV-TAm | 2 | 23 | 96:4 |
||
| 4 | 33 | 93:7 |
All | All | |
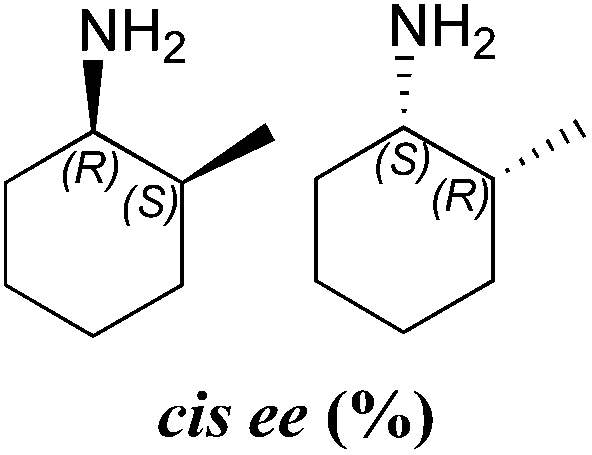
| 24 | 47 | 88:12 |
>99 (1S,2R) | >99 (1S,2S) | |
| 48 | 58 | 88:12 |
|||
| Pp-TAm | 2 | 31 | 61:36 |
||
| 4 | 49 | 54:46 |
All | All | |
| 24 | 90 | 42:58 |
>99 (1S,2R) | >99 (1S,2S) | |
| 48 | 90 | 43:57 |
|||
| ArRMut11 | 2 | 6 | 20:80 |
||
| 4 | 20 | 40:60 |
All | All | |
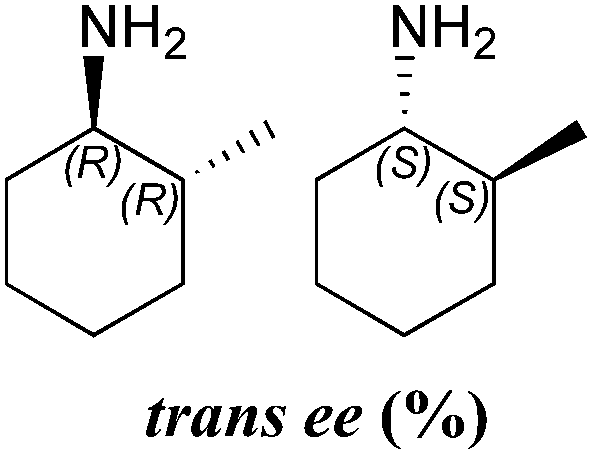
| 24 | 76 | 38:62 |
>99 (1R,2S) | >99 (1R,2R) | |
| 48 | 91 | 36:64 |
|||
The absolute configurations of the 3a stereoisomers were determined using known (R)- and (S)-selective TAms (ArRMut11 and CV-TAm respectively) with (2R)-3 and racemic 3. The amine products 3a from the four reactions were then correlated to the isomeric amine products by chiral GC-analysis to establish the stereochemical outcome of the reactions.
In all cases the amine was formed in exceptionally high stereoselectivities (>99% ee) while the variable cis:trans ratios resulted from the ability of the ω-TAms to distinguish between the stereocentre at the α-methyl position. Racemic samples of 3 were used in all experiments, other than when establishing absolute configurations. For reactions where high conversions and/or high diastereomeric ratios were observed, some racemisation at the α-carbon of 3 will have occurred at pH 8 used with CV-TAm and Pp-TAm and pH 10 with ArRMut11. Such dynamic asymmetric transaminations involving α-substituted ketones have received little attention in the literature to date. Recently they have been described with an α-substituted ketone possessing a large α-phenethylether group.33 However here we have the much less sterically differentiating methyl group at the α-position where notably for CV-TAm excellent ees and high diastereoselectivities were observed. The change in cis/trans product ratio over time probably reflects the consumption of the prefered isomer of 3 at shorter reaction times. Notably, for CV-TAm a high preference was observed for the (2R)-methyl group.
In light of the high stereoselectivities for CV-TAm, docking calculations were performed in order to gain insights into the stereopreference observed. Calculated binding affinities of all four PLP-imine quinonoid intermediates of the reaction were determined and results are summarised in Table 5.
Interestingly, the best calculated relative affinity (∼−8 kcal mol−1) was observed with the (R)-configured ligand, which is preferentially transformed by the CV-TAm (Table 5, entries 1 and 2). In contrast, the minor product of the transamination reaction (1S,2S)-3a had a lower calculated affinity (−6.4 kcal mol−1, entries 3 and 4). Thus the trend generally matched the observed experimental data. For more insights, both equatorial quinonoid intermediates (R)-(entry 1) and (S)-(entry 3) were evaluated in more detail after modelling into the active site using the holo structure of CV-TAm (4AH3).48 As shown in Fig. 2, the position of the six-membered ring varied depending on the methyl group stereochemistry. Moreover, the docked structures show that the (2R)-quinonoid (green), better fits the space available in the binding pocket (indicated in grey). However, apart from slightly better positioning of the (2R)- vs. the (2S)-quinonoid no additional steric factors could be determined to explain the stereochemistries observed. In general, the residue Lys288 is known to play a crucial role in the catalytic mechanism of TAms.48,49 When no substrate is in the active site it forms a Schiffs base with the PLP cofactor, and during the reaction the amine donor replaces the Lys288, which is released as a consequence and changes its position. It is therefore possible that the dynamic repositioning of Lys288 further influences the stereopreference observed.
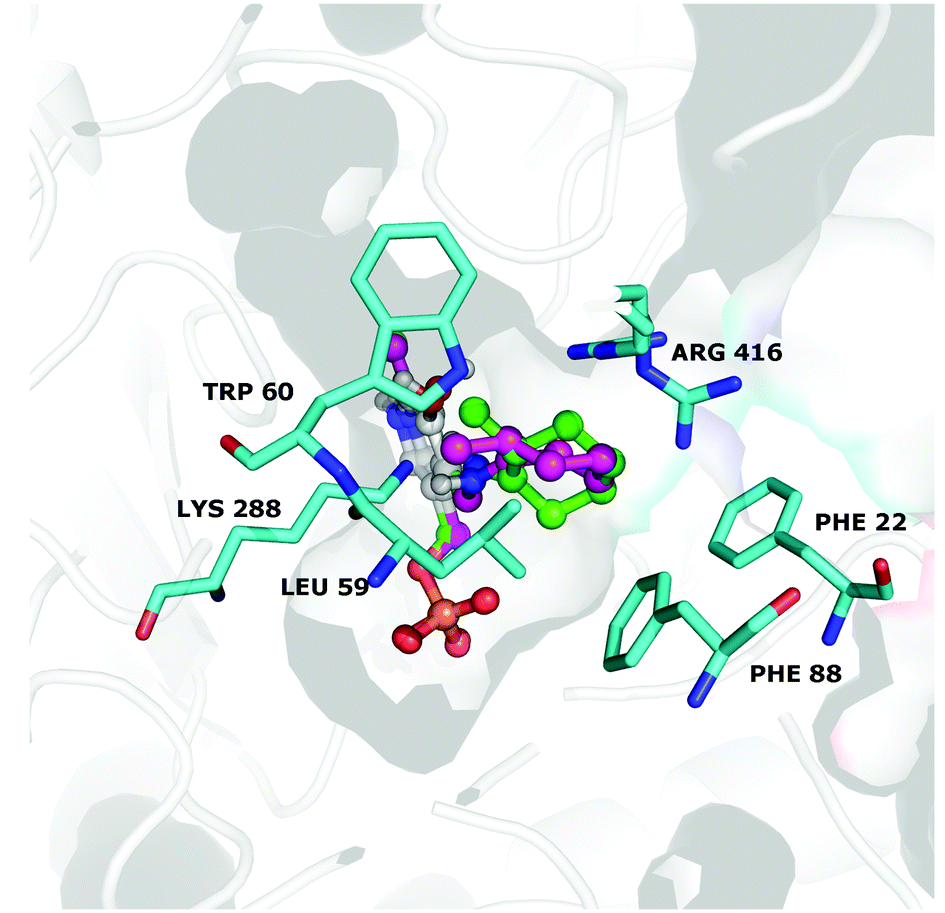 | ||
| Fig. 2 Docking of (2S) (pink) and (2R) (green) quinonoid intermediate into the active site of CV-TAm (4AH3). The binding pocket is indicated as grey shadow, and amino acids involved in the transamination reaction are labelled. | ||
Conclusions
Several (S)- and (R)-ω-transaminases have been investigated for the transamination of a range of cyclic ketones. In a preliminary screen suitable TAms were identified to transform the substrates of interest and the amine donor 2-propylamine 12 was accepted by all five selected ω-TAms for further study. The amination of cyclohexanone 1, 2-methylcyclohexanone 3, and α-tetralone 8 were studied in more detail: yields of up to 94% were achieved with selected transaminases. When using 2-methylcyclohexanone 3 enantio- and diastereoselectivities were investigated. The highest selectivities were obtained when using CV-TAm: (1S)-2-methylcyclohexylamine was formed with complete selectivity at the amine position and up to 24:1 diastereoselectivity for the cis (1S,2R) isomer. This high reaction selectivity is extremely interesting as it enables the cyclic amine 3a containing two-defined chiral centres to be formed in one step from racemic starting material. Such dynamic asymmetric transaminations with α-ketones have received little attention to date, and the high selectivities achieved by CV-TAm with α-substituted substrates will be explored in future work with CV-mutants.
Experimental
General
All starting materials were obtained from commercial suppliers and used as received unless otherwise stated. DNA-modifying enzymes were obtained from Thermo scientific (Germany) or New England Biolabs (USA). The enzymes used for shifting the equilibrium were commercially available from: L-lactate dehydrogenase from rabbit muscle (Sigma-Aldrich, Austria), glucose dehydrogenase (X-zyme, Germany), and formate dehydrogenase from Candida boidinii (Codexis, USA). The L-alanine dehydrogenase was prepared as reported previously.46 (R)- and (S)-8a are commercially available. (R)-3 was prepared as previously described.50Cloning of TAm genes in suitable expression vectors
A synthetic gene for the previously reported ω-TAm from Arthrobacter sp. variant (ArRMut11)1 was designed, as codon-optimised genes (DNA 2.0, U.S.A.), and subsequently cloned into the expression vector pET29a (Invitrogen, Germany) using standard techniques. Additionally, plasmids containing codon-optimised genes of the ω-TAms from Chromobacterium violaceum,37Vibrio fluvialis,38Pseudomonas putida (Pp-TAm),36 and Klebsiella pneumoniae39 were used. In addition, the ω-TAm from Mycobacterium vanbaalenii Mvan4516 (Mv-TAm) (gene bank accession no. 119958286; ABM15291)40 was amplified from genomic DNA using the primers listed below and standard PCR procedures. Restriction sites were introduced to enable the cloning in the desired expression vector pET29a (Table 6).| Name | Sequence | Restriction site |
|---|---|---|
| Restriction sites used for cloning are underlined. | ||
| Kp_forward |
 ACACTGGACGATCTCGC ACACTGGACGATCTCGC |
NdeI |
| Kp_reverse |
 TTCGCTAAAAAATGTTTC TTCGCTAAAAAATGTTTC |
XhoI |
| Mv_forward | ATATA GGCATTG ACACCGGAACCTCGAATCTC GGCATTG ACACCGGAACCTCGAATCTC |
NdeI |
| Mv_reverse | ATA TCAG TACTGGATCGCTTCGATCAG TCAG TACTGGATCGCTTCGATCAG |
NotI |
Heterologous enzyme expression and preparation
The ω-TAms from Chromobacterium violaceum and Vibrio fluvialis were expressed as described previously.37,38 All other ω-TAms were expressed in E. coli BL21(DE3)pLysS in lysogeny broth medium containing ampicillin (100 mg L−1). Cultures were grown at 37 °C until an OD600 of 0.5-0.7 was reached. Enzyme expression was induced by addition of isopropyl-β-D-thiogalactopyranoside (0.5 mM), and the temperature was reduced to 25 °C. After 16–20 h cells were harvested by centrifugation and stored at −20 °C. To prepare cell free crude extract cells (20% v/w) were suspended in HEPES buffer (100 mM, pH 7.5) containing pyridoxal-5-phosphate (PLP) (0.5 mM), and disrupted by ultra sonication (2 × 1 min, 40% output). The crude extract was cleared by centrifugation (20 min, 16.000g) and either directly used or freeze dried and stored at −20 °C.General procedure for TAm screening
For the HPLC-based TAm screening (R)- or (S)-α-methylbenzylamine (MBA) 11 (25 mM) and pyridoxal-5-phosphate (PLP) (0.5 mM) were dissolved in HEPES buffer (100 mM, pH 8). E. coli crude extract (12.5 μL) containing the overexpressed ω-TAm were added to 212.5 μL of this solution. The reaction was started by the addition of 25 μL of substrate in DMSO (final concentration 10 mM, 10 vol%). After an incubation for 21 h at 30 °C and 180 rpm the reaction was stopped by the addition of 250 μL of acetonitrile containing 0.2% TFA. Denaturated protein was removed by centrifugation, and the supernatant was analysed by HPLC (Agilent) using a Discovery®Bio Wide Pore C18 column (Supelco, 25 × 4.6 mm, 10 μm beads) with UV detection at 254 nm. Concentrations of acetophenone were determined using a linear gradient: 30%–60% B over 10 min (A = water, B = acetonitrile, both containing 0.1% TFA). The acetophenone produced eluted at a retention time of 8.6 min.Determination of initial rates of the five selected TAms
Initial rates were determined using the HPLC-based method described above using cyclohexanone 1 as the substrate. Cyclohexanone (10 mM), (R)- or (S)-MBA 11 (10 mM) and PLP (1 mM) were solved in sodium phosphate buffer (100 mM, pH 8). The reaction was started by addition of 100 μL of enzyme solution (crude cell extract or freeze dried cells). Samples were taken after 5 min, 10 min, 15 min, and 30 min, and the linear slope was used for the calculation of initial rates and specific activities (Table 7).| TAm | Crude cell extract [U mg−1 total protein] | Freeze dried cell extract [U mg−1 total protein] |
|---|---|---|
| n.d. – not determined. | ||
| CV-TAm | 0.58 | 0.32 |
| Kp-TAm | n.d. | 0.008 |
| Pp-TAm | 0.13 | 0.11 |
| Mv-TAm | 0.006 | 0.006 |
| ArRMut11 | 0.39 | 0.24 |
General procedure for the TAm reactions using different equilibrium shifting systems
Transamination reactions using the (a) AlaDH/FDH, (b) LDH/GDH or (c) 2-propylamine IPA 12 system were performed as follows: (a) substrate (20 mM), D- or L-alanine (200 mM), ammonium formate (60 mM), PLP (1 mM), NAD+ (0.5 mM), formate dehydrogenase (11 U) and L-alanine dehydrogenase (12 U) were dissolved in sodium phosphate buffer (1 mL, 100 mM, pH 8); (b) substrate (20 mM), D- or L-alanine (200 mM), glucose (60 mM), PLP (1 mM), NAD+ (0.5 mM), glucose dehydrogenase (30 U) and L-lactate dehydrogenase (90 U) were dissolved in sodium phosphate buffer (1 mL, 100 mM, pH 8); (c) substrate (20 mM), IPA 12 (200 mM), and PLP (1 mM) were dissolved in sodium phosphate buffer (1 mL, 100 mM, pH 8 or 10). All biotransformations were started by the addition of lyophilised E. coli crude cell extract containing the overexpressed ω-TAm corresponding to an activity of 0.5 U in the amination of cyclohexanone (CV-TAm, Pp-TAm and ArRMut11 see Table 1), while the maximum amount of 10 mg was added in the case of the Kp-TAm and Mv-TAm. The mixture was incubated at 30 °C or 45 °C and 800 rpm in a thermoshaker (Eppendorf, Germany).Samples were taken at different time points, and the reaction was stopped by addition of 10 vol% of NaHCO3, and extracted with ethyl acetate (2 × 500 μL). Conversions to the amine were measured by GC (Agilent 7890 A system equipped with a FID detector). The biotransformations of cyclohexanone 1 and 2-methylcyclohexanone 3 were monitored using an Agilent DB-1701 column (30 m, 0.25 mm, 0.25 μm) using the following temperature programmes: A 60 °C, hold for 5 min, 15 °C min−1 to 150 °C; retention times cyclohexylamine 1a 6 min and cyclohexanone 1 7.9 min. B 60 °C, hold for 5 min, 5 °C min−1 to 80 °C, 60 °C min−1 to 250 °C; retention times trans-3a 5.9 min, cis-3a 6.4 min and 3 8.4 min. Conversions for α-tetralone 8 were determined using an Agilent HP5 (30 m, 0.32 mm, 0.25 μm) and the following programme: 120 °C, 5 °C min−1 to 160 °C, 50 °C min−1 to 300 °C, retention times 1-aminotetraline 8a 4.4 min and α-tetralone 8 4.7 min.
Determination of enantiomeric purity
The enantiomeric exessess of amines 3a and 8a were determined by GC using a modified β-cyclodextrin column (CP-Chirasil-DEX CB, 25 m, 0.32 mm, 0.25 μm) after derivatisation to the corresponding trifuoroacetamides. For the derivatisation trifluoroacetic anhydride (5 μL) was added to the extracted sample, and after an incubation (800 rpm) at 30 °C the reaction was quenched with NaHCO3 and the ee was analysed. Temperature programme for 3a: 100 °C hold 2 min, 2 °C min−1 to 135 °C, 25 °C min−1 to 180 °C. Retention times: (1S,2R)-3a 5.5 min, (1R,2S)-3a 5.7 min, (1S,2S)-3a 6.7 min and (1R,2R)-3a 7 min. Temperature programme for 8a: 120 °C, 5 °C min−1 to 160 °C, 20 °C min−1 to 180 °C. Retention times: (S)-8a 8.2 min and (R)-8a 8.5 min.Docking studies
For docking experiments the holo crystal structure of the ω-TAm from C. violaceum was used (pdb: 4AH3). The apo-structure of 4AH3 as well as the structure of the quinonoid intermediate51 of all conformers were generated with Maestro and the energy optimisation of the ligands was performed using the MacroModel “Minimization” followed by a “Conformational Search” (all Schrödinger LLC). The two conformations with the lowest energy were used for the docking experiments. Optimisation of the protein structure after removal of the bound PLP was performed with autodock tools v 1.5.6. Docking calculations were performed using AutoDock Vina52 with a x = 24/y = 24/z = 24 grid box centred on x = 4.8, y = −0.5, z = 7.4. Lowest energy clusters were selected and visualised using pymol v0.99rc6.Acknowledgements
This work has been supported by a postdoctoral fellowship of the German academic exchange service (DAAD) to N. R., the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET- Funding Program. H. L. was funded by the Austrian Science Fund (FWF, project W9-01 DK Molecular Enzymology).Notes and references
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- P. Omvik, P. Lundjohansen and H. Haugland, Cardiovasc. Drugs Ther., 1993, 7, 125–132 CrossRef CAS.
- M. R. Farlow and J. L. Cummings, Am. J. Med., 2007, 120, 388–397 CrossRef CAS PubMed.
- M. Fuchs, D. Koszelewski, K. Tauber, W. Kroutil and K. Faber, Chem. Commun., 2010, 46, 5500–5502 RSC.
- J. A. J. Schmitt, J. G. Ramaekers, M. J. Kruizinga, M. P. J. van Boxtel, E. F. P. M. Vuurman and W. J. Riedel, J. Psychopharmacol., 2002, 16, 207–214 CrossRef CAS PubMed.
- M. Van Ameringen, J. Oakman, C. Mancini, B. Pipe and H. Chung, J. Clin. Psychopharmacol., 2004, 24, 42–48 CrossRef PubMed.
- Z. Han, S. G. Koenig, H. Zhao, X. P. Su, S. P. Singh and R. P. Bakale, Org. Process Res. Dev., 2007, 11, 726–730 CrossRef CAS.
- R. L. Watts, J. Jankovic, C. Waters, A. Rajput, B. Boroojerdi and J. Rao, Neurology, 2007, 68, 272–276 CrossRef CAS PubMed.
- R. Webster, A. Boyer, M. J. Fleming and M. Lautens, Org. Lett., 2010, 12, 5418–5421 CrossRef CAS PubMed.
- C. Sonesson, T. Barf, J. Nilsson, D. Dijkstra, A. Carlsson, K. Svensson, M. W. Smith, I. J. Martin, J. N. Duncan, L. J. King and H. Wikstrom, J. Med. Chem., 1995, 38, 1319–1329 CrossRef CAS.
- Z. P. Zhuang, M. P. Kung, W. Clarke, S. Maayani, M. Mu and H. F. Kung, Chirality, 1995, 7, 452–458 CrossRef CAS PubMed.
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS PubMed.
- Y. Kim, J. Park and M. J. Kim, ChemCatChem, 2011, 3, 271–277 CrossRef CAS PubMed.
- L. K. Thalen, D. B. Zhao, J. B. Sortais, J. Paetzold, C. Hoben and J. E. Backvall, Chem. – Eur. J., 2009, 15, 3403–3410 CrossRef CAS PubMed.
- A. N. Parvulescu, P. A. Jacobs and D. E. De Vos, Adv. Synth. Catal., 2008, 350, 113–121 CrossRef CAS PubMed.
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef CAS.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef PubMed.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed.
- M. S. Malik, E. S. Park and J. S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS PubMed.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS.
- D. Zhu and L. Hua, Biotechnol. J., 2009, 4, 1420–1431 CrossRef CAS PubMed.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- M. D. Truppo and N. J. Turner, Org. Biomol. Chem., 2010, 8, 1280–1283 CAS.
- C. Molinaro, P. G. Bulger, E. E. Lee, B. Kosjek, S. Lau, D. Gauvreau, M. E. Howard, D. J. Wallace and P. D. O'Shea, J. Org. Chem., 2012, 77, 2299–2309 CrossRef CAS PubMed.
- I. K. Mangion, B. D. Sherry, J. J. Yin and F. J. Fleitz, Org. Lett., 2012, 14, 3458–3461 CrossRef CAS PubMed.
- B. Wang, H. Land and P. Berglund, Chem. Commun., 2013, 49, 161–163 RSC.
- R. C. Simon, C. S. Fuchs, H. Lechner, F. Zepeck and W. Kroutil, Eur. J. Org. Chem., 2013, 3397–3402 CrossRef CAS PubMed.
- D. Pressnitz, C. S. Fuchs, J. H. Sattler, T. Knaus, P. Macheroux, F. G. Mutti and W. Kroutil, ACS Catal., 2013, 3, 555–559 CrossRef CAS.
- E. Siirola, F. G. Mutti, B. Grischek, S. F. Hoefler, W. M. F. Fabian, G. Grogan and W. Kroutil, Adv. Synth. Catal., 2013, 355, 1703–1708 CrossRef CAS PubMed.
- N. Richter, R. C. Simon, W. Kroutil, J. M. Ward and H. C. Hailes, Chem. Commun., 2014, 50, 6098–6100 RSC.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- C. Sayer, R. J. Martinez–Torres, N. Richter, M. N. Isupov, H. C. Hailes, J. Littlechild and J. M. Ward, FEBS J., 2014, 281, 2240–2253 CrossRef CAS PubMed.
- J. Limanto, E. R. Ashley, J. Yin, G. L. Beutner, B. T. Grau, A. M. Kassim, M. M. Kim, A. Klapers, Z. Liu, H. R. Strotman and M. D. Truppo, Org. Lett., 2014, 16, 2716–2719 CrossRef CAS PubMed.
- E. Busto, R. C. Simon, B. Grischek, V. Gotor-Fernandez and W. Kroutil, Adv. Synth. Catal., 2014, 356, 1937–1942 CrossRef CAS PubMed.
- C. K. Chung, P. G. Bulger, B. Kosjek, K. M. Belyk, N. Rivera, M. E. Scott, G. R. Humphrey, J. Limanto, D. C. Bachert and K. M. Emerson, Org. Process Res. Dev., 2014, 18, 215–227 CrossRef CAS.
- B. R. Lichman, E. D. Lamming, T. Pesnot, J. M. Smith, H. C. Hailes and J. M. Ward, Green Chem., 2015, 17, 852–855 RSC.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS PubMed.
- J. S. Shin, H. Yun, J. W. Jang, I. Park and B. G. Kim, Appl. Microbiol. Biotechnol., 2003, 61, 463–471 CrossRef CAS PubMed.
- R. J. Martinez-Torres, A. Bour, I. N. Taylor, H. C. Hailes and J. M. Ward, in preparation.
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- K. Smithies, M. E. B. Smith, U. Kaulmann, J. L. Galman, J. M. Ward and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 570–574 CrossRef CAS PubMed.
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99–107 CrossRef CAS.
- S. Schätzle, F. Steffen-Munsberg, A. Thontowi, M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2439–2445 CrossRef PubMed.
- G. Shin, S. Mathew, M. Shon, B. G. Kim and H. Yun, Chem. Commun., 2013, 49, 8629–8631 RSC.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS PubMed.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, N. G. Turrini, J. H. Sattler, A. Lerchner, A. Skerra and W. Kroutil, Eur. J. Org. Chem., 2012, 1003–1007 CrossRef CAS PubMed.
- J.-S. Park, M. S. Malik, J.-Y. Dong and J.-S. Shin, ChemCatChem, 2013, 12, 3538–3542 CrossRef PubMed.
- C. Sayer, M. N. Isupov, A. Westlake and J. A. Littlechild, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 564–576 CrossRef CAS PubMed.
- M. S. Humble, K. E. Cassimjee, M. Hakansson, Y. R. Kimbung, B. Walse, V. Abedi, H. J. Federsel, P. Berglund and D. T. Logan, FEBS J., 2012, 279, 779–792 CrossRef CAS PubMed.
- M. Hall, C. Stueckler, H. Ehammer, E. Pointner, G. Oberdorfer, K. Gruber, B. Hauer, R. Stuermer, W. Kroutil, P. Macheroux and K. Faber, Adv. Synth. Catal., 2008, 350, 411–418 CrossRef CAS PubMed.
- M. S. Humble, K. E. Cassimjee, V. Abedi, H. J. Federsel and P. Berglund, ChemCatChem, 2012, 4, 1167–1172 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
| This journal is © The Royal Society of Chemistry 2015 |