
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An investigation of nitrile transforming enzymes in the chemo-enzymatic synthesis of the taxol sidechain†
Birgit
Wilding
a,
Alicja B.
Veselá
b,
Justin J. B.
Perry
c,
Gary W.
Black
c,
Meng
Zhang
c,
Ludmila
Martínková
b and
Norbert
Klempier
*a
aacib GmbH (Austrian Centre of Industrial Biotechnology), c/o Institute of Organic Chemistry, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria. E-mail: klempier@tugraz.at; Fax: +43 316 873 32402; Tel: +43 316 873 32445
bInstitute of Microbiology of the Academy of Sciences of the Czech Republic, v.v.i., Vídeňská 1083, CZ-14220 Prague, Czech Republic
cIndustrial Biotechnology Research Group, Department of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne NE1 8ST, United Kingdom
First published on 24th June 2015
Abstract
Paclitaxel (taxol) is an antimicrotubule agent widely used in the treatment of cancer. Taxol is prepared in a semisynthetic route by coupling the N-benzoyl-(2R,3S)-3-phenylisoserine sidechain to the baccatin III core structure. Precursors of the taxol sidechain have previously been prepared in chemoenzymatic approaches using acylases, lipases, and reductases, mostly featuring the enantioselective, enzymatic step early in the reaction pathway. Here, nitrile hydrolysing enzymes, namely nitrile hydratases and nitrilases, are investigated for the enzymatic hydrolysis of two different sidechain precursors. Both sidechain precursors, an openchain α-hydroxy-β-amino nitrile and a cyanodihydrooxazole, are suitable for coupling to baccatin III directly after the enzymatic step. An extensive set of nitrilases and nitrile hydratases was screened towards their activity and selectivity in the hydrolysis of two taxol sidechain precursors and their epimers. A number of nitrilases and nitrile hydratases converted both sidechain precursors and their epimers.
Introduction
Paclitaxel (taxol) is a complex natural compound used in anticancer therapy against a variety of cancers, such as ovarian, gastric, head and neck, non-small lung, prostate and breast cancer.1–4 Taxol was first isolated from the bark of the pacific yew Taxus brevifolia, following an initiative of the US National Cancer Institute (NCI), screening for antineoplastic activity of new substances from various origins.1,5Taxol acts as microtubule stabilizer, binding to tubulin in polymerized microtubules, disrupting the cell cycle, ultimately leading to cell death.6–8 Despite its known limitations, e.g., poor solubility, toxicities and emerging drug resistance, taxol is still widely used in cancer therapy. New administration forms, formulations and taxane analogues have been designed to overcome selectivity, efficacy, toxicity and drug resistance issues.9–11 Numerous efforts have been made to determine the structure activity relationship of taxol.12 In recent years, the role of the (2R,3S)-N-benzoyl-3-phenylisoserine C-13 sidechain had been confirmed as essential for the biological activity of taxol (Fig. 1).13
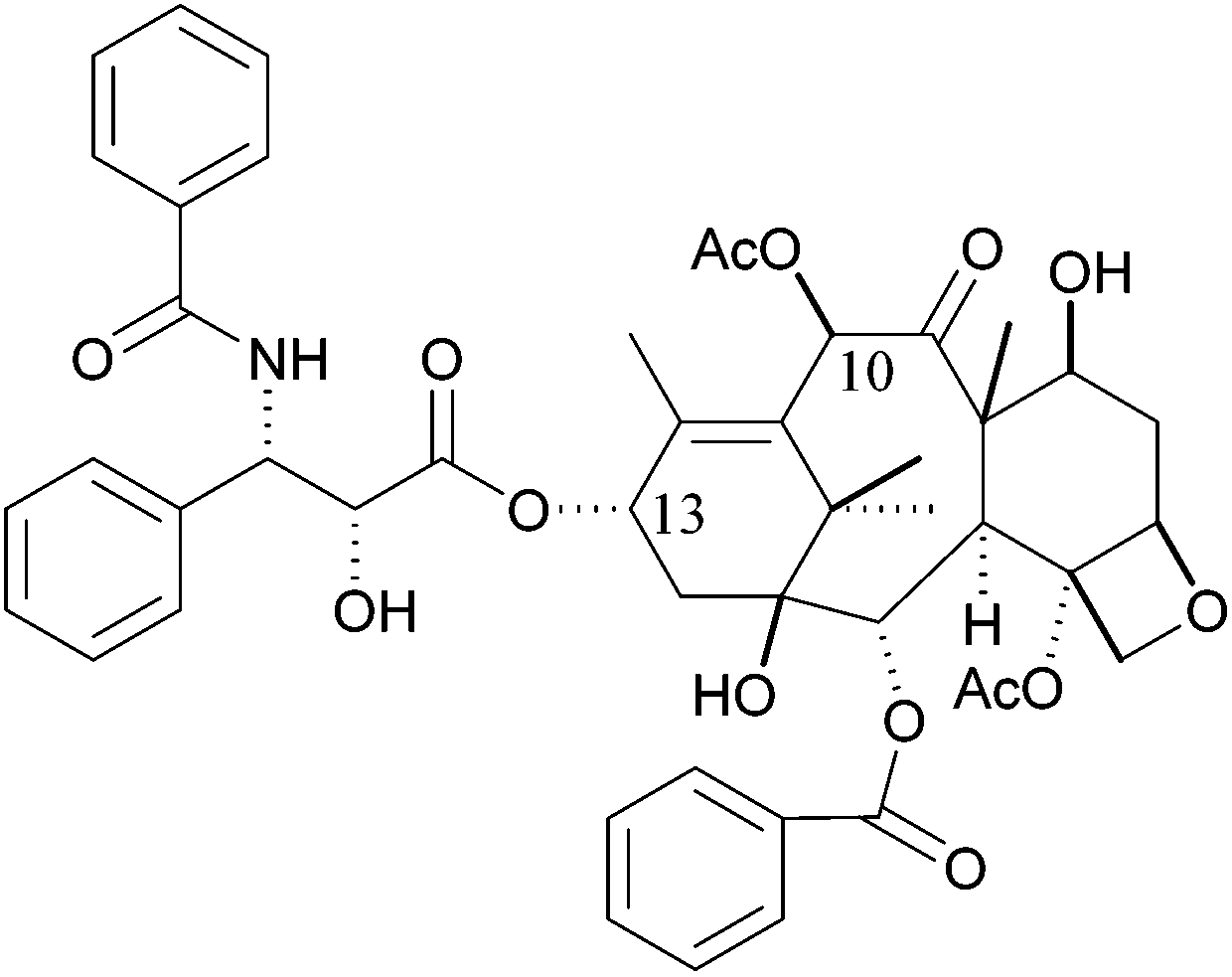 | ||
| Fig. 1 Structure of Paclitaxel (taxol), consisting of the baccatin III core structure and the (2R,3S)-N-benzoyl-3-phenylisoserine C-13 sidechain. | ||
Various sidechain precursors have been prepared in asymmetric chemical syntheses14 and in chemoenzymatic approaches15 using acylases,16 lipases17 and reductases.18
In this work, nitrile transforming enzymes, namely nitrilases and nitrile hydratases, are investigated for the synthesis of the taxol sidechain. Nitrilases (EC 3.5.5.1) and nitrile hydratases (EC 4.2.1.84) are attractive biocatalysts for the fine chemicals and pharmaceutical industries. Nitrilases catalyse the cleavage of nitriles to the corresponding carboxylic acids and ammonia and have been shown to catalyse the hydrolysis of a variety of nitriles, including the enantioselective synthesis of β-amino acids from β-amino nitriles.19 However, several nitrilases were reported to convert nitriles to both, acid and amide products (Scheme 1).20 Nitrile hydratases are Fe- or Co-metalloenzymes that catalyse the hydration of nitriles to their corresponding amides.21 To the best of our knowledge, nitrilases have not yet been investigated in the synthesis of the taxol sidechain. Previously, a nitrile hydratase from Debaryomyces hansenii DSM 3428 was used in whole cell experiments for the hydration of (±)-trans-3-phenyloxirane-2-carbonitrile. The reaction proceeded with low enantioselectivity (ee-value <36%), despite stopping the reaction shortly before 50% conversion was achieved.15b
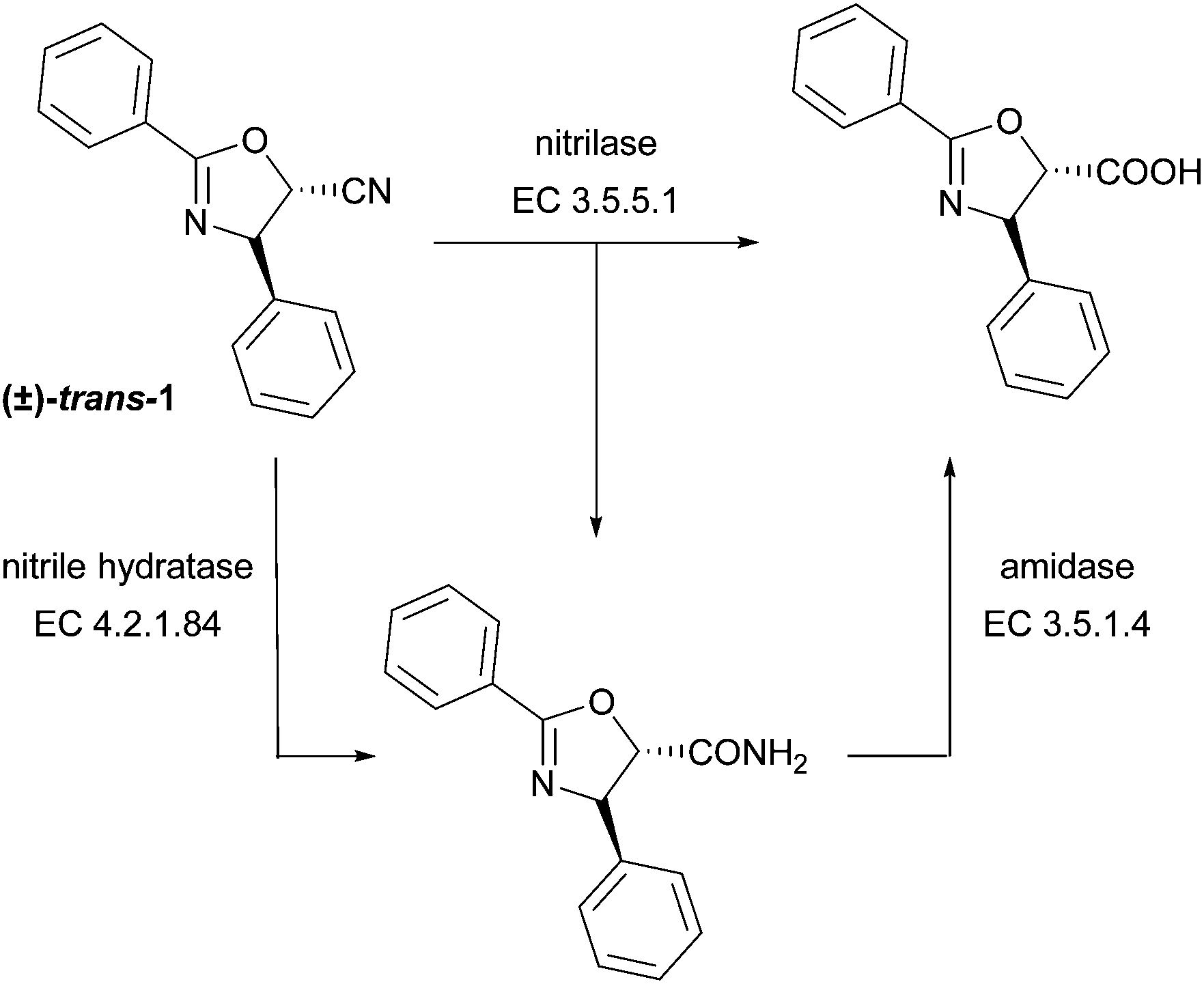 | ||
| Scheme 1 Enzymatic conversion of the taxol sidechain precursor (±)-trans-1 to the corresponding carboxylic acid and/or carboxamide. Nitrilase and nitrile hydratase catalysed reactions were investigated for the taxol sidechain precursors (±)-trans-1 and (±)-syn-2 and their epimers (±)-cis-1, and (±)-anti-2. | ||
Here, two different taxol sidechain precursors were prepared in chemical synthesis, an openchain α-hydroxy-β-amino nitrile and a cyanodihydrooxazole. A set of 24 nitrilases and four nitrile hydratases was investigated for the biotransformation of these taxol sidechain precursors and their epimers. The enzymatic hydrolysis of these nitrile containing sidechain precursors applies the stereoselective, enzyme-catalysed reaction as the last step in the synthesis.
Results and discussion
Two different taxol sidechain precursors were prepared in chemical synthesis, as depicted in Scheme 2. The corresponding acids and amides were prepared in chemical synthesis as reference materials for the biotransformation reactions.22 In the first synthetic step, benzaldehyde was transformed to a mixture of (±)-cis- and (±)-trans-3-phenyloxirane-2-carbonitrile in a Darzens reaction.23 The epimers were separated by column chromatography and used separately to synthesise the dihydrooxazoles (±)-trans-1 and (±)-cis-1 in a Ritter-type reaction.24 Ring opening under acidic conditions gave the openchain precursors (±)-syn-2 and (±)-anti-2.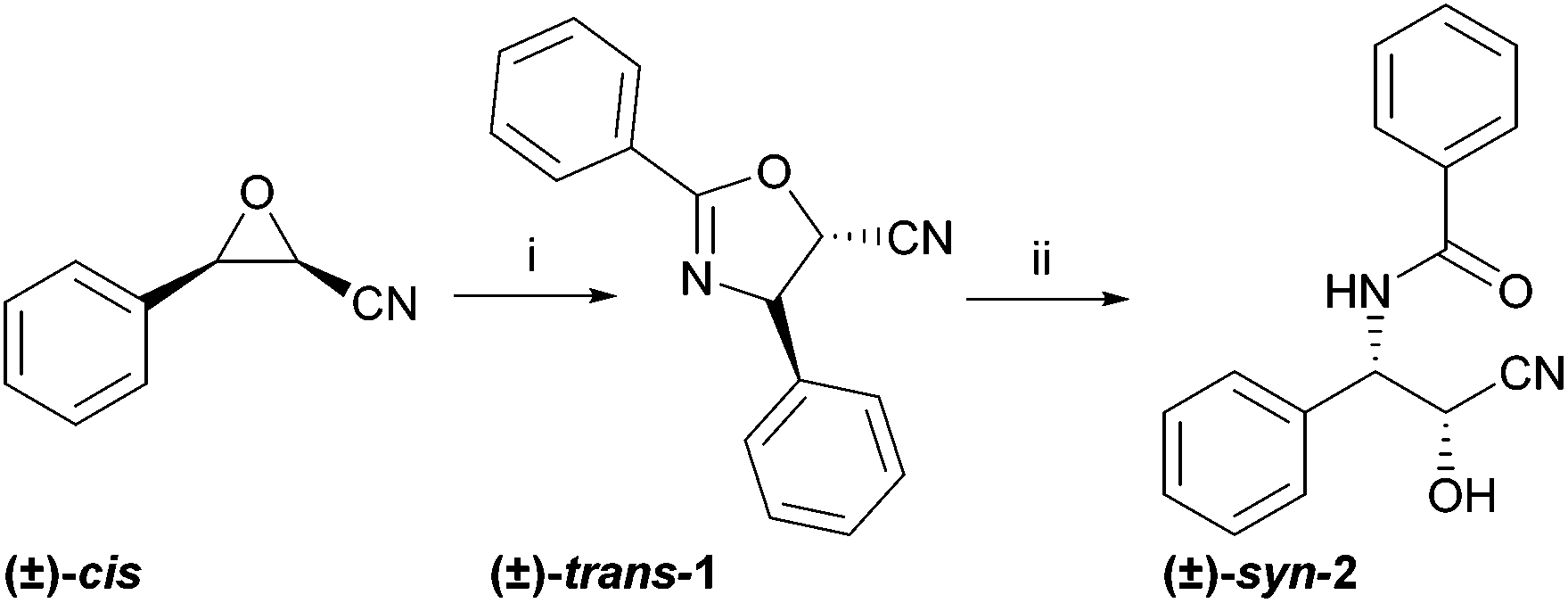 | ||
| Scheme 2 Synthesis of the taxol sidechain precursors (±)-trans-1 and (±)-syn-2. The epimers (±)-cis-1 and (±)-anti-2 were prepared analogously; (i) benzonitrile, BF3*Et2O, (ii) 1 M aq. HCl, methanol, 60 °C. (±)-cis- and (±)-trans-3-Phenyloxirane-2-carbonitrile were prepared from benzaldehyde as described in the ESI.† | ||
The sidechain precursors (±)-trans-1 and (±)-syn-2 and their epimers were investigated as substrates for a set of different nitrile hydratases and nitrilases (Scheme 1 and 2). Both sidechain precursors, (±)-trans-1 and the cyanohydrin (±)-syn-2 were stable under the reaction conditions of the biotransformation reactions, as confirmed in blank reactions.25 All biotransformation reactions (nitrilase and nitrile hydratase catalysed reactions) were evaluated in a HPLC-(MS) based screening, using acid and amide references prepared in chemical synthesis.22 Commercially available, easy to use nitrile hydratases from Prozomix, Ltd (PRO-E0257 to PRO-E0259)26 were used. Nitrilases included 18 commercially available nitrilases from two different suppliers, namely Codexis, Inc. (NIT-101 to NIT-114)27 and Prozomix, Ltd (PRO-E0260 to PRO-E0264)26 and six fungal nitrilases overexpressed in E. coli.28
The openchain precursor (±)-syn-N-(2-cyano-2-hydroxy-1-phenylethyl)benzamide, (±)-syn-2, and its epimer (±)-anti-N-(2-cyano-2-hydroxy-1-phenylethyl)benzamide, (±)-anti-2, were readily hydrolysed by three out of four nitrile hydratases tested, as summarised in Table 1. Conversions for (±)-anti-2 were higher than for (±)-syn-2. Examination of the amino acid sequences of the nitrile hydratases26 indicated that Co-type nitrile hydratases (PRO-E0257, PRO-E0258, PRO-E0259), but not the Fe-type nitrile hydratase (PRO-E0256) catalysed this reaction.
| Nitrile hydratase | Conversion of (±)-syn-2 to amide [%] | Conversion of (±)-anti-2 to amide [%] |
|---|---|---|
| PRO-E0256 | 0 | 0 |
| PRO-E0257 | 5.8 | 15.8 |
| PRO-E0258 | 12.5 | 42.0 |
| PRO-E0259 | 37.1 | 53.7 |
Less than one third of the commercially available nitrilases catalysed the hydrolysis of (±)-syn-N-(2-cyano-2-hydroxy-1-phenylethyl)benzamide, (±)-syn-2. Of these five nitrilases, NIT-106 and NIT-114 gave carboxamide, 23% and 15% conversion (after 15 hours) respectively, as the only product. Two other nitrilases, NIT-111 and PRO-E0260, yielded the desired carboxylic acid as product, however also gave significant amounts of amide as the by-product, as depicted in Fig. 2. A single nitrilase, PRO-E0263, yielded the desired carboxylic acid and only minor amounts (less than 10%) of the amide by-product. (±)-syn-2 was not hydrolysed by any of the fungal nitrilases tested.
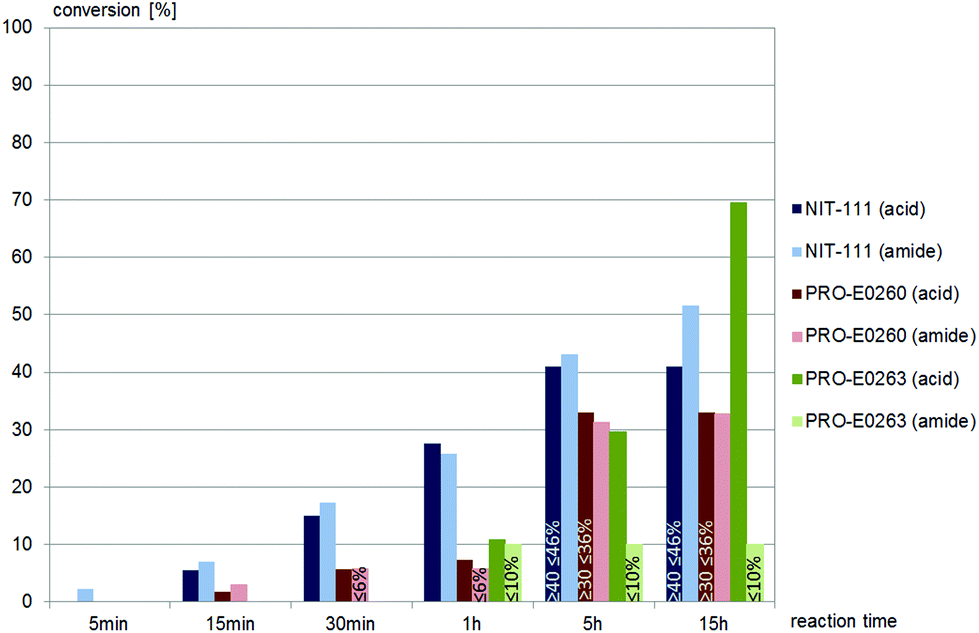 | ||
| Fig. 2 Screening results of (±)-syn-2, (±)-syn-N-(2-cyano-2-hydroxy-1-phenylethyl)benzamide, 0.4 mM, with commercially available nitrilases (4 mg mL−1 commercial enzyme preparation in buffer). NIT-101 to NIT-105, NIT-108, NIT-112, NIT-113, PRO-E0261, PRO-E0262 and PRO-E0264 gave no conversion, NIT-106 and NIT-114 gave amide as only product. None of the fungal nitrilases tested hydrolysed (±)-syn-2. | ||
(±)-anti-N-(2-Cyano-2-hydroxy-1-phenylethyl)benzamide, (±)-anti-2, was accepted by similar nitrilases as (±)-syn-2, as depicted in Fig. 3. NIT-106 gave the amide as the main product, NIT-111 and PRO-E0260 gave almost equal amounts of the acid and amide throughout the reaction time, as depicted in Fig. 3. PRO-E0263 gave the best results for the hydrolysis of (±)-anti-2, yielding the carboxylic acid as the only product. (±)-anti-2 was not hydrolysed by any of the fungal nitrilases tested.
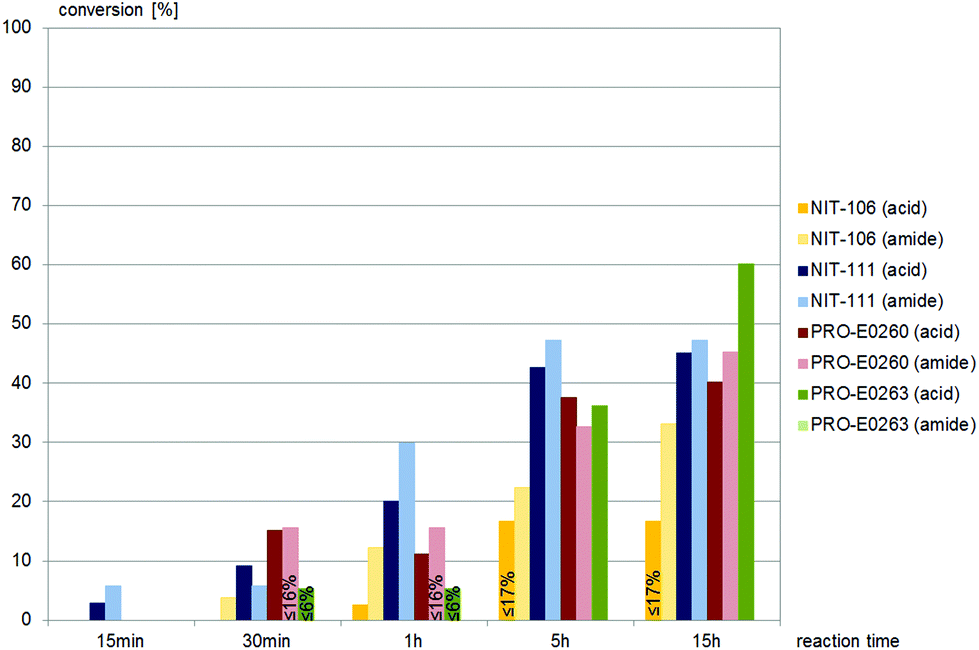 | ||
| Fig. 3 Screening results of (±)-anti-2, (±)-anti-N-(2-cyano-2-hydroxy-1-phenylethyl)benzamide, 0.4 mM, with commercially available nitrilases. NIT-101–NIT-105, NIT-108, NIT-112 to NIT-114, PRO-E0261, PRO-E0262 and PRO-E0264 gave no conversion. None of the fungal nitrilases tested hydrolysed (±)-anti-2. | ||
Nitrilases PRO-E0260 and PRO-E0264 are nitrilases from Bradyrhizobium japonicum. Nitrilase PRO-E0260 is an arylacetonitrilase and exhibits its highest activity for (R,S)-mandelonitrile but did not show enantioselectivity.29 PRO-E0260 hydrolysed (±)-syn-2 and (±)-anti-2, however, gave almost equal amounts of the acid and amide as products. PRO-E0264 has little activity towards branched nitriles, such as mandelonitrile, its preferential substrates being, e.g. hydrocinnamonitrile and heptanenitrile.30 No activity of PRO-E0264 was observed towards (±)-syn-2 and (±)-anti-2.
The dihydrooxazole (±)-trans-1, (±)-trans-2,4-diphenyl-4,5-dihydrooxazole-5-carbonitrile, was chosen as the second sidechain precursor. Here, the enantioselective α-hydroxy-β-amino nitrile moiety is protected in a cyclic precursor. The corresponding carboxylic acid of (±)-trans-1 can be directly coupled to baccatin III. The final cleavage of the dihydrooxazole ring occurs subsequently under defined acidic conditions.31
The dihydrooxazoles (±)-cis-1 and (±)-trans-1 were hydrolysed by all nitrile hydratases tested. Good conversions and ee-values were achieved for the hydration of (±)-cis-1 (Table 2). Biotransformations of (±)-trans-1 gave higher, almost quantitative, conversions with all nitrile hydratases, even when reaction time and concentration of enzyme were significantly decreased. Quantitative conversions, however, indicate poor enantioselectivity of these nitrile hydratase catalysed reactions.
| Enzyme | Conversion of (±)-cis-1 to amide [%]; ee-valuea [%] | Conversion of (±)-trans-1 to amidea [%] |
|---|---|---|
| a (±)-cis- 1: 200 μL commercial enzyme preparation, reaction time 21 h, (±)-trans-1: 50 μL commercial enzyme preparation.26 Total volume 500 μL. Results for additional enzyme concentrations and reaction times are available in the ESI. | ||
| PRO-E0256 | 31.7; 65.2 | 91.7 |
| PRO-E0257 | 51.6; 83.4 | 93.7 |
| PRO-E0258 | 63.2; 87.6 | 96.9 |
| PRO-E0259 | 74.4; 90.9 | 100 |
Only two nitrilases, of the 15 commercial nitrilases and six fungal nitrilases tested, hydrolysed (±)-cis-1, and both gave amide as the major product (Fig. 4). In contrast to (±)-cis-1, the sidechain precursor (±)-trans-1 was hydrolysed by half of the commercially available nitrilases and all the fungal nitrilases tested (Fig. 5–7).
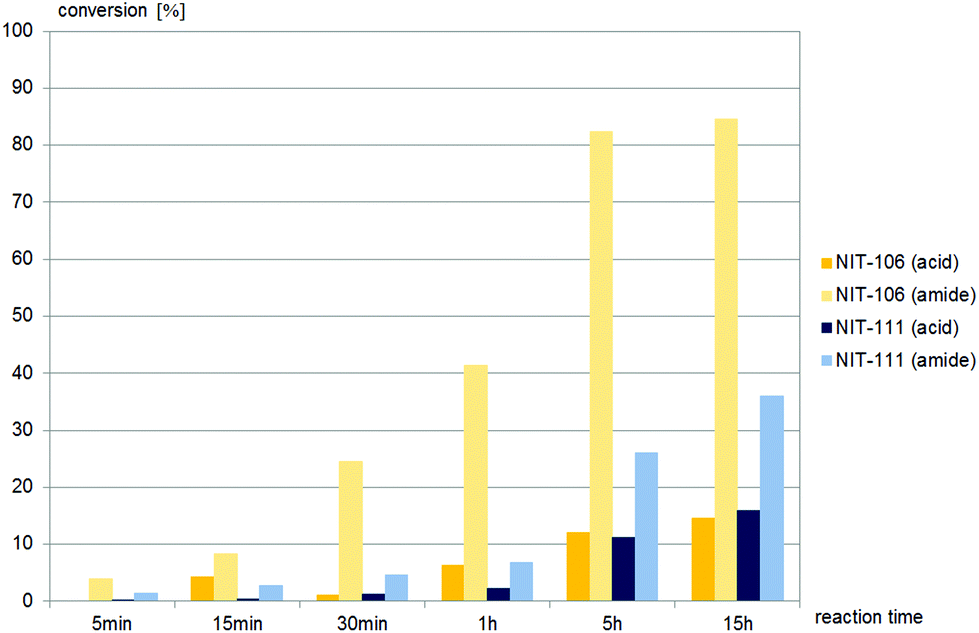 | ||
| Fig. 4 Screening results of (±)-cis-1, (±)-cis-2,4-diphenyl-4,5-dihydrooxazole-5-carbonitrile, 0.4 mM, with commercially available nitrilases (4 mg mL−1 commercial enzyme preparation in buffer). None of the fungal nitrilases tested hydrolysed (±)-cis-1. | ||
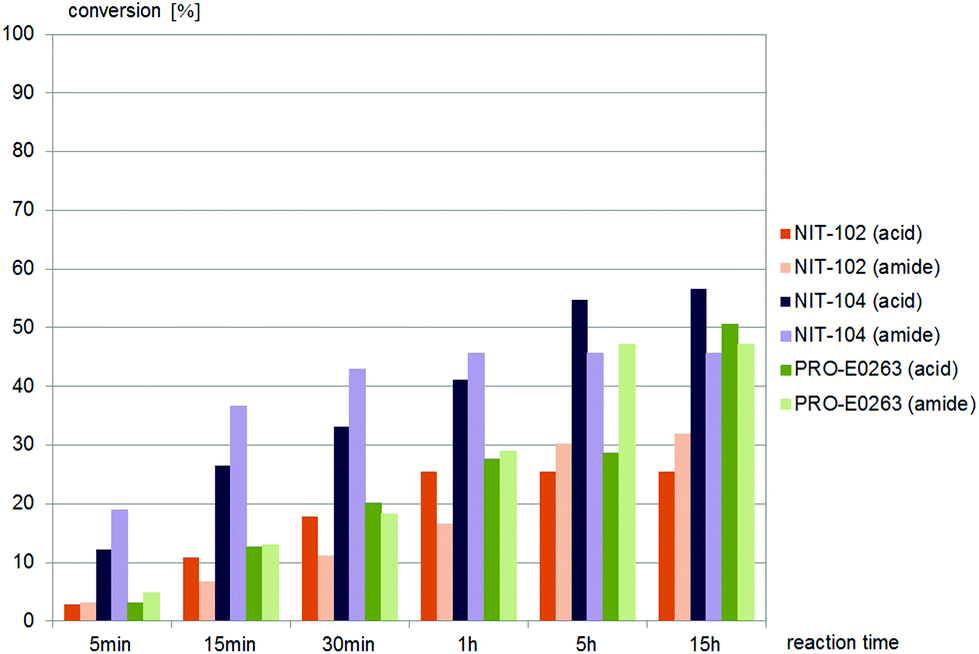 | ||
| Fig. 5 Screening results of (±)-trans-1, (±)-trans-2,4-diphenyl-4,5-dihydrooxazole-5-carbonitrile, 0.4 mM, with commercially available nitrilases (4 mg mL−1 commercial enzyme preparation in buffer). NIT-102, NIT-104, PRO-E0263, giving similar amounts of acid and amide as products. | ||
 | ||
| Fig. 6 Screening results of (±)-trans-1, (±)-trans-2,4-diphenyl-4,5-dihydrooxazole-5-carbonitrile, 0.4 mM, with commercially available nitrilases giving the acid as the main product (4 mg mL−1 commercial enzyme preparation in buffer). NIT-101, NIT-103, NIT-109, NIT-110, NIT-112, NIT-113, PRO-E0261, PRO-E0262 and PRO-E0264 gave no conversion, NIT-102, NIT-104 and PRO-E0263 gave similar amounts of acid and amide products, as summarized in Fig. 5. | ||
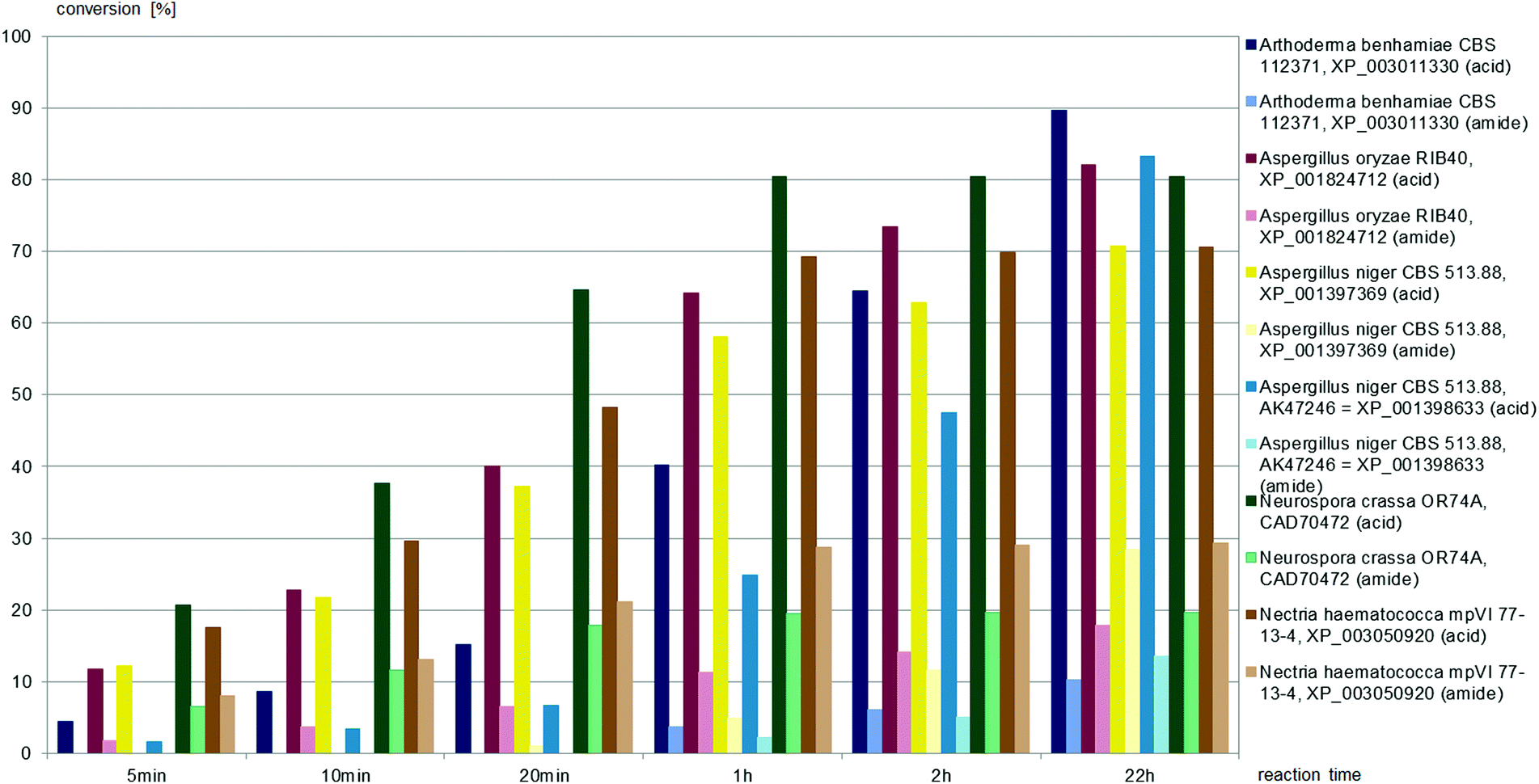 | ||
| Fig. 7 Screening results of (±)-trans-2,4-diphenyl-4,5-dihydrooxazole-5-carbonitrile (1.0 mM) with fungal nitrilases (9.5 mL cell suspension of appropriate optical density in 100 mL buffer). | ||
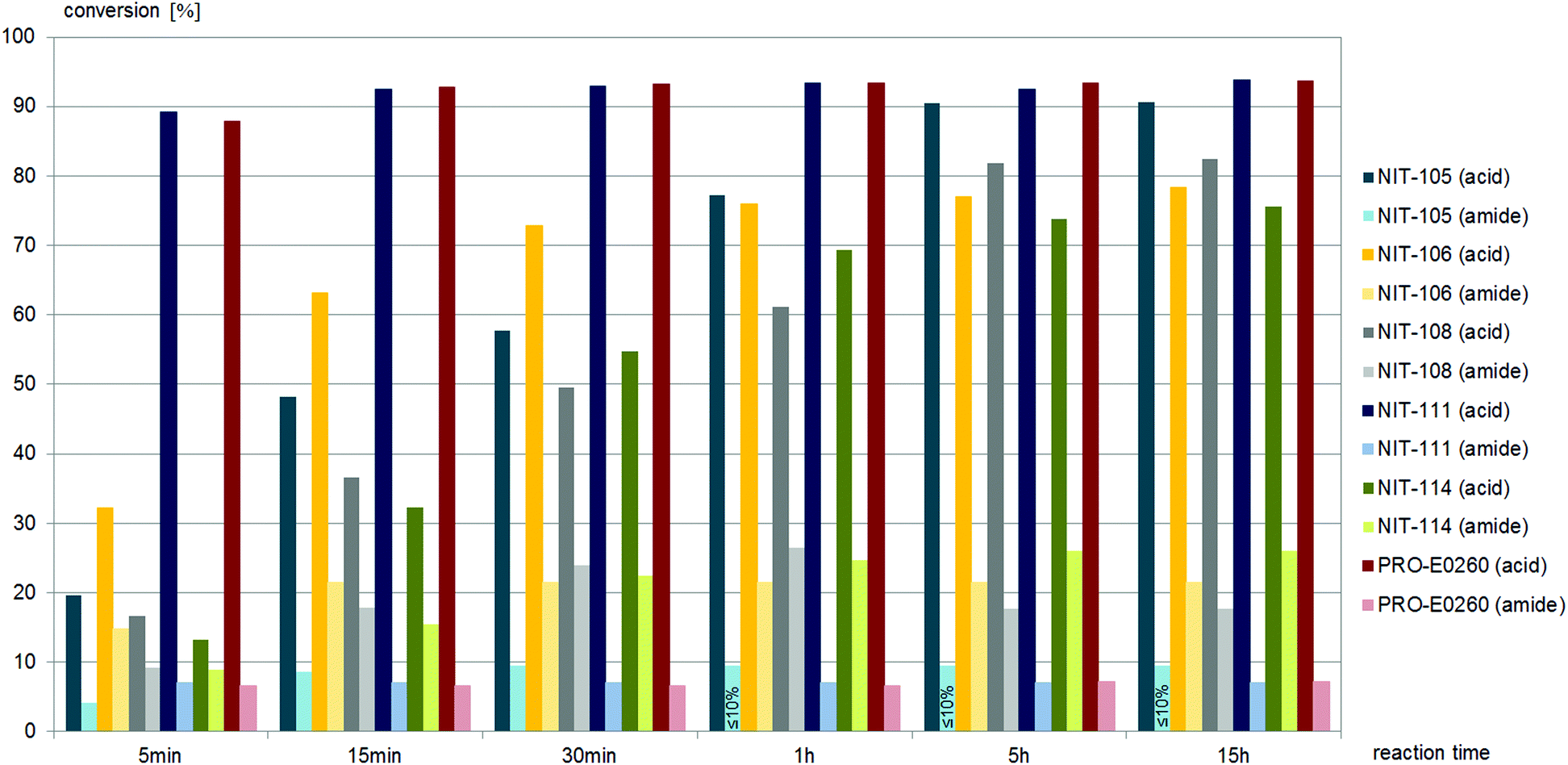
Nine different commercially available nitrilases accepted (±)-trans-2,4-diphenyl-4,5-dihydrooxazole-5-carbonitrile, (±)-trans-1 as the substrate. Reactions giving similar amounts of acid and amide are summarised in Fig. 5, while reactions giving carboxylic acid as main product are summarised in Fig. 6. NIT-106, NIT-108 and NIT114 gave 15–25% of amide by-product after 15 hours reaction time, NIT-105, NIT-111 and PRO-E0260 gave only minor amounts of less than 10% of amide by-product.
(±)-trans-2,4-Diphenyl-4,5-dihydrooxazole-5-carbonitrile, (±)-trans-1, was hydrolysed to the carboxylic acid by all six fungal nitrilases, as depicted in Fig. 7. The nitrilases from Aspergillus oryzae, Neurospora crassa, and Nectria haematococca produced both carboxylic acid and amide throughout the reaction time. The nitrilases from Arthoderma benhamiae and Aspergillus niger hydrolysed (±)-trans-1 to the carboxylic acid at the beginning of the reaction, amide was only formed after approximately 25% conversion to the acid.
All fungal nitrilases used here were previously characterised as arylacetonitrilases with preference for phenylacetonitrile and (R,S)-mandelonitrile as substrates. Their similar substrate specificities correspond with considerable identities of their amino acid sequences (mostly over 50%). Nevertheless, differences have previously been observed between their specific activities, enantioselectivities and chemoselectivities for (R,S)-mandelonitrile.28,32
The ee-values obtained in the nitrilase catalysed reactions of (±)-trans-1 were below 80%.33 The moderate ee-values might be explained by a number of reasons. Racemisation and/or epimerisation of the compounds might occur during or after the biotransformation. Stopping the reaction by precipitating the enzyme might not have been efficient enough, as the protein might not have been quantitatively precipitated. Reactions could be most efficiently stopped by using immobilized enzyme which can be easily removed from the reaction mixture. In commercially available enzymes, additives in the enzyme preparation might influence enantioselectivity and ratio of the acid and amide products. The additive dithiothreitol (DTT) is added to nitrilases to prevent disulphide bond formation of the catalytically active cysteine, though it has been previously proven to catalyse the non-stereoselective hydrolysis of nitriles to amides.34 In recent examples, the presence of organic solvents has been shown to enhance activity and stereoselectivity in nitrilase catalysed biotransformations.35 The influence of organic solvents on enzymatic nitrile hydrolysis is poorly studied so far, especially compared to other hydrolytic enzymes, such as lipases and esterases.36 Influences on the stereoselectivity of the nitrilase and nitrile hydratase catalysed reactions need to be further investigated to achieve a reliable nitrile transforming biocatalyst for the synthesis of the taxol sidechain.
A preparative scale biotransformation of (±)-trans-1 was carried out with with whole cells of E. coli expressing the nitrilase from Neurospora crassa OR74A. Approximately 50% conversion of (±)-trans-1 were achieved after three hours. However, the enantioselectivity of the reaction was not satisfactory, with an er37 of 1/1.6 (4S,5R)-2,4-diphenyl-4,5-dihydro-1,3-dihydro-1,3-oxazole-5-carboxylic acid to enantiomer.
Conclusions
In this work, an extensive set of nitrilases and nitrile hydratases was screened towards their activity and selectivity in the hydrolysis of two taxol sidechain precursors and their epimers. Both sidechain precursors were designed to utilize the enzymatic step as final step in the synthesis. A number of nitrilases and nitrile hydratases catalysed the biotransformation of both sidechain precursors and their epimers.All nitrilases and nitrile hydratases tested showed similar substrate specificity towards the taxol sidechain precursor (±)-syn-N-(2-cyano-2-hydroxy-1-phenylethyl)benzamide (±)-syn-2, and its epimer (±)-anti-2. The openchain compounds (±)-syn-2 and (±)-anti-2 were converted by all Co-type nitrile hydratases tested, while the Fe-type nitrile hydratase PRO-E0256 did not convert (±)-syn-2 or (±)-anti-2. Three commercially available nitrilases were found that hydrolyse the openchain precursor (±)-syn-2. Nitrilase PRO-E0263 gave less than 10% of amide by-product. The same nitrilases also hydrolysed (±)-anti-2. Here, PRO-E0263 produced the desired carboxylic acid as only product from (±)-anti-2.
(±)-cis- and (±)-trans-2,4-Diphenyl-4,5-dihydrooxazole-5-carbonitrile, (±)-cis-1 and (±)-trans-1, were excellent substrates for all Co- and Fe-nitrile hydratases tested, giving the corresponding amides in 30–100% yield. The substrate specificity of nitrilases towards (±)-trans-1 and its epimer (±)-cis-1 differed quite drastically. Only two of 23 nitrilases accepted (±)-cis-1 as substrate, the amide was the major product formed. The epimer (±)-trans-1, however, was converted by 18 different nitrilases, where three nitrilases gave similar amounts of acid and amide as product, and the remaining 15 gave the desired carboxylic acid as main product. Three commercially available nitrilases (NIT-105, NIT-111, PRO-E0260) gave less than 10% of amide by-product, four of the six fungal nitrilases (Arthoderma benhamiae, Aspergillus niger, Neurospora crassa, Aspergillus oryzae) gave less than 20% of amide by-product.
Fungal nitrilases, first of all arylacetonitrilases, have almost been neglected until recently due to their low production in wild-type strains, but genome mining has enabled the discovery of the first enzymes of this type. Their biocatalytic potential was confirmed with a new type of nitrile substrates in this work, and indicated further screening of this enzyme group would be promising.
This work presents the first investigation of nitrilases as tools for the chemo-enzymatic synthesis of the taxol sidechain. Nitrile hydratases (especially Co-type nitrile hydratases) and arylacetonitrilases (both, bacterial and fungal) were found to be suitable catalysts for one or both taxol sidechain precursors tested. The stereoselectivity of the enzyme catalysed reactions will need to be further investigated and improved for a possible application of these nitrile transforming enzymes in an enantioselective, chemo-enzymatic synthesis of the taxol sidechain.
Experimental
General methods
Thin layer chromatography was carried out with precoated aluminium silica gel 60 F254 plates, column chromatography with Merck Silica Gel 60 (0.040–0.063 mm). 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III with autosampler (1H NMR 300.36 MHz, 13C NMR 75.53 MHz). Chemical shifts for 1H NMR are reported in ppm relative to Me4Si as internal standard. Assignments were supported by HSQC and/or HMBC measurements. For all screening reactions, including blank reactions, multiple parallel determinations (at least two) were run. HPLC-MS analysis was carried out on an Agilent 1200 series using a Merck Purospher STAR® (RP-18e, 5 μm, LiChroCART® 250-4) column (flow 0.9 mL min−1, injection 10 μL) or on a Waters 600 with a PDA detector 996 using a Chromolith® SpeedROD RP-18 endcapped 50-4.6 column (flow 2 mL min−1, injection 10 μL), and 0.1% v/v acetic acid in water and acetonitrile as eluents. Determination of ee-values was performed by HPLC analysis on an Agilent 1100 series using a Chiralpak AD-H (Daicel Chemical Industries, Ltd, 0.46 cm × 25 cm) column, ethanol as eluent, flow 0.55 mL min−1, column oven temperature 40 °C or a Chiralpak AGP column (Daicel Chemical Industries, Ltd, 150 × 4 mm, 5 μm) and 90% acetate buffer (100 mM, pH 4.4), 10% acetonitrile as eluents, flow 0.9 mL min−1, column oven temperature 25 °C or a Chiralpak AGP column (Daicel Chemical Industries, Ltd, 150 × 4 mm, 5 μm) 10% iso-propanol and 90% citrate buffer (50 mM, pH 4.4), flow 0.9 mL min−1, column oven 22 °C.25,33Substrate synthesis
Biotransformation reactions
Acknowledgements
The authors wish to thank Jasmin Resch and Carina Hasenöhrl for skilful assistance in substrate synthesis and screening reactions. Financial support from project P504/11/0394 (Czech Science Foundation) is gratefully acknowledged.Notes and references
- (a) K. C. Nicolaou, W.-M. Dai and R. K. Guy, Angew. Chem., Int. Ed. Engl., 1994, 33, 15–44 CrossRef PubMed; (b) K. C. Nicolaou and R. K. Guy, Angew. Chem., Int. Ed. Engl., 1995, 34, 2079–2090 CrossRef CAS PubMed.
- H. Oettle, Cancer Treat. Rev., 2014, 40, 1039–1047 CrossRef PubMed.
- D. Lorusso, F. Petrelli, A. Coinu, F. Raspagliesi and S. Barni, Gynecol. Oncol., 2014, 133, 117–123 CrossRef CAS PubMed.
- Q. Chu, M. Vincent, D. Logan, J. A. Mackay and W. K. Evans, Lung Cancer, 2005, 50, 355–374 CrossRef PubMed.
- V. Barsch, Das Taxol Buch, Georg Thieme Verlag, Stuttgart, 2004 Search PubMed.
- S. B. Horwitz, J. Fant and P. B. Schiff, Nature, 1979, 277, 665–667 CrossRef PubMed.
- S. Sharma, C. Lagisetti, B. Poliks, R. M. Coates, D. G. I. Kingston and S. Bane, Biochem., 2013, 52, 2328–2336 CrossRef CAS PubMed.
- M. Magnani, G. Maccari, J. M. Andreu, J. F. Díaz and M. Botta, FEBS J., 2009, 276, 2701–2712 CrossRef CAS PubMed.
- (a) R. A. Jibodh, J. S. Lagas, B. Nuijen, J. H. Beijnen and J. H. M. Schellens, Eur. J. Pharmacol., 2013, 717, 40–46 CrossRef CAS PubMed; (b) V. A. de Weger, J. H. Beijnen and J. H. M. Schellens, Anti-Cancer Drugs, 2014, 25, 488–494 CrossRef CAS PubMed.
- (a) J. A. Yared and K. H. R. Tkaczuk, Drug Des., Dev. Ther., 2012, 6, 371–384 CAS; (b) I. Ojima, R. Geney, I. M. Ungureanu and D. Li, IUBMB Life, 2002, 53, 269–274 CrossRef CAS PubMed; (c) G. A. Orr, P. Verdier-Pinard, H. McDaid and S. Band Horwitz, Oncogene, 2003, 22, 728–7295 CrossRef PubMed.
- Recent examples include: (a) S. Zhao, W. Wang, Y. Huang, Y. Fu and Y. Cheng, Med. Chem. Commun., 2014, 5, 1658–1663 RSC; (b) A. G. Cheetham, P. Zhang, Y.-A. Lin, R. Lin and H. Cui, J. Mater. Chem. B, 2014, 2, 7316–7326 RSC; (c) E. Bouvier, S. Thirot, F. Schmidt and C. Monneret, Org. Biomol. Chem., 2003, 1, 3343–3352 RSC; (d) X. Hu, J. Li, W. Lin, Y. Huang, X. Jing and Z. Xie, RSC Adv., 2014, 4, 38405–38411 RSC; (e) M. V. Trmcic, R. V. Matovic, G. I. Tovilovic, B. Z. Ristic, V. S. Trajkovic, Z. B. Ferjancic and R. N. Saicic, Org. Biomol. Chem., 2012, 10, 4933–4942 RSC.
- (a) D. G. I. Kingston, Phytochemistry, 2007, 68, 1844–1854 CrossRef CAS PubMed; (b) Y.-F. Wang, Q.-W. Shi, M. Dong, H. Kiyota, Y.-C. Gu and B. Cong, Chem. Rev., 2011, 111, 7652–7709 CrossRef CAS PubMed and references cited therein; (c) K. C. Nicolaou and R. A. Valiulin, Org. Biomol. Chem., 2013, 11, 4154–4163 RSC; (d) W.-S. Fang and X.-T. Liang, Mini-Rev. Med. Chem., 2005, 5, 1–12 CrossRef CAS.
- (a) O. Expósito, M. Bonfill, E. Moyano, M. Onrubia, M. H. Mirjalili, R. M. Cusidó and J. Palazóna, Anti-Cancer Agents Med. Chem., 2009, 9, 109–121 CrossRef; (b) F. Guéritte-Voegelein, D. Guénard, F. Lavelle, M.-T. Le Goff, L. Mangatal and P. Potier, J. Med. Chem., 1991, 34, 992–998 CrossRef; (c) C. Colderch, Y. Tang, J. Klett, S.-E. Zhang, Y.-T. Ma, W. Shaorong, R. Matesanz, B. Pera, A. Canales, J. Jíménez-Barbero, A. Morreale, J. F. Díaz, W.-S. Fang and F. Gago, Org. Biomol. Chem., 2013, 11, 3046–3056 RSC; (d) S.-H. Chen, M. Xue, S. Huang, B. H. Long, C. A. Fairchild, W. C. Rose, J. F. Kadow and D. Vyas, Bioorg. Med. Chem. Lett., 1997, 7, 3057–3062 CrossRef CAS; (e) M. Xue, B. H. Long, C. Fairchild, K. Johnston, W. C. Rose, J. F. Kadow, D. M. Vyas and S.-H. Chen, Bioorg. Med. Chem. Lett., 2000, 10, 1327–1331 CrossRef CAS; (f) E. J. Roh, D. Kim, C. O. Lee, S. U. Choi and C. E. Song, Bioorg. Med. Chem., 2002, 10, 3145–3151 CrossRef CAS.
- Selected references: (a) J.-N. Denis, A. E. Greene, A. A. Serra and M. J. Luche, J. Org. Chem., 1986, 51, 46–50 CrossRef CAS; (b) J.-N. Denis, A. Correa and A. E. Greene, J. Org. Chem., 1990, 55, 1957–1959 CrossRef CAS; (c) L. Deng and E. N. Jacobsen, J. Org. Chem., 1992, 57, 4320–4323 CrossRef CAS; (d) Z.-M. Wang, H. C. Kolb and K. B. Sharpless, J. Org. Chem., 1994, 59, 5104–5105 CrossRef CAS; (e) M. E. Bunnage, S. G. Davies and C. J. Goodwin, J. Chem. Soc., Perkin Trans. 1, 1994, 2385–2391 RSC; (f) R. P. Srivastava, J. K. Zjawiony, J. R. Peterson and J. D. McChesney, Tetrahedron: Asymmetry, 1994, 5, 1683–1688 CrossRef CAS; (g) Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristi, Wiley-VCH, Weinheim, 1996 Search PubMed; (h) S. H. Kang, C. M. Kim and J.-H. Youn, Tetrahedron Lett., 1999, 40, 3581–3582 CrossRef CAS; (i) S.-H. Lee, J. Yoon, K. Nakamura and Y.-S. Lee, Org. Lett., 2000, 2, 1243–1246 CrossRef CAS; (j) Z. Zhou, X. Mei, J. Chang and D. Feng, Synth. Commun., 2001, 31, 3609–3615 CrossRef CAS; (k) J. M. Andrés, M. A. Martínez, R. Pedrosa and A. Pérez-Encabo, Tetrahedron: Asymmetry, 2001, 12, 347–253 CrossRef; (l) M. V. Voronkov, A. V. Gontcharov and Z.-M. Wang, Tetrahedron Lett., 2003, 44, 407–409 CrossRef CAS; (m) Z. Zhou and X. Mei, Synth. Commun., 2003, 33, 723–728 CrossRef CAS; (n) J.-M. Lee, H.-S. Lim, K.-C. Seo and S.-K. Chung, Tetrahedron: Asymmetry, 2003, 14, 3639–3641 CrossRef CAS PubMed; (o) J. C. Borah, S. Gogoi, J. Boruwa, B. Kalita and N. C. Barua, Tetrahedron Lett., 2004, 45, 3689–3691 CrossRef CAS PubMed; (p) W.-J. Liu, B.-D. Lv and L.-Z. Gong, Angew. Chem., Int. Ed., 2009, 48, 6503–6506 CrossRef CAS PubMed; (q) P. Dziedzic, P. Schyman, M. Kullberg and A. Córdova, Chem. – Eur. J., 2009, 15, 4044–4048 CrossRef CAS PubMed; (r) L. Munoz-Hernández and J. A. Soderquist, Org. Lett., 2009, 11, 2571–2574 CrossRef PubMed; (s) Y. Qian, X. Xu, L. Jiang, D. Prajapati and W. Hu, J. Org. Chem., 2010, 75, 7483–7486 CrossRef CAS PubMed; (t) J.-L. Jiang, M. Yao and C.-D. Lu, Org. Lett., 2014, 16, 318–321 CrossRef CAS PubMed.
- (a) R. N. Patel, Annu. Rev. Microbiol., 1998, 98, 361–395 CrossRef PubMed; (b) M. Zagozda and J. Plenkiewicz, Tetrahedron: Asymmetry, 2008, 19, 1455–1460 CrossRef CAS PubMed.
- Selected references: (a) G. Cardillo, L. Gentilucci, A. Tolomelli and C. Tomasini, J. Org. Chem., 1998, 63, 2351–2353 CrossRef CAS; (b) G. Cardillo, A. Tolomelli and C. Tomasini, Eur. J. Org. Chem., 1999, 155–161 CrossRef CAS.
- Selected references: (a) J. C. Borah, J. Boruwa and N. C. Barua, Curr. Org. Synth., 2007, 4, 175–199 CrossRef CAS; (b) E. Forró and F. Fülöp, Tetrahedron: Asymmetry, 2010, 21, 637–639 CrossRef PubMed; (c) R. Brieva, J. Z. Crich and C. J. Sih, J. Org. Chem., 1993, 58, 1068–1075 CrossRef CAS; (d) H. Hönig, P. Seufer-Wasserthal and H. Weber, Tetrahedron, 1990, 46, 3841–3850 CrossRef; (e) H. Hamamoto, V. A. Mamedov, M. Kitamoto, N. Hayashi and S. Tsuboi, Tetrahedron: Asymmetry, 2000, 11, 4485–4407 CrossRef CAS; (f) R. N. Patel, A. Banerjee, R. Y. Ko, J. M. Howell, W.-S. Li, F. T. Comezoglu, R. A. Partyka and L. Szarka, Biotechnol. Appl. Biochem., 1994, 20, 23–33 CAS; (g) D.-M. Gou, Y.-C. Liu and C.-S. Chen, J. Org. Chem., 1993, 58, 1287–1289 CrossRef CAS; (h) C. J. Shi, in Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristi, Wiley-VCH, Weinheim, 1996, ch. 20, pp 433–441 Search PubMed; (i) D. Lee and M.-J. Kim, Tetrahedron Lett., 1998, 39, 2163–2166 CrossRef CAS; (j) P. G. M. Wuts, R. l. Gu and J. M. Northuis, Tetrahedron: Asymmetry, 2000, 11, 2117–2123 CrossRef CAS; (k) T. Mandai, T. Oshitari and M. Susowake, Synlett, 2002, 1665–1668 CrossRef CAS PubMed; (l) N. Anand, M. Kapoor, S. Koul, S. C. Taneja, R. L. Sharma and G. M. Qazi, Tetrahedron: Asymmetry, 2004, 15, 3131–3138 CrossRef CAS PubMed; (m) D. Castagnolo, S. Armaroli, F. Corelli and M. Botta, Tetrahedron: Asymmetry, 2004, 15, 941–949 CrossRef CAS PubMed; (n) T. Oshitari and T. Mandai, Synlett, 2003, 2374–2376 CAS; (o) E. Forró and F. Fülöp, Eur. J. Org. Chem., 2010, 3074–3079 CrossRef PubMed.
- Selected references: (a) R. N. Patel, A. Banerjee, J. M. Howell, C. G. McNamee, D. Brozozowski, D. Mirfakhrae, V. Nanduri, J. K. Thottathil and L. J. Szarka, Tetrahedron: Asymmetry, 1993, 4, 2069–2084 CrossRef CAS; (b) J. Kearns and M. M. Kayser, Tetrahedron Lett., 1994, 35, 2845–2848 CrossRef CAS; (c) M. M. Kayser, M. D. Mihovilovic, J. Kearns, A. Feicht and J. D. Stewart, J. Org. Chem., 1999, 64, 6603–6608 CrossRef CAS PubMed; (d) J. A. R. Rodrigues, H. M. S. Milagre, C. D. F. Milagre and P. J. S. Moran, Tetrahedron: Asymmetry, 2005, 16, 3099–3106 CrossRef CAS PubMed; (e) I. Rimoldi, M. Pellizzoni, G. Facchetti, F. Molinari, D. Zerla and R. Gandolfi, Tetrahedron: Asymmetry, 2011, 22, 2110–2116 CrossRef CAS PubMed.
- Reviews: (a) R. Thuku, D. Brady, M. Benedik and B. Swell, J. Appl. Microbiol., 2009, 106, 703–727 CrossRef CAS PubMed; (b) L. Martínková and V. Kȓen, Curr. Opin. Chem. Biol., 2010, 14, 130–137 CrossRef PubMed; (c) J.-S. Gong, Z.-M. Lu, H. Li, J.-S. Shi, Y.-M. Zhou and Z.-H. Xu, Microb. Cell Fact., 2012, 11, 142–160 CrossRef CAS PubMed; (d) J.-S. Gong, Z.-M. Lu, H. Li, J.-S. Shi, Y.-M. Zhou and Z.-H. Xu, Appl. Microbiol. Biotechnol., 2013, 97, 6603–6611 CrossRef CAS PubMed. Selected articles: (e) D. Williamson, K. Dent, B. Weber, A. Varsani, J. Frederick, R. Thuku, R. Cameron, J. Heerden, D. Cowan and B. Sewell, Appl. Microbiol. Biotechnol., 2010, 88, 143–153 CrossRef CAS PubMed; (f) J. E. Raczynska, C. E. Vorgias, G. Antranikian and W. Rypniewski, J. Struct. Biol., 2011, 173, 294–302 CrossRef CAS PubMed; (g) M. Winkler, O. Kaplan, V. Vejvoda, N. Klempier and L. Martínková, J. Mol. Catal. B: Enzym., 2009, 59, 243–247 CrossRef CAS PubMed; (h) D. Brady, A. Beeton, J. Zeevaart, C. Kgaje, F. van Rantwijk and R. A. Sheldon, Appl. Microbiol. Biotechnol., 2004, 64, 76–85 CrossRef CAS PubMed; (i) M. Winkler, L. Martínkova, A. C. Knall, S. Krahulec and N. Klempier, Tetrahedron, 2005, 61, 4249–4260 CrossRef CAS PubMed.
- (a) B. C. Fernandes, C. Mateo, C. Kiziak, A. Churma, J. Wacker, F. van Rantwijk, A. Stolz and R. Sheldon, Adv. Synth. Catal., 2006, 348, 2597–2603 CrossRef CAS PubMed; (b) D. Williamson, K. Dent, B. Weber, A. Varsani, J. Frederick, R. Thuku, R. Cameron, J. Heerden, D. Cowan and B. Sewell, Appl. Microbiol. Biotechnol., 2010, 88, 143–153 CrossRef CAS PubMed.
- Selected references: (a) C. D. Mathew, T. Nagasawa, M. Kobayashi and H. Yamada, Appl. Environ. Microbiol., 1988, 54, 1030–1032 CAS; (b) S. Prasad and T. C. Bhalla, Biotechnol. Adv., 2010, 28, 725–741 CrossRef CAS PubMed; (c) W. Huang, J. Jia, J. Cummings, M. Nelson, G. Schneider and Y. Lindqvist, Structure, 1997, 5, 691–699 CrossRef CAS; (d) S. van Pelt, M. Zhang, L. Otten, J. Holt, D. Sorotkin, F. van Rantwijk, G. W. Black, J. J. Perry and R. Sheldon, Org. Biomol. Chem., 2011, 9, 3011–3019 RSC.
- Procedures for the synthesis of the acid and amide reference materials and characterization data are available in the ESI.†.
- Selected references: (a) A. Jonczyk and T. Zomerfeld, Tetrahedron Lett., 2003, 44, 2359–2361 CrossRef CAS; (b) F. W. Bachelor and R. K. Bansal, J. Org. Chem., 1969, 34, 3600–3604 CrossRef CAS; (c) G. Liu, D. Zhang, J. Li, G. Xu and J. Sun, Org. Biomol. Chem., 2013, 11, 900–904 RSC; (d) W.-J. Liu, B.-D. Lv and L.-Z. Gong, Angew. Chem., Int. Ed., 2009, 48, 6503–6506 CrossRef CAS PubMed.
- Selected references: (a) B. D. Feske, I. A. Kaluzna and J. D. Stewart, J. Org. Chem., 2005, 70, 9654–9657 CrossRef CAS PubMed; (b) X. Deng, J. Su, Y. Zhao, L.-Y. Peng, Y. Li, Z.-J. Yao and Q.-S. Zhao, Eur. J. Med. Chem., 2011, 46, 4238–4244 CrossRef CAS PubMed; (c) M. V. Voronkov, A. V. Gontcharov, Z.-M. Wang, P. F. Richardson and H. C. Kolb, Tetrahedron, 2004, 60, 9043–9048 CrossRef CAS PubMed; (d) F. Benfatti, G. Cardillo, L. Gentilucci, A. Tolomelli, M. Monari and F. Piccinelli, Adv. Synth. Catal., 2007, 349, 1256–1264 CrossRef CAS PubMed; (e) J. L. García Ruano and C. García Paredes, Tetrahedron Lett., 2000, 41, 5357–5361 CrossRef; (f) F. Li, Z.-H. Wang, L. Zhao and F.-E. Chen, Synlett, 2011, 2883–2885 CAS.
- Representative HPLC chromatograms are available in the ESI.†.
- Additional information on the commercial enzyme preparations by Prozomix, Ltd. can be found in the ESI† or online at http://www.prozomix.com/.
- Additional information on the commercial enzyme preparations by Codexis, Inc. can be found in the ESI† or online at http://www.codexis.com/.
- O. Kaplan, A. B. Vesela, A. Petříčková, F. Pasquarelli, M. Pičmanová, A. Rinágelová, T. C. Bhalla, M. Pátek and L. Martínková, Mol. Biotechnol., 2013, 54, 996–1003 CrossRef CAS PubMed.
- D. Zhu, C. Mukherjee, E. R. Biehl and L. Hua, J. Biotechnol., 2007, 129, 645–650 CrossRef CAS PubMed.
- D. Zhu, C. Mukherjee, Y. Yang, B. E. Rios, D. T. Gallagher, N. N. Smith, E. R. Biehl and L. Hua, J. Biotechnol., 2008, 133, 327–333 CrossRef CAS PubMed.
- (a) D. G. I. Kingston, A. G. Chaudhary, A. A. L. Gunatilaka and M. L. Middleton, Tetrahedron Lett., 1994, 35, 4483–4484 CrossRef CAS; (b) E. Baloglu and D. G. I. Kingston, J. Nat. Prod., 1999, 62, 1068–1071 CrossRef CAS PubMed.
- (a) A. Petříčková, O. Sosedov, S. Baum, A. Stolz and L. Martínková, J. Mol. Catal. B: Enzym., 2012, 77, 74–80 CrossRef PubMed; (b) A. B. Veselá, A. Petříčková, P. Weyrauch and L. Martínková, Biocatal. Biotransform., 2013, 31, 49–56 CrossRef.
- ee-values/er-values (see ref. 37) were determined by chiral HPLC, methods and representative chromatograms are available in the ESI.†.
- M. Winkler, A. Glieder and N. Klempier, Chem. Commun., 2006, 1298–1300 RSC.
- Selected references: (a) N. Layh and A. Willets, Biotechnol. Lett., 1998, 20, 329–331 CrossRef CAS; (b) P. Kaul and U. C. Banerjee, J. Ind. Microbiol. Biotechnol., 2008, 35, 713–720 CrossRef CAS PubMed; (c) Z.-J. Zhang, J. Pan, J.-F. Liu, J.-H. Xu, Y.-C. He and Y.-Y. Liu, J. Biotechnol., 2011, 152, 24–29 CrossRef CAS PubMed; (d) C. Vergne-Vaxelaire, F. Bordier, A. Fossey, M. Besnard-Gonnet, A. Debard, A. Mariage, V. Pellouin, A. Perret, J.-L. Petit, M. Stam, M. Salanoubat, J. Weissenbach, V. De Berardinis and A. Zaparucha, Adv. Synth. Catal., 2013, 355, 1763–1779 CrossRef CAS PubMed; (e) A. B. Veselá, A. Křenková and L. Martínková, BioTech2014, Prague, Czech Republic, 2014, Poster: production of (R)-mandelic acid by nitrilases from filamentous fungi – comparison of the co-solvent and fed-batch setup Search PubMed.
- Review: G. Carrea and S. Riva, Angew. Chem., Int. Ed., 2000, 39, 2226–2254 CrossRef CAS . Selected references: (a) C. B. Faulds, M. Pérez-Boada and A. T. Martínez, Bioresour. Technol., 2011, 102, 4962–4967 CrossRef CAS PubMed; (b) Z. Guo, M. K. Y. Wong, M. R. Hickey, B. P. Patel, X. Qian and A. Goswami, Org. Process Res. Dev., 2014, 18, 774–780 CrossRef CAS; (c) H. J. Park, J. C. Joo, K. Park, Y. H. Kim and Y. J. Yoo, J. Biotechnol., 2013, 163, 346–352 CrossRef CAS PubMed; (d) J. Kuper, T. Seng, D. Roccatano, M. Wilmanns and U. Schwaneberg, J. Am. Chem. Soc., 2007, 129, 5786–5787 CrossRef CAS PubMed; (e) T. Gerhards, U. Mackfeld, M. Bocola, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed; (f) T. Koudelakova, R. Chaloupkova, J. Brezovsky, Z. Prokop, E. Sebestova, M. Hesseler, M. Khabiri, M. Plevaka, D. Kulik, I. Kuta Smatanova, P. Rezacova, R. Ettrich, U. T. Bornscheuer and J. Damborsky, Angew. Chem., Int. Ed., 2013, 52, 1959–1963 CrossRef CAS PubMed.
- R. E. Gawley, J. Org. Chem., 2006, 71, 2411–2416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all compounds, experimental procedures for acid and amide reference materials, representative HPLC chromatograms of biotransformation reactions from non-chiral and chiral HPLC measurements, biotransformations with cosolvents, information on commercial enzyme preparations. See DOI: 10.1039/c5ob01191d |
| This journal is © The Royal Society of Chemistry 2015 |