
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A Sn atom-economical approach toward arylstannanes: Ni-catalysed stannylation of aryl halides using Bu3SnOMe†
Kimihiro
Komeyama
*,
Ryota
Asakura
and
Ken
Takaki
Department of Applied Chemistry, Graduate School of Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima City, Hiroshima 739-8527, Japan. E-mail: kkome@hiroshima-u.ac.jp
First published on 8th July 2015
Abstract
Stannylation of carbon–halogen bonds is one of the most promising and straightforward approaches for the preparation of organostannane compounds. Although a wide variety of methods are now available, all protocols require the use of highly nucleophilic organometals or wasteful stannyl sources like distannanes. Here, we report a new nickel-catalysed stannylation of aryl and alkenyl-halides using Bu3SnOMe as a stannyl source to afford aryl and vinyl-stannanes, respectively. This method enables the stannylation of not only bromides, but also chlorides and triflates to furnish functionalized aryl- and alkenyl-stannanes without the release of wasteful and toxic stannyl byproducts.
Arylstannanes are useful synthetic intermediates because of their versatility in the construction of aryl-C,1 -NR2,2 -F,3 and -OCF3
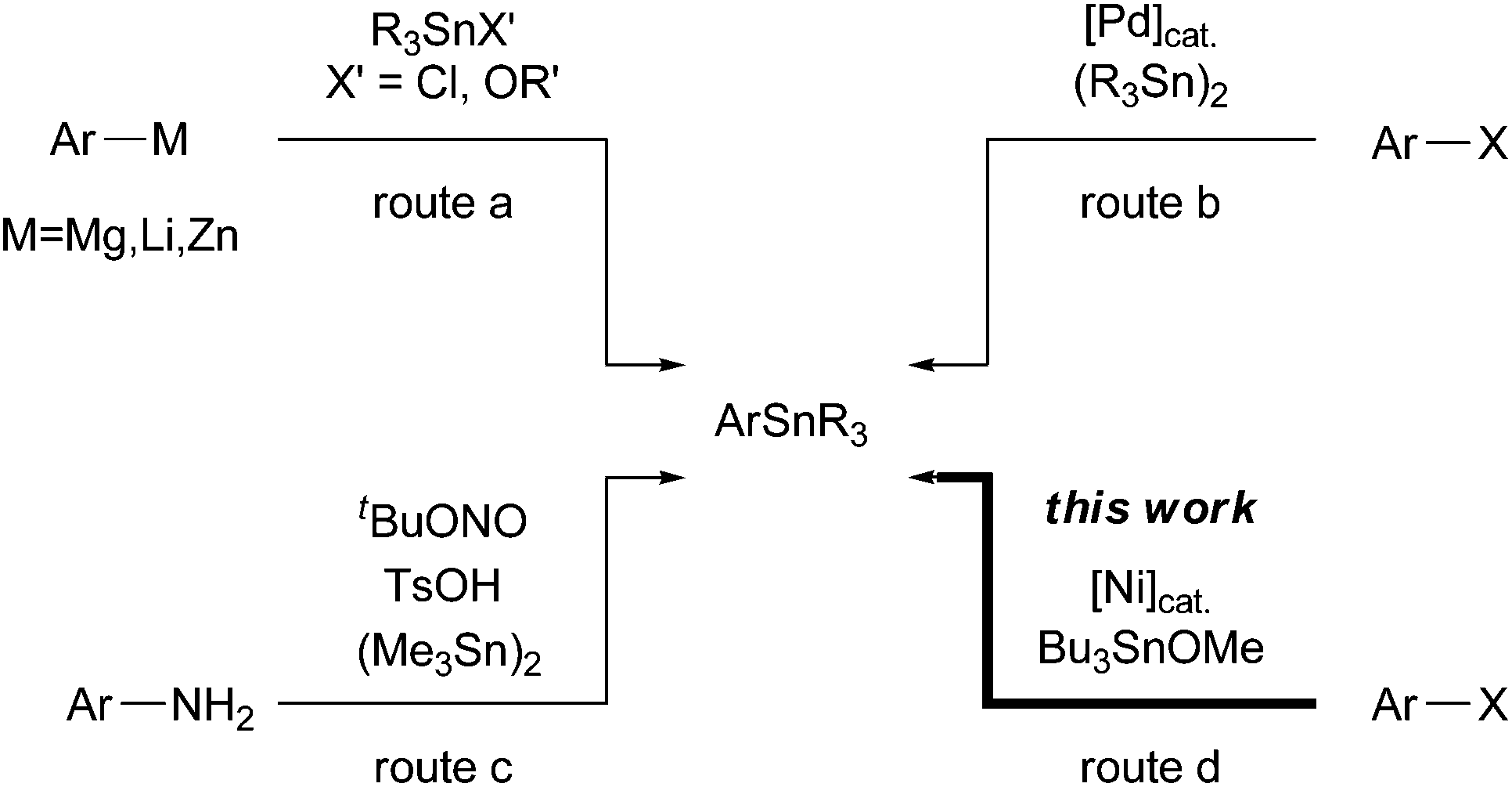
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 bonds. The most promising route to afford arylstannanes relies on the trapping of arylmetal species (Mg, Li, and Zn)3a,5 using trialkylstannyl electrophiles R3SnX′ (route a, Scheme 1). However, these protocols have some drawbacks: poor functional group tolerance and/or delicate conditions for the preparation of arylmetal species. In contrast, the Pd-catalysed stannylation of aryl halides (Scheme 1b)6 and the recently proposed Sandmeyer-type reaction of anilines (Scheme 1c)7 using hexaalkyl distannanes have been demonstrated as alternative procedures. Although both of the above methods are useful and powerful for functionalized arylstannane synthesis, the release of highly toxic stannyl byproducts is unavoidable. These disadvantages drastically reduce the efficiency of organostannane chemistry in both academia and industrial pursuits.
4 bonds. The most promising route to afford arylstannanes relies on the trapping of arylmetal species (Mg, Li, and Zn)3a,5 using trialkylstannyl electrophiles R3SnX′ (route a, Scheme 1). However, these protocols have some drawbacks: poor functional group tolerance and/or delicate conditions for the preparation of arylmetal species. In contrast, the Pd-catalysed stannylation of aryl halides (Scheme 1b)6 and the recently proposed Sandmeyer-type reaction of anilines (Scheme 1c)7 using hexaalkyl distannanes have been demonstrated as alternative procedures. Although both of the above methods are useful and powerful for functionalized arylstannane synthesis, the release of highly toxic stannyl byproducts is unavoidable. These disadvantages drastically reduce the efficiency of organostannane chemistry in both academia and industrial pursuits.
 | ||
| Scheme 1 Representative synthetic method for arylstannanes. | ||
On the other hand, a few transition metal-catalysed reactions involve nucleophilic stannylation processes using trialkylstannyl alkoxides ROSn(alkyl)3 as terminal electrophiles. For instance, the interception of alkynylzinc8 and alkenylcopper intermediates9 generated in situ using stannyl alkoxides quickly transforms into the corresponding alkynyl or alkenyl stannane compounds, respectively.10 However, these catalytic processes have never been applied to the stannylation of ubiquitous carbon (sp2)–halogen bonds because of their low reactivity for zinc and copper complexes. In this paper, we report a Ni-catalysed stannylation of aryl halides using Bu3SnOMe in the presence of manganese powder (Scheme 1d). This stannylation process could be an ideal and straightforward approach to afford aryl or vinyl stannanes from both organohalides and organotriflates. The proposed process possesses the following advantages: (1) available substrates, (2) a broad scope of functional groups, and (3) Sn atom-economy without the release of wasteful toxic inorganic stannyl residues.
Initially, we explored the suitable reaction conditions for the stannylation of aryl bromides 1a and 1b using stannyl electrophile as model substrates based on the previous work by Tsuji and Fujihara (Table 1).11 When 1a was treated with Bu3SnOMe (1.2 equiv.) in the presence of NiBr2 (10 mol%), 2,2′-bipyridine (bpy, 10 mol%), and Mn powder (2.0 equiv. treated with 20 mol% of chlorotrimethylsilane), arylstannane 2a was afforded in 72% yield along with biaryl 3a in 14% yield (entry 1).12 The preformed [NiBr2(bpy)] complex exhibited higher catalytic performance to afford 2a in 91% yield (entry 2).13 The 2,2′-bipyridine ligand and Ni catalyst were crucial (entries 3–7). The replacement of Mn or 1a with Zn or 4-iodoanisole, respectively, induced the homocoupling reaction (entries 8 and 9). In contrast, the optimized conditions (entry 2) were not sufficient for the stannylation of electron-poor aryl bromides like 1b; instead, the homocoupling mainly proceeded due to the high reactivity of low-valent Ni toward the aryl halides14 and/or the poor nucleophilicity of the generated aryl nickel.15 Fortunately, the low chemoselectivity was improved by replacing the bpy ligand with electron-donating PPh3 (entry 10). Further improvement was achieved by increasing ligand loading (30 mol%) and by adding Et4NI11,16 (entries 11–13). In this stannylation, other stannyl electrophiles such as Bu3SnCl, Bu3SnOtBu, and Bu3SnOAc also participated, leading to 2a in 74%, 69%, and 54% yields, respectively (Table S1 in the ESI†). Additionally, hexane, toluene, tetrahydrofuran, 1,4-dioxane, and acetonitrile were not suitable solvents; most of the substrates remained unchanged.
|
|
|||||
|---|---|---|---|---|---|
| Yielda/% | |||||
| Entry | 1 | [Ni]cat. | Ligand (x mol%) | 2 | 3 |
| a Determined by GC yield with tridecane as the internal standard. Parenthetical value indicates isolated yield. b tbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. c Zn was used instead of Mn. d 4-Iodoanisole was used instead of bromide 1a. e 25 °C, 4 h. f 25 °C, 18 h. g Et4NI (20 mol%) was added. | |||||
| 1 | 1a | NiBr2 | bpy (10) | 72 | 14 |
| 2 | 1a | NiBr2(bpy) | — | 91 (85) | 9 |
| 3 | 1a | NiBr2 | — | 0 | 0 |
| 4 | 1a | NiBr2 | PPh3 (20) | 21 | 7 |
| 5 | 1a | NiBr2 | dppe (10) | 0 | 0 |
| 6 | 1a | NiBr2 | tbpyb (10) | 10 | 70 |
| 7 | 1a | — | bpy (20) | 0 | 0 |
| 8c | 1a | NiBr2(bpy) | — | 23 | 60 |
| 9d,e | 1a | NiBr2(bpy) | — | 60 | 39 |
| 10f | 1b | NiBr2(bpy) | — | 22 | 68 |
| 11f | 1b | NiBr2 | PPh3 (20) | 75 | 12 |
| 12f | 1b | NiBr2 | PPh3 (30) | 86 | 6 |
| 13f,g | 1b | NiBr2 | PPh3 (30) | 96 (90) | 4 |
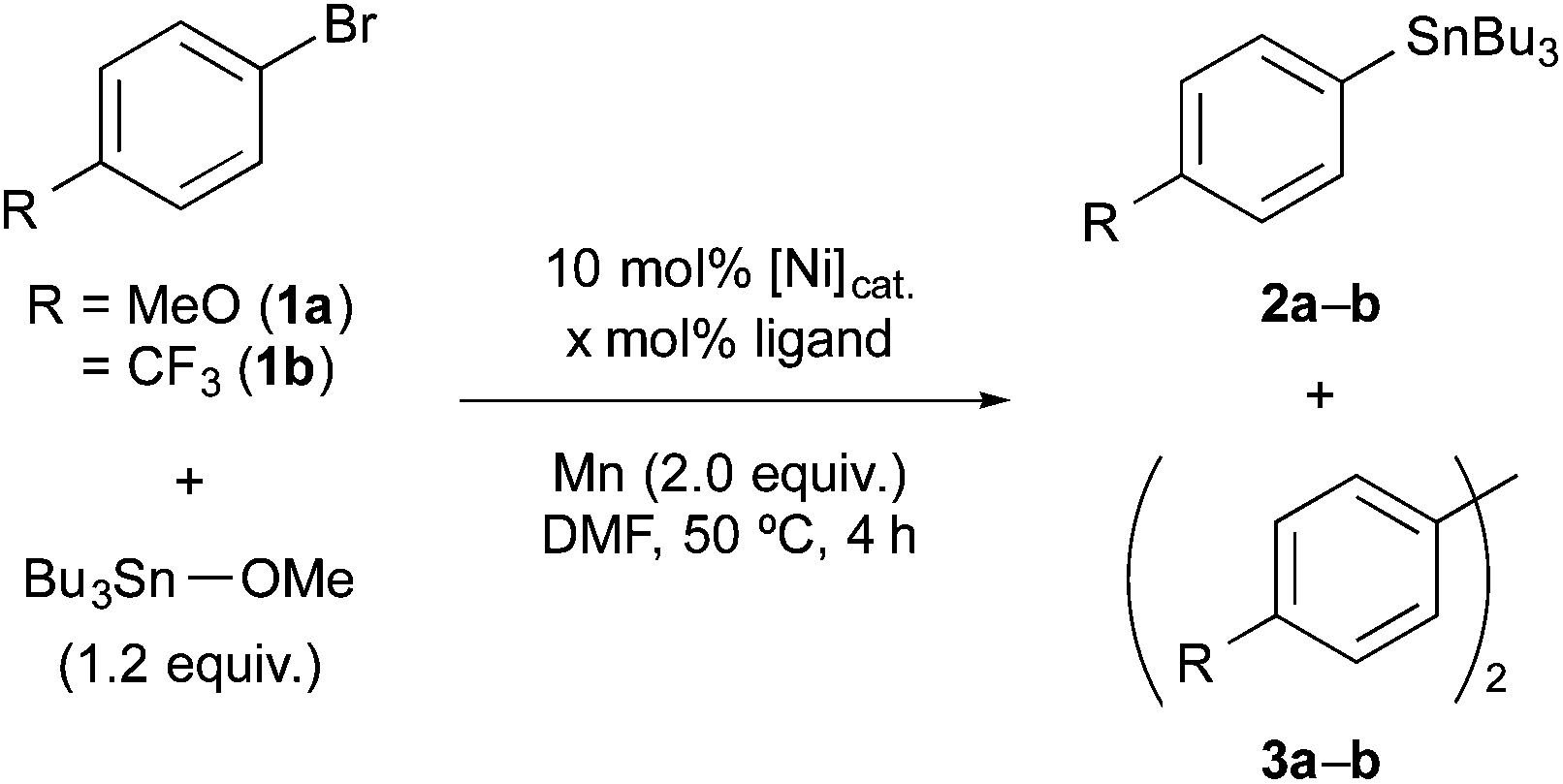
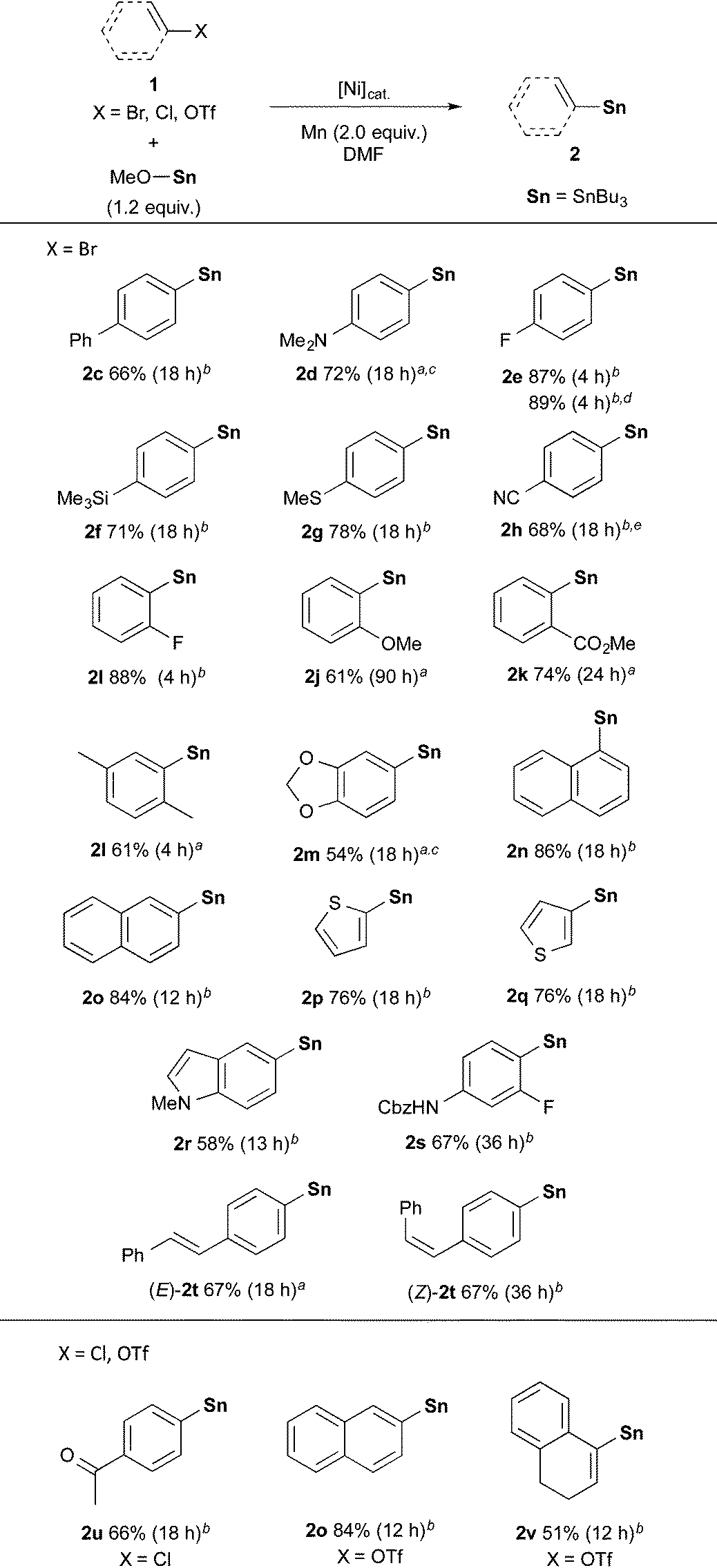
With the optimized conditions in hand (entries 2 and 13, Table 1), we next investigated the substrate scope in the Ni-catalysed stannylation by employing various aryl or vinyl halides with Bu3SnOMe (Table 2). Aryl bromides containing electron-donating (1d) and -withdrawing substituents (1e–1g) at the para-position were well tolerated, giving rise to the corresponding stannylated products (2c–2g) in good yields. In addition, the stannylation was successfully carried out on a gram-scale synthesis, giving rise to 2e in 2.05 g (89% yield). For the efficient stannylation of 4-cyano bromobenzene (1h), bidentate phosphine ligands bearing large bite angles were effective (Table S2 in the ESI†). ortho and meta-substituents (1i–1o) also participated in the stannylation, leading to the corresponding stannylated products (2i–2o). Heteroaryl bromides (1p–1r) also underwent this transformation to afford the stannylation products in good-to-high yields. Slightly acidic N–H bonds did not prevent the reaction and afforded 4-amino-2-fluorophenyl stannane (2s), which is an intermediate in torezolid synthesis.17 In addition, the stereochemistry of (E)- and (Z)-olefinic moieties (1t) was maintained during the stannylations. Furthermore, the present stannylation is active not only for bromides, but also for chloride 1u and triflates 1o and 1v, yielding the corresponding stannylated products 2u, 2o, and 2v, respectively.
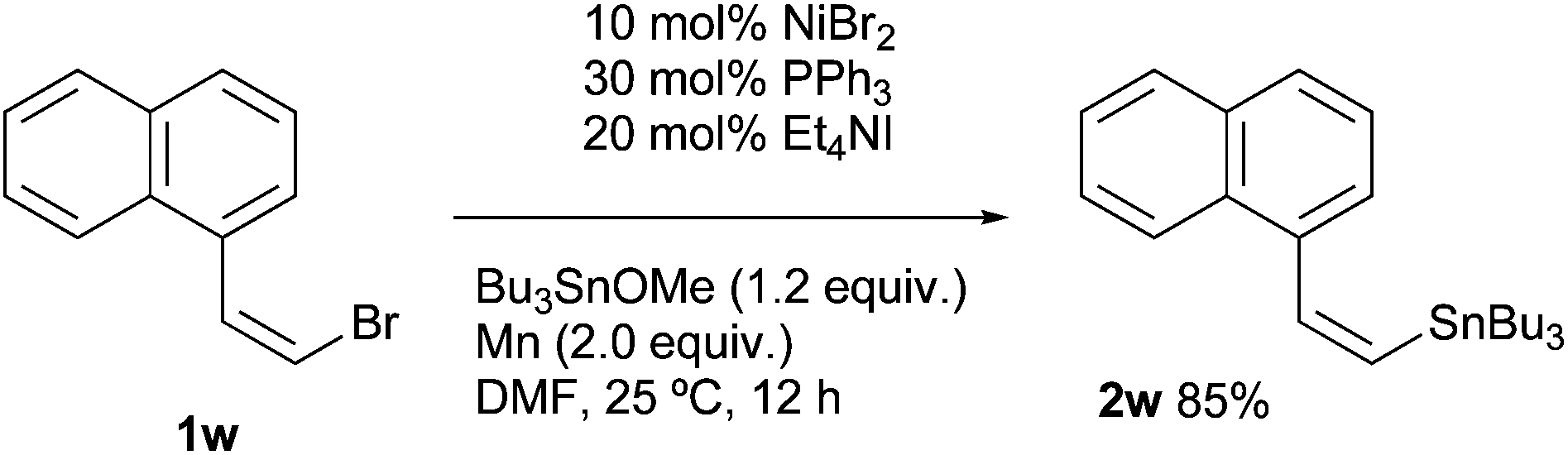 | (1) |
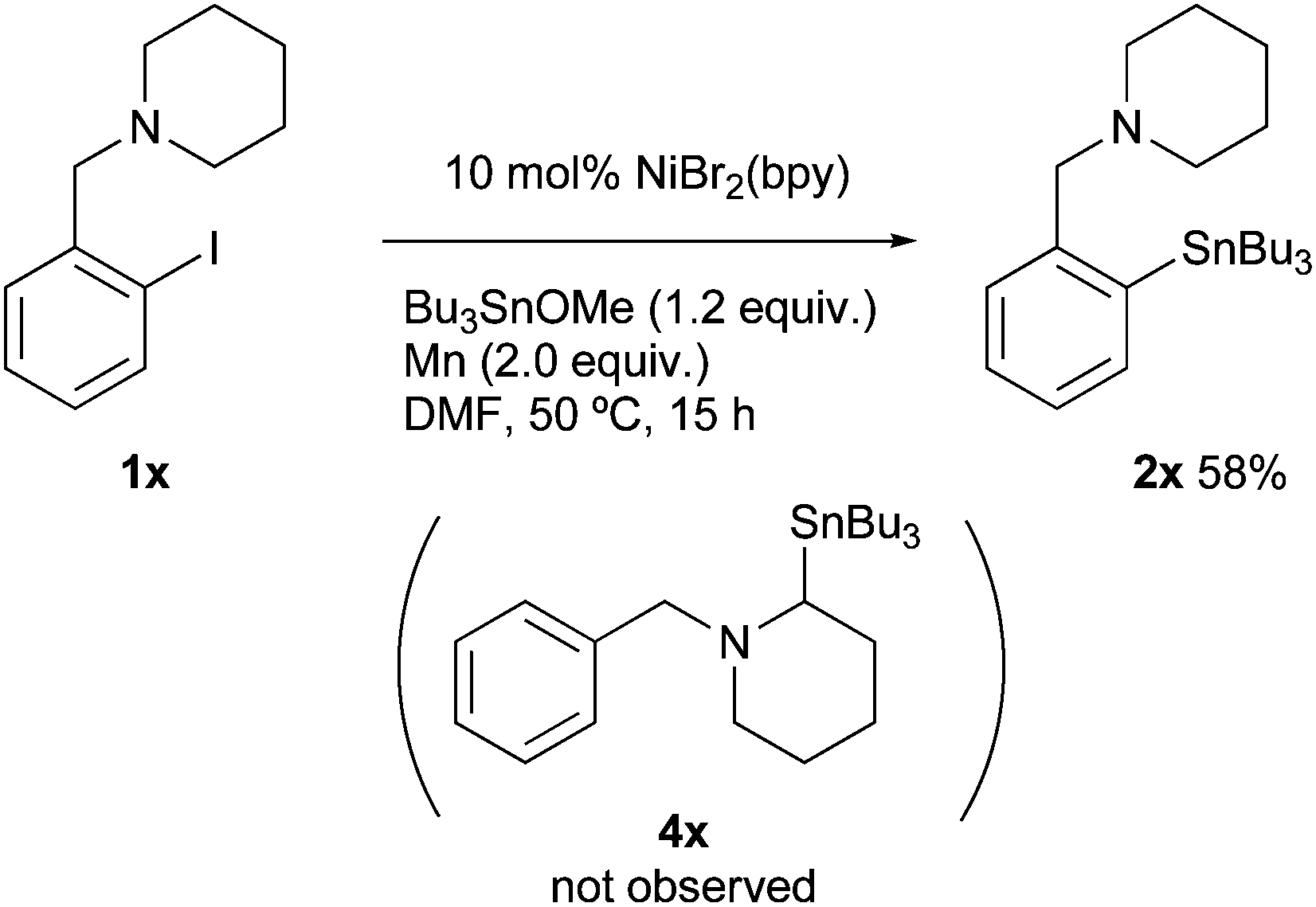 | (2) |
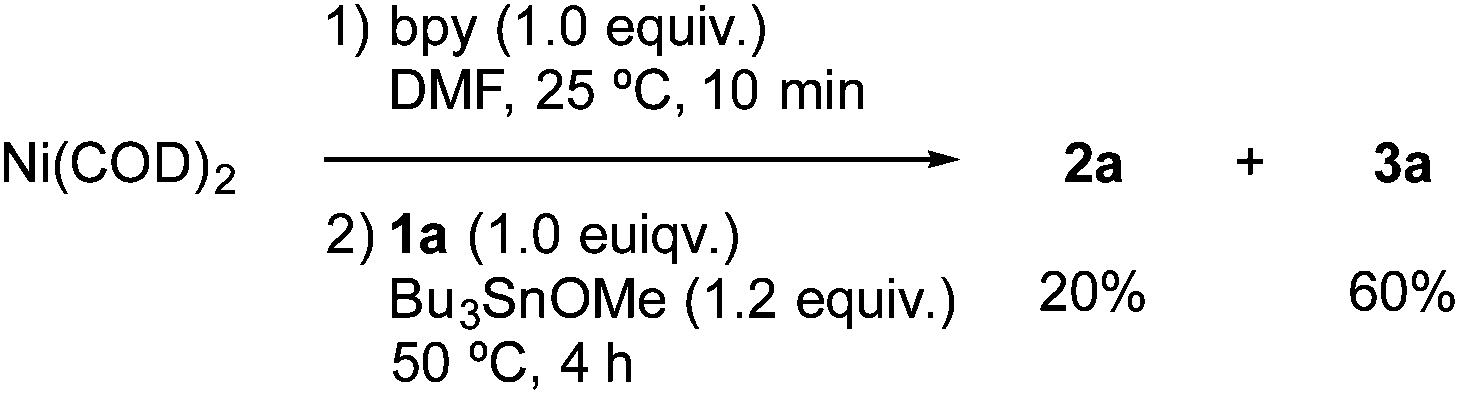 | (3) |
A stereocontrol study was conducted using (Z)-1-(bromovinyl)naphthalene (1w), as shown in eqn (1). The reaction of 1w with Bu3SnOMe exclusively yielded (Z)-vinyl stannane 2w with complete retention of the stereo-integrity.18 In addition, it is known that the aryl radical possessing a (dialkylamino)methyl group at the ortho-position, derived from the halogen atom abstraction of 1-(2-iodobenzyl)piperidine (1x), rapidly undergoes 1,5-hydrogen atom transfer to afford an α-amino alkyl radical.19 This radical might be converted to alkyl stannane 4x through the formation of alkyl nickel species via recombination between Ni and the alkyl radical.20 However, the reaction of 1w provided simple arylstannane 2x (eqn (2)). Furthermore, the addition of a hydrogen atom donor like 9,10-dihydroanthracene21 into the reaction media did not fully block the stannylation; 45–62% yields of 2a were obtained even if excess scavenger (2.0–3.0 equiv.) was present in the reaction media.22 These findings imply that the primary pathways for oxidative addition of Ni into organohalides 1 do not generate free organic radicals.
The stoichiometric reaction of NiBr2(bpy) with 1a and Bu3SnOMe in the presence of various amounts of Mn powder (Scheme 2) provided some important information about the mechanism. As the loading of Mn was increased, the yield of stannylated product 2a increased, while that of the homocoupling product 3a decreased. Particularly note that the lower loading (0.9–1.1 equiv.) of Mn powder mainly produced the homocoupling product 3a. A similar product distribution was observed in the reaction of Ni(COD)2/bpy with 1a and Bu3SnOMe in the absence of an Mn reductant (eqn (3)). These results could indicate that monovalent Ni is an active intermediate in the Ni-catalysed stannylation. Thus, the present stannylation could be initiated by the generation of [Ni]0 complex A from the reduction of the initial Ni(II) with Mn powder (Scheme 3). The oxidative addition of aryl halides (Ar–X: 1) to A afforded Ar–[Ni]II–X B. Although the divalent Ni intermediate B might be slightly active in the interception of Bu3SnOMe (Scheme 2 and eqn (3)), disproportionation of B would spontaneously occur to afford [Ni]IIX2 and [Ni]IIAr2C,14a,23 which would lead to the homocoupling product 3 and Avia reductive elimination. In contrast, in the presence of excess Mn, B could be preferentially reduced to [Ni]I–Ar D, which would be more active than B for interception because of the higher nucleophilicity of monovalent Ni.15,16 This would afford the stannylated product 2 as well as [Ni]I–OMe E, followed by the regeneration of A with Mn.
 | ||
| Scheme 2 Stoichiometric reaction of NiBr2(bpy) with 1a (1.0 equiv.) and Bu3SnOMe (1.2 equiv.) in the presence of Mn powder (x equiv.). | ||
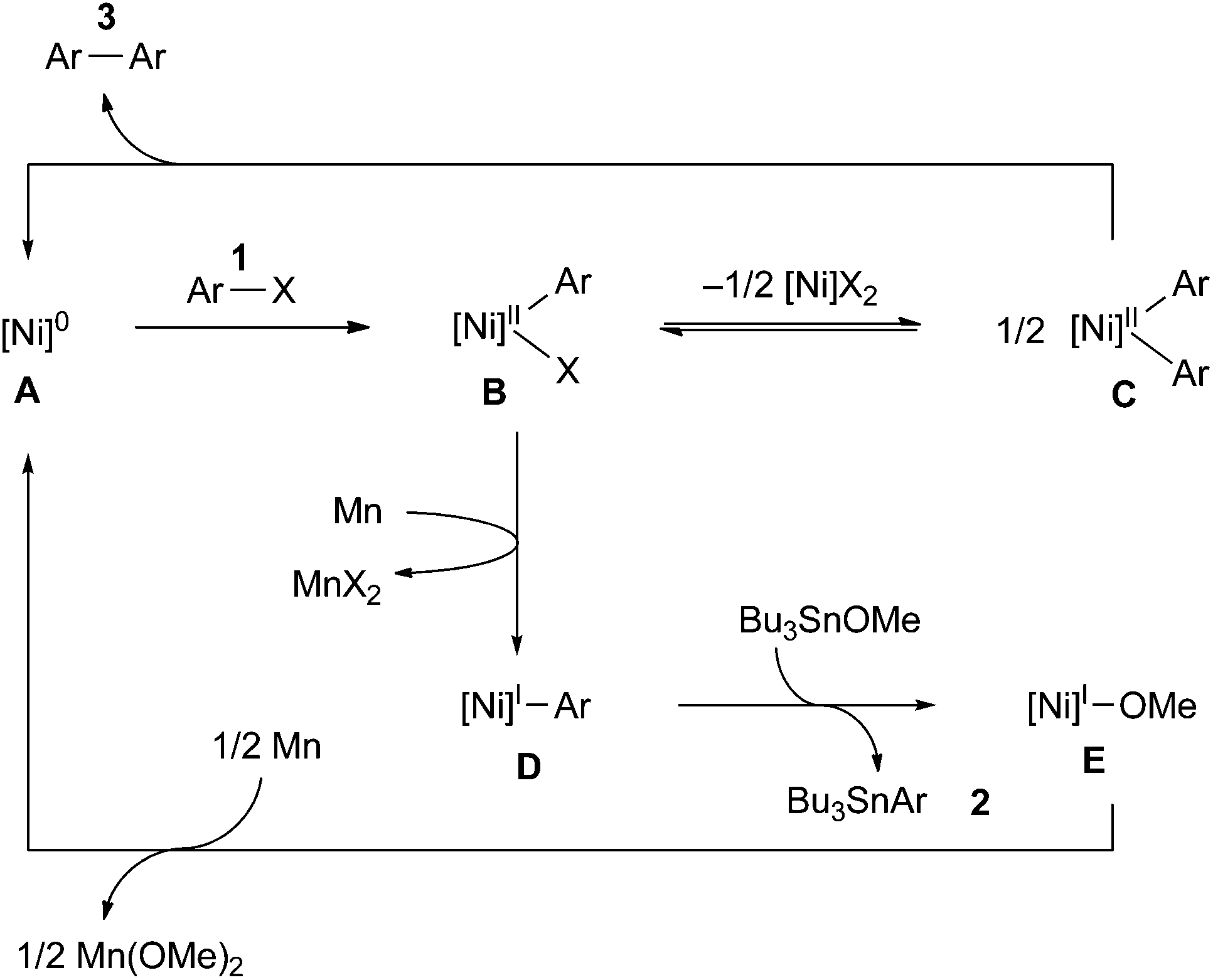 | ||
| Scheme 3 Plausible reaction pathway for the Ni-catalysed stannylation of aryl halides. | ||
Conclusions
In conclusion, we have demonstrated a simple and atom-economical stannylation using stannyl electrophiles catalysed by Ni complexes in the presence of an Mn reductant. This stannylation can be tolerated by a diverse set of functional groups on aryl halides and does not release wasteful stannyl residues. Preliminary mechanistic studies suggest that aryl Ni(I) species are intermediates in this transformation. Further mechanistic studies and synthetic applications of this transmetalation process of the C–Ni bond are underway.Acknowledgements
This work was partially supported by a Grant-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. K. K. acknowledges financial support from the Electronic Technology Research Foundation of Chugoku. We also acknowledge the Natural Science Center for Basic Research and Development (N-BARD) in Hiroshima University for HRMS analysis.Notes and references
- (a) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef PubMed; (b) M. Kosugi and K. Fugami, Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley, New York, 2002, p. 263 Search PubMed.
- P. Y. S. Lam, G. Vincent, D. Bonne and C. G. Clark, Tetrahedron Lett., 2002, 43, 3091 CrossRef CAS.
- (a) T. Furuya, A. E. Strom and T. Ritter, J. Am. Chem. Soc., 2009, 131, 1662 CrossRef CAS PubMed; (b) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 4648 CrossRef CAS PubMed.
- C. Huang, T. Liang, S. Harada, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 13308 CrossRef CAS PubMed.
- (a) L. Adam Shih-Yuan and D. Wen-Chin, Tetrahedron Lett., 1996, 37, 495 CrossRef; (b) T. Hayashi and M. Ishigedani, Tetrahedron, 2001, 57, 2589 CrossRef CAS; (c) P. Knochel and R. D. Singer, Chem. Rev., 1993, 93, 2117 CrossRef CAS; (d) C. Gosmini and J. Périchon, Org. Biomol. Chem., 2005, 3, 216 RSC.
- (a) D. Azarian, S. S. Dua, C. Eaborn and D. R. Walton, J. Organomet. Chem., 1976, 117, C55 CrossRef CAS; (b) H. Azizian, C. Eaborn and A. Pidcock, J. Organomet. Chem., 1981, 215, 49 CrossRef CAS.
- D. Qiu, L. Jin, S. Wang, S. Tang, X. Wang, F. Mo, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 11581 CrossRef CAS PubMed.
- K. Kiyokawa, N. Tachikake, M. Yasuda and A. Baba, Angew. Chem., Int. Ed., 2011, 50, 10393 CrossRef CAS PubMed.
- (a) Y. Takemoto, H. Yoshida and K. Takaki, Chem. – Eur. J., 2012, 18, 14841 CrossRef CAS PubMed; (b) T. Wakamatsu, K. Nagao, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2013, 52, 11620 CrossRef CAS PubMed.
- Interception of the carbon–silver bond using other stannyl electrophiles was reported. See: J. Liu, X. Xie and Y. Liu, Chem. Commun., 2013, 49, 11794 RSC.
- T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106 CrossRef CAS PubMed.
- A possibility that the biaryl 3a was formed through the Stille coupling of 1a with the generated 2a was completely ruled out in the reaction of 1a with PhSnBu3 under the identical conditions. D. A. Powell, T. Maki and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 510 CrossRef CAS PubMed.
- The reaction of 4-anisyl chloride under the identical conditions (Table 1, entry 2) afforded 2a in 33% yield, along with 3a in 32%.
- (a) T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319 CrossRef CAS; (b) V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS.
- J.-X. Hu, H. Wu, C.-Y. Li, W.-J. Sheng, Y.-X. Jia and J.-R. Gao, Chem. – Eur. J., 2011, 17, 5234 CrossRef CAS PubMed.
- (a) M. Zembayashi, K. Tamao, J.-I. Yoshida and M. Kumada, Tetrahedron Lett., 1977, 18, 4089 CrossRef; (b) M. Iyoda, H. Otsuka, K. Sato, N. Nisato and M. Oda, Bull. Chem. Soc. Jpn., 1990, 63, 80 CrossRef CAS; (c) V. Percec, J.-Y. Bae, M. Zhao and D. H. Hill, J. Org. Chem., 1995, 60, 176 CrossRef CAS.
- W. B. Im, S. H. Choi, J.-Y. Park, S. H. Choi, J. Finn and S.-H. Yoon, Eur. J. Med. Chem., 2011, 46, 1027 CrossRef CAS PubMed.
- J. J. Hirner and S. A. Blum, Organometallics, 2011, 30, 1299 CrossRef CAS.
- (a) V. Snieckus, J. C. Cuevas, C. P. Sloan, H. Liu and D. P. Curran, J. Am. Chem. Soc., 1990, 112, 896 CrossRef CAS; (b) M. Murakami, M. Hayashi and Y. Ito, J. Org. Chem., 1992, 57, 793 CrossRef CAS; (c) N. Yoshikai, A. Mieczkowski, A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2010, 132, 5568 CrossRef CAS PubMed.
- (a) H. Yin, C. Zhao, H. You, K. Lin and H. Gong, Chem. Commun., 2012, 48, 7034 RSC; (b) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146 CrossRef CAS PubMed; (c) H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022 RSC; (d) Y. Peng, X.-B. Xu, J. Xiao and Y.-W. Wang, Chem. Commun., 2013, 50, 472 RSC; (e) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (f) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2014, 6, 1115 RSC.
- (a) B. L. Tran, B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 2555 CrossRef CAS PubMed; (b) S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799 CrossRef CAS PubMed.
- Decreasing the solubility of Bu3SnOMe by the addition of 9,10-dihydroanthracene would cause low yields. Indeed, as the amount of the scavenger increases (1.0–3.0 equiv.), the homocoupling reaction exclusively occurred to form 3a in 16–31% yields. See Table S3 in the ESI.†.
- A. Nakamura and S. Otsuka, Tetrahedron Lett., 1974, 15, 463 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data for the screening of reaction conditions, experimental procedures and characterization for new compounds. See DOI: 10.1039/c5ob01096a |
| This journal is © The Royal Society of Chemistry 2015 |