
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of conformationally restricted adenine bicycloribonucleosides†
Hubert
Hřebabecký
a,
Eliška
Procházková
a,
Michal
Šála
a,
Pavla
Plačková
a,
Eva
Tloušťová
a,
Ona
Barauskas
b,
Yu-Jen
Lee
b,
Yang
Tian
b,
Richard
Mackman
b and
Radim
Nencka
*a
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i., Gilead Sciences & IOCB Research Centre, 166 10 Prague 6, Czech Republic. E-mail: nencka@uochb.cas.cz
bGilead Sciences, Inc., 333 Lakeside Drive, Foster City, CA 94404, USA
First published on 22nd July 2015
Abstract
We prepared a novel series of conformationally restricted bicyclonucleosides and nucleotides. The synthetic approach employed a ring closing metathesis to provide access to both 6 and 7 membered saturated and unsaturated rings linking the 3′ to 5′ methylene groups of the sugar. The bicyclonucleosides were also transformed to the corresponding phosphoramidate prodrugs by an innovative one-pot protocol of boronate ester protection, coupling of the phosphoryl chloridate and deprotection of the boronate. A similar strategy was also employed for the synthesis of the corresponding monophosphates as crucial intermediates for the synthesis of selected triphosphates. The biological properties of the nucleosides and monophosphate prodrugs were assessed for antiviral and cytostatic activities in cell based assays whilst the triphosphates were evaluated in enzymatic assays. The lack of significant effects suggests that the linkage of the 3′ to 5′ via a ring system and the subsequent conformational restriction of the ribose ring to the South conformation are incompatible with the kinases and polymerases that recognize nucleosides and their metabolites.
Introduction
Nucleosides and their phosphorylated metabolites (nucleotides) are vital components of living organisms. The recognition of nucleosides by proteins e.g. kinases involved in the activation of the mono, di- and triphosphates, and the multiple roles of the triphosphate metabolites in providing energy for biological processes, and carrying genetic information, makes understanding the protein–nucleoside/tide interactions valuable. One component of this interaction is the three dimensional conformation of the nucleoside/tide species in solution which has been well studied. The sugar ring of nucleosides/tides rapidly interconverts between the so called North (3′-endo) and South (2′-endo) conformations in solution (Fig. 1). In addition to the sugar ring conformation, the orientation of the 4′–5′ bond (γ angle) and the orientation of the nucleobase (χ angle) are also essential features.1,2 Although a hydroxy group (or a phosphate group) at 5′ position can rotate around the axis, the energetically preferred conformations are plus synclinal (+sc) and antiperiplanar (ap), which are equally populated in solutions of purine nucleosides, whereas +sc conformation is preferred in nucleotides.1 | ||
| Fig. 1 North and South conformation of natural nucleosides and examples of derivatives locked in these conformations. | ||
During interaction with their protein targets, nucleosides/tides presumably adopt a specific binding conformation. Given the flexibility of the sugar ring these interactions with proteins may favor one or more conformations. For example, Marquez and co-workers elegantly showed that deoxynucleosides/tides locked in South and North conformations can be efficiently recognized by different enzymes.3–6 Carbocyclic nucleoside derivatives N-MCT and S-MCT were designed to ‘lock’ the ring in the North and South conformation, respectively. They discovered that the North conformation effectively binds to polymerases3–5 whereas the South is recognized by HSV-1 thymidine kinase.6
Recently, we have prepared a different pair of carbocyclic North/South adenosines7,8 (as well as their analogues with other nucleobases), primarily inspired by LNA9–11 monomers. The North derivatives were largely inactive against a panel of viruses (suggesting limited recognition by viral polymerases if one assumes the triphosphates were generated in the cells utilized), but phosphatidylinositol 4-kinase IIα bound the North-type adenosine derivative with affinity significantly exceeding that of adenosine.7 Nucleoside triphosphate binding proteins are probably able to discriminate between nucleotides with different γ angles; however, there is no direct evidence of this to date. Taken together, our understanding of conformational preferences of nucleoside/tide binding proteins is still rather limited yet fundamental to the design of selective therapeutics to address the abnormalities in the pathways that lead to disease.
In this study novel bicyclic adenosine nucleoside derivatives were designed that combine both the conformational restriction of the sugar moiety in the South orientation as well as provide for a defined γ angle (either +sc or ap). Furthermore, to interrogate whether the phosphorylated species can interact with proteins we have prepared monophosphate prodrugs in an attempt to deliver the monophosphate species into cells and potentially the triphosphate metabolite through activation by intracellular kinases. Finally, selected triphosphates were prepared and evaluated for their recognition properties toward a variety of viral polymerases.
Results and discussion
Design and synthesis of a new series of bicyclic nucleoside and nucleotide derivatives
The design of the bicyclic adenosine derivatives was inspired by the work of Leumann et al., where bicyclic and tricyclic deoxynucleoside derivatives were utilized as monomers for oligonucleotide synthesis with duplex stabilizing properties.12–15 In contrast to Leumann's work, we were primarily interested in the preparation of ribonucleosides/tides with six and seven membered, saturated and unsaturated, rings bridging between the 3′ and 5′ position of the ribo-sugar. To access these ring systems we envisaged utilizing a ring closing metathesis approach. Allose derivative 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 was selected as the starting material in the forward synthesis (Scheme 1). Deprotection and oxidative cleavage of the 5,6-diol was performed with periodic acid in ethyl acetate17 to provide the 5-carboxaldehyde 2, which was then used directly in the next reaction with vinylmagnesium bromide or allylmagnesium bromide to provide dienes 3 and 6, respectively. The dienes 3 and 6 were then treated with the 2nd generation Grubbs’ catalyst in dichloromethane to generate the 6- and 7-membered ring intermediates as mixtures of diastereoisomers. The diastereoisomers were separated by chromatography on silica gel to yield compounds 4a (32%), 4b (18%), 7a (26%), and 7b (30%). The yields are calculated from starting allose derivative 1. Selective benzoylation of the compounds provided monobenzoates 5a (92%), 5b (94%), 8a (89%), and 8b (74%).
16 was selected as the starting material in the forward synthesis (Scheme 1). Deprotection and oxidative cleavage of the 5,6-diol was performed with periodic acid in ethyl acetate17 to provide the 5-carboxaldehyde 2, which was then used directly in the next reaction with vinylmagnesium bromide or allylmagnesium bromide to provide dienes 3 and 6, respectively. The dienes 3 and 6 were then treated with the 2nd generation Grubbs’ catalyst in dichloromethane to generate the 6- and 7-membered ring intermediates as mixtures of diastereoisomers. The diastereoisomers were separated by chromatography on silica gel to yield compounds 4a (32%), 4b (18%), 7a (26%), and 7b (30%). The yields are calculated from starting allose derivative 1. Selective benzoylation of the compounds provided monobenzoates 5a (92%), 5b (94%), 8a (89%), and 8b (74%).
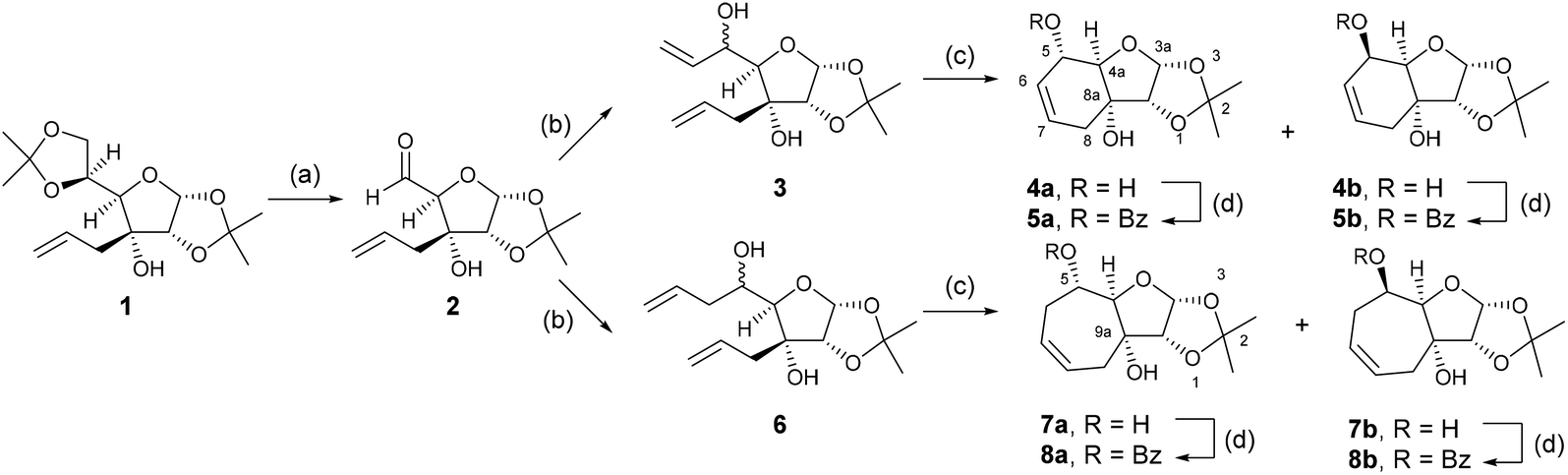 | ||
| Scheme 1 Synthesis of sugar derivatives 5a, 5b and 7a, 7b. Reagents and conditions: (a) H5IO6, EtOAc, r.t., 5 h; (b) vinyl or allylmagnesium bromide, THF, 2 h at 0 °C and overnight at r.t.; (c) Grubbs’ catalyst 2nd generation, DCM, 40 °C, 4 h, 32% of 4a, 18% of 4b, 26% of 7a and 30% of 7b from 1; (d) benzoyl chloride, pyridine, r.t., overnight, 92% of 5a, 94% of 5b, 89% of 8a and 74% of 8b. | ||
Compounds 5a, 5b, 8a, and 8b were then converted to anomeric mixtures of acetates by treatment with aqueous trifluoroacetic acid followed by acetylation with acetic acid anhydride in acetonitrile, in the presence of 4-dimethylaminopyridine (Scheme 2). The sugar derivatives were then subjected to a Vorbrüggen reaction18 with N6-benzoyladenine in acetonitrile and Lewis acid catalyst, tin(IV) chloride, to afford the protected nucleoside analogues, 9a (58%), 9b (55%), 12a (63%), and 12b (76%), respectively. Methanolysis with methanolic sodium methoxide yielded the free nucleosides 10a (68%), 10b (92%), 13a (80%), and 13b (66%), respectively.
 | ||
| Scheme 2 Synthesis of free nucleoside analogues 10a, 10b, 11a, 11b, 13a, 13b, 14a, 14b. Reagents and conditions: (a) 1. 80% aq. TFA, r.t., 3 h; 2. Ac2O, CH3CN, DMAP, r.t., 16 h; 3. N6-benzoyladenine, CH3CN, SnCl4, r.t., 3 h, 58% of 9a, 55% of 9b, 63% of 12a, 76% of 12b; (b) 0.1 M methanolic CH3ONa, r.t., 16 h, 58% of 10a, 92% of 10b, 63% of 13a, 66% of 13b; (c) Pd(OH)2/C, H2, DMF, r.t., 16 h, 66% of 11a, 70% of 11b, 78% of 14a, 75% of 14b. | ||
Product of N9-alkylation of all compounds was confirmed by heteronuclear correlation 1H,13C-HMBC experiment. In HMBC, crosspeaks corresponding to interactions between proton H2 from the pseudosugar moiety and carbon atoms C4′ and C8′ from adenine part were found. Based on this fact, N9-alkylated product was confirmed. In the case of the N7 isomer, crosspeaks corresponding to H2-C8 and H2-C5 should be found, but in our compounds, they were not observed. The configuration at carbon atom C2 (α, β) of the studied compounds was confirmed by 1H,1H-ROESY experiments, where the interactions of close-in-space protons are visible as crosspeaks. For this purpose, interaction of H2 with 3a-OH was observed. Crosspeaks corresponding to interactions of H2 with 3-OH and 3a-OH were found and β anomer was confirmed. As examples, a part of 1H,13C-HMBC and 1H,1H-ROESY of compound 10a is shown in ESI S23.†
Hydrogenation of the unsaturated nucleosides on Pd(OH)2/C in DMF led to saturated analogues 11a (66%), 11b (70%), 14a (78%), and 14b (75%), respectively.
The unambiguous transformation of the prepared nucleosides to monophosphate nucleotide prodrugs (Pro-Tides) required protection of the hydroxyl groups at positions 3 and 3a (Scheme 3). Typically an isopropylidene would suffice, but significant problems were encountered during acidic removal of the protecting group. A cyclic phenyl boronate was found to be the ideal protection. The unprotected nucleosides were treated with phenylboronic acid in boiling pyridine, which was slowly distilled off to azeotropically remove water. Subsequent reaction of the protected products with N-[chloro(phenoxy)phosphoryl]-L-alaninate in tetrahydrofuran in the presence of 1-methylimidazole5 afforded the target Pro-Tides in 50–76% yields over two steps as 1:1 mixture of diastereomers (determined by NMR spectroscopy) due to the chiral phosphorus atom (Scheme 3).19
 | ||
| Scheme 3 Preparation of protected nucleotide analogue 16a and 16b. Reagents and conditions: (a) phenyl boronic acid, pyridine, heating; (b) N-[chloro(phenoxy)phosphoryl]-L-alaninate, 1-methylimidazole, THF, r.t., overnight, 54% of 16a, 55% of 16b. Structures of Pro-Tides 17a, 17b, 18a, 18b, 19a, and 19b. | ||
In order to investigate the effect of the nucleosides 13a and 13b on the enzymatic level, we had to prepare the triphosphates that would mimic ATP in its South conformation. We achieved this goal by sequential synthesis of the monophosphates, followed by the addition of pyrophosphate to generate triphosphates. The yield of phosphates generated using a similar procedure to that described for the prodrugs via the cyclic boronate was generally very low (26% of 23a and 27% of 23b), therefore, we resorted to the isopropylidene group. Isopropylidene derivatives 22a and 22b were prepared by a treatment of 13a or 13b with 2,2-dimethoxypropane in dimethylformamide in the presence of conc. H2SO4. Protected nucleosides 22a and 22b were converted to phosphate with POCl3 in trimethyl phosphate and then deprotected with 50% aqueous trifluoroacetic acid. The crude product was purified and converted to the sodium salt providing nucleotide 23a in 38% and 23b in 39% yield, respectively.
Triphosphates 24a and 24b were prepared from the triethylammonium salts, obtained by cation-exchange of the sodium salts 23a and 23b on Dowex 50 (Et3NH+).20 The triethylammonium salts were treated with imidazole, triphenylphosphine, triethylamine and 2,2′-dipyridyl disulfide in a mixture of DMSO and DMF at ambient temperature overnight. Reaction of the imidazolide intermediates that were generated with tributylammonium pyrophosphate led to the formation of the crude triphosphates which were then purified by HPLC (Scheme 4).
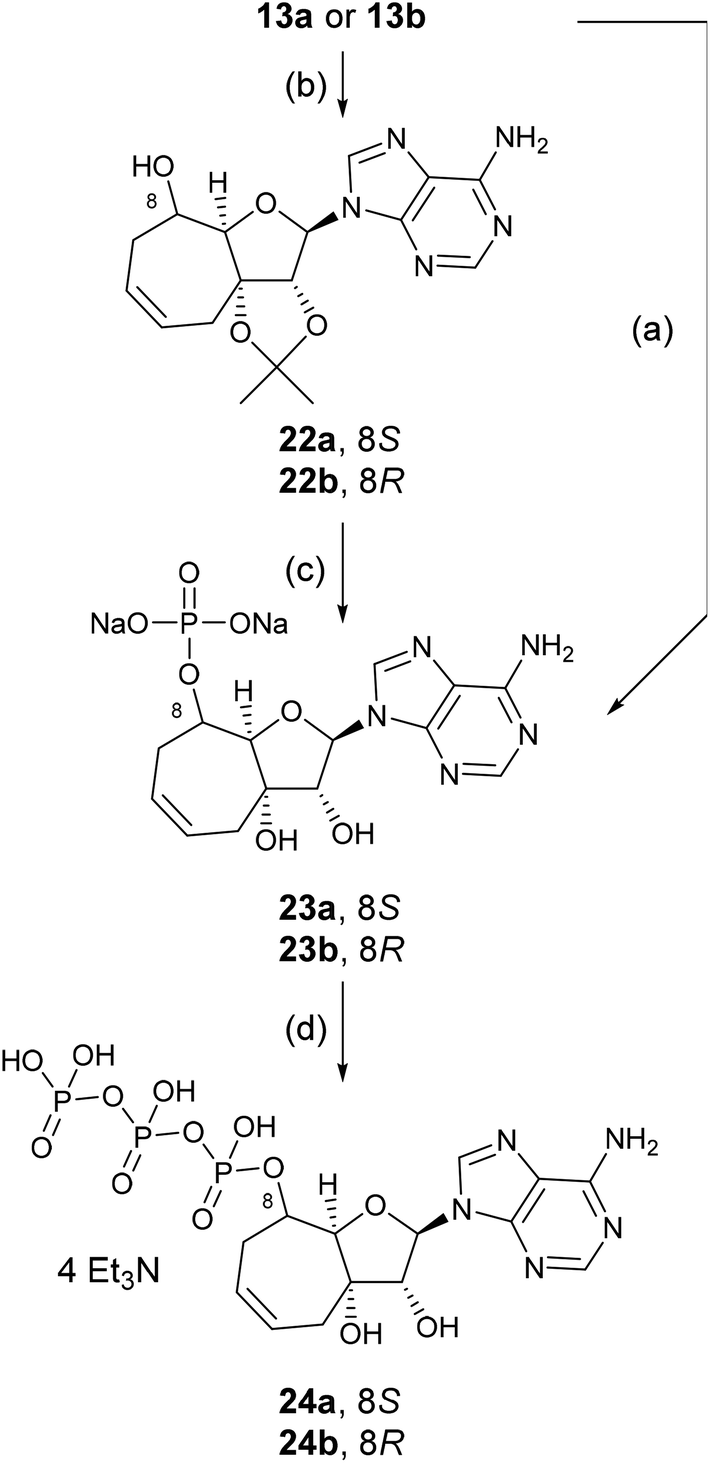 | ||
| Scheme 4 Preparation of triphosphate analogues 24a and 24b. Reagents and conditions: (a) 1. phenyl boronic acid, pyridine; 2. POCl3, PO(OCH3)3, r.t., 4 h; 3. H2O, 0 °C, 26% of 23a, 27% of 23b; (b) 1. 2,2,-dimethoxypropane, DMF, H2SO4, 79% of 22a, 69.5% of 22b; (c) 1. POCl3, PO(OCH3)3, r.t., 4 h; 2. H2O, 0 °C; 3. 50% aqueous TFA, r.t., 48 h, 38% of 23a, 39% of 23b; (d) 1. Dowex 50 (Et3NH+); 2. imidazole, PPh3, Et3N, 2,2′-dipyridyl disulfide, DMF-DMSO, r.t., overnight; 3. (nBu3NH)2P2H2O7, DMSO, r.t., overnight. | ||
Conformational analysis of compound 13b
Due to the additional seven-membered ring in compound 13b, the flexibility of the ribose ring is significantly reduced. Experimental J-coupling values obtained from 1H NMR spectrum were compared with values calculated by two different approaches. First, the geometry of compound 13b was optimized by DFT starting from four initial conformations. Torsion angles Φ0 and Φ3 with pucker amplitude ΦMAX = 40° according to Altona and Sundaralingam2 were set and four geometries with phase angles P = 0°, 90°, 180° and 270° were optimized. For these calculations, we used both possible conformations of the nucleobase (syn and anti), since our ROESY and 13C NMR studies indicates that both these conformers are present in solution and the anti conformer is the major one (Fig. 2).21 The results of torsion angle calculations are summarized in Table 1.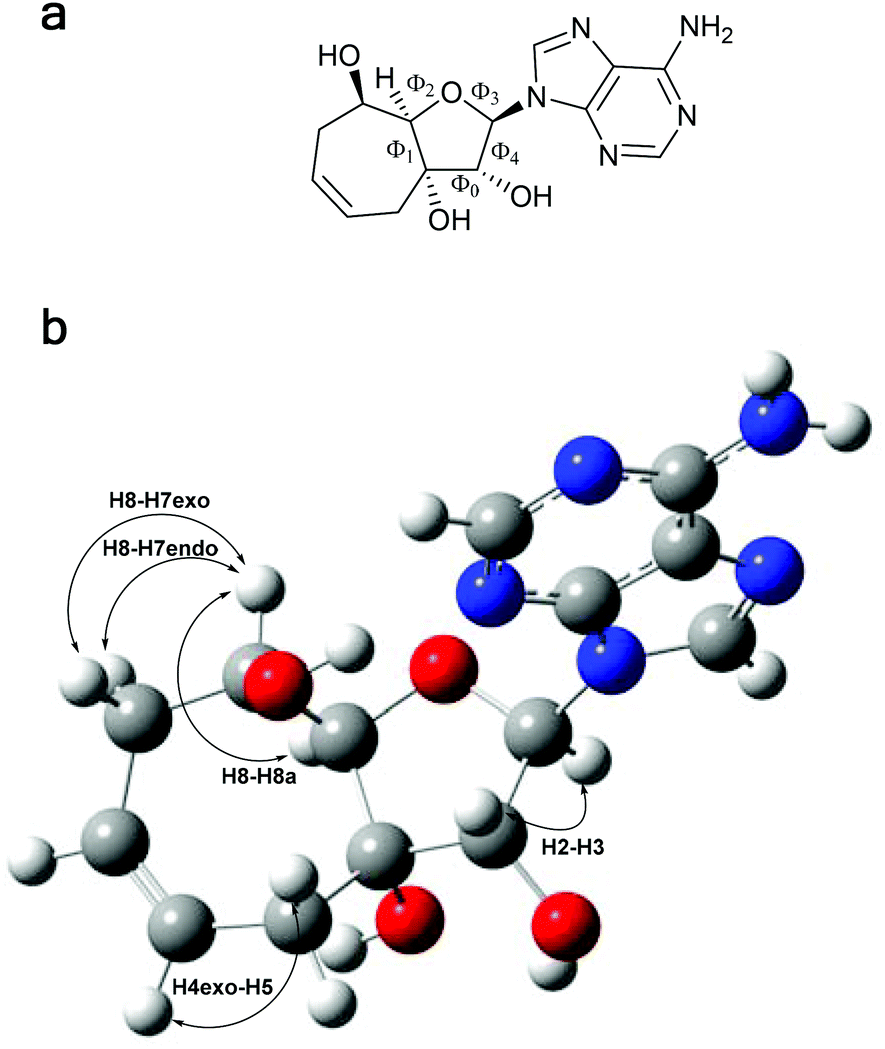 | ||
| Fig. 2 (a) The chemical structure of syn conformer of compound 13b and the torsion angles Φ0–Φ4. (b) Optimized geometry of compound 13b (B3LYP/6-31+g(d,p)) and the spin-spin interactions described in the Table 2. | ||
| Torsion angles | syn (P = 163°) | anti (P = 153°) |
|---|---|---|
| Φ 0 | −31.2° | −32.2° |
| Φ 1 | 21.0° | 18.0° |
| Φ 2 | −1.0° | 5.1° |
| Φ 3 | −19.7° | −26.2° |
| Φ 4 | 31.9° | 36.4° |
The obtained torsion angles were used as input for the J-couplings calculated by empirical Haasnoot–Altona equation22 (JALTONA in Table 2), which describes the dependence of vicinal coupling constant (3J) values on the torsion angle between the coupled nuclei. J-couplings were also calculated by the DFT method (JDFT in Table 2).
Both theoretical approaches provided similar values, which fit well with the experimentally obtained J-couplings (JEXPER in Table 2). From the obtained torsion angles, we can conclude that compound 13b is present in a solution in South conformation (2E). The orientation of the nucleobase affects the conformation of the ribose moiety marginally and both the conformers possess similar phase angle P (Table 1).
In the studied compounds, configuration at carbon atom C5 (5a, 5b, 8a, 8b), C7 (9a, 9b, 10a, 10b, 11a, 11b, 18a, 18b) or C8 (12a, 12b, 13a, 13b, 14a, 14b, 17a, 17b, 19a, 19b) was investigated by J-coupling analysis of interactions H5-H5a, H7-H7a or H8-H8a, respectively, and confirmed by DFT calculations in combination with Karplus-like equation. It is based on the fact that the value of vicinal 3J-coupling depends on the torsion angle between coupled nuclei. In general, based on the geometry optimization (DFT method), it can be said that torsion angle H8a-C8a-C8-H8 (or H7a-C7a-C7-H7) in S-isomers is close to 180°, whereas the torsion angle in R-isomers is close to 90°. Using the Karplus equation containing cosine functions, it is clear that the corresponding J-coupling constant of S-isomers will be higher than that in the case of R-isomers. As an example, in compound 12a (8S isomer), the vicinal coupling constant 3J(8a–8) = 7.9 Hz was found, whereas, in compound 12b (8R isomer), 3J(8a–8) = 1.2 Hz was found. In the same way, R/S configuration was determined in all studied compounds. Furthermore, it was found that the chemical shift of carbon atom C5, C7 or C8 of S-isomers is higher than that in the case of R-isomers. As an example, in S-isomer 12a, δC-8 = 72.65 ppm, whereas in R-isomer 12bδC-8 = 70.34 ppm. These results were supported by 1H,1H-ROESY experiments. As an example, a part of the ROESY spectrum of compound 10b, where the 7R-isomer was found, is shown in ESI S26.† There is a crosspeak corresponding to interaction between H3 and 7-OH protons which are close in space. This crosspeak confirms configuration at carbon atom C7. On the other hand, in the case of compound 5a, where the 5S-isomer was found, it was possible to observe ROESY crosspeak between 8a-OH and ortho protons from the benzoyl moiety, see ESI S15 and S26.†
Biological evaluation of nucleoside derivatives and their monophosphate prodrugs in cell based assays
All the locked nucleoside derivatives (10a, 10b, 11a, 11b, 13a, 13b, 14a, and 14b) were screened for cytostatic activity on several cell lines (HepG2, HL60, HeLa S3, CCRF-CEM) and all lacked cytostatic activity. These compounds were also screened for antiviral activity in a panel of viruses including HCV (1a, 1b, and 2a) (Huh-7 cells), HIV (MT4 cells), HRV (HeLa cells) and RSV (HEp-2 cells) and no activity was observed up to 50 μM of testing. Subsequently the phosphoramidate prodrugs were also evaluated in the same suite of assays and, in general, no activities up to assay limits were observed. The phosphoramidate prodrug 17b exerted modest antiviral activity against HCV 1a (EC50 = 23 μM) which suggests that this nucleoside monophosphate may have interactions with the enzymes responsible for the activation of the triphosphate and subsequent inhibition of the viral polymerase, or alternatively targets other proteins that can impact the replication of HCV.Enzymatic decomposition of prodrugs 17a and 17b monitored by 31P NMR spectroscopy
To further understand the lack of biological activity for the majority of the monophosphate prodrugs and confirm that the weakly active prodrug compound 17b was being efficiently converted to the parent monophosphate inside the HCV replicon cells, we evaluated the prodrug breakdown process using carboxypeptidase Y. It is widely reported that the Pro-Tide prodrug breakdown is initiated by carboxypeptidases.23 The starting phosphoramidate derivative 17b has two signals in the proton decoupled 31P NMR spectra corresponding to the two P diastereoisomers. After the enzyme addition (Fig. 3b), two new phosphorus signals for intermediate 20b24 appeared which over several hours resolved to a single 31P NMR signal for product 21b. Similar progress of the reaction was observed after chemical hydrolysis of the starting phosphoramidate in triethylamine solution to yield the same final intermediate 21b. For compound 17a, similar progress of both enzymatic and chemical hydrolytic reaction was observed.
 | ||
| Fig. 3 (a) Proposed breakdown pathway to phosphoramidate intermediate. (b) Enzymatic decomposition of phosphoramidate derivative 17b monitored by 31P NMR spectroscopy. | ||
The final reaction mixture containing the product 21b, methanol and phenol, was confirmed by comparing the 13C NMR spectra of the reaction mixture before and after the removal of MeOH and PhOH by HPLC (ESI Fig. S3†). The data with carboxypeptidase Y suggests that on the timescale of the cell based assays (3–5 days) the phosphoramidate prodrugs are being efficiently cleaved by esterases even though the position 8 (or 7 in the case of the 6-membered ring) is sterically hindered. Whilst the data does not prove the intracellular formation of the monophosphate since further intracellular breakdown is necessary e.g. the P–N bond cleavage by HINT enzymes to form the monophosphate, it does support that the breakdown process is not limited by the esterase cleavage and encouraged the preparation of the triphosphate species for evaluation.
Evaluation of triphosphates 24a and 24b in enzymatic assays
The triphosphate of nucleoside 17b, i.e.24b was tested for inhibition of the HCV RNA-dependent RNA polymerase25 and found to be a weak inhibitor with IC50 = 143 μM. Also, this triphosphate compound did not inhibit polymerase from influenza virus and human rhinovirus 16 (IC50 were higher than 200 μM in both cases). Both triphosphates 24a and 24b were also evaluated in enzymatic assays of our current interest including phosphatidylinositol 4-kinases (PI4 K IIIα, PI4 K IIIβ, PI4 K IIα) and adenylate cyclases from Bordetella pertussis and Anthrax edema factor respectively. However, none of these enzymes were inhibited by these triphosphates. Altogether the data suggest that the triphosphates are not able to effectively interact with target proteins, which can be attributed either to unfavorable South conformation or sterically demanding bridging ring.Conclusions
We report on a novel series of bicyclic ribonucleosides locked in South conformation through linking of the 3′ and 5′ methylene via ring closing metathesis methodology.The efficient preparation of phosphoramidate prodrugs was achieved by employing a one-pot procedure involving a cyclic boronate protection of the cis-diol which can be easily removed after the installation of the prodrug during work-up conditions.
All the bicyclic nucleoside derivatives and their prodrugs were screened in several cell-based assays in order to assess phenotypic engagement with cellular proteins. The lack of activity for the majority suggests that the conformationally restricted sugars in the South orientation, and/or the additional atoms of the ring appended to the natural ribose sugar are not compatible with protein binding sites. In one example, where modest activity was observed 17b, evaluation of the triphosphate analog suggested that the effect was likely non-specific. The results reported suggest that additional tools will be required to investigate the conformational aspects of sugars that favor nucleoside/tide interactions with proteins of interest.
Experimental
General
Melting points were determined on a Büchi B-540 apparatus. NMR spectra (δ, ppm; J, Hz) were measured on a Bruker Avance II-600 and/or Bruker Avance II-500 instruments (600.1 or 500.0 MHz for 1H and 150.9 or 125.7 MHz for 13C) in hexadeuterated dimethyl sulfoxide and referenced to the solvent signal (δ 2.50 and 39.70 ppm, respectively). Mass spectra were measured on a LTQ Orbitrap XL (Thermo Fisher Scientific) using electrospray ionization (ESI). Column chromatography was performed on Silica gel 60 (Fluka) and thin-layer chromatography (TLC) on Silica gel 60 F254 foils (Merck). Solvents were evaporated at 2 kPa and bath temperature 30–60 °C; the compounds were dried at 13 Pa and 50 °C. Elemental analyses were obtained on a Perkin–Elmer CHN Analyzer 2400, Series II Sys (Perkin–Elmer). The elemental compositions for all compounds were within ±0.4% of the calculated values. For all the tested compounds satisfactory elemental analysis was obtained supporting >95% purity. Optical rotation was measured on polarimeter Autopol IV (Rudolph Research Analytical) at 589 nm wavelength in chloroform or methanol.Preparation of compounds 4a, 4b, 7a, and 7b
Periodic acid (7.07 g, 31.0 mmol) was added to a solution of 3-allyl-1,2:5,6-di-O-isopropylidene-α-D-allofuranose16 (1, 8.41 g, 28.0 mmol) in ethyl acetate (320 mL) and the mixture was stirred at room temperature under argon for 5 h, then filtered and evaporated. The residue was co-evaporated with toluene (2 × 100 mL) and dried under vacuum at 40 °C for 5 h. To a solution of thus obtained aldehyde in THF (200 mL) cooled to 0 °C was added dropwise 1 M vinylmagnesium bromide solution in THF or 1 M allylmagnesium bromide solution in diethyl ether (80 mL), the mixture was stirred at 0 °C for 2 h and at room temperature overnight, cooled to 0 °C, treated with water (10 ml) and evaporated. The residue was triturated with ethyl acetate (200 ml), the solids were filtered off and the solvent was removed in vacuo. The 2nd generation Grubbs catalyst (392 mg, 0.462 mmol) was added to a solution of the residue in dichloromethane and the solution was stirred at 40 °C under argon for 4 h, then at room temperature overnight and evaporated. Chromatography of the residue on silica gel (1 kg) in toluene–ethyl acetate (3:2) afforded compounds 4a and 4b or 7a and 7b. For yields, physical, analytical, and spectral data see ESI S1–S3.†
Selective benzoylation of 4a, 4b, 7a, and 7b
A solution of benzoyl chloride (0.67 mL, 5.77 mmol) in pyridine (9 ml) was added dropwise at room temperature within 20 min to a solution of dihydroxy derivative 4a, 4b, 7a, and 7b (5 mmol) in pyridine (20 mL) and the mixture was left overnight. Then water (2 mL) was added and, after 10 min, pyridine was evaporated. The residue was partitioned between ethyl acetate (50 mL) and water (20 mL). The organic phase was separated and washed with 5% hydrochloric acid (20 mL), water (20 mL), and 10% aqueous KHCO3 (3 × 20 mL), dried over anhydrous Na2SO4 and evaporated. Crystallization of the residue from cyclohexane afforded benzoates 5a, 5b, 8a or 8b. For yields, physical, analytical, and spectral data see ESI S3–S4.†Synthesis of protected nucleosides 9a, 9b, 12a, and 12b
A solution of monobenzoate 5a, 5b, 8a or 8b (4 mmol) in 80% aqueous trifluoroacetic acid (12 mL) was left at room temperature for 3 h then evaporated and co-distilled with toluene (3 × 20 mL). Acetic anhydride (2.27 mL, 24.0 mmol) was added to a solution of the residue and dimethylaminopyridine (488 mg, 3.99 mmol) in acetonitrile (15 mL) and the mixture was left at room temperature overnight. Then methanol (∼2 mL) was added and the solvents were evaporated. A solution of the residue in ethyl acetate (25 mL) was washed with water (20 mL) and then with 10% aqueous potassium hydrogen carbonate (2 × 20 mL), dried over anhydrous sodium sulfate and evaporated. To a stirred solution of the residue in acetonitrile (17 mL) was added benzoyladenine (1.05 g, 4.39 mmol) and then dropwise 1 M solution of tin(IV) chloride in dichloromethane (6 mL). The obtained solution was left at room temperature for 3 h and poured into a saturated aqueous solution of potassium hydrogen carbonate (70 mL). The mixture was extracted with ethyl acetate (2 × 100 mL) and the collected extracts were dried over sodium sulfate and evaporated. The residue was chromatographed on silica gel (200 g) in ethyl acetate–toluene (4:1). For yields, physical, analytical, and spectral data see ESI S4–S6.†
Deprotection of 9a, 9b, 12a, and 12b
A solution of the protected nucleoside 9a, 9b, 12a or 12b (2 mmol) in 0.1 M methanolic sodium methoxide (15 mL) was set at room temperature overnight. Water (0.5 mL) was then added and crystalline product 10a, 10b, 13a or 13b was filtered off, washed with methanol and ether. For yields, physical, analytical, and spectral data see ESI S6–S8.†Preparation of saturated nucleosides 11a, 11b, 14a, and 14b
Palladium hydroxide on carbon (20 wt%, 15 mg) was added to a solution of 10a, 10b, 13a or 13b (0.5 mmol) in dimethylformamide (2.5 ml) and the mixture was stirred under hydrogen at room temperature overnight. The catalyst was filtered off and the filtrate was evaporated. The solution of the residue in methanol (10 ml) was decolorized with charcoal and evaporated. The residue was crystalized from methanol. For yields, physical, analytical, and spectral data see ESI S8–S9.†Preparation of protected nucleotide analogues 16a, 16b, 17a, 17b, 18a, 18b, 19a, and 19b
Free nucleoside analogue 10a, 10b, 11a, 11b, 13a, 13b, 14a or 14b (0.5 mmol) was co-distilled with pyridine (3 × 5 mL) and the residue was dissolved in pyridine (8 mL). To the solution was added phenylboronic acid (65 mg, 0.533 mmol), and pyridine (∼6 mL) was distilled off under atmospheric pressure within 4 h and the remaining pyridine was evaporated under vacuum. A stirred solution of the residue in THF (2.5 mL) was treated under argon with a solution of N-[chloro(phenoxy)phosphoryl]-L-alaninate6 (417 mg, 1.50 mmol) in THF (2.5 mL) and 1-methylimidazole (250 μL, 3.14 mmol). The mixture was stirred at room temperature overnight and then evaporated. A solution of the residue in chloroform (10 mL) was washed with water (2 × 10 mL), dried over sodium sulfate and evaporated. Chromatography on silica gel in ethyl acetate–acetone–ethanol–water (100:15:6:4) and co-evaporation with methanol afforded the product as a solid foam.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 173.74 (d, JCO–P = 5.0, CO). ESI MS, m/z (%): 570.4 (31) [M + Na + H], 569.4 (100) [M + Na], 547.4 (12) [M + H]. HRMS (ESI): calcd for C23H28O8N6P [M + H] 547.17007; found: 547.17013.
O B), 173.80 (d, JCO–P = 4.1, CO A). ESI MS, m/z (%): 570.2 (30) [M + Na + H], 569.2 (100) [M + Na], 547.2 (41) [M + H]. HRMS (ESI): calcd for C23H27O8N6NaP [M + Na] 569.15202; found: 569.15175, calcd for C23H28O8N6P [M + H] 547.17007; found: 547.16996.
O B), 173.89 (d, JCO,P = 4.6, CO A). 31P NMR (202 MHz, CD3COCD3): δ 2.95, 3.96. HRMS (ESI): calcd for C24H29O8N6NaP [M + Na] 583.16767; found: 583.16760, calcd for C24H30O8N6P [M + H] 561.18572; found: 561.18578.
O B), 173.33 (d, JCO–P = 4.7, CO A). 31P NMR (202 MHz, CD3COCD3): δ 3.73, 3.96. ESI MS, m/z (%): 584.3 (32) [M + Na + H], 583.3 (100) [M + Na], 561.3 (56) [M + H]. HRMS (ESI): calcd for C24H30O8N6NaP [M + Na] 561.18572; found: 561.18557.
O), 173.75 (d, JCO–P = 5.2, CO). ESI MS, m/z (%): 572.2 (30) [M + Na + H], 571.2 (100) [M + Na], 549.2 (29) [M + H]. HRMS (ESI): calcd for C23H29O8N6NaP [M + Na] 571.16767; found: 569.16739, calcd for C23H30O8N6P [M + H] 549.18572; found: 549.18571.
O). ESI MS, m/z (%): 572.4 (28) [M + Na + H], 571.4 (100) [M + Na], 549.3 (12) [M + H]. HRMS (ESI): calcd for C23H29O8N6NaP [M + Na] 571.16767; found: 571.16749.
O), 173.85 (d, JCO–P = 5.3, CO). HRMS (ESI): calcd for C24H31O8N6NaP [M + Na] 585.18332; found: 585.18308, calcd for C24H32O8N6P [M + H] 563.20137; found: 563.20133.
O). ESI MS, m/z (%): 586.2 (33) [M + Na + H], 585.2 (100) [M + Na], 563.2 (18) [M + H]. HRMS (ESI): calcd for C24H32O8N6P [M + H] 563.20137; found: 563.20136.
O), 173.74 (d, JCO–P = 5.0, CO). ESI MS, m/z (%): 570.4 (31) [M + Na + H], 569.4 (100) [M + Na], 547.4 (12) [M + H]. HRMS (ESI): calcd for C23H28O8N6P [M + H] 547.17007; found: 547.17013.
O B), 173.80 (d, JCO–P = 4.1, CO A). ESI MS, m/z (%): 570.2 (30) [M + Na + H], 569.2 (100) [M + Na], 547.2 (41) [M + H]. HRMS (ESI): calcd for C23H27O8N6NaP [M + Na] 569.15202; found: 569.15175, calcd for C23H28O8N6P [M + H] 547.17007; found: 547.16996.
O B), 173.89 (d, JCO,P = 4.6, CO A). 31P NMR (202 MHz, CD3COCD3): δ 2.95, 3.96. HRMS (ESI): calcd for C24H29O8N6NaP [M + Na] 583.16767; found: 583.16760, calcd for C24H30O8N6P [M + H] 561.18572; found: 561.18578.
O B), 173.33 (d, JCO–P = 4.7, CO A). 31P NMR (202 MHz, CD3COCD3): δ 3.73, 3.96. ESI MS, m/z (%): 584.3 (32) [M + Na + H], 583.3 (100) [M + Na], 561.3 (56) [M + H]. HRMS (ESI): calcd for C24H30O8N6NaP [M + Na] 561.18572; found: 561.18557.
O), 173.75 (d, JCO–P = 5.2, CO). ESI MS, m/z (%): 572.2 (30) [M + Na + H], 571.2 (100) [M + Na], 549.2 (29) [M + H]. HRMS (ESI): calcd for C23H29O8N6NaP [M + Na] 571.16767; found: 569.16739, calcd for C23H30O8N6P [M + H] 549.18572; found: 549.18571.
O). ESI MS, m/z (%): 572.4 (28) [M + Na + H], 571.4 (100) [M + Na], 549.3 (12) [M + H]. HRMS (ESI): calcd for C23H29O8N6NaP [M + Na] 571.16767; found: 571.16749.
O), 173.85 (d, JCO–P = 5.3, CO). HRMS (ESI): calcd for C24H31O8N6NaP [M + Na] 585.18332; found: 585.18308, calcd for C24H32O8N6P [M + H] 563.20137; found: 563.20133.
O). ESI MS, m/z (%): 586.2 (33) [M + Na + H], 585.2 (100) [M + Na], 563.2 (18) [M + H]. HRMS (ESI): calcd for C24H32O8N6P [M + H] 563.20137; found: 563.20136.
Preparation of isopropylidene derivatives 22a and 22b
2,2-Dimethoxypropane (5 mL) and conc. H2SO4 (120 μL) were added to a solution of 13a or 13b (319 mg, 1.0 mmol) in dimethyl formamide (6 mL) and the mixture was stirred at ambient temperature for 24 h. It was then was neutralized with pulverized KHCO3 and evaporated. The residue was triturated with water, the solid was filtered off, washed with water, dried and chromatographed on silica gel (40 g) in ethyl acetate–acetone–ethanol–water (400:30:12:8).
Preparation of phosphates 23a and 23b
Method A: To a stirred solution of 22a or 22b (179 mg, 0.5 mmol) in trimethyl phosphate (1.2 mL) cooled to 0 °C was added dropwise POCl3 (213 μL, 2.25 mmol). The mixture was stirred at ambient temperature for 4 h and then cooled to 0 °C. Water was added (0.8 mL) and the mixture was neutralized with aqueous ammonia and applied on a Dowex 50 (H+) column (30 mL). The column was eluted with water (200 mL) and then with 2.5% aqueous ammonia. UV absorbing fractions were collected and evaporated. A solution of the residue in 50% aqueous trifluoroacetic acid (2 mL) was kept at room temperature for 48 h, then evaporated and twice co-evaporated with water. A solution of the residue in water was neutralized with aqueous ammonia and adsorbed on silica gel, which was placed on a top of silica gel column (40 mL). Elution with 2-propanol–water–conc. aqueous ammonia (15:4:2) afforded crude 23a or 23b, which were re-purified on Dowex 50 (H+) column (15 mL) by elution with water (100 mL) and 2.5% aqueous ammonia. UV absorbing fractions were collected, concentrated and converted to sodium salt using Dowex 50 (Na+) column (30 mL) by the elution of the column with water and subsequent evaporation of UV absorbing fractions.
Method B: Nucleoside analogue 13a or 13b (160 mg, 0.5 mmol) was co-distilled with pyridine (3 × 5 mL) and dissolved in pyridine (8 mL). To the solution was added phenylboronic acid (65 mg, 0.53 mmol), pyridine (∼6 mL) was distilled off at atmospheric pressure within 4 h and the remaining pyridine was evaporated under vacuum. To a stirred solution of the residue in trimethyl phosphate (1.2 mL) cooled to 0 °C was added dropwise POCl3 (213 μL, 2.25 mmol). The mixture was stirred at ambient temperature for 4 h. The reaction mixture was worked up as in Method A. 60 mg (26%) of 23a and 62 mg (27%) of nucleotide analogue 23b were obtained.
Preparation of triphosphates 24a and 24b
A solution of phosphate 23a or 23b in water (0.2 mL) was placed on top of a Dowex 50 (Et3NH+) column (10 mL) and the column was eluted with water. UV absorbing fractions were collected and evaporated. A mixture of the residue, imidazole (51 mg, 0.75 mmol) and triphenylphosphine (131 mg, 0.5 mmol) was co-distilled with DMF (3 × 3 mL). To a solution of the residue in DMSO (0.25 mL) and DMF (0.5 mL) were added triethylamine (24.5 μL, 0.175 mmol) and 2,2′dipyridyl disulfide (110 mg, 0.5 mmol) and the mixture was stirred at room temperature under an argon atmosphere overnight. The mixture was then added dropwise at 0 °C to a mixture of acetone (15 mL), diethylether (15 mL) and saturated solution of NaClO4·H2O in acetone (0.15 mL). The precipitate was centrifuged at 0 °C and washed with a mixture of diethylether–acetone (1:1, 2 × 20 mL) and ether (2 × 20 mL), both at 0 °C. The residue was dried over P2O5 under vacuum overnight. Dry imidazolide was dissolved in 0.5 M solution of (nBu3NH)2P2H2O7 in DMSO (0.3 mL) and the solution was left at room temperature overnight. Crude triphosphate was purified by reverse phase HPLC.
Carboxypeptidase Y assay
The phosphoramidate derivative 17a or 17b (5.0 mg) was dissolved in acetone-d6 (200 μL) and then 400 μL of Trizma (tris(hydroxymethyl)aminomethane) buffer (pH 7.4) was added at room temperature (25 °C). 31P NMR spectrum (202 MHz with H3PO4 as external standard) of this sample was measured to obtain 31P chemical shifts of the starting phosphoramidate. Next, carboxypeptidase Y (0.3 mg dissolved in 200 μL of Trizma buffer) was added and 31P NMR spectra were recorded at various times after the addition (25 °C).Chemical hydrolysis
The studied compound 17a or 17b (5.4 mg) was dissolved in 500 μL of triethylamine aqueous solution (3% Et3N in D2O) and 31P NMR spectra (202 MHz with H3PO4 as external standard) were recorded forthwith at room temperature (25 °C) and then at various times after the sample preparation.Screening of antiviral activity
The anti-rhinovirus activity of test compounds was measured in H1-HeLa cells. Compounds were prepared in replicate 3-fold serial dilutions in DMSO (384-well format), and 0.1 μL of these dilutions were transferred acoustically to assay plates with an Echo instrument. H1-HeLa cells in RPMI medium supplemented with 10% heat inactivated FBS and antibiotics were premixed in batch with a mixture of HRV1A, HRV14, and HRV16 at a TCID90 of 4X for each strain. After three days of incubation at 33 °C, virus-induced cytopathic effects were determined by a Cell-titer Glo viability assay (Promega, Madison, WI).Anti HCV activity of compounds was determined in multiplex assay using HUH 7-lunet stably replicating I389luc-ubi-neo/NS3-3′/ET genotype 1b replicon and 2aLucNeo-25 cell line encoding a genotype 2a JFH-1 replicon. Cells were maintained in Dulbecco medium (DMEM) supplemented with GlutaMAX (Invitrogen), 10% FBS (not heat-inactivated), 1 mg mL−1 G-418, Pen-Strep, and non-essential amino acids. Cells were plated into 384-well assay plates with 1600 cells per well and treated with serial dilutions of compounds. Following three days of incubation, the activity of Renilla luciferase was quantified using the Dual-Glo luciferase assay system from Promega (Promega, Madison, WI).
Inhibition of the activity of PI4Ks
The lipid kinase activity was determined by ADP-Glo Kinase Assay (Promega) measuring the amount of ADP produced during the kinase reaction.26 Reactions were carried out in a total volume of 5 μl and contained PI4 K enzyme (final concentrations for PI4 K IIIβ was 4 ng μL−1, for PI4 K IIIα was 2 ng μL−1, and for PI4 K IIα was 2 ng μL−1) in kinase buffer (20 mM TRIS pH 7.5; 5 mM MgCl2; 0.2% Triton-X; 0.1 mg mL−1 BSA; 2 mM DTT), PI/PS (lipid kinase substrate) in kinase buffer (final concentration = 50 μM), inhibitors (10 mM stock solutions in DMSO were diluted with kinase buffer to a final concentration dependent on inhibitor's activity, e.g. 400–0.01; 300–0.0001; 150–0.00001 μM for PI4 K IIIβ), and the reaction was started by adding ATP in kinase buffer (final concentration 100 μM). This reaction was carried out for 60 min at 25 °C and the amount of hydrolyzed ATP was measured according to the manufacturer's protocol (add ADP/Glo Reagent to terminate the kinase reaction and deplete the remaining ATP, then add Kinase Detection Reagent to simultaneously convert ADP to ATP and allow the newly synthesized ATP to be measured using a luciferase/luciferin reaction). Luminescence was measured using a TECAN infinite M 1000 spectrophotometer.Acknowledgements
This study was supported by the Czech Science Foundation (grant no. P207/12/P625 (M. Š.) and no. 15-09310S (P. P. and R. N.)), the Ministry of the Interior of the Czech Republic (VG20102015046) and Gilead Sciences, Inc. (Foster City, CA, USA). The work of Eliška Procházková (NMR spectroscopy) was supported by the Czech Science Foundation (grant No. 13-24880S). Subvention for the development of research organization (RVO: 61388963) is also acknowledged.Notes and references
- G. M. Blackburn, M. J. Gait, D. Loakes and D. M. Williams, Nucleic Acids in Chemistry and Biology, 3rd Edn, 2006, pp. 13–75 Search PubMed.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS.
- V. E. Marquez, A. Ezzitouni, P. Russ, M. A. Siddiqui, H. Ford, R. J. Feldman, H. Mitsuya, C. George and J. J. Barchi, J. Am. Chem. Soc., 1998, 120, 2780–2789 CrossRef CAS.
- V. E. Marquez, M. A. Siddiqui, A. Ezzitouni, P. Russ, J. Y. Wang, R. W. Wagner and M. D. Matteucci, J. Med. Chem., 1996, 39, 3739–3747 CrossRef CAS PubMed.
- V. E. Marquez, S. H. Hughes, S. Sei and R. Agbaria, Antiviral Res., 2006, 71, 268–275 CrossRef CAS PubMed.
- V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M. C. Nicklaus and R. Agbaria, J. Am. Chem. Soc., 2004, 126, 543–549 CrossRef CAS PubMed.
- M. Dejmek, M. Šála, H. Hřebabecký, M. Dračínský, E. Procházková, D. Chalupska, M. Klíma, P. Plačková, M. Hájek, G. Andrei, L. Naesens, P. Leyssen, J. Neyts, J. Balzarini, E. Boura and R. Nencka, Bioorg. Med. Chem., 2015, 23, 184–191 CrossRef CAS PubMed.
- M. Dejmek, H. Hřebabecký, M. Šála, M. Dračínský, E. Procházková, P. Leyssen, J. Neyts, J. Balzarini and R. Nencka, Bioorg. Med. Chem., 2014, 22, 2974–2983 CrossRef CAS PubMed.
- S. Kauppinen, B. Vester and J. Wengel, Drug Discovery Today Technol., 2005, 2, 287–290 CrossRef PubMed.
- R. N. Veedu and J. Wengel, Chem. Biodiversity, 2010, 7, 536–542 CAS.
- M. Šála, M. Dejmek, E. Procházková, H. Hřebabecký, J. Rybáček, M. Dračínský, P. Novák, S. Rosenbergová, J. Fukal, V. Sychrovský, I. Rosenberg and R. Nencka, Org. Biomol. Chem., 2015, 13, 2703–2715 Search PubMed.
- C. J. Leumann, Bioorg. Med. Chem., 2002, 10, 841–854 CrossRef CAS.
- A. Stauffiger and C. J. Leumann, Eur. J. Org. Chem., 2009, 1153–1162 CrossRef CAS PubMed.
- P. Silhar and C. J. Leumann, Bioorg. Med. Chem., 2010, 18, 7786–7793 CrossRef CAS PubMed.
- M. Hollenstein and C. J. Leumann, ChemBioChem, 2014, 15, 1901–1904 CrossRef CAS PubMed.
- S. Hanessian and D. J. Ritson, J. Org. Chem., 2006, 71, 9807–9817 CrossRef CAS PubMed.
- M. J. Robins, S. F. Wnuk, X. D. Yang, C. S. Yuan, R. T. Borchardt, J. Balzarini and E. De Clercq, J. Med. Chem., 1998, 41, 3857–3864 CrossRef CAS PubMed.
- (a) U. Niedballa and H. Vorbrüggen, J. Org. Chem., 1974, 39, 3654–3660 CrossRef CAS; (b) K. S. Toti, M. Derudas, C. McGuigan, J. Balzarini and S. Van Calenbergh, Eur. J. Med. Chem., 2011, 46, 3704–3713 CrossRef CAS PubMed; (c) P. P. Seth, G. Vasquez, C. A. Allerson, A. Berdeja, H. Gaus, G. A. Kinberger, T. P. Prakash, M. T. Migawa, B. Bhat and E. E. Swayze, J. Org. Chem., 2010, 75, 1569–1581 CrossRef CAS PubMed; (d) A. E. Hakansson and J. Wengel, Bioorg. Med. Chem. Lett., 2001, 11, 935–938 CrossRef CAS.
- During the preparation of this manuscript, Mayes et al. also reported utilization of similar technique for installation of different types of phosphoramidate prodrugs and according to our experience this procedure can be applied to a variety of other nucleoside substrates with results significantly exceeding the traditional approaches. B. A. Mayes, J. Arumugasamy, E. Baloglu, D. Bauer, A. Becker, N. Chaudhuri, G. M. Latham, J. Li, S. Mathieu, F. P. McGarry, E. Rosinovsky, A. Stewart, C. Trochet, J. Wang and A. Moussa, Org. Process Res. Dev., 2014, 18, 717–724 CrossRef CAS.
- (a) A. R. Kore and B. Srinivasan, Curr. Org. Synth., 2013, 10, 903–934 CrossRef CAS; (b) M. Hocek, J. Org. Chem., 2014, 79, 9914–9921 CrossRef CAS PubMed; (c) M. Hollenstein, Molecules, 2012, 17, 13569–13591 CrossRef CAS PubMed; (d) K. Burgess and D. Cook, Chem. Rev., 2000, 100, 2047–2059 CrossRef CAS PubMed.
- In ROESY spectrum of compound 13b, it is possible to observe crosspeaks H8′–H2, H8′–H3, H8′–3OH indicating that both conformers (syn and anti) are present in solution, see in ESI S33.† Furthermore, hydrogen-coupled 13C NMR spectra were measured to obtain heteronuclear coupling constants between H2 and C4′/C8′, which also support the conclusion that there is a mixture of both conformers in solution. syn/anti Conformation has been also investigated using DFT calculations of shielding constants in combination with experimentally obtained chemical shifts. There is a significant difference between calculated chemical shift (B3LYP/6-31g(d)) of carbon atom C2 in syn and anti conformer (92.2 and 85.4 ppm, respectively). Experimentally obtained chemical shift of C2 (measured in DMSO-d6) is equal to 86.8 ppm indicating that anti conformer is predominant.
- C. A. G. Haasnoot, F. A. A. M. de Leeuw and C. Altona, Tetrahedron, 1980, 36, 2783–2792 CrossRef CAS.
- C. McGuigan, P. Murziani, M. Slusarczyk, B. Gonczy, J. Vande Voorde, S. Liekens and J. Balzarini, J. Med. Chem., 2011, 54, 7247–7258 CrossRef CAS PubMed.
- In situ structure determination of intermediate 20b was performed during approximately the first two hours of the reaction, when the concentration of the intermediate was above 50%. The structure determination is based on the in situ analysis of C–P spin–spin interactions across 1–3 bonds visible in 13C NMR spectra as a line splitting. It is clear that the methylester was hydrolyzed; the intensity of the signals corresponding to the methyl of the methylester group (two signals = two diastereoisomers around 52.55 ppm) decreased whereas the intensity of methanol single signal (52.45 ppm) increased. The phenyl moiety is still attached to the molecule, signals corresponding to the carbon atoms in ortho position are still two doublets (two, because of the two diastereoisomers and doublets because of the C–P interaction). After several hours, the phenyl ring was cleaved and the stable final product was obtained. The two doublets around 120.3 ppm, corresponding to ortho carbons, disappeared and one singlet signal around 118.9 ppm corresponding to ortho carbons of phenol appeared. After the phenol release, there is no chiral center at the phosphorus atom, thus no two diastereoisomers exist. It is visible, for instance, in 13C NMR spectra of carbon atom 8a, where the two doublets (87.8 ppm) corresponding to the two diastereoisomers of the intermediate disappeared and one doublet (88.6 ppm) coming from carbon atom 8a of the final product appeared (ESI Fig. S1 and S2†).
- J. Y. Feng, G. Cheng, J. Perry, O. Barauskas, Y. Xu, M. Fenaux, S. Eng, N. Tirunagari, B. Peng, M. Yu, Y. Tian, Y.-J. Lee, G. Stepan, L. L. Lagpacan, D. Jin, M. Hung, K. S. Ku, B. Han, K. Kitrinos, M. Perron, G. Birkus, K. A. Wong, W. Zhong, C. U. Kim, A. Carey, A. Cho and A. S. Ray, Antimicrob. Agents Chemother., 2014, 58, 1930–1942 CrossRef PubMed.
- I. Mejdrová, D. Chalupská, M. Kögler, M. Šála, P. Plačková, A. Baumlová, H. Hřebabecký, E. Procházková, M. Dejmek, R. Guillon, D. Strunin, J. Weber, G. Lee, G. Birkus, H. Mertlíková-Kaiserová, E. Boura and R. Nencka, J. Med. Chem., 2015, 58, 3767–3793 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00987a |
| This journal is © The Royal Society of Chemistry 2015 |