
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A divergent approach to benzylisoquinoline-type and oxoaporphine alkaloids via regioselective direct ring metalation of alkoxy isoquinolines†
Benedikt
Melzer
and
Franz
Bracher
*
Department of Pharmacy – Center for Drug Research, Ludwig-Maximilians University, Butenandtstr. 5-13, D-81377 Munich, Germany. E-mail: Franz.Bracher@cup.uni-muenchen.de; Fax: +49-89-2180-77802; Tel: +49-89-2180-77301
First published on 9th June 2015
Abstract
Methoxy- and benzyloxy-substituted isoquinolines are regioselectively metalated at C-1 with the Knochel–Hauser base, subsequent trapping with aromatic aldehydes gives aryl(isoquinolin-1-yl)carbinols as building blocks for divergent syntheses of different types of benzylisoquinoline alkaloids. Photochemical cyclization of ortho-bromo analogues under reductive conditions gives oxoaporphine alkaloids. Nine benzylisoquinoline alkaloids and two oxoaporphine alkaloids were obtained in two or three steps from appropriate isoquinolines.
Introduction
Benzylisoquinoline alkaloids are a large group of plant secondary metabolites which includes about 2500 known structures. Besides simple benzylisoquinolines, more complex tetracyclic ring systems (aporphines, protoberberines, cularines, pavines) and the pentacyclic morphinane-type alkaloids belong to this class. All these alkaloids share a common biosynthetic origin, with (S)-norcoclaurine, a metabolite formed by condensation of dopamine and 4-hydroxyphenylacetaldehyde, as the first common intermediate. Manifold biological activities have been reported for alkaloids from this class, among them narcotic, spasmolytic, dopaminergic, ion-channel modulating, and cytotoxic properties. A timely review of structures, biosynthesis and pharmacology of benzylisoquinoline alkaloids is provided by Hagel and Facchini.1Benzylisoquinoline alkaloids in the narrower sense bear up to three oxygen functions (hydroxy, methoxy, methylenedioxy) on the carbocyclic part of the isoquinoline, and one or more oxygen functions at the benzylic ring; the benzylic carbon is a methylene group in most cases, but can also be a carbinol (or its methyl ether) or a carbonyl group (Fig. 1).
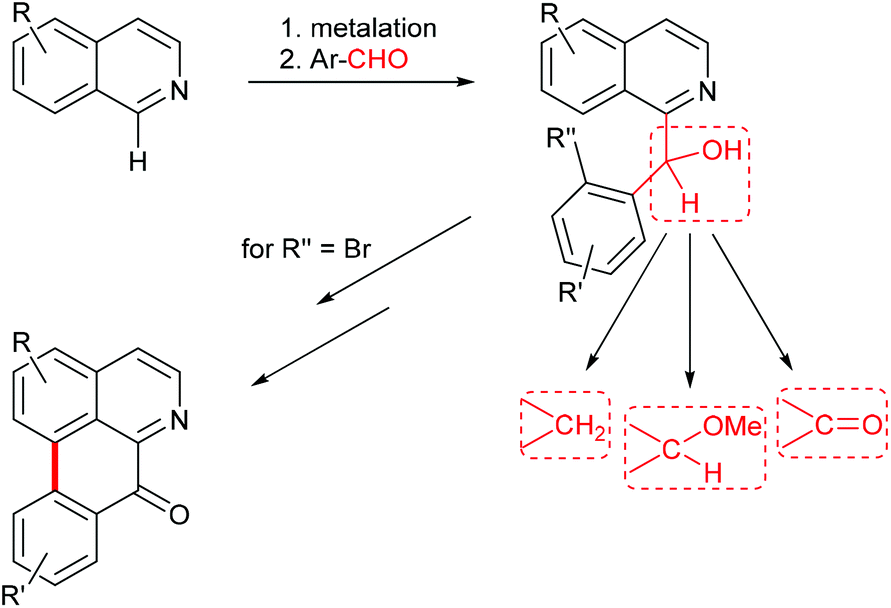 | ||
| Fig. 1 New approach to benzylisoquinoline and oxoaporphine alkaloids via regioselective metalation of substituted isoquinolines. | ||
Classical synthetic approaches to the benzylisoquinoline alkaloids are inspired by the biosynthesis and include acid-mediated cyclizations of arylacetamides (Bischler-Napieralski) or arylacetaldimines of phenylethylamines (Pictet-Spengler), followed by a dehydrogenation step.2 Alternatively, isoquinolines can be benzylated at C-1 via N-benzoyl-1,2-dihydroisoquinoline-1-carbonitriles (Reissert synthesis3), but this method suffers from the need to use stoichiometric amounts of toxic cyanide. For the preparation of alkaloids containing a C-1 substituent other than methylene, laborious variants of these methodologies must be applied.4–9 A Pomeranz-Fritsch approach to benzylisoquinolines through cyclization of complex 2-aryl arylethylaminoacetaldehyde acetals was found to have a very narrow scope.10,11 A convenient access to 1-benzoylisoquinolines has been developed recently, utilizing direct acylation of isoquinolines at C-1 with benzoyl radicals.12,13 Other approaches start from 1-iodoisoquinolines, and consist of either nucleophilic substitution with deprotonated arylacetonitriles, followed by oxidation,14 or zinc insertion followed by copper-catalyzed coupling with an aroyl chloride.15
In continuation of our recent work on the synthesis of aromatic alkaloids using direct ring metalations of heterocycles as the crucial step,16,17 we intended to work out a novel, flexible approach to benzylisoquinoline alkaloids. This work was inspired by two single reports of Knochel on the direct metalation of isoquinoline18 and 6,7-dimethoxyisoquinoline19 at C-1 with the hindered amide base TMPMgCl·LiCl. Quenching with iodine, followed by Pd-catalyzed Negishi cross-coupling of the 1-iodoisoquinoline with a benzylzinc reagent gave the benzylisoquinoline alkaloid papaverine (9), whereas attempted direct cross-coupling of the magnesiated isoquinoline with a benzyl chloride did not provide the alkaloid.19 We envisaged to trap 1-magnesiated isoquinoline building blocks with appropriately substituted benzaldehydes to obtain aryl(isoquinolin-1-yl)methanols, which in turn would open an access to benzylisoquinolines, benzoylisoquinolines, and 1′-methoxy-substituted benzylisoquinolines in one single step (hydrogenolysis of the benzylic hydroxy group, oxidation, or etherification) each. This should represent a divergent approach to four common subtypes of benzylisoquinoline alkaloids. Further, carbinols prepared from ortho-bromo-substituted benzaldehydes should open an access to oxoaporphine alkaloids via intramolecular biaryl synthesis, utilizing either photochemical,14,20–22 radical23 or Pd-catalyzed24,25 reactions. The oxoaporphines are of high pharmaceutical relevance due to their antibacterial, antifungal, anticancer and other biological activities26,27 (Fig. 1).
Results and discussion
We started our investigations with 6,7-dimethoxyisoquinoline (1a), since the metalation at C-1 with the Knochel–Hauser base (TMPMgCl·LiCl) had been reported previously by the Knochel group.19 Metalation with 1.5 equivalents of the base over 4 h at room temperature, followed by reaction with various benzaldehydes 2 (1.5 equiv.) at 0 °C gave the expected racemic secondary alcohols 3a–d in moderate yields (37–53%) (Scheme 1). One of these alcohols (3b) is the racemate of the alkaloid papaverinol (isolated from Papaver somniferum and other plants; previously synthesized by either oxidation of papaverine (9)28 or reduction of papaveraldine (4)29), alcohol 3a is the dimethyl ether of the alkaloid annocherin A (7).30 Side-reactions were not observed, and significant amounts (up to 49%) of the starting material 1a were recovered. Modifications of the reaction conditions were examined in order to improve the yields. The use of just 1.1 equivalents of TMPMgCl·LiCl led to lower yields of the secondary alcohols. However, the use of 2.0 equivalents TMPMgCl·LiCl did not increase the yields. Also longer reaction times for the metalation step up to 16 h did not result in higher yields. Attempted metalation with the “frustrated Lewis pair” TMPMgCl·BF3·LiCl, a system which was applied to the regioselective metalation of pyridines and quinolines before,31 failed completely due to spontaneous formation of a precipitate.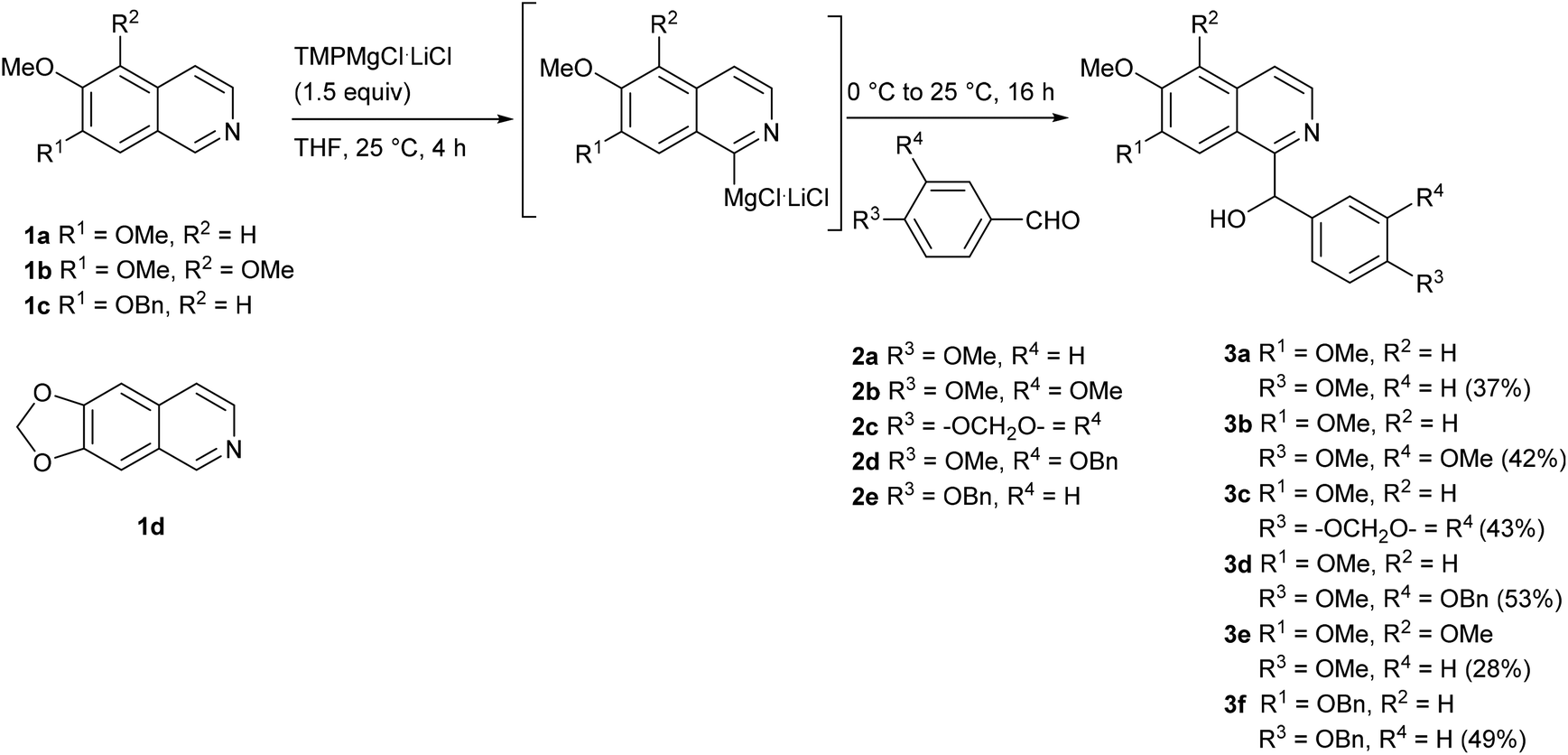 | ||
| Scheme 1 Synthesis of secondary alcohols 3 from alkoxy-substituted isoquinolines 1. | ||
In order to explore the scope of this methodology, especially with view on alkaloids with other substitution patterns in the isoquinoline part, we expanded our method to the metalation of readily available isoquinolines 1b–d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32–34 (Scheme 1). The metalation of 5,6,7-trimethoxyisoquinoline (1b) with TMPMgCl·LiCl (1.5 equiv.) at room temperature for 4 h, followed by reaction with 4-methoxybenzaldehyde (2a) afforded the secondary alcohol 3e in 28% yield. The direct metalation of 7-benzyloxy-6-methoxyisoquinoline (1c) under the same reaction conditions and reaction with 4-(benzyloxy)benzaldeyhde (2e) provided the secondary alcohol 3f in 49% yield. Unfortunately, any attempts to perform a controlled ring metalation of 6,7-methylenedioxyisoquinoline (1d) failed. An iodine quenching after a metalation experiment gave a poorly separable mixture of iodinated products, from which <10% of impure 8-iodo derivative was isolated. We further investigated whether better yields can be obtained by activation of the aromatic aldehydes with a Lewis acid (BF3), but independent of the sequence of addition of the components spontaneous precipitation was observed, and not even traces of the desired carbinols were obtained.
32–34 (Scheme 1). The metalation of 5,6,7-trimethoxyisoquinoline (1b) with TMPMgCl·LiCl (1.5 equiv.) at room temperature for 4 h, followed by reaction with 4-methoxybenzaldehyde (2a) afforded the secondary alcohol 3e in 28% yield. The direct metalation of 7-benzyloxy-6-methoxyisoquinoline (1c) under the same reaction conditions and reaction with 4-(benzyloxy)benzaldeyhde (2e) provided the secondary alcohol 3f in 49% yield. Unfortunately, any attempts to perform a controlled ring metalation of 6,7-methylenedioxyisoquinoline (1d) failed. An iodine quenching after a metalation experiment gave a poorly separable mixture of iodinated products, from which <10% of impure 8-iodo derivative was isolated. We further investigated whether better yields can be obtained by activation of the aromatic aldehydes with a Lewis acid (BF3), but independent of the sequence of addition of the components spontaneous precipitation was observed, and not even traces of the desired carbinols were obtained.
Having the carbinols 3a–f in hand, divergent syntheses were accomplished, leading to benzylisoquinoline alkaloids bearing other functional groups at the benzylic C-1′ position.
Oxidation of the secondary alcohols 3b and 3e with manganese(IV) oxide in refluxing dichloromethane for 6 h afforded the natural products papaveraldine (4; isolated from Papaver somniferum and other plants; first total synthesis by oxidation of papaverine (9)35) in 68% and thalimicrinone (5; isolated from Thalictrum minus var. microphyllum;36 first total synthesis utilizing a Reissert-type reaction5) in 98% yield, respectively (Scheme 2).
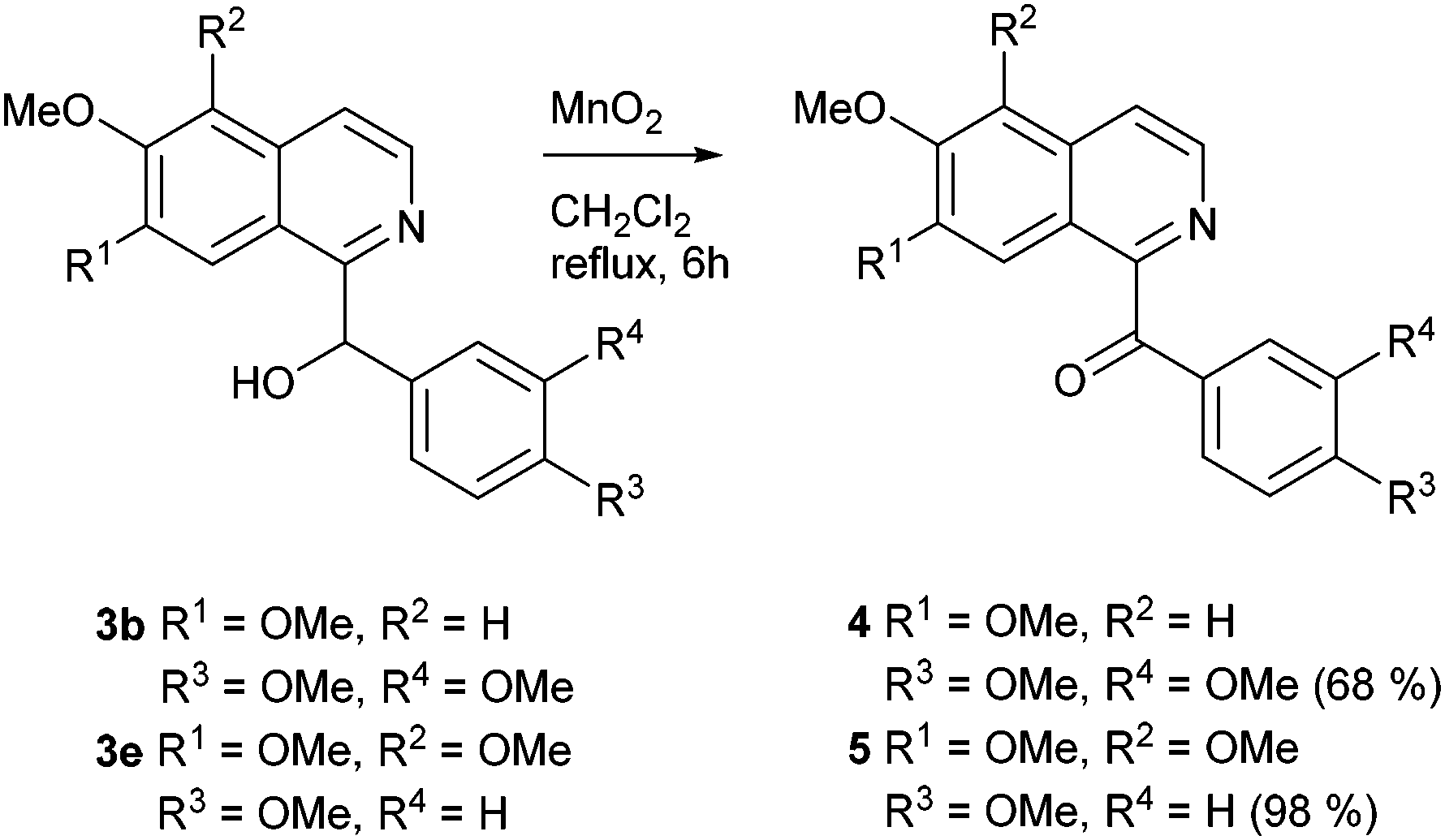 | ||
| Scheme 2 Synthesis of the benzoylisoquinoline alkaloids papaveraldine (4) and thalimicrinone (5). | ||
Deprotonation of the secondary alcohols 3b and 3c using sodium hydride in dry DMF and subsequent reaction with iodomethane over 2 h furnished the racemic O-methylated alkaloids setigerine (6a; isolated from Papaver setigerum DC;37 first total synthesis from papaverinol (3b), see ref. 38) and setigeridine (6b; isolated from Papaver setigerum DC;37 first total synthesis see ref. 9) in yields of 77% and 91%, respectively. Analogous O-methylation of 3f under the same conditions led to 6c, the central intermediate in the synthesis of annocherin B (8), in 90% yield (Scheme 3).
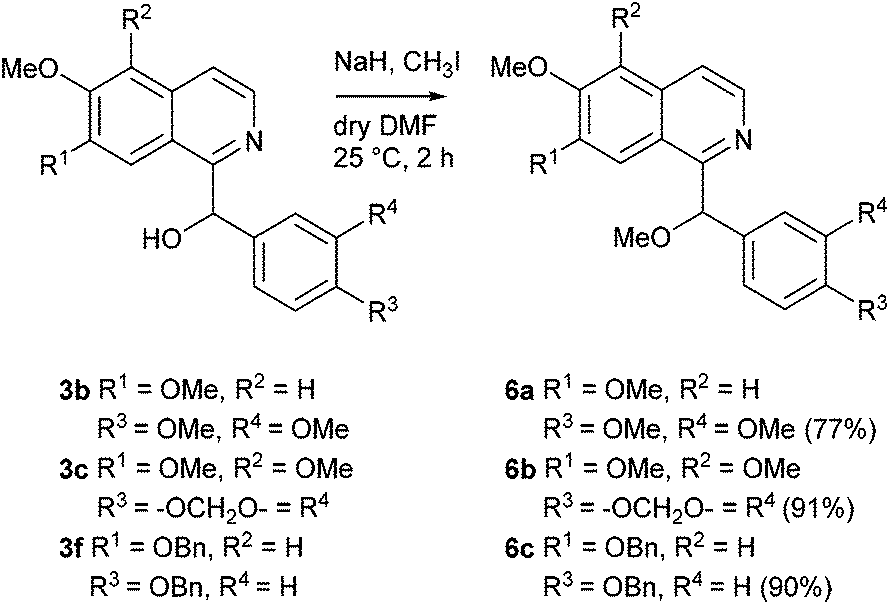 | ||
| Scheme 3 O-Methylation of the alcohols 3b,c,f to setigerine (6a), setigeridine (6b), and 6c. | ||
By hydrogenolytic removal of both O-benzyl protective groups in 3f and 6c in methanol over Pd/C catalyst (room temperature for 24 h, 90 and 66% yields) the first total syntheses of the racemic alkaloids annocherin A (7) and annocherin B (8; both isolated from Annona cherimola39) were completed. The preparation of 7 was accomplished in just two steps (overall yield 44%) starting from 7-benzyloxy-6-methoxyisoquinoline (1c), annocherin B (8) was synthesised in three steps with an overall yield of 29% (Scheme 4).
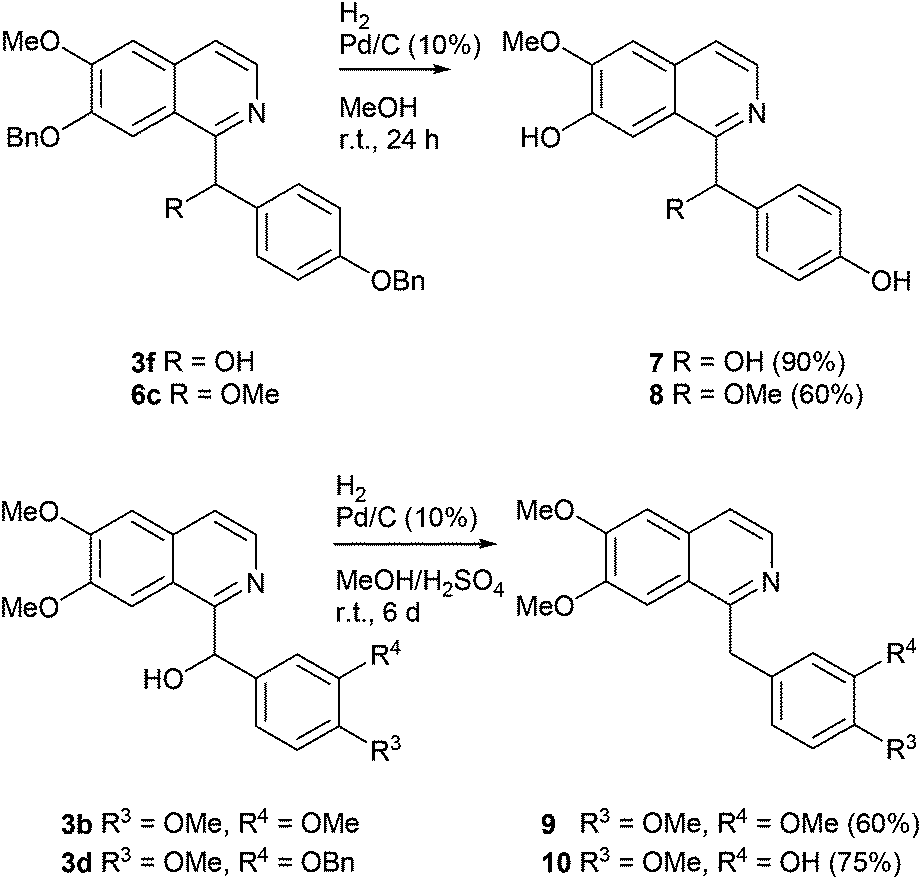 | ||
| Scheme 4 Synthesis of the alkaloids annocherin A (7), annocherin B (8), papaverine (9), and palaudine (10) by multiple hydrogeneolyses. | ||
Deoxygenation of the benzhydrol-type alcohol papaverinol (3b) was found to be less feasible, and needed careful optimization of the reaction conditions. Finally, we found that hydrogenation over Pd/C (10%) in a methanol/sulphuric acid mixture at room temperature for 6 d gives the benzylisoquinoline alkaloid papaverine (9; isolated from Papaver somniferum and other plants; first total synthesis by Pictet and Gams40) in 60% yield. It was also possible to simultaneously remove the O-benzyl protective group and deoxygenate the benzylic position of compound 3d under these conditions to yield the alkaloid palaudine (10; isolated from Papaver somniferum;41 first total synthesis by O-demethylation of papaverine42) in 75% yield (Scheme 4).
Having established a general access to variously substituted benzylisoquinolines, we wished to extend this methodology to the total synthesis of oxoaporphine alkaloids. As mentioned in the introduction (Fig. 1), intermediates bearing ortho-bromobenzyl residues can be applied to intramolecular aryl–aryl coupling reactions, and on the basis of literature data the photochemical approach appeared most promising. Formally, the pertinent 1-(2-bromobenzoyl)isoquinolines appear to be the best substrates, but previous investigations revealed that the corresponding carbinols are much more susceptible to this cyclization, an oxidation of the carbinol to the keto group obviously takes place after completed cyclization under the workup conditions.20–22 Chuang et al.14 even reported a “reductive photocyclization” of 1-(2-bromobenzoyl)isoquinolines to oxoaporphines, comprising an in situ reduction of the starting ketones to the carbinols, followed by photocyclization and aerial re-oxidation during workup. These insights made our above-mentioned approach highly attractive, since it provides a direct access to the carbinols as the most promising cyclization substrates.
Fortunately, the ring metalation/aldehyde quenching protocol could be applied to ortho-bromobenzaldehydes without any problems. Reaction of C-1 magnesiated 6,7-dimethoxyisoquinoline with 2-bromobenzaldeyhde (11a) furnished carbinol 12a in 69% yield, with 6-bromovertraldehyde (11b) the carbinol 12b was obtained in 35% yield. A first attempt of a photocyclization of 12a in methanol (concentration 2.5 mM) in a photoreactor (mercury vapour lamp, 125 W, 3 h) gave the expected oxoaporphine alkaloid lysicamine (13; isolated from Lysichiton camtschatcense Schott var. japonicum Makino;43 first total synthesis starting from nuciferine44) in only 10% yield, accompanied by numerous by-products. Longer reaction times led to even more by-products. However, photocyclization in the presence of NaBH4 (1.5 equivalents) proceeded well and provided lysicamine (13) in 53% yield after 3 h reaction time. Obviously, it is suitable not only to use the carbinol as a starting material, but also to suppress the formation of keto forms of educt and product during the irradiation process, and then rely on spontaneous oxidation during workup. Photo-induced cyclization of alcohol 12b under the same conditions yielded the oxoaporphine alkaloid oxoglaucine (14; isolated from Liriodendron tulipifera, L.45 first total synthesis utilizing a Pschorr cyclization starting from nitropapaveraldine46) in 59% in just 1 h (Scheme 5).
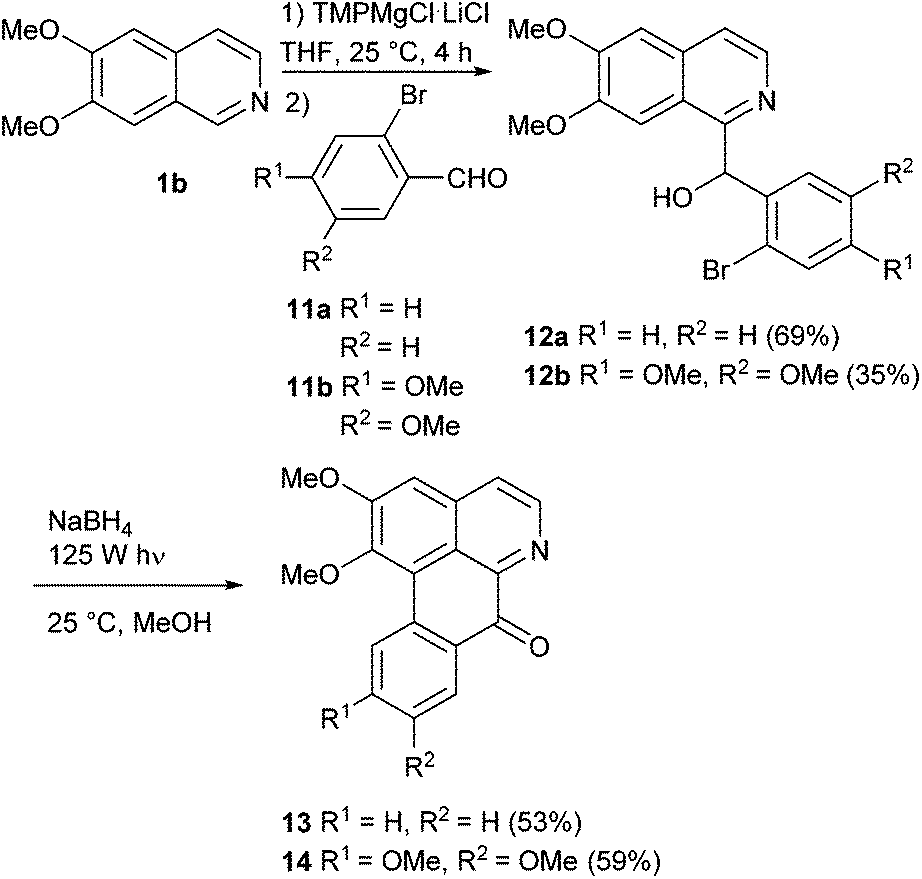 | ||
| Scheme 5 Synthesis of the oxoaporphine alkaloids lysicamine (13) and oxoglaucine (14). | ||
In conclusion, we worked out a new protocol for the synthesis of aryl(isoquinolin-1-yl)carbinols by direct regioselective metalation of alkoxy isoquinolines, followed by reaction with aromatic aldehydes. These carbinols are versatile intermediates for divergent syntheses of benzylisoquinoline alkaloids and oxoaporphines. Eleven alkaloids were synthesized using this protocol in two (for 1′-methoxy compounds three) steps starting from readily accessible 1-unsubstituted alkoxy isoquinolines.
Experimental section
General information
All reactions were performed under nitrogen atmosphere with flame-dried glassware, unless otherwise stated. Solvents used were of HPLC grade or p.a. grade and/or purified according to standard procedures. Photochemical reactions were conducted using a HPK 125W high pressure mercury vapor lamp from Heraeus Noblelight. Melting points were determined by open tube capillary method with a Büchi melting point B-450 apparatus. IR measurements were carried out with a Perkin-Elmer FTIR Paragon 1000 spectrometer. NMR spectra were recorded with Jeol J NMR GX (400 or 500 MHz) and Avance III HD Bruker BioSpin (400 or 500 MHz) spectrometers with residual non-deuterated solvent as internal standard. Spectra were recorded in deuterated solvents and chemical shifts are reported in parts per million (ppm). J values are given in Hertz. Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet. Signal assignments were carried out based on 1H, 13C, HMBC, HMQC and COSY spectra. NMR spectra were analyzed with the NMR software MestReNova, Version 5.1.1-3092 (Mestrelab Research S.L.) HRMS were performed by electron impact (EI) at 70 eV with a Thermo Finnigan MAT 95 or a Jeol GCmate II spectrometer or by electrospray ionization (ESI) with a Thermo Finnigan LTQ FT Ultra Fourier Transform Ion Cyclotron resonance mass spectrometer. Chromatographic purification of products was performed by using flash column chromatography on Merck silica gel 60 (0.015–0.040 mm) as stationary phase.General procedure A (preparation of secondary alcohols 3a–f/12a–b)
A dry and nitrogen flushed 25 mL Schlenk tube, equipped with a magnetic stirring bar, was charged with the appopriate isoquinoline 1a,b (1.00 mmol) in THF (4 mL) or 1c (2.00 mmol) in dry THF (6 mL). TMPMgCl·LiCl (1.50 equiv., 1.0 M in THF/toluene) was added to this solution dropwise over 2 min. The reaction mixture was stirred at room temperature for 4 h. After cooling to 0 °C the corresponding benzaldehyde 2a–e/11a–b (1.50 equiv.), dissolved in dry THF (2 mL), was added dropwise to the reation mixture. The mixture was allowed to warm to room temperature within 16 h. Then the mixture was quenched with satd. aqueous NH4Cl solution (4 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure B (oxidation of secondary alcohols)
To a solution of the appropriate secondary alcohol 3b/3e in dichloromethane (3 mL) was added manganese(IV) oxide (10 equiv.). The resulting suspension was heated to reflux for 6 h. After cooling to room temperature, the mixture was filtered through a pad of celite, and the celite was washed with dichloromethane (20 mL). The combined organic layers were concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure C (methylation of secondary alcohols)
To a suspension of sodium hydride (1.5 equiv., 60% in mineral oil) in dry DMF (1–2 mL) was added a solution of the appropriate secondary alcohol 3b/3c/3f (1.0 equiv.) in dry DMF (1–2 mL). The reaction mixture was stirred under nitrogen atmosphere at room temperature for 30 min, before iodomethane (3.0 equiv.) was added. Stirring was continued for 2 h and then water (15 mL) was added to the mixture. The reaction mixture was extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (2 × 20 mL), dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure D (hydrogenolysis of diaryl methanols)
To a solution of 3b/3d in MeOH (2–10 mL) was added conc. H2SO4 (0.1–0.5 mL) and 10% Pd/C (0.070 g–0.100 g). The mixture was stirred under hydrogen atmosphere at room temperature for 6 d. Then the catalyst was filtered off through a pad of celite and the resulting solution was concentrated under reduced pressure. Satd. aqueous NaHCO3 solution (20 mL) was added to the residue, followed by extraction with dichloromethane (3 × 20 mL). The combined organic layers were dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure E (Hydrogenolysis of benzyl ethers)
To a solution of 3f/3c in MeOH (10 mL) was added 10% Pd/C (0.100 g). The mixture was stirred under hydrogen atmosphere at room temperature for 24 h. Then the catalyst was filtered off through a pad of celite and the resulting solution was concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure F (Photocyclization to give oxoaporphines)
A stirred solution of NaBH4 (0.019 g, 0.500 mmol) and the secondary alcohol 12a (0.124 g, 0.330 mmol) in MeOH (132 mL) or 12b (0.130 g, 0.300 mmol) in MeOH (120 mL) was irradiated in a photo reactor equipped with a medium pressure mercury vapor lamp (125 W) at room temperature for the indicated time. The solvent was then evaporated and the residue was dissolved in dichloromethane (200 mL). The solution was washed with water (3 × 50 mL), the organic layer was dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (dichloromethane/methanol = 97:3).
:1 + 2% triethylamine) to give 3a (0.119 g, 37%) as a pale yellow solid. mp 143–146 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.40 (d, J = 5.6 Hz, 1H), 7.50 (d, J = 5.6 Hz, 1H), 7.26 (d, J = 8.7 Hz, 2H), 7.07 (s, 1H), 7.06 (s, 1H), 6.82 (d, J = 8.7 Hz, 2H), 6.39 (br s, 1H), 6.15 (s, 1H), 3.98 (s, 3H), 3.80 (s, 3H), 3.75 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) = 159.3, 156.9, 152.8, 149.9, 139.0, 136.1, 133.7, 129.1 (2C), 121.0, 119.9, 114.3 (2C), 105.4, 103.4, 72.4, 56.2, 56.0, 55.4; HRMS (ESI): m/z (%) = 326.1390 [M + H]+ (calcd for C19H19NO4: 325.1314); IR (KBr pellet): ν (cm−1) = 2935, 1829, 1621, 1567, 1504, 1487, 1435, 1418, 1252, 1202, 1160, 1101, 1039.
:1 + 2% triethylamine) to give 3b (0.150 g, 42%) as a yellow solid. mp 135–137 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.36 (d, J = 5.6 Hz, 1H), 7.47 (d, J = 5.6 Hz, 1H), 7.08 (s, 1H), 7.03 (s, 1H), 6.89 (dd, J = 8.2, 2.0 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H), 6.76 (d, J = 8.2 Hz, 1H), 6.42 (br s, 1H), 6.11 (s, 1H), 3.95 (s, 3H), 3.79 (s, 3H), 3.78 (s, 3H), 3.73 (s, 3H); 13C NMR (101 MHz, CDCl3: δ (ppm) = 156.7, 152.8, 150.0, 149.5, 148.9, 139.0, 136.4, 133.7, 121.1, 120.3, 119.9, 111.1, 110.8, 105.4, 103.4, 72.7, 56.2, 56.0, 56.0, 55.9; HRMS (ESI): m/z (%) = 356.1491 [M + H]+ (calcd for C20H21NO5: 355.1420); IR (KBr pellet): ν (cm−1) = 3342, 2941, 2839, 1620, 1593, 1569, 1513, 1472, 1454, 1403, 1273, 1257, 1237, 1161, 1135, 1065, 1021.
:1 + 2% triethylamine) to give 3c (0.144 g, 43%) as a yellow solid. mp 74–76 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.38 (d, J = 5.6 Hz, 1H), 7.49 (d, J = 5.6 Hz, 1H), 7.09 (s, 1H), 7.05 (s, 1H), 6.94 (dd, J = 7.9, 1.7 Hz, 1H), 6.74 (d, J = 7.9 Hz, 1H), 6.64 (d, J = 1.7 Hz, 1H), 6.41 (br s, 1H), 6.10 (s, 1H), 5.87 (d, J = 1.4 Hz, 1H), 5.84 (d, 3.98 J = 1.4 Hz, 1H), 3.98 (s, 3H), 3.81 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) = 156.5, 152.8, 150.0, 148.2, 147.4, 139.0, 137.8, 133.6, 121.6, 121.0, 119.9, 108.2, 107.9, 105.4, 103.3, 101.1, 72.6, 56.1, 56.0; HRMS (ESI): m/z (%) = 340.1179 [M + H]+ (calcd for C19H17NO5: 339.1107); IR (KBr pellet): ν (cm−1) = 3331, 3011, 2902, 1622, 1571, 1509, 1486, 1273, 1237, 1160, 1038, 976, 859.
:1 + 2% triethylamine) to give 3d (0.460 g, 53%) as a pale yellow solid. mp 137–138 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.30 (d, J = 5.6 Hz, 1H), 7.41 (d, J = 5.6 Hz, 1H), 7.22–7.13 (m, 5H), 6.97 (s, 1H), 6.90 (s, 1H), 6.85 (dd, J = 8.2, 2.0 Hz, 1H), 6.74 (d, J = 8.2 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 6.25 (br s, 1H), 6.00 (s, 1H), 4.96 (d, J = 12.3 Hz, 1H), 4.90 (d, J = 12.3 Hz, 1H), 3.91 (s, 3H), 3.75 (s, 3H), 3.62 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) = 156.6, 152.7, 149.9, 149.6, 148.5, 139.0, 137.0, 136.3, 133.6, 128.5 (2C), 127.8, 127.4 (2C), 121.0, 120.9, 119.9, 113.7, 111.8, 105.3, 103.3, 72.6, 71.1, 56.1 (2C), 55.9; HRMS (ESI): m/z (%) = 432.1805 [M + H]+ (calcd for C26H25NO5: 431.1733); IR (KBr pellet): ν (cm−1) = 3317, 2951, 2835, 1510, 1403, 1395, 1263, 1236, 1156, 1138, 1068, 1026.
:1) to give 3e (0.100 g, 28%) as a pale orange amorphous solid. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.42 (d, J = 5.8 Hz, 1H), 7.85 (d, J = 5.8 Hz, 1H), 7.25 (d, J = 8.8 Hz, 2H), 6.91 (s, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.34 (br s, 1H), 6.14 (s, 1H), 4.02 (s, 3H), 3.96 (s, 3H), 3.79 (s, 3H), 3.75 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) = 159.3, 157.0, 153.6, 147.2, 143.9, 138.5, 135.9, 129.1 (2C), 128.9, 122.2, 115.1, 114.3 (2C), 99.7, 72.5, 61.7, 61.3, 56.0, 55.4; HRMS (EI): m/z (%) = 355.1407 (calcd for C20H21NO5: 355.1420); IR (NaCl film): ν (cm−1) = 2938, 2835, 1611, 1588, 1510, 1490, 1476, 1396, 1251, 1122, 1061, 1035, 962, 833.
:1 + 2% triethylamine) to give 3f (0.233 g, 49%) as a pale yellow solid. mp 107–109 °C; 1H NMR (500 MHz, CD2Cl2): δ (ppm) = 8.38 (d, J = 5.6 Hz, 1H), 7.53 (d, J = 5.6 Hz, 1H), 7.41–7.38 (m, 6H), 7.38–7.34 (m, 3H), 7.30 (m, 1H), 7.18–7.14 (m, 3H), 7.13 (s, 1H), 6.86 (d, J = 8.5 Hz, 2H), 6.14 (d, J = 5.6 Hz, 1H), 6.04 (d, J = 5.6 Hz, 1H), 5.05 (d, J = 11.8 Hz, 1H), 5.01 (s, 2H), 4.95 (d, J = 11.8 Hz, 1H), 3.96 (s, 3H); 13C NMR (126 MHz, CD2Cl2): δ (ppm) = 158.9, 157.5, 153.7, 149.6, 139.5, 137.6, 136.9, 136.7, 134.2, 129.3 (2C), 129.2 (2C), 129.0 (2C), 128.8, 128.5, 128.2 (2C), 128.0 (2C), 121.2, 120.2, 115.5 (2C), 106.0, 105.5, 72.5, 71.3, 70.5, 56.5; HRMS (EI): m/z (%) = 477.1943 (calcd for C31H27NO4: 477.1940); IR (KBr pellet): ν (cm−1) = 3403, 3034, 2932, 1606, 1509, 1275, 1237, 1162, 1056, 1009, 743, 695.
:1 + 2% triethylamine) to give 4 (0.080 g, 68%) as a white solid. mp 206–208 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.45 (d, J = 5.5 Hz, 1H), 7.70 (d, J = 1.9 Hz, 1H), 7.64 (d, J = 5.5 Hz, 1H), 7.54 (s, 1H), 7.42 (dd, J = 8.4, 1.9 Hz, 1H), 7.14 (s, 1H), 6.86 (d, J = 8.4 Hz, 1H), 4.05 (s, 3H), 3.95 (s, 6H), 3.94 (s, 3H); 13C NMR (126 MHz, CDCl3: δ (ppm) = 194.1, 154.0, 153.0, 153.3, 151.2, 149.2, 140.2, 134.1, 130.1, 127.0, 123.0, 121.4, 112.1, 110.1, 105.0, 104.2, 56.3, 56.3, 56.2, 56.2; HRMS (EI): m/z (%) = 353.1292 (calcd for C20H19NO5: 353.1263); IR (KBr pellet): ν (cm−1) = 3424, 3007, 2970, 2933, 1656, 1593, 1582, 1504, 1460, 1433, 1270, 1229, 1140, 1025, 860, 749.
:2) to give 5 (0.059 g, 98%) as a pale yellow solid. mp 151–153 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.48 (d, J = 5.6 Hz, 1H), 7.98 (d, J = 5.6 Hz, 1H), 7.95 (d, J = 8.9 Hz, 2H), 7.36 (s, 1H), 6.96 (d, J = 8.9 Hz, 2H), 4.08 (s, 3H), 4.03 (s, 3H), 3.93 (s, 3H), 3.88 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) = 194.0, 164.2, 154.7, 154.2, 146.9, 144.4, 139.7, 133.4 (2C), 129.9, 129.3, 124.0, 116.7, 113.9 (2C), 100.5, 61.8, 61.4, 56.3, 55.7; HRMS (EI): m/z (%) = 353.1264 (calcd for C20H19NO5: 353.1263); IR (KBr pellet): ν (cm−1) = 2944, 1651, 1601, 1479, 1280, 1256, 1163, 1127, 1028, 934, 832.
:5) to give 6a (0.065 g, 77%) as a pale yellow solid. mp 145–147 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.41 (d, J = 5.6 Hz, 1H), 7.71 (s, 1H), 7.47 (d, J = 5.6 Hz, 1H), 7.05 (d, J = 1.9 Hz, 1H), 7.04 (s, 1H), 6.95 (dd, J = 8.3, 1.9 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 5.85 (s, 1H), 3.98 (s, 3H), 3.87 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 3.46 (s, 3H); 13C NMR (126 MHz, CDCl3: δ (ppm) = 157.2, 152.5, 149.6, 149.0, 148.3, 140.7, 134.2, 133.6, 122.1, 119.8, 118.7, 110.8, 109.7, 105.2, 104.5, 87.2, 57.5, 56.1, 56.0 (3C); HRMS (EI): m/z (%) = 369.1575 (calcd for C21H23NO5: 369.1576); IR (KBr pellet): ν (cm−1) = 3439, 2933, 1512, 1476, 1461, 1428, 1269, 1249, 1225, 1155, 1140, 1108, 1049, 1026, 855.
:1) to give 6b (0.048 g, 91%) as a brown amorphous solid. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.39 (d, J = 5.6 Hz, 1H), 7.69 (s, 1H), 7.46 (d, J = 5.6 Hz, 1H), 7.03 (s, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.92 (s, 1H), 6.71 (d, J = 8.1 Hz, 1H), 5.87 (d, J = 2.0 Hz, 2H), 5.81 (s, 1H), 3.98 (s, 3H), 3.87 (s, 3H), 3.45 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) = 157.1, 152.5, 149.6, 147.7, 146.8, 140.7, 135.0, 134.1, 122.1, 119.8, 119.7, 108.0, 107.2, 105.2, 104.5, 101.0, 87.3, 57.5, 56.0 (2C); HRMS (EI): m/z (%) = 353.1265 (calcd for C20H19NO5: 353.1263); IR (KBr pellet): ν (cm−1) = 2935, 1829, 1621, 1567, 1504, 1487, 1435, 1418, 1252, 1202, 1160, 1101, 1039.
:2:2) afforded 7 (0.128 g, 90%) as a yellow solid. mp 166–168 °C; 1H NMR (400 MHz, MeOD): δ (ppm) = 8.23 (d, J = 5.7 Hz, 1H), 7.57 (d, J = 5.7 Hz, 1H), 7.43 (s, 1H), 7.22 (s, 1H), 7.18 (d, J = 8.6 Hz, 2H), 6.71 (d, J = 8.6 Hz, 2H), 6.19 (s, 1H), 3.97 (s, 3H); 13C NMR (101 MHz, MeOD): δ (ppm) = 158.6, 158.1, 153.9, 149.2, 138.6, 135.2, 135.0, 129.5 (2C), 123.1, 121.2, 116.2 (2C), 108.6, 106.4, 74.8, 56.4; HRMS (EI): m/z (%) = 297.0999 (calcd for C17H15NO4: 297.1001); IR (KBr pellet): ν (cm−1) = 3410, 3180, 2377, 2297, 1611, 1514, 1274, 1229, 1194, 1051, 977, 855, 842.
:1) afforded 8 (0.033 g, 66%) as a brown solid. mp 178–180 °C; 1H NMR (400 MHz, MeOD): δ (ppm) = 8.19 (d, J = 5.7 Hz, 1H), 7.68 (s, 1H), 7.56 (d, J = 5.7 Hz, 1H), 7.21–7.19 (m, 3H), 6.70 (d, J = 8.7 Hz, 2H), 5.81 (s, 1H), 3.96 (s, 3H), 3.38 (s, 3H); 13C NMR (101 MHz, MeOD): δ (ppm) = 158.1, 157.8, 154.0, 149.2, 139.2, 135.3, 132.5, 129.3 (2C), 123.9, 121.4, 116.0 (2C), 108.9, 106.4, 86.4, 57.4, 56.4; HRMS (EI): m/z (%) = 311.1164 (calcd for C18H17NO4: 311.1158); IR (NaCl film): ν (cm−1) = 2923, 1611, 1594, 1509, 1479, 1453, 1431, 1345, 1259, 1231, 1195, 1167, 1095, 856, 752.
:1 + 5% triethylamine) to give 9 (0.057 g, 60%) as a white solid. mp 145–147 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.36 (d, J = 5.6 Hz, 1H), 7.41 (d, J = 5.6 Hz, 1H), 7.33 (s, 1H), 7.03 (s, 1H), 6.83–6.79 (m, 2H), 6.75 (d, J = 8.7 Hz, 1H), 4.52 (s, 2H), 3.98 (s, 3H), 3.89 (s, 3H), 3.80 (s, 3H), 3.75 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) = 157.9, 152.5, 149.9, 149.1, 147.6, 141.2, 133.5, 132.4, 123.0, 120.6, 118.8, 112.0, 111.3, 105.4, 104.3, 56.1, 56.0, 55.9 (2C), 42.4; HRMS (ESI): m/z (%) = 339.1471 [M + H]+ (calcd for C20H21NO4: 339.1470); IR (KBr pellet): ν (cm−1) = 2939, 2835, 1617, 1589, 1564, 1506, 1435, 1416, 1260, 1234, 1203, 1158, 1140, 1029, 985.
:1) to give 12a (0.258 g, 69%) as a white solid. mp 107–108 °C; 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.40 (d, J = 5.6 Hz, 1H), 7.66–7.62 (m, 1H), 7.56 (d, J = 5.6 Hz, 1H), 7.13–7.09 (m, 3H), 7.04 (s, 1H), 6.90–6.85 (m, 1H), 6.66 (s, 1H), 3.94 (s, 3H), 3.83 (s, 3H); 13C NMR (101 MHz, CD2Cl2): δ (ppm) = 156.8, 153.6, 151.0, 143.2, 139.2, 134.1, 133.4, 130.2, 130.0, 128.7, 124.5, 121.4, 120.5, 105.8, 103.4, 71.6, 56.7, 56.5; HRMS (ESI): m/z (%) = 374.0389 [M + H]+ (calcd for C18H17BrNO3: 373.0314); IR (KBr pellet): ν (cm−1) = 3509, 3347, 3057, 2933, 1623, 1572, 1510, 1480, 1437, 1401, 1325, 1276, 1237, 1202, 1161, 1074, 1020, 974.
:2) to give 12b (0.152 g, 35%) as a white solid. mp 185–186 °C; 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.39 (d, J = 5.6 Hz, 1H), 7.55 (d, J = 5.6 Hz, 1H), 7.13 (s, 1H), 7.09 (s, 1H), 7.06 (s, 1H), 6.56 (s, 1H), 6.35 (s, 1H), 6.29 (s, 1H), 3.94 (s, 3H), 3.88 (s, 3H), 3.80 (s, 3H), 3.49 (s, 3H); 13C NMR (101 MHz, CD2Cl2): δ (ppm) = 157.1, 153.6, 151.0, 150.2, 149.9, 139.2, 135.1, 134.0, 121.4, 120.4, 115.6, 114.3, 112.2, 105.8, 103.6, 71.5, 56.8, 56.6, 56.4, 56.2; HRMS (ESI): m/z (%) = 433.0507 (calcd for C20H20BrNO5: 433.0525); IR (KBr pellet): ν (cm−1) = 3293, 2937, 1619, 1597, 1570, 1508, 1434, 1405, 1271, 1254, 1235, 1200, 1155, 1030, 858.
References
- J. M. Hagel and P. J. Facchini, Plant Cell Physiol., 2013, 54, 547 CrossRef PubMed.
- M. Shamma, The isoquinoline alkaloids: chemistry and pharmacology, Academic Press, 1972 Search PubMed.
- J. R. Kershaw and B. C. Uff, Chem. Commun., 1966, 331 RSC.
- H.-Y. Cheng and R. W. Doskotch, J. Nat. Prod., 1980, 43, 151 CrossRef CAS.
- S. Al-Khalil and P. L. Schiff Jr., J. Nat. Prod., 1985, 48, 989 CrossRef CAS.
- J. Jacobs, N. van Tuyen, P. Markusse, C. V. Stevens, L. Maat and N. De Kimpe, Tetrahedron, 2009, 63, 1188 CrossRef.
- E. Awuah and A. Capretta, J. Org. Chem., 2010, 75, 5627 CrossRef CAS PubMed.
- R. Shankar, S. S. More, M. V. Madhubabu, N. Vembu and U. K. Syam Kumar, Synlett, 2012, 1013 CAS.
- M. V. Madhubabu, R. Shankar, R. Akula, U. K. Syam Kumar and M. V. Basaveswara Rao, Der Pharma Chemica, 2014, 6, 50 CAS.
- A. J. Birch, A. H. Jackson and P. V. R. Shannon, J. Chem. Soc., Perkin Trans. 1, 1974, 2190 RSC.
- K. Kido and Y. Watanabe, Heterocycles, 1980, 14, 1151 CrossRef CAS.
- K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082 CrossRef CAS PubMed.
- Y. Siddaraju, M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856 CrossRef CAS PubMed.
- T.-H. Chuang, C.-F. Li, H.-Z. Lee and Y.-C. Wen, J. Org. Chem., 2013, 78, 4974 CrossRef CAS PubMed.
- A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040 CrossRef CAS PubMed . This protocol could not be applied to 2-bromobenzoyl chloride, see ref. 14.
- B. Melzer, A. Plodek and F. Bracher, J. Org. Chem., 2014, 79, 7239 CrossRef CAS PubMed.
- A. Plodek, M. König and F. Bracher, Eur. J. Org. Chem., 2015, 1302 CrossRef CAS.
- A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958 CrossRef CAS PubMed.
- A. Metzger, M. A. Schade and P. Knochel, Org. Lett., 2008, 10, 1107 CrossRef CAS PubMed.
- S. M. Kupchan and P. F. O'Brien, J. Chem. Soc., Chem. Commun., 1973, 915 RSC.
- L. Castedo, J. M. Saá, R. Suau and C. Viilaverde, Heterocycles, 1980, 14, 1131 CrossRef CAS.
- S. V. Kessar, Y. P. Gupta, V. S. Yadav, M. Narula and T. Mohammed, Tetrahedron Lett., 1980, 21, 3307 CrossRef CAS.
- K. Orito, S. Uchiito, Y. Satoh, T. Tatsuzawa, R. Harada and M. Tokuda, Org. Lett., 2000, 2, 307 CrossRef CAS PubMed.
- G. D. Cuny, Tetrahedron Lett., 2004, 45, 5167 CrossRef CAS.
- S. Chaudhary, S. Pecic, O. LeGendre and W. W. Harding, Tetrahedron Lett., 2009, 50, 2437 CrossRef CAS PubMed.
- C. Stévigny, C. Bailly and J. Quetin-Leclercq, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 173 CrossRef.
- Y. Liu, J. Liu, D. Di, M. Li and Y. Fen, Curr. Top. Med. Chem., 2013, 13, 2116 CrossRef CAS PubMed.
- J.-L. Ferron and P. L'Ecuyer, Can. J. Chem., 1955, 33, 97 CrossRef CAS.
- L. Stuchlik, Monatsh. Chem., 1900, 21, 813 CrossRef CAS.
- J. A. Weisbach, J. L. Kirkpatrick, E. Macko and B. Douglas, J. Med. Chem., 1968, 11, 760 CrossRef CAS PubMed . The authors erroneously claimed 3a to be a natural product.
- M. Jaric, B. A. Haag, A. Unsinn, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 5451 CrossRef CAS PubMed.
- D. L. Boger, C. E. Brotherton and M. D. Kelley, Tetrahedron, 1981, 37, 3977 CrossRef CAS.
- T. R. Suess and F. R. Stermitz, J. Nat. Prod., 1981, 44, 688 CrossRef CAS.
- E. Reimann and H. Renz, Arch. Pharm., 1993, 326, 253 CrossRef CAS.
- G. Goldschmiedt, Monatsh. Chem., 1885, 6, 954 CrossRef.
- K. H. C. Baser, J. Nat. Prod., 1982, 45, 704 CrossRef CAS.
- J. Slavik and L. Slavikova, Collect. Czech. Chem. Commun., 1996, 61, 1047 CrossRef CAS.
- S. Mahboobi, H. Pongratz and W. Wiegrebe, Pharmazie, 1997, 52, 399 CAS.
- C.-Y. Chen, F.-R. Chang, W.-B. Pan and Y.-C. Wu, Phytochemistry, 2001, 56, 753 CrossRef CAS PubMed.
- A. Pictet and A. Gams, Ber. Dtsch. Chem. Ges., 1909, 42, 2943 CrossRef CAS.
- E. Brochmann-Hanssen and K. Hirai, J. Pharm. Sci., 1968, 57, 940 CrossRef CAS PubMed.
- A. Brossi and S. Teitel, J. Org. Chem., 1970, 35, 1684 CrossRef CAS PubMed.
- N. Katsui, K. Sato, S. Tobinaga and N. Takeuchi, Tetrahedron Lett., 1966, 50, 6257 CrossRef CAS PubMed.
- M. Tomita, Y. Tsang-Hsiung, H. Furukawa and Y. Hui-Mei, Yakugaku Zasshi, 1962, 82, 1574 CAS.
- M. A. Buchanan and E. E. Dickey, J. Org. Chem., 1960, 25, 1389 CrossRef CAS.
- J. Cohen, W. von Rosenthal and W. I. Taylor, J. Org. Chem., 1961, 26, 4143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00926j |
| This journal is © The Royal Society of Chemistry 2015 |