
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-headed nucleotides introducing thymine nucleobases in the major groove of nucleic acid duplexes†
Michael
Dalager
a,
Nicolai K.
Andersen
a,
Pawan
Kumar
a,
Poul
Nielsen
*a and
Pawan K.
Sharma
*b
aNucleic Acid Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, 5230 Odense M, Denmark. E-mail: pouln@sdu.dk
bDepartment of Chemistry, Kurukshetra University, Kurukshetra 136 119, India. E-mail: pksharma@kuk.ac.in
First published on 29th May 2015
Abstract
Four different double-headed nucleosides each combining two thymine nucleobases with different linkers were synthesised. The 5-position of 2′-deoxyuridine was connected to the N1-position of a thymine through either m- or p-disubstituted phenyl or phenylacetylene linkers by the use of Suzuki or Sonogashira couplings. When introduced into oligonucleotides, the thermal stability of dsDNA and DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RNA duplexes were determined and structural information was obtained from CD- and fluorescence spectroscopy. Also the recognition of abasic sites was studied. In general, the more stable duplexes were obtained with m- rather than p-substitution and with phenylacetylene rather than phenyl linkers.
:RNA duplexes were determined and structural information was obtained from CD- and fluorescence spectroscopy. Also the recognition of abasic sites was studied. In general, the more stable duplexes were obtained with m- rather than p-substitution and with phenylacetylene rather than phenyl linkers.
Introduction
Nucleic acids are excellent scaffolds for the design of supramolecular structures. The programmable structure of the DNA duplex formed by the Watson–Crick base pairs is the basis for the design of both structural and functional elements.1–4 Furthermore, the convenient automated synthesis of DNA allows intensive and selective modification. Hereby intriguing applications in DNA nanotechnology and the development of therapeutics are possible.1–4 Recently, we and others have investigated a series of modified nucleotides called double-headed nucleotides, with two nucleobases attached to the same nucleotide.5–22 These have been used to introduce additional nucleobases into DNA duplexes or other secondary structures. The recognition potential from the additional nucleobases has been explored and interesting structural motifs have been found. Recently, we introduced a series of double-headed nucleotides where the additional bases are linked to the 5-position of pyrimidine nucleotides through a propyne linker (Fig. 1, BT and BC), and all eight possible combinations were made (i.e.TT, CT, AT, GT, TC, CC, AC and GC).21 These were introduced into oligonucleotides including fully modified sequences that when mixed with complementary DNA, forms very stable duplexes with a full array of additional nucleobases in the major groove. The thermal stability of these duplexes was increased as compared to unmodified duplexes, mainly due to the stacking from the increased π-system formed by the propyne linkers. Hereby the potential has been obtained for forming true double-coding DNA with the internal code as well as with an external code in the major groove. However, the array of nucleobases in the major groove can take several conformations due to the flexibility in the same propyne linkers.21 Herein, we introduce four new double-headed nucleotide analogues where the propyne linker of TT is replaced with more rigid linkers separating the two thymine nucleobases. A phenyl group is introduced in the linker with m- or p-substitution either with or without an acetylene spacer giving the monomers X, Y, Z and W, respectively (Fig. 1). The influence of these linkers on duplex stability and structure is evaluated. | ||
| Fig. 1 Double-headed nucleotides. B = nucleobase. T = thymin-1-yl. | ||
Results and discussion
Synthesis
The 5′-DMTr-protected 2′-deoxy-5-iodouridine 123,24 was converted into the 2′-deoxy-5-ethynyluridine derivative 2 following standard procedures,25 and 1 and 2 were hereafter used as starting materials for the preparation of the four nucleotide monomers (Scheme 1). N3-Benzoylated thymine 326,27 was coupled to 4-iodobenzenboronic acid to afford the known N1-(4-iodophenyl)thymine derivative 428 and further converted to the corresponding boronic ester 528 using methodology developed by Gothelf and co-workers.28,29 By using similar procedures, also the m-substituted isomers 6 and 7 were obtained in good yields. By the use of Sonogashira cross couplings, 6 and 4 were reacted with the nucleoside 2 to obtain the double-headed nucleotides 8 and 9, respectively, in good yields. Suzuki couplings were applied to combine 7 and 5 with the nucleoside 1 to give the double-headed nucleosides 10 and 11, respectively. Using a standard phosphitylation protocol, the four nucleosides 8–11 were converted to the phosphoramidites 12–15 (Scheme 1). These were hereafter incorporated into oligonucleotides giving the nucleotide monomers X, Y, Z and W, respectively. Standard automated DNA-synthesis was applied with 1H-tetrazole as the activator. Extended coupling times of 20 or 30 min were used for the modified phosphoramidites in order to get coupling yields >90% in all cases. | ||
| Scheme 1 Reagents and conditions: (a) ref. 28; (b) ref. 28; (c) m-iodobenzeneboronic acid, Cu(OAc)2, pyridine, 70%; (d) octamethyl-2,2′-bis(1,3,2-dioxaborolane), Pd(OAc)2, KOAc, DMF, 48%; (e) i. Pd(PPh3)4, CuI, Et3N, DMF, ii. NH3, MeOH, 8 72%, 9 71%; (f) Pd(PPh3)4, THF, H2O, NaOH, MeOH, 10 59%, 11 76%; (g) NC(CH2)2OP(N(iPr)2)Cl, (iPr)2NEt, CH2Cl2, 12 77%, 13 89%, 14 97%, 15 54%; (h) DNA synthesis. DMTr = 4,4′-dimethoxytrityl. | ||
Hybridization studies
The four monomers were incorporated in three different 9-mer sequence contexts replacing thymidines in the unmodified oligonucleotide sequences ON1 and ON6 (Table 1). First, each monomer was introduced centrally in ON1 giving ON2–ON5. Hereafter, the monomers were placed in the oligo-T stretch of ON6 to give ON7–ON10, and finally, three consecutive incorporations of each monomer in ON6 gave the oligonucleotides ON11–ON14. The oligonucleotides were mixed in a medium salt buffer with complementary DNA and RNA sequences as well as in a high salt buffer with a DNA containing a central abasic site. The melting temperatures (Tm) of the resulting duplexes were determined from UV melting curves (Table 1). The concentrations of each strand were determined based on extinction coefficients, which for each of the four monomers X–W were determined from UV absorption. Circular dichroism (CD) spectra were recorded for each duplex in order to verify the overall helical structure of the duplexes. Finally, fluorescence emission spectroscopy was performed by single wavelength excitation at the UV absorption maxima and a scan of emission wavelengths. Fluorescence emission spectra were recorded for all oligonucleotides and duplexes.| Entry | Sequence | T m (ΔTm)a/°C | ||
|---|---|---|---|---|
| 3′-d(CAC TAT ACG) | 3′-r(CAC UAU ACG) | 3′-d(CAC TΦT ACG)b | ||
| a Melting temperatures, Tm's, obtained from the maxima of the first derivatives of the melting curves (A260vs. temperature) recorded in a buffer containing 2.5 mM Na2HPO4, 5.0 mM NaH2PO4, 100 mM NaCl, 0.1 mM EDTA, pH 7.0 using 1.5 μM concentrations of each strand. All determinations are averages of at least duplicates within 0.5 °C. ΔTm = change in Tm's relative to the unmodified reference duplex. b Measured in a buffer containing 700 mM instead of 100 mM NaCl. Φ corresponds to an abasic site, and X, Y, Z and W corresponds to the incorporation of 12, 13, 14 and 15, respectively. | ||||
| ON1 | 5′-d(GTG ATA TGC) | 31.0 | 28.5 | 8.0 |
| ON2 | 5′-d(GTG AXA TGC) | 28.0 (−3.0) | 26.5 (−2.0) | 19.5 (+11.5) |
| ON3 | 5′-d(GTG AYA TGC) | 28.0 (−3.0) | 27.0 (−1.5) | 13.0 (+5.0) |
| ON4 | 5′-d(GTG AZA TGC) | 26.0 (−5.0) | 22.0 (−6.5) | 21.5 (+13.5) |
| ON5 | 5′-d(GTG AWA TGC) | 25.0 (−6.0) | 21.0 (−7.5) | 8.5 (+0.5) |
| 3′-d(CAC AAA ACG) | 3′-r(CAC AAA ACG) | 3′-d(CAC AΦA ACG)b | ||
| ON6 | 5′-d(GTG TTT TGC) | 33.0 | 32.0 | 15.0 |
| ON7 | 5′-d(GTG TXT TGC) | 31.5 (−1.5) | 29.0 (−3.0) | 19.0 (+4.0) |
| ON8 | 5′-d(GTG TYT TGC) | 31.5 (−1.5) | 31.0 (−1.0) | 16.5 (+1.5) |
| ON9 | 5′-d(GTG TZT TGC) | 30.5 (−2.5) | 26.0 (−6.0) | 19.0 (+4.0) |
| ON10 | 5′-d(GTG TWT TGC) | 28.5 (−4.5) | 24.0 (−8.0) | 19.0 (+4.0) |
| ON11 | 5′-d(GTG XXX TGC) | 30.0 (−1.0) | 36.5 (+1.5) | 16.0 (+0.3) |
| ON12 | 5′-d(GTG YYY TGC) | 26.5 (−2.2) | 32.0 (0.0) | 17.0 (+0.7) |
| ON13 | 5′-d(GTG ZZZ TGC) | 26.5 (−2.2) | 20.5 (−3.8) | 13.0 (−0.7) |
| ON14 | 5′-d(GTG WWW TGC) | 19.0 (−4.7) | 19.0 (−4.3) | 11.0 (−1.3) |
When introduced into the first sequence, ON1, the single incorporations of the four monomers in ON2–5 show a general trend of destabilization of the duplexes formed with complementary DNA; monomers X and Y both leading to ΔTm's of −3 °C, decreasing to −5 °C and −6 °C for Z and W, respectively (Table 1). The same was observed when the oligonucleotides were targeted with an RNA complement, albeit with a slightly lower destabilizing effect for single incorporations of X and Y, with ΔTm's of −2 °C and −1.5 °C, respectively, increasing to a significant destabilizing effect for Z and W, with ΔTm's −6.5 °C and −7.5 °C, respectively. These results clearly show that the alkyne containing monomers are much better accommodated in the duplexes which is in accordance with other 5-ethynyl- versus 5-phenylpyrimidine nucleotide monomers studied.30,31 When introduced in a DNA duplex with a central abasic site opposite the modification, a significant stabilizing effect is observed, with monomers X and Z reaching ΔTm's of +11.5 °C and +13.5 °C, monomer Y having a more modest stabilization of +5.0 °C and monomer W barely any stabilization with a ΔTm of +0.5 °C. These data indicate that the aromatic moieties of the two monomers with m-configured linkers (X and Z) might intercalate in the abasic site of the complementary strand. Similar effects have been observed before with nucleobases having aromatic substituents.32
All the duplexes were studied by CD-spectroscopy (CD-spectra can be found in the ESI, Fig. S1–S9†). CD-curves can display distinct characteristics for A- and B-type duplex structures, and as expected, the unmodified dsDNA duplex demonstrates clear B-type characteristic, i.e. two positive bands at 220 and 280 nm and a negative band at 250 nm, whereas the unmodified DNA:RNA duplex demonstrates an A/B-hybrid type, i.e. a large positive band at 270 nm and two smaller negative bands at 240 and 210 nm. The CD-spectra of ON2–5 with the DNA-complement were almost perfect B-type duplex spectra (Fig. S1†), whereas the spectra of ON2–5 with the RNA-complement demonstrate the expected characteristics of the A/B-hybrid duplex type although with lower intensities of the bands (Fig. S2†). Also the spectra of dsDNA with an abasic site demonstrates clear B-type characteristic, however, for ON4 containing monomer Z, the negative band at 250 is shifted to 260 nm and intensity is increased (Fig. S3†).
All the four monomers were found to be quite fluorescent although X and Y more than Z and W (Fig. S10†). Fluorescence emission spectra were measured for the single strands ON2–5 as well as for the duplexes formed between ON2–5 and the three complements (Fig. 2 and S11†). For ON2, a broad emission band around 400 nm is observed in all cases, and although the single strand is showing the highest intensity only small degrees of quenching is indicated with the duplexes (Fig. 2). Hereby no structural conclusion can be drawn in this case, as this indicates that the fluorophore is to some degree quenched by the surroundings in both the single strand and in the duplexes. A similar picture is observed for ON3 (Fig. S11†) and for ON4, although the emission is in general very low in the latter case (Fig. S11†). For ON5, the emission is more intense for the duplex formed with an abasic site than for the two other duplexes and for the single strand (Fig. S11†), and this might indicate that the double-functional nucleobase is not situated in the abasic site where it would be expected to be quenched but rather protruding out in the major groove. This is in accordance with the lower melting temperature of this duplex compared to the corresponding duplexes formed with ON2–4. Nevertheless, this does not prove intercalation for X or Z as indicated by the Tm's.
 | ||
| Fig. 2 Selected fluorescence emission spectra for ON's containing monomer X. (a) ON2, and (b) ON7. Excitation wavelength = 255 nM. | ||
Hereafter, the duplexes formed with ON6–10 were evaluated. With complementary DNA, the modifications are generally less destabilising when incorporated between thymidine neighbours than between adenine neighbours with ΔTm's from −1.5 °C to −4.5 °C for ON7–10 as compared to −3.0 to −6.0 °C for ON2–5. As before, X and Y are demonstrating smaller decreases in Tm than Z and W. With complementary RNA, more or less similar destabilisation was observed in the two sequence contexts. With the abasic site in the complementary DNA, larger differences were observed and even the unmodified duplex formed by ON6 is significantly more stable than the corresponding duplex formed by ON1. On the other hand, the further increase in thermal stability obtained by introducing the four monomers X–W are significantly smaller for ON7–10 as compared to ON2–5 and with less variation (ΔTm's from +1.5 to +4.0 °C). The CD-curves of the duplexes formed by ON6–10 demonstrate the same trends as with ON1–5. The duplexes formed with complementary DNA and RNA show signature B-type and A/B-type helix characteristics, respectively (Fig. S4–S6†). Fluorescence emission spectra were measured for all the oligonucleotides ON7–10 and their corresponding duplexes and again broad emission bands around 400 nm were observed with varying intensity (Fig. 2 and S12†). For ON7, the emission is more intense for the duplexes with DNA and RNA than for the single strand (Fig. 2) indicating that the additional nucleobase is placed in the major groove of the duplexes with less interaction with other nucleobases. For ON8 the same is seen with an even higher intensity for the duplex with DNA, and for ON10, both the duplexes with DNA and RNA show a very high emission. For ON9, like with ON4 also containing monomer Z, the intensity is very low in all cases.
Finally, the duplexes formed with ON11–14 containing three consecutive incorporations of each modified monomer X, Y, Z and W were evaluated. For the duplexes formed with complementary DNA, the decreases in thermal stability were more or less additive with the ΔTm's for each modification being similar to what was obtained for ON7–10. The most stable duplex was formed by ON11 containing three incorporations of monomer X, and the least stable was formed by ON14 with three incorporations of monomer W. For the duplexes with complementary RNA, a significant compensation in thermal stability was observed, probably due to a stacking effect of consecutive aromatic moieties. Hence ON11 forms a duplex that is stabilised as compared to the unmodified duplex with 4.5 °C corresponding to 1.5 °C for each modification, and ON12 forms a duplex with the same stability as the unmodified duplex. The corresponding duplexes formed by ON13 and ON14 were significantly less stable following the line that Z and W are not as well accommodated in the duplexes as X and Y. The DNA duplexes with abasic sites were in general unstable with only small effects from the three modified nucleotides. The CD-spectra for the duplexes in this series display more deviations from the standard picture, as might be expected due to higher degree of modification (Fig. S7–S9†). For the dsDNA duplexes, the B-type indicative positive band at 280 nm has moved toward 270 nm and is less intensive. In one case, ON13:DNA, this band is fully neutralized. This might indicate that the duplex structure has moved towards an A/B-intermediate type as seen before with consecutive incorporations of 5-modified pyrimidine nucleotides.31,33 The modified DNA:RNA duplexes formed by ON11–14, on the other hand, are showing the same clear A/B-type structures as with the single modified duplexes formed by ON7–10. With the abasic site in the dsDNA, the picture is also the same as with ON7–10. The fluorescence emission spectra for single strands and duplexes of ON11–15 (Fig. S13†) are in general showing less intensity for the broad 400 nm emission band as compared to ON7–10. This indicates some degree of quenching due to aromatic contacts. Exceptions to this are the two duplexes ON13:DNA and ON14:DNA which demonstrate higher emissions than the corresponding single strands ON13 and ON14 and the corresponding duplexes with similar modifications, Z and W, ON9:DNA and ON10:DNA, respectively.
Modelling studies
Molecular dynamics simulations were applied in order to model the structural impacts of the modified monomers. In Fig. 3 are shown the minimized structures for three of the duplexes formed with complementary DNA. It is indicated that monomer X is participating in stacking in the major groove but also that hydrogen bonding to the Hoogsteen face of an adenine in the +2 position might take place. This indicates some flexibility in the duplex and that the m-configuration of the linker allows X to accommodate in different ways in the major groove. The monomer Y on the other hand, is clearly protruding out of the groove and does not seem to form any important interactions in the major groove despite stacking of the acetylene with the adjacent base. In the duplex containing the monomer Z, the additional thymine seems to be bending upwards in the major groove, and stacking interactions seem to be poor.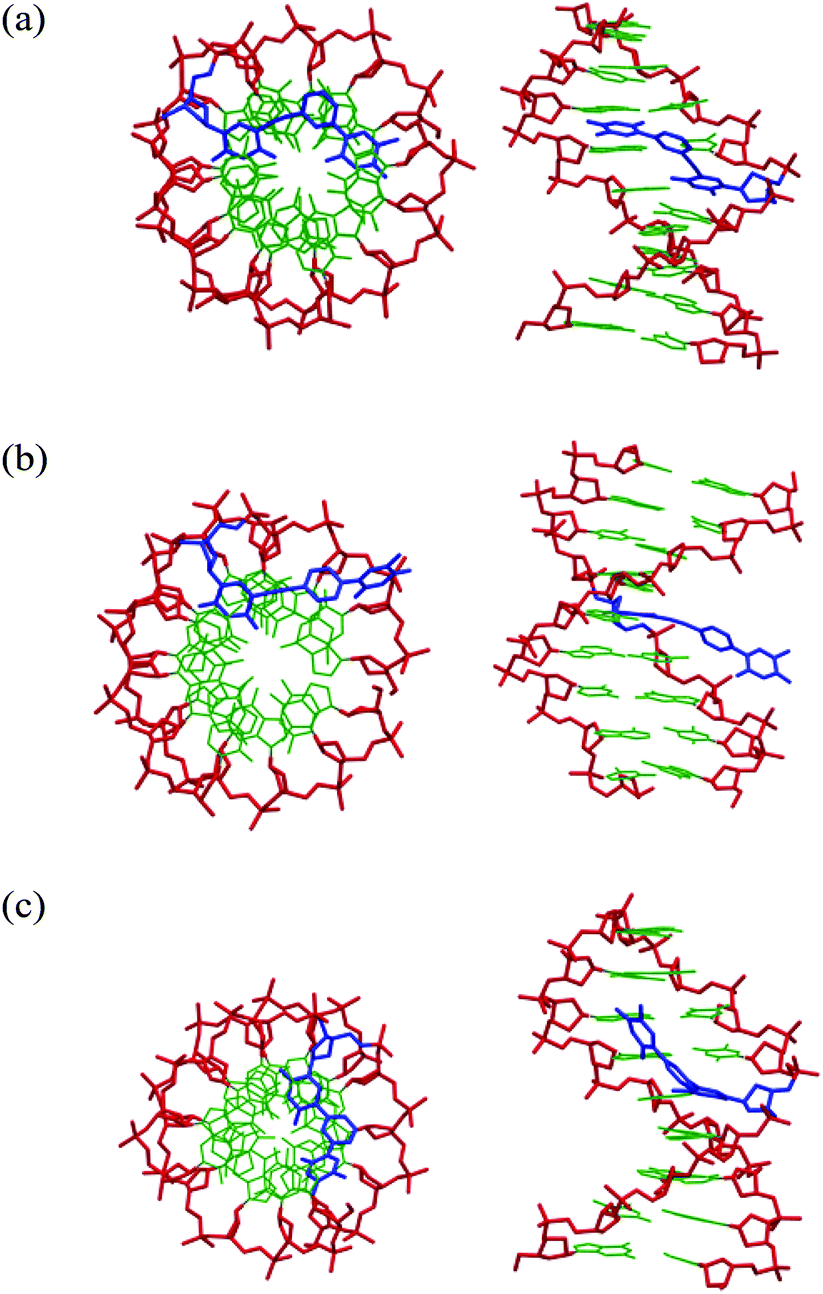 | ||
| Fig. 3 Minimized structures from selected molecular dynamics simulations. Top views and side views of (a) ON2:DNA, (b) ON3:DNA and (c) ON4:DNA. The sugar-phosphate backbone is colored red, Watson–Crick nucleobases green and modified monomers blue. | ||
Discussion
The four phosphoramidites leading to the double-headed nucleotide monomers X, Y, Z and W were all conveniently synthesized via organometallic coupling methods and there are no major differences in their synthetic availability. On the other hand, it is very clear from the hybridization data that the two monomers containing an acetylene linker between the pyrimidine nucleoside and the additional thymine-phenyl group, X and Y, are much better accommodated into the duplexes than Z and W, where the phenyl group directly attaches to the 5-position of the thymine leading to detrimental effects on duplex stability. Consecutive incorporations of pyrimidine nucleotides with 5-substituents are often showing a compensation in the destabilization due to stacking effects,31,33,34 and also this effect is clearly only relevant with 5-acetylenes (X and Y) as compared to 5-phenyls (Z and W) and only in the DNA:RNA duplexes. In the case of X (ON11) this even leads to a stabilised duplex, and the reason for this is probably as indicated from the modelling experiment that the additional thymine in X can participate in stacking in the major groove whereas in Y, it is probably protruding out of the groove. In general, however, all of the four new monomers are showing lower duplex stabilities in both dsDNA and in DNA:RNA duplexes than the propyne containing monomers BT and BC (Fig. 1).21 This indicates that the phenyl group is not an optimal spacer, which can be due to either the hydrophobicity, the rigidity as well as the overall geometry of the double-headed nucleotide. Taking into account also the synthetic availability, the design introduced by BT and BC seems to be the optimal for obtaining double-coding DNA with a full array of different nucleobases in the major groove without compromising the duplex stability. However, it might be relevant to prepare other nucleobase combinations based on the design of X to see if stable fully modified duplexes can be obtained as it was possible for BT/BC-series.
The new monomers, especially X and Y, might find relevance based on their fluorescent properties. Hence both of them are highly fluorescent and the fluorescence emission spectra demonstrate that the emission varies in different single strand and duplex contexts. In the present study, however, we were not able to use this for structural information neither did the monomers demonstrate properties as fluorescent probes for abasic sites. Monomer X did stabilise an abasic site significantly, however, the fluorescent signal was not clear and any conclusion about intercalation in the abasic site cannot be drawn. On the other hand, the fluorescence might be important when the monomers are introduced in other nanostructures.
Conclusions
Four new double-headed nucleotide monomers were conveniently synthesised and incorporated into DNA. Additional nucleobases are hereby positioned in the major groove of dsDNA and DNA:RNA duplexes with minor destabilisation for two of the monomers (X and Y) and even stabilisation for monomer X when three consecutive incorporations are applied. The monomers have interesting fluorescent properties and in combination with other double-headed nucleotides they might find applications as new building blocks in nucleic acid nanotechnology.
Experimentals
All commercial reagents were used as supplied except DCM, which was distilled before use. Anhydrous solvents and reagents were dried over activated molecular sieves, 4 Å for DCM, Pyridine and Toluene, and 3 Å for MeCN, DMF and THF. All moisture sensitive reactions were carried out using anhydrous solvents and Argon or Nitrogen atmosphere. ESI-TOF Mass spectra were recorded on a Thermo Finnigan TSQ 700 spectrometer. 1H and 13C NMR were recorded at ambient temperature on a Bruker AVANCE III 400, 400 MHZ (1H) and 100 MHz (13C). Chemical shifts are given in parts per million referenced to the deuterated solvent residual peak (CDCl3, 7.26 ppm and 77.16 ppm; DMSO-d6, 2.50 ppm and 39.52 ppm). Assignment of NMR spectra was based on 2D spectra and follows standard nucleoside convention. Signals from additional thymines are marked as H-6′′, C-2′′ etc.Preparation of 3-benzoyl-1-(3-iodophenyl)-5-methylpyrimidine-2,4(1H,3H)-dione (6)
To a stirred suspension of dry Cu(OAc)2 (1.099 g, 6.052 mmol), N3-Benzoylthymine 3 (928 mg, 4.035 mmol), m-iodobenzeneboronic acid (2,0 g, 8.070 mmol), and activated 3 Å molecular sieves (1.6 g) in anhydrous CH2Cl2 (40 mL) was added pyridine (652 μL, 8.07 mmol). The mixture was stirred vigorously at room temperature for 70 h in the presence of air. The reaction mixture was added DCM (100 mL), filtered through a pad of Celite, washed with a solution of EDTA (2.02 g) in water (100 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–90% EtOAc in petroleum ether) to give the product 6 (1.217 g, 70%) as a white solid. Rf = 0.70 (4:1 EtOAc/petroleum ether); 1H NMR (400 MHz, DMSO-d6) δ 8.08 (dd, J = 8.4, 1.2 Hz, 2H), 7.97 (t, J = 1.8 Hz, 1H), 7.89 (d, J = 1.0 Hz, 1H), 7.86–7.72 (m, 2H), 7.67–7.48 (m, 3H), 7.32 (t, J = 8.0 Hz, 1H), 1.89 (d, J = 1.0 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.47, 163.01, 148.81, 142.08, 139.37, 137.27, 135.54, 135.44, 131.17, 130.95, 130.48, 129.41, 126.70, 109.21, 94.26, 11.64; HRMS-ESI m/z 454.9856 [M + Na]+; calcd (C18H13IN2O3Na) 454.9864.
Preparation of 3-benzoyl-5-methyl-1-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)pyrimidine-2,4(1H,3H)-dione (7)
To a stirred solution of compound 6 (1.0 g, 2.315 mmol), Pd(OAc)2 (15.6 mg, 69 μmol) and KOAc (682 mg, 6.944 mmol) in DMF (20.0 mL) was added bis(pinacolato)diboron (882 mg, 3.472 mmol) and the mixture was stirred at 60 °C for 3 h. The mixture was added Et2O (50 mL) and washed with water (3 × 25 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–30% EtOAc in DCM) to give the product 7 (480 mg, 48%) as a white solid. Rf = 0.65 (9:1 DCM/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.01–7.93 (m, 2H), 7.84 (ddd, J = 6.6, 1.7, 1.1 Hz, 1H), 7.77 (m, 1H), 7.64 (m, 1H), 7.55–7.42 (m, 4H), 7.30 (q, J = 1.1 Hz, 1H), 2.01 (d, J = 1.2 Hz, 3H), 1.35 (s, 12H); 13C NMR (101 MHz, CDCl3) δ 168.95, 163.35, 149.39, 140.67, 138.01, 135.31, 135.06, 132.12, 131.84, 130.60, 129.51, 129.25, 129.06, 111.14, 84.43, 25.00, 12.47; HRMS-ESI m/z 455.1755 [M + Na]+; calcd (C24H25BN2O5Na) 455.1749.
Preparation of 5′-O-dimethoxytrityl-5-(3-(thymin-1-yl)phenyl)ethynyl-2′-deoxyuridine (8)
To a degassed and stirred solution of compound 6 (500 mg, 1.155 mmol), compound 2 (500 mg, 0.902 mmol) and Et3N (377 μL, 2.708 mmol) in DMF (15 mL) was added tetrakis(triphenylphosphine)palladium(0) (134 mg, 0.116 mmol) and CuI (44 mg, 0.231 mmol). The reaction mixture was stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure and the residue was dissolved in EtOAc, washed with brine (10 mL) and water (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–15% MeOH in DCM) to give the intermediate (567 mg, 73%) as a white solid. Rf 0.38 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H, H-6), 7.95 (dd, J = 8.3, 1.1 Hz, 2H, Ph), 7.62 (t, J = 7.5 Hz, 1H, Ph), 7.46 (dd, J = 13.8, 7.6 Hz, 4H, Ph), 7.33 (dd, J = 8.8, 1.1 Hz, 4H, Ph), 7.29–7.17 (m, 4H, Ph), 7.13 (t, J = 7.3 Hz, 1H, Ph), 7.02 (dt, J = 7.5, 1.3 Hz, 1H, Ph), 6.98 (d, J = 0.8 Hz, 1H, H-6′′), 6.77 (dd, J = 9.0, 2.6 Hz, 4H, Ph), 6.73 (t, J = 1.7 Hz, 1H), 6.37 (dd, J = 7.3, 6.0 Hz, 1H, H-1′), 4.50 (m, 1H, H-3′), 4.08 (m, 1H, H-4′), 3.66 (s, 6H, 2 × OCH3), 3.52 (dd, J = 10.8, 2.2 Hz, 1H, H-5′a), 3.27 (dd, J = 10.8, 3.1 Hz, 1H, H-5′b), 2.49 (ddd, J = 13.7, 5.7, 2.9 Hz, 1H, H-2′a), 2.33 (dd, J = 13.7, 6.5 Hz, 1H, H-2′b), 1.96 (d, J = 0.8 Hz, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 168.83 (COPh), 163.26 (C-4′′), 162.26 (C-4), 158.76 (Ph), 149.90 (C-2′′), 149.04 (C-2), 144.56 (Ph), 142.99 (C-6), 140.36 (C-6′′), 138.03, 135.81, 135.62, 135.12, 131.91, 131.77, 130.57, 130.06, 129.27, 129.04, 128.77, 128.22, 128.10, 127.18, 126.65, 124.51, 113.52 (Ph), 111.18 (C-5), 100.07 (C-5′′), 91.93 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 87.29 (CPh3), 86.87 (C-4′), 85.94 (C-1′), 82.35 (CC), 72.19 (C-3′), 63.59 (C-5′), 55.35 (2 × OCH3), 41.96 (C-2′), 12.46 (CH3); HRMS-ESI m/z 960.4420 [M + Et3NH]+; calcd (C56H58N5O10) 960.4184. A solution of this intermediate (547 mg, 0.638 mmol) in saturated ammonia in methanol (30 mL) was stirred overnight at room temperature. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (0–20% MeOH in DCM) to give the product 8 (472 mg, 98%) as a white solid. Rf 0.32 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H, H-6), 7.44 (d, J = 7.4 Hz, 2H, Ph), 7.33 (d, J = 8.8 Hz, 4H, Ph), 7.26–7.17 (m, 4H, Ph), 7.11 (t, J = 7.3 Hz, 1H, Ph), 7.04 (dd, J = 5.3, 3.6 Hz, 1H, Ph), 6.82 (d, J = 1.2 Hz, 1H, H-6′′), 6.77 (dd, J = 8.8, 3.6 Hz, 4H, Ph), 6.67 (d, J = 1.1 Hz, 1H, Ph), 6.38 (m, 1H, H-1′), 4.47 (m, 1H, 3′), 4.08 (m, 1H, H-4′), 3.67 (s, 6H, 2 × OCH3), 3.53 (m, 1H, H-5′a), 3.27 (dd, J = 10.8, 3.1 Hz, 1H, H-5′b), 2.50 (ddd, J = 13.3, 5.6, 2.7 Hz, 1H, H-2′a), 2.30 (m, 1H, H-2′b), 1.89 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.22 (C-4′′), 162.77 (C-4), 158.6 (Ph), 150.15 (C-2), 149.97 (C-2′′), 144.56 (Ph), 142.82 (C-6), 140.45 (C-6′′), 138.26, 135.68, 135.53, 131.55, 129.97, 129.93, 128.92, 128.77, 128.05, 127.96, 127.01, 126.41, 124.40, 113.37 (Ph), 110.91 (C-5), 99.97 (C-5′′), 91.80 (CC), 87.13 (CPh3), 86.75 (C-4′), 85.82 (C-1′), 82.46 (CC), 71.98 (C-3′), 63.48 (C-5′), 55.23 (2 × OCH3), 41.84 (C-2′), 12.23 (CH3); HRMS-ESI m/z 777.2527 [M + Na]+; calcd (C43H38N4O9Na) 777.2531.
C), 87.29 (CPh3), 86.87 (C-4′), 85.94 (C-1′), 82.35 (CC), 72.19 (C-3′), 63.59 (C-5′), 55.35 (2 × OCH3), 41.96 (C-2′), 12.46 (CH3); HRMS-ESI m/z 960.4420 [M + Et3NH]+; calcd (C56H58N5O10) 960.4184. A solution of this intermediate (547 mg, 0.638 mmol) in saturated ammonia in methanol (30 mL) was stirred overnight at room temperature. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (0–20% MeOH in DCM) to give the product 8 (472 mg, 98%) as a white solid. Rf 0.32 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H, H-6), 7.44 (d, J = 7.4 Hz, 2H, Ph), 7.33 (d, J = 8.8 Hz, 4H, Ph), 7.26–7.17 (m, 4H, Ph), 7.11 (t, J = 7.3 Hz, 1H, Ph), 7.04 (dd, J = 5.3, 3.6 Hz, 1H, Ph), 6.82 (d, J = 1.2 Hz, 1H, H-6′′), 6.77 (dd, J = 8.8, 3.6 Hz, 4H, Ph), 6.67 (d, J = 1.1 Hz, 1H, Ph), 6.38 (m, 1H, H-1′), 4.47 (m, 1H, 3′), 4.08 (m, 1H, H-4′), 3.67 (s, 6H, 2 × OCH3), 3.53 (m, 1H, H-5′a), 3.27 (dd, J = 10.8, 3.1 Hz, 1H, H-5′b), 2.50 (ddd, J = 13.3, 5.6, 2.7 Hz, 1H, H-2′a), 2.30 (m, 1H, H-2′b), 1.89 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.22 (C-4′′), 162.77 (C-4), 158.6 (Ph), 150.15 (C-2), 149.97 (C-2′′), 144.56 (Ph), 142.82 (C-6), 140.45 (C-6′′), 138.26, 135.68, 135.53, 131.55, 129.97, 129.93, 128.92, 128.77, 128.05, 127.96, 127.01, 126.41, 124.40, 113.37 (Ph), 110.91 (C-5), 99.97 (C-5′′), 91.80 (CC), 87.13 (CPh3), 86.75 (C-4′), 85.82 (C-1′), 82.46 (CC), 71.98 (C-3′), 63.48 (C-5′), 55.23 (2 × OCH3), 41.84 (C-2′), 12.23 (CH3); HRMS-ESI m/z 777.2527 [M + Na]+; calcd (C43H38N4O9Na) 777.2531.
Preparation of 5′-O-dimethoxytrityl-5-(4-(thymin-1-yl)phenyl)ethynyl-2′-deoxyuridine (9)
To a degassed and stirred solution of compound 4 (420 mg, 0.972 mmol), compound 2 (468 mg, 0.845 mmol) and Et3N (353 μL, 2.536 mmol) in DMF (15 mL) was added tetrakis(triphenylphosphine)palladium(0) (112 mg, 0.097 mmol) and CuI (37 mg, 0.194 mmol). The reaction mixture was stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure and the residue was dissolved in EtOAc, washed with brine (10 mL) and water (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–15% MeOH in DCM) to give the intermediate (641 mg, 88%) as a white solid. Rf 0.38 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, H-6), 7.99–7.92 (m, 2H, Ph), 7.62 (dt, J = 9.2, 7.8 Hz, 1H, Ph), 7.49 (t, J = 7.8 Hz, 2H, Ph), 7.42 (d, J = 7.4 Hz, 2H, Ph), 7.32 (d, J = 8.8 Hz, 4H, Ph), 7.25–7.18 (m, 4H, Ph, H-6′′),7.17–7.08 (m, 3H, Ph), 7.00 (d, J = 8.6 Hz, 2H, Ph), 6.77 (dd, J = 8.9, 3.7 Hz, 4H, Ph), 6.34 (t, J = 6.5 Hz, 1H, H-1′), 4.54 (m, 1H, H-3′), 4.11 (m, 1H, H-4′), 3.66 (s, 6H, 2 × OCH3), 3.46 (m, 1H, H-5′a), 3.27 (dd, J = 10.7, 3.1 Hz, 1H, H-5′b), 2.52 (m, 1H, H-2′a), 2.29 (m, 1H, H-2′b), 1.98 (d, J = 0.9 Hz, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 168.88 (COPh), 163.19 (C-4′′), 161.69 (C-4), 158.73 (Ph), 149.40 (C-2′′), 149.10 (C-2), 144.57 (Ph), 143.01 (C-6), 140.22 (C-6′′), 137.79, 135.68, 135.64, 135.21, 132.64, 131.66, 130.60, 130.04, 129.29, 128.18, 128.03, 127.19, 125.89, 123.22, 113.49 (Ph), 111.56 (C-5), 100.11 (C-5′′), 92.44 (CC), 87.20 (CPh3), 86.92 (C-4′), 86.06 (C-1′), 81.94 (CC), 72.38 (C-3′), 63.62 (C-5′), 55.34 (2 × OCH3), 41.85 (C-2′), 12.49 (CH3); HRMS-ESI m/z 960.4409 [M + Et3NH]+; calcd (C56H58N5O10) 960.4184. A solution of this intermediate (620 mg, 0.723 mmol) in saturated ammonia in methanol (30 mL) was stirred overnight at room temperature. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (0–15% MeOH in DCM) to give the product 9 (440 mg, 81%) as a white solid. Rf 0.32 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3 + CD3OD) δ 8.32 (s, 1H, H-6), 7.46 (d, J = 6.7 Hz, 2H, Ph), 7.42–7.31 (m, 4H, Ph), 7.26 (t, J = 7.4 Hz, 2H, Ph), 7.20–7.08 (m, 4H, Ph, H-6′′), 7.06–6.94 (m, 2H, Ph), 6.79 (dd, J = 5.7, 3.0 Hz, 4H, Ph), 6.34 (t, J = 5.6 Hz, 1H, H-1′), 4.50 (m, 1H, H-3′), 4.13 (m, 1H, H-4′), 3.70 (s, 6H, 2 × OCH3), 3.48 (m, 1H, H-5′a), 3.28 (m, 1H, H-5′b), 2.50 (m, 1H, H-2′a), 2.30 (dd, J = 13.3, 5.9 Hz, 1H, H-2′b), 1.95 (s, 3H, CH3); 13C NMR (101 MHz,CDCl3 + CD3OD) δ 164.75 (C-4′′), 162.52 (C-4), 158.56 (Ph), 150.38 (C-2′′), 149.84 (C-2), 144.48 (Ph), 142.80 (C-6), 140.41 (C-6′′), 138.00, 135.66, 132.45, 129.94, 129.91, 128.00, 127.95, 127.02, 125.78, 123.07, 113.31 (Ph), 111.36 (C-5), 99.98 (C-5′′), 92.25 (CC), 87.01 (CPh3), 86.95 (C-4′), 86.01 (C-1′), 81.78 (CC), 71.64 (C-3′), 63.54 C-5′), 55.16 (2 × OCH3), 41.71 (C-2′), 12.09 (CH3); HRMS-ESI m/z 777.2529 [M + Na]+; calcd (C43H38N4O9Na) 777.2531.
Preparation of 5′-O-dimethoxytrityl-5-(3-(thymin-1-yl)phenyl)-2′-deoxyuridine (10)
A stirred solution of compound 7 (500 mg, 1.157 mmol), compound 1 (633 mg, 0.965 mmol), and tetrakis(triphenylphosphine)palladium(0) (112 mg, 0.096 mmol) in a degassed mixture of THF and H2O (2:1, 90 mL) was added MeOH (23 mL) and NaOH (772 mg, 19.29 mmol). The mixture was stirred at 60 °C for 65 h. The resulting solution was carefully neutralized with a 1 M aqueous solution of HCl and extracted with EtOAc (150 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–20% MeOH in DCM) to give the product 10 (418 mg, 59%) as a white solid. Rf 0.35 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H, H-6), 7.46–7.34 (m, 2H), 7.30 (dd, J = 6.4, 2.6 Hz, 2H), 7.23–7.15 (m, 5H), 7.15–7.06 m, 3H), 6.98 (d, J = 7.8 Hz, 1H), 6.69 (d, J = 8.0 Hz, 4H), 6.38 (t, J = 6.6 Hz, 1H, H-1′), 6.26 (s, 1H, H-6′′), 4.38 (m, 1H, H-3′), 4.00 (m, 1H, H-4′), 3.72 (s, 6H, 2 × OCH3), 3.45 (m, 1H, H-5′a), 3.10 (m, 1H, H-5′b), 2.40 (m, 1H, H-2′a), 2.27 (m, 1H, H-2′b), 1.68 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.11 (C-4′′), 162.01 (C-4), 158.49 (Ph), 150.09 (C-2′′), 149.97 (C-2), 144.72 (Ph), 140.33 (C-6′′), 138.57 (Ph), 137.61 (C-6), 135.74, 135.50, 133.67, 129.81, 129.32, 128.72, 127.83, 127.80, 126.96, 126.49, 125.39 (Ph), 114.46 (C-5), 113.14 (Ph), 110.57 (C-5′′), 86.49 (CPh3), 86.22 (C-4′), 85.32 (C-1′), 71.82 (C-3′), 63.25 (C-5′), 55.27 (2 × OCH3), 41.35 (C-2′), 12.13 (CH3); HRMS-ESI m/z 753.2535 [M + Na]+; calcd (C41H38N4O9Na) 753.2531.
Preparation of 5′-O-dimethoxytrityl-5-(4-(thymin-1-yl)phenyl)-2′-deoxyuridine (11)
A stirred solution of compound 5 (175 mg, 0.405 mmol), compound 1 (222 mg, 0.338 mmol), and tetrakis(triphenylphosphine)palladium(0) (39 mg, 0.034 mmol) in a degassed mixture of THF and H2O (2:1, 40 mL) was added MeOH (11 mL) and NaOH (270 mg, 6.75 mmol). The mixture was stirred at 60 °C for 65 h. The resulting solution was carefully neutralized with a 1 M aqueous solution of HCl and extracted with EtOAc (150 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–8% MeOH in DCM) to give the product 11 (224 mg, 76%) as a white solid. Rf 0.35 (9:1 DCM/MeOH); 1H NMR (400 MHz, CDCl3 + CD3OD) δ 7.94 (s, 1H, H-6), 7.40 (s, 2H, NH), 7.37–7.30 (m, 4H, Ph), 7.28–7.09 (m, 7H, Ph), 6.96 (s, 1H, H-6′′), 6.96–6.90 (m, 2H, Ph), 6.72 (dd, J = 8.9, 2.4 Hz, 4H, Ph), 6.38 (dd, J = 7.8, 5.8 Hz, 1H, H-1′), 4.47 (m, 1H, H-3′), 4.12 (d, J = 2.8 Hz, 1H, H-4′), 3.76 (s, 6H, OCH3), 3.45 (dd, J = 10.6, 2.8 Hz, 1H, H-5′a), 3.24 (dd, J = 10.6, 3.6 Hz, 1H, H-5′b), 2.52 (ddd, 1H, J = 14.0, 5.6, 2.4 Hz, H-2′a), 2.32 (ddd, 1H, J = 14.0, 7.6, 6.4 Hz, H-2′b), 1.93 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3 + CD3OD) δ 165.21 (C-4′′), 162.82 (C-4), 158.73 (Ph), 150.68 (C-2′′), 150.50 (C-2), 144.62 (Ph), 141.02 (C-6′′), 138.07 (Ph), 137.82 (C-6), 135.79, 135.74, 132.97, 130.16, 129.50, 128.24, 128.09 (Ph), 127.24, 126.21 (Ph), 114.65 (C-5), 113.35 (Ph), 111.18 (C-5′′), 86.91 (CPh3), 86.84 (C-4′), 85.86 (C-1′), 71.78 (C-3′), 63.76 (C-5′), 55.39 (2 × OCH3), 41.57 (C-2′), 12.18 (CH3); HRMS-ESI m/z 753.2525 [M + Na]+; calcd (C41H38N4O9Na) 753.2531.
Preparation of 3′-O-(P-2-cyanoethyl-N,N-diisopropylaminophosphinyl)-5′-O-dimethoxytrityl-5-(3-(thymin-1-yl)phenyl)ethynyl-2′-deoxyuridine (12)
To a stirred solution of compound 8 (350 mg, 0.464 mmol) in DCM (12 mL) was added N,N-diisopropylethylamine (318 μL, 1.857 mmol) followed by 2-cyanoethyl-N,N-diisopropylphosphoramidochloridite (311 μL, 1.393 mmol). The reaction mixture was stirred at room temperature for 3 h. The reaction was quenched by the addition of MeOH (0.5 mL) and then added EtOAc (20 mL). The mixture was washed with a saturated aqueous solution of NaHCO3 (3 × 5 mL) and brine (3 × 5 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–100% EtOAc in petroleum ether) to give the product 12 (340 mg, 77%) as a white foam. Rf 0.32 (9:1 EtOAc/petroleum ether); 31P NMR (162 MHz, CDCl3) δ 149.12, 148.62; HRMS-ESI m/z 977.3624 [M + Na]+; calcd (C52H55N6O10PNa) 977.3610.
Preparation of 3′-O-(P-2-cyanoethyl-N,N-diisopropylaminophosphinyl)-5′-O-dimethoxytrityl-5-(4-(thymin-1-yl)phenyl)ethynyl-2′-deoxyuridine (13)
To a stirred solution of compound 9 (300 mg, 0.398 mmol) in DCM (10 mL) was added N,N-diisopropylethylamine (272 μL, 1.592 mmol) followed by 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (266 μL, 1.194 mmol). The reaction mixture was stirred at room temperature for 2 h. The reaction was quenched by the addition of MeOH (0.5 mL) and then added EtOAc (20 mL). The mixture was washed with a saturated aqueous solution of NaHCO3 (3 × 5 mL) and brine (3 × 5 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–100% EtOAc in petroleum ether) to give the product 13 (337 mg, 89%) as a white foam. Rf 0.32 (9:1 EtOAc/petroleum ether); 31P NMR (162 MHz, CDCl3) δ 149.07, 148.63; HRMS-ESI m/z 993.4046 [M + K]+; calcd (C52H55N6O10PK) 993.3354.
Preparation of 3′-O-(P-2-cyanoethyl-N,N-diisopropylaminophosphinyl)-5′-O-dimethoxytrityl-5-(3-(thymin-1-yl)phenyl)-2′-deoxyuridine (14)
To a stirred solution of compound 10 (288 mg, 0.395 mmol) in DCM (10 mL) was added N,N-diisopropylethylamine (270 μL, 1.578 mmol) followed by 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (264 μL, 1.183 mmol). The reaction mixture was stirred at room temperature for 2 h. The reaction was quenched by the addition of MeOH (0.5 mL) and then added EtOAc (20 mL). The mixture was washed with a saturated aqueous solution of NaHCO3 (3 × 5 mL) and brine (3 × 5 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–100% EtOAc in petroleum ether) to give the product 14 (356 mg, 97%) as a white foam. Rf 0.30 (9:1 EtOAC/petroleum ether); 31P NMR (162 MHz, CDCl3) δ 149.16, 148.63; HRMS-ESI m/z 953.3633 [M + Na]+; calcd (C50H55N6O10PNa) 953.3610.
Preparation of 3′-O-(P-2-cyanoethyl-N,N-diisopropylaminophosphinyl)-5′-O-dimethoxytrityl-5-(4-(thymin-1-yl)phenyl)-2′-deoxyuridine (15)
To a stirred solution of compound 11 (149 mg, 0.204 mmol) in DCM (10 mL) was added N,N-diisopropylethylamine (140 μL, 0.816 mmol) followed by 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (137 μL, 0.612 mmol). The reaction mixture was stirred at room temperature for 2 h. The reaction was quenched by the addition of MeOH (0.5 mL) and then added EtOAc (20 mL). The mixture was washed with a saturated aqueous solution of NaHCO3 (3 × 5 mL) and brine (3 × 5 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (0–100% EtOAc in petroleum ether) to give the product 15 (103 mg, 54%) as a white foam. Rf 0.30 (9:1 EtOAc/petroleum ether); 31P NMR (162 MHz, CDCl3) δ 149.10, 148.67; HRMS-ESI m/z 931.3790 [M + H]+; calcd (C50H56N6O10) 931.2711.
Synthesis of oligodeoxynucleotides
Oligonucleotide synthesis was carried out on a PersSeptive Biosystems EXPEDITE 8900 Nucleic Acid Synthesis System, by the phosphoramidite approach. Synthesis of oligonucleotides were carried out on a 0.2 μmol or 1.0 μmol scale using phosphoramidites 12, 13, 14 and 15 as well as the corresponding commercial 2-cyanoethyl phosphoramidites of the natural 2′-deoxynucleotides. The synthesis followed regular protocol for the DNA synthesizer. For the amidites 12, 14 and 15, a coupling time of 20 minutes were used, while for 13 a prolonged coupling time of 30 minutes was used. 1H-Tetrazole was used as activator in all cases. Coupling for all 2-cyanoethyl phosphoramidites were >90%. The 5′-O-DMT-ON oligonucleotides were cleaved from the solid support by treatment with concentrated aqueous ammonia at 55 °C for 16 h. Purities >90% were confirmed by ion-exchange HPLC. With purities <90% the ON's were purified by reverse-phase HPLC on a Water 600 system, using an Xterra prep MS C18 (10 μm, 7.8 × 150 mm) and 2.50mL min−1. flow setting; buffer: triethylammonium acetate (0.05 M); 0–70% buffer, 38 min; 70–100% buffer, 7 min; 100% buffer, 10 min All oligonucleotides were detritylated by treatment with 80% aqueous acetic acid for 20 min, quenched with sodium acetate (3 M) and sodium perchlorate (5 M), and finally precipitated from acetone at −18 °C for 16 h.Hybridization experiments
Micro molar extinction coefficients for the modified monomers were determined from the UV spectra of the detritylated monomers (made in analytical scale from 8, 9, 10 and 11) and a dilution experiment (Xε260 = 24.65, Yε260 = 12.30, Zε260 = 11.02, Wε260 = 10.65 cm−1 mM−1). Extinction coefficients of the modified oligonucleotides were estimated by a standard procedure, and corrected with the micro molar extinction coefficients for the modified monomers. UV melting experiments were carried out on a Perkin-Elmer Lambda20 UV/VIS Spectrometer. Samples were dissolved in a medium salt buffer (100 mM NaCl, 10 mM NaH2PO4, 5 mM Na2HPO4, 0.1 mM EDTA; pH 7) for normal melting temperature experiments, and a high salt buffer (700 mM NaCl, 10 mM NaH2PO4, 5 mM Na2HPO4, 0.1 mM EDTA; pH 7) for abasic site melting temperature experiments, in both cases using 1.5 μM concentrations of the complementary oligonucleotides. The increase in absorbance at 260 nm is recorded as a function of time, while temperature is increased linearly using a Peltier temperature programmer. Samples are annealed at 90 °C prior to measurements. For normal melting experiments a temperature range of 10 °C–75 °C was applied, while for abasic site experiments, a temperature range of 2 °C – 50 °C was applied. The melting temperature was calculated as the maximum peak of the first order derivatives of the temperature/absorbance curves. All melting temperature determinations are averages of duplicate measurements within ±1.0 °C.Circular dichroism
Circular dichroism spectra were obtained on a Jasco J815 CD spectrometer. Samples were dissolved in medium or high salt buffer, and concentrations of the complementary sequences were 1.5 μM for regular duplexes and 3 μM for abasic duplexes. Samples are annealed at 90 °C and spectra were recorded at 5 °C for normal duplexes and 2 °C for abasic duplexes. Background spectra were recorded and subtracted from all measurements.Fluorescence spectroscopy
UV absorption spectra (280–800 nm) were measured on a Perkin Elmer lambda35 UV/VIS spectrometer, using quartz optical cells with a path length of 1.0 cm. Fluorescence emission spectra (280–500 nm) were recorded on a Perkin Elmer LS 55 using quartz optical cells with a path length of 1.0 cm, and an excitational wavelength of 255 nm. Samples were dissolved to 1.5 μM using medium or high salt buffer. All double stranded samples were annealed at 90 °C prior to measurements. Background spectra were recorded and subtracted from all measurements.Molecular modeling
:DNA helix was built in the MacroModel V.9.1 suite of programs35,36 and modified with the appropriate C-5 thymine modification. The model structure was subjected to an torsional rotation MCMM structure search.39 In the MCMM structure search the rotation around C1′/N1, C5/C1′′ and either C7′′/N1′′ or C6′′/N1′′. Bonds are given a 180° rotational freedom and during the MCMM 1000 structures are generated. The obtained structures are subjected to a multiple minimization to reduce the number of structures generated into a number of local and global minima. The global minima structure generated is then used in the subsequent MD simulation.
Acknowledgements
The project was supported by Nucleic Acid Center and The Danish National Research Foundation, The Danish Councils for Independent Research | Technology and Production Sciences (FTP) and Natural Science (FNU), and The Villum Kann Rasmussen Foundation.Notes and references
- M. Endo and H. Sugiyama, ChemBioChem, 2009, 10, 2420 CrossRef CAS PubMed.
- F. C. Simmel, Chem. Unserer Zeit, 2013, 47, 164 CrossRef CAS PubMed.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636 RSC.
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 937 CrossRef CAS PubMed.
- C. Switzer, T. P. Prakash and Y. Ahn, Bioorg. Med. Chem. Lett., 1996, 6, 815 CrossRef CAS.
- T. Wu, M. Froeyen, G. Schepers, K. Mullens, J. Rozenski, R. Busson, A. Van Aershot and P. Herdewijn, Org. Lett., 2004, 6, 51 CrossRef CAS PubMed.
- S. L. Pedersen and P. Nielsen, Org. Biomol. Chem., 2005, 3, 3570 CAS.
- T. Wu, K. Nauwelaerts, A. Van Aershot, M. Froeyen, E. Lescrinier and P. Herdewijn, J. Org. Chem., 2006, 71, 5423 CrossRef CAS PubMed.
- M. S. Christensen, C. M. Madsen and P. Nielsen, Org. Biomol. Chem., 2007, 5, 1586 CAS.
- C. Andersen, P. K. Sharma, M. S. Christensen, S. I. Steffansen, C. M. Madsen and P. Nielsen, Org. Biomol. Chem., 2008, 6, 3983 CAS.
- M. S. Christensen, A. Bond and P. Nielsen, Org. Biomol. Chem., 2008, 6, 81 CAS.
- T. Umemoto, J. Wengel and A. S. Madsen, Org. Biomol. Chem., 2009, 7, 1793 CAS.
- K. I. Shaikh, C. S. Madsen, L. J. Nielsen, A. S. Jørgensen, H. Nielsen, M. Petersen and P. Nielsen, Chem. – Eur. J., 2010, 16, 12904 CrossRef CAS PubMed.
- P. Kielkowski, R. Pohl and M. J. Hocek, J. Org. Chem., 2011, 76, 3457 CrossRef CAS PubMed.
- A. S. Jørgensen, K. I. Shaikh, G. Enderlin, E. Ivarsen, S. Kumar and P. Nielsen, Org. Biomol. Chem., 2011, 9, 1381 Search PubMed.
- C. S. Madsen, S. Witzke, P. Kumar, K. Negi, P. K. Sharma, M. Petersen and P. Nielsen, Chem. – Eur. J., 2012, 18, 7434 CrossRef CAS PubMed.
- C. S. Madsen, L. J. Nielsen, N. S. Pedersen, A. Lauritsen and P. Nielsen, RSC Adv., 2013, 3, 10696 RSC.
- P. Kumar, P. K. Sharma, C. S. Madsen, M. Petersen and P. Nielsen, ChemBioChem, 2013, 14, 1072 CrossRef CAS PubMed.
- P. Kumar, C. S. Madsen and P. Nielsen, Bioorg. Chem. Med. Lett., 2013, 23, 6847 CrossRef CAS PubMed.
- S. Kumar, S. I. Steffansen, N. Albæk and P. Nielsen, Tetrahedron, 2014, 70, 583 CrossRef CAS PubMed.
- P. Kumar, A. F. Sorinas, L. J. Nielsen, M. Slot, K. Skytte, A. S. Nielsen, M. D. Jensen, P. K. Sharma, B. Vester, M. Petersen and P. Nielsen, J. Org. Chem., 2014, 79, 8020 CrossRef CAS PubMed.
- P. Kumar, P. K. Sharma and P. Nielsen, J. Org. Chem., 2014, 79, 11534 CrossRef CAS PubMed.
- R. D. Sheardy and N. C. Seeman, J. Org. Chem., 1986, 51, 4301 CrossRef CAS.
- M. Ahmadian, P. Zhang and D. E. Bergstrom, Nucleic Acid Res., 1998, 26, 3127 CrossRef CAS PubMed.
- D. J. Hurley and Y. Tor, J. Am. Chem. Soc., 1998, 120, 2194 CrossRef CAS.
- K. A. Cruickshank, J. Jiricny and C. B. Reese, Tetrahedron Lett., 1984, 25, 681 CrossRef CAS.
- O. R. Ludek and C. Meier, Eur. J. Org. Chem., 2006, 941 CrossRef CAS PubMed.
- M. F. Jacobsen, C. S. Andersen, M. M. Knudsen and K. V. Gothelf, Org. Lett., 2007, 9, 2851 CrossRef CAS PubMed.
- M. F. Jacobsen, M. M. Knudsen and K. V. Gothelf, J. Org. Chem., 2006, 71, 9183 CrossRef CAS PubMed.
- P. Kumar, N. Chandak, P. Nielsen and P. K. Sharma, Bioorg. Med. Chem., 2012, 20, 3843 CrossRef CAS PubMed.
- P. Kumar, M. Hornum, L. J. Nielsen, G. Enderlin, N. K. Andersen, C. Len, G. Hervé, G. Sartori and P. Nielsen, J. Org. Chem., 2014, 79, 2854 CrossRef CAS PubMed.
- S. Nakano, Y. Uotani, K. Uenishi, M. Fujii and N. Sugimoto, Nucleic Acids Res., 2005, 33, 7111 CrossRef CAS PubMed.
- P. Kočalka, N. K. Andersen, F. Jensen and P. Nielsen, ChemBioChem, 2007, 8, 2106 CrossRef PubMed.
- M. Kaura, P. Kumar and P. J. Hrdlicka, Org. Biomol. Chem., 2012, 10, 8575 CAS.
- P. D. J. Grootenhuis and P. A. Kollman, J. Am. Chem. Soc., 1989, 111, 4046 CrossRef CAS.
- MacroModel, version 9.1, Schrödinger, LLC, New York, NY, 2005 Search PubMed.
- W. C. Still, A. Tempcyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127 CrossRef CAS.
- A. Pérez, I. Marchán, D. Svozil, J. Sponer, T. E. Cheatham, C. A. Laughton and M. Orozco, J. Biophys., 2007, 3817 CrossRef PubMed.
- G. Chang, W. C. Guida and W. C. Still, J. Am. Chem. Soc., 1989, 111, 4379 CrossRef CAS.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765 CrossRef CAS.
- S. J. Weiner, P. A. Kollman, D. T. Nguyen and D. A. Case, J. Comput. Chem., 1986, 7, 230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MALDI-TOF data for oligonucleotides. CD-spectra. Fluorescence emission spectra. Selected NMR-spectra. See DOI: 10.1039/c5ob00872g |
| This journal is © The Royal Society of Chemistry 2015 |