
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid and scalable synthesis of innovative unnatural α,β or γ-amino acids functionalized with tertiary amines on their side-chains†
Séverine
Schneider
a,
Hussein
Ftouni
a,
Songlin
Niu
a,
Martine
Schmitt
a,
Frédéric
Simonin
b and
Frédéric
Bihel
*a
aLaboratoire d'Innovation Thérapeutique, UMR7200, CNRS, Université de Strasbourg, faculté de pharmacie, 74, route du Rhin, 67401 Illkirch, France. E-mail: fbihel@unistra.fr
bUniversity of Strasbourg, CNRS, UMR7242, ESBS, 67412 Illkirch Graffenstaden, France
First published on 18th May 2015
Abstract
We report a selective ruthenium catalyzed reduction of tertiary amides on the side chain of Fmoc-Gln-OtBu derivatives, leading to innovative unnatural α,β or γ-amino acids functionalized with tertiary amines. Rapid and scalable, this process allowed us to build a library of basic unnatural amino acids at the gram-scale and directly usable for liquid- or solid-phase peptide synthesis. The diversity of available tertiary amines allows us to modulate the physicochemical properties of the resulting amino acids, such as basicity or hydrophobicity.
Introduction
Although peptides are the keystones of most vital processes, only a few peptide-based drugs were marketed in the 20th century. Among other criteria, low bioavailability and metabolic instability led to a global rejection of peptides as drug candidates by the pharmaceutical industry. However, in the last decade, new therapeutic strategies were promoted, and more than 100 peptide-based drugs have already reached the market.1 In order to circumvent the inherent problems of bioavailability and biostability of peptides, protein-like backbones called peptidomimetics were developed in order to mimic native peptides, but with better pharmacological properties. Unnatural amino acids were then generated from natural amino acids through chemical modifications of this backbone (amine alkylation, backbone extension, cyclization or isosteric replacements).2 Another great area of interest was the design of “non-natural” side chains, allowing the improvement of interactions between the peptide and its targeted protein. Regarding this aspect, a large diversity of side chains were developed, but non-natural amino acids bearing various aromatic side chains were more extensively reported.2d,3 In contrast, side chains bearing a basic amino group mimicking ornithine, lysine or arginine derivatives were less well reported. Most of the lysine analogs resulted from a reductive amination between aldehydes and Fmoc-Lys-OH affording monoalkylated or symmetric dialkylated lysine derivatives.4 Through an alkylation of glycine in the solid-phase, Scott et al. reported an interesting synthesis of non-natural racemic amino acids bearing the pyrrolidine ring.5 Indeed, introduction of azacycles through reductive amination is still challenging.6 For the first time, we propose in this paper a versatile synthesis of non-natural basic amino acids, bearing a high diversity of cyclic or acyclic tertiary amines on the side chain. Protected by a Fmoc group in the N-terminus, these new non-natural amino acids are directly available for liquid or solid phase peptide synthesis.Results and discussion
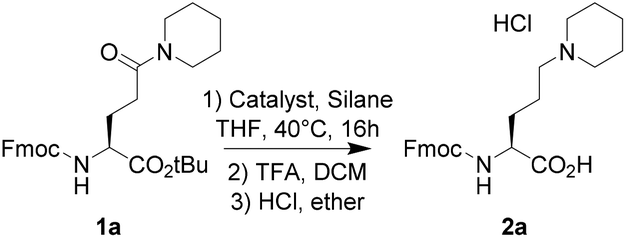
Metal-catalyzed reductions of carboxamides in combination with silanes were extensively studied in recent years with simple amides such as benzamide derivatives.7 Our challenge was to look for the selective reduction of the amide function of the glutamate derivative 1 (Table 1), bearing two other reducible groups (ester and carbamate) and one chiral center. Starting from the commercially available Fmoc-L-Glu-OtBu, piperidine was coupled on the side chain to afford amide derivative 1a in a quantitative yield. We next evaluated three known catalytic systems (Table 1), involving Zn,7f Ir,7k or Ru,7r for reduction of the carboxamide function in a selective manner towards both urethane (Fmoc) and tert-butyl ester functions. As compound 1a was poorly soluble in toluene, THF was preferred as the solvent. Zinc-mediated reduction was not observed (entry 1), whereas [Ir(COD)Cl]2 and Ru3(CO)12 in combination with Et2SiH2 or tetramethyldisiloxane (TMDS) produced amine 2a in high yields at only 40 °C (entries 2–4 and 7). As the main goal of this study was to propose a gram-scale synthesis of non-natural basic amino acids, the catalytic system Ru3(CO)12/TMDS was selected as it is cheaper than the system associated with an iridium-based catalyst and Et2SiH2. In 2013, Reeves et al. reported that 1% Ru3(CO)12 in the presence of 4 eq. of TMDS at 50 °C in toluene was efficient enough to reduce a large set of amides.7r In our case, the reduction of the glutamine derivative 1a required at least 3 mol% of the catalyst in THF at 40 °C (Table 1, compare entries 7 & 8). Easy to handle, the reaction was carried out under air. The key-step of this procedure appeared to be the purification stage. Indeed, after the completion of the reduction step, the resulting tert-butyl ester was recovered in poor yields (<30%) after normal or reversed phase chromatography, although no side product was observed. A treatment of this tert-butyl ester with trifluoroacetic acid afforded compound 2a as a TFA salt. Precipitation in ether of this salt led to medium yield (≈50%). Subsequently, the trifluoroacetate counter-ion was exchanged with HCl 1 M in diethyl ether, affording a white precipitate 2a as a hydrochloride salt in 81% yield. As the optimization step was performed on a 100 mg scale, we next extended the scope of the reaction to the gram-scale with a large set of tertiary amines (Table 2). Under these conditions, compounds 2 were globally obtained in good to excellent yields after precipitation in ether. The reaction was compatible with the hindered amine (2d) and allowed the introduction of other chiral centers (2c, 2d). Several functional groups were tolerated, such as tertiary amines (2e, 2g), 2-aminopyrimidine (2f), ester (2h), and alcohol (2i). A series of amines bearing an aromatic ring was also easily synthesized (2j–o). Although aniline derivatives were well tolerated (2l, 2n), benzimidazole derivative 2m was not recovered as its precursor amide 1m was poorly stable. Acyclic amines (2n–q) were also easily obtained in high yields. Interestingly, after coupling the Bn-NH-NH-Boc on Fmoc-L-Glu-OtBu, we were able to reduce the hydrazide function to produce the corresponding hydrazine derivative 2r as a non-natural analog of lysine. In spite of the protonated state of the terminal amine, Fmoc cleavage was observed, explaining the modest yield of 30% for compound 2r. With yields inferior to 80%, compounds 2g, 2h, and 2r were purified by flash chromatography to remove the byproducts. All the other compounds were easily recovered after precipitation with purity higher than 98%. Starting from Fmoc-L-Asp-OtBu, we were able to obtain compound 4, exhibiting a shorter side-chain than 2a. Next, we explored the possibility of reducing the carboxamide function on the backbone. Starting from commercially available Fmoc-L-Asp(OtBu)-OH or Fmoc-L-Glu(OtBu)-OH, piperidine was coupled to the C-terminus part to afford compounds 5 and 6. Ruthenium-based reduction of the resulting carboxamide led to the β,γ-diamino acid 7 and the γ,δ-diamino acid 8 in excellent yields (Scheme 1).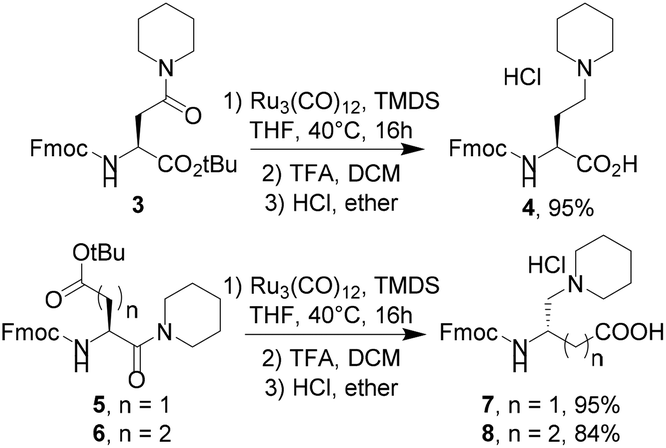 | ||
| Scheme 1 Ru-catalyzed synthesis of nor-ornithine analogs and β,γ and γ,δ-diamino acids. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Silane | Equiv. | Yieldb (%) |
a Reaction conditions: (1) 1a (0.2 mmol), catalyst, silane, THF (1 mL), 40 °C, 16 h; (2) TFA/DCM (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v – 2 mL); (3) HCl 1 N in ether (5 mL).
b Determined by HPLC using caffeine as external standard. :v – 2 mL); (3) HCl 1 N in ether (5 mL).
b Determined by HPLC using caffeine as external standard.
|
|||||
| 1 | Zn(OAc)2 | 3 | (EtO)3SiH | 8 | 0 |
| 2 | [Ir(COD)Cl]2 | 3 | Et2SiH2 | 8 | 74 |
| 3 | [Ir(COD)Cl]2 | 3 | TMDS | 8 | 59 |
| 4 | Ru3(CO)12 | 3 | Et2SiH2 | 8 | 74 |
| 5 | Ru3(CO)12 | 3 | (EtO)3SiH | 8 | 60 |
| 6 | Ru3(CO)12 | 3 | iPr3SiH | 8 | 0 |
| 7 | Ru3(CO)12 | 3 | TMDS | 8 | 80 |
| 8 | Ru3(CO)12 | 1 | TMDS | 8 | 40 |
| 9 | Ru3(CO)12 | 5 | TMDS | 8 | 80 |
| 10 | Ru3(CO)12 | 3 | TMDS | 2 | 60 |
| 11 | Ru3(CO)12 | 3 | TMDS | 4 | 78 |
| 12 | Ru3(CO)12 | 3 | TMDS | 6 | 81 |

The absence of the need for purification by chromatography allowed us to easily produce large quantity of these unnatural amino acids, with 98% minimum purity measured by HPLC and NMR. However, neither of these analytical techniques allows the detection of ruthenium. Using ICP-mass spectrometry, we were able to quantify 2.1% (w/w) of ruthenium after final precipitation. Using resin-bound dimercaptotriazine as a scavenger in a solution of 2a in a mixture of DCM/MeOH [9:1], we were able to remove up to 90% of the ruthenium, leading to a final 0.2% (w/w) of residual ruthenium. We next evaluated whether this whole synthetic procedure may induce racemization of the Cα of the amino acids formed. To answer this question, both enantiomers L-2a and D-2a were coupled to H-L-Phe-NH2 using several standard coupling reagents (BOP, COMU, HATU). By using HPLC, we were able to evaluate the racemization ratio as lower than 1% (ESI†). By repeating this last coupling reaction, we determined that the resulting traces of racemization were occurring during the coupling reaction with H-Phe-NH2, and not during the reductive process.
This very efficient synthesis allows a rapid and scalable access to a large variety of unnatural N-Fmoc α-, β- or γ-amino acids. These residues can be easily introduced into the peptide sequence as mimetics of natural basic amino acids such as ornithine, lysine or arginine. However, these new unnatural amino acids give access to a larger range of basicity and hydrophobicity, depending on the tertiary amine chosen on the side chain. To highlight this concept, we compared the hydrophobicity (logD) and the basicity (pKa) for the 20 unnatural amino acids synthesized in this paper and compared them with ornithine, lysine and arginine (Chart 1). To be as close as possible to a peptide structure, calculations were performed with each amino acid under the form Ac-Xaa-NH-Me. Ornithine, lysine and arginine form a very hydrophilic cluster (logD < −4) with basicity ranging from 9.6 to 11.2. In contrast, the 20 unnatural amino acids show extended ranges of logD (from −3.4 to +0.5) and pKa (from 4.7 to 10.3). By introducing these unnatural amino acids into peptide sequences in replacement of basic amino acids such as lysine, ornithine or arginine, we will be able to modulate the physicochemical properties of the resulting peptidomimetics. A first application for these novel non-natural basic amino acids was demonstrated in a liquid-phase synthesis of compounds 9a and 9b (Scheme 2), which were recently reported as the first orally active antagonists of NPFF receptors to prevent opioid-induced hyperalgesia in rodents.9 Starting from compounds 2a and 2j, compounds 9a and 9b were easily synthesized using standard peptide synthesis. This new strategy appears to be much more efficient and less time-consuming than the previously reported method.9 More importantly, the availability of the building block 2 allows a fast optimization of the N- and C-terminus in a convergent manner, which is not possible with the reported synthesis.9 Compounds 9 were initially developed as a peptidomimetic of the corresponding arginine residue.9 Using standard solid-phase synthesis, we pushed the concept further by replacing the arginine moiety of the neuropeptide FF by the residue 2j in order to generate a peptidomimetic analog of the peptide NPFF (Fig. 1). Using 2j immediately after precipitation in ether and using the Fmoc-strategy on a Rink-amide resin, we were able to synthesize peptide 10 with a good purity by HPLC after cleavage. The analysis of the crude peptide by ICP-MS led us to quantify the residual ruthenium as less than 20 ppm. Affinities of 10 for both NPFF1 and NPFF2 receptors were measured and compared to the endogenous peptide NPFF. Although showing lower affinities than NPFF,10 compound 10 still shows a significant affinity for NPFF2R, but binds poorly to NPFF1R. These results highlight how these new basic amino acids can be used to generate peptidomimetics with interesting pharmacological profiles.
 | ||
| Chart 1 LogD7.4 and pKa for Ac-Xaa-NHMe. Calculator plugin (Marvin 6.0.2, 2013, http://www.chemaxon.com). LogD was predicted at a pH 7.4 following the method reported by Klopman et al.8 | ||
 | ||
| Scheme 2 Application of the non-natural amino acid 2a to the synthesis of 9, an orally-active antagonist of opioid-induced hyperalgesia. | ||
 | ||
| Fig. 1 Application of the non-natural amino acid 2j to the synthesis of an unnatural analog of the endogenous neuropeptide NPFF 10. | ||
Conclusion
We have reported an efficient and selective metal-catalyzed amide reduction allowing the rapid production at the gram-scale, of original unnatural basic α-, β- or γ-amino acids, readily available for liquid- or solid-phase peptide synthesis. These new amino acids will constitute important building blocks for the synthesis of future peptidomimetics. The versatility of the process allows the introduction of a very large diversity of basic tertiary amines exhibiting various physicochemical properties, potentially offering a wide spectrum of uses (cell-penetrating peptide, peptidomimetic, etc.).Experimental
General procedure for the reduction of amides into the corresponding amines 2
TMDS (6.5 mL, 35.7 mmol, 8 equiv.) was added to Ru3(CO)12 (0.144 g, 0.223 mmol, 0.05 equiv.) under air. The mixture was stirred for 5 minutes, then a solution of compound 1 (4.46 mmol, 1 equiv.) in 24 mL of THF was added, and the reaction mixture was stirred for 16 hours under air at 40 °C. Volatiles were removed in vacuo. The reaction mixture was diluted with DCM (24 mL), and cooled to 0 °C in an ice bath. Trifluoroacetic acid (20 mL) was slowly added at 0 °C. After addition, the reaction mixture was stirred at room temperature for 4 hours. Volatiles were removed under reduced pressure, and the resulting mixture was diluted in diethyl ether (10 mL). A solution of HCl (1 M) in diethyl ether (48 mL) was slowly added to the mixture, and the reaction mixture was stirred at room temperature for 5 minutes, affording a white powder after filtration. The powder was next solubilized in DCM with 10% MeOH. To remove residual ruthenium, dimercaptotriazine on silica (SiliaBond DMT from Silicycle, 0.5 mmol g−1 of resin, 1.8 mmol) was added and the mixture was stirred for 1 h at room temperature, and filtered and rinsed with DCM. Compounds 2 were recovered after evaporation and lyophilization. Only compounds 2g, 2h and 2r were required to be purified by chromatography using a reversed-phase C18 column.Acknowledgements
We would like to thank Clara Van Hoey for her help in the binding assay.Notes and references
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- (a) I. Avan, C. D. Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575–3594 RSC; (b) J. Gante, Angew. Chem., Int. Ed., 1994, 33, 1699–1720 CrossRef PubMed; (c) A. Grauer and B. Konig, Eur. J. Org. Chem., 2009, 5099–5111 CrossRef CAS PubMed; (d) A. Perdih and M. S. Dolenc, Curr. Org. Chem., 2011, 15, 3750–3799 CrossRef CAS.
- (a) S. J. Zuend, M. P. Coughlin, M. P. Lalonde and E. N. Jacobsen, Nature, 2009, 461, 968–U223 CrossRef CAS PubMed; (b) W. L. Scott, Z. N. Zhou, J. G. Martynow and M. J. O'Donnell, Org. Lett., 2009, 11, 3558–3561 CrossRef CAS PubMed; (c) J. He, S. H. Li, Y. Q. Deng, H. Y. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J. Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed.
- (a) Z. P. Huang, J. T. Du, X. Y. Su, Y. X. Chen, Y. F. Zhao and Y. M. Li, Amino Acids, 2007, 33, 85–89 CrossRef CAS PubMed; (b) A. F. Armstrong, N. Oakley, S. Parker, P. W. Causey, J. Lemon, A. Capretta, C. Zimmerman, J. Joyal, F. Appoh, J. Zubieta, J. W. Babich, G. Singh and J. F. Valliant, Chem. Commun., 2008, 5532–5534, 10.1039/B810706h; (c) D. Groff, P. R. Chen, F. B. Peters and P. G. Schultz, ChemBioChem, 2010, 11, 1066–1068 CrossRef CAS PubMed; (d) R. Wieneke, A. Bernecker, R. Riedel, M. Sumper, C. Steinem and A. Geyer, Org. Biomol. Chem., 2011, 9, 5482–5486 RSC; (e) N. Uhlig and C. J. Li, Org. Lett., 2012, 14, 3000–3003 CrossRef CAS PubMed; (f) R. J. Hopkinson, L. J. Walport, M. Munzel, N. R. Rose, T. J. Smart, A. Kawamura, T. D. W. Claridge and C. J. Schofield, Angew. Chem., Int. Ed., 2013, 52, 7709–7713 CrossRef CAS PubMed.
- W. L. Scott, M. J. O'Donnell, F. Delgado and J. Alsina, J. Org. Chem., 2002, 67, 2960–2969 CrossRef CAS PubMed.
- K. Lu, W. J. Ye, L. Zhou, L. B. Collins, X. Chen, A. Gold, L. M. Ball and J. A. Swenberg, J. Am. Chem. Soc., 2010, 132, 3388–3399 CrossRef CAS PubMed.
- (a) M. Igarashi and T. Fuchikami, Tetrahedron Lett., 2001, 42, 1945–1947 CrossRef CAS; (b) S. Hanada, T. Ishida, Y. Motoyama and H. Nagashima, J. Org. Chem., 2007, 72, 7551–7559 CrossRef CAS PubMed; (c) H. Sasakuma, Y. Motoyama and H. Nagashima, Chem. Commun., 2007, 4916–4918, 10.1039/B711743d; (d) N. Sakai, K. Fujii and T. Konakahara, Tetrahedron Lett., 2008, 49, 6873–6875 CrossRef CAS PubMed; (e) S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032–15040 CrossRef CAS PubMed; (f) S. Das, D. Addis, S. L. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 4971–4971 CrossRef CAS; (g) S. Das, S. L. Zhou, D. Addis, S. Enthaler, K. Junge and M. Beller, Top. Catal., 2010, 53, 979–984 CrossRef CAS; (h) S. Das, D. Addis, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 12186–12192 CrossRef CAS PubMed; (i) S. Laval, W. Dayoub, L. Pehlivan, E. Metay, A. Favre-Reguillon, D. Delbrayelle, G. Mignani and M. Lemaire, Tetrahedron Lett., 2011, 52, 4072–4075 CrossRef CAS PubMed; (j) Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. – Eur. J., 2011, 17, 1768–1772 CrossRef CAS PubMed; (k) C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304–11307 CrossRef CAS PubMed; (l) S. Das, B. Join, K. Junge and M. Beller, Chem. Commun., 2012, 48, 2683–2685 RSC; (m) S. Park and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 640–653 CrossRef CAS PubMed; (n) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem. – Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed; (o) T. Dombray, C. Helleu, C. Darcel and J. B. Sortais, Adv. Synth. Catal., 2013, 355, 3358–3362 CrossRef CAS PubMed; (p) J. A. Fernandez-Salas, S. Manzini and S. P. Nolan, Chem. Commun., 2013, 49, 9758–9760 RSC; (q) Y. H. Li, J. A. M. de la Torre, K. Grabow, U. Bentrup, K. Junge, S. L. Zhou, A. Bruckner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 11577–11580 CrossRef CAS PubMed; (r) J. T. Reeves, Z. L. Tan, M. A. Marsini, Z. X. S. Han, Y. B. Xu, D. C. Reeves, H. Lee, B. Z. Lu and C. H. Senanayakea, Adv. Synth. Catal., 2013, 355, 47–52 CrossRef CAS PubMed; (s) M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231–2234 CrossRef CAS PubMed; (t) W. L. Xie, M. D. Zhao and C. M. Cui, Organometallics, 2013, 32, 7440–7444 CrossRef CAS; (u) T. X. Zhang, Y. Zhang, W. X. Zhang and M. M. Luo, Adv. Synth. Catal., 2013, 355, 2775–2780 CrossRef CAS PubMed.
- G. Klopman, J. Y. Li, S. M. Wang and M. Dimayuga, J. Chem. Inf. Comput. Sci., 1994, 34(4), 752–781 CrossRef CAS.
- F. Bihel, J. P. Humbert, S. Schneider, I. Bertin, P. Wagner, M. Schmitt, E. Laboureyras, B. Petit-Demouliere, E. Schneider, C. Mollereau, G. Simonnet, F. Simonin and J. J. Bourguignon, ACS Chem. Neurosci., 2015, 6, 438–445 CrossRef CAS PubMed.
- K. Elhabazi, J. P. Humbert, I. Bertin, M. Schmitt, F. Bihel, J. J. Bourguignon, B. Bucher, J. A. J. Becker, T. Sorg, H. Meziane, B. Petit-Demouliere, B. Ilien and F. Simonin, Neuropharmacology, 2013, 75, 164–171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General reaction procedures for Cpds 1, 9 and 10 and compound characterization; racemization study. See DOI: 10.1039/c5ob00828j |
| This journal is © The Royal Society of Chemistry 2015 |