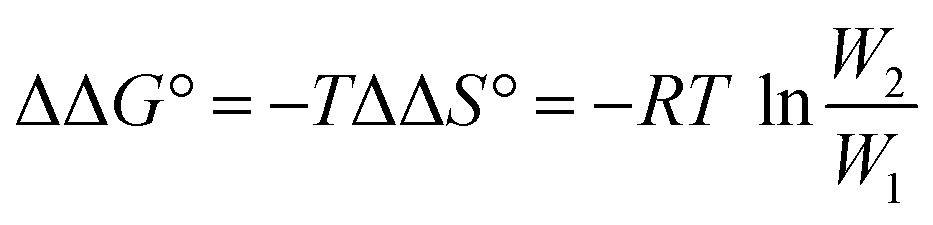Influence of receptor flexibility on intramolecular H-bonding interactions†
Received
22nd April 2015
, Accepted 10th June 2015
First published on 1st July 2015
Abstract
Atropisomers of a series of zinc tetraphenyl porphyrins were synthesized and used as supramolecular receptors. Rotation around the porphyrin-meso phenyl bonds is restricted by installing ortho-chlorine substituents on the phenyl groups. The chlorine substituents allowed chromatographic separation of atropisomers, which did not interconvert at room temperature. The porphyrin meso phenyl groups were also equipped with phenol groups, which led to the formation of intramolecular H-bonds when the zinc porphyrins were bound to pyridine ligands equipped with ester or amide side arms. Binding of the pyridine ligands with the conformationally locked chloroporphyrins was compared with the corresponding unsubstituted porphyrins, which are more flexible. The association constants of 150 zinc porphyrin–pyridine complexes were measured in two different solvents, toluene and 1,1,2,2-tetrachloroethane (TCE). These association constants were then used to construct 120 chemical double mutant cycles to quantify the influence of chlorine substitution on the free energy of intramolecular H-bonds formed between the phenol side arms of the porphyrins and the ester or amide side arms of the pyridine ligands. Conformational restriction leads to increases in the stability of some complexes and decreases in the stability of others with variations in the free energy contribution due to intramolecular H-bonding of −5 to +6 kJ mol−1.
Introduction
Many dynamic biological processes are governed by receptor-ligand recognition and molecular self-assembly through multiple non-covalent interactions.1 If we want to be able to apply these strategies from Nature in the design of artificial functional complexes, a basic understanding of the role of receptor flexibility is essential. An increase or decrease in the conformational mobility of a receptor can influence the thermodynamics of binding due to changes in conformational entropy.2 The difficulties associated with evaluating receptor flexibility is a problem for the quantitative estimation of the overall binding affinity of multivalent complexes that make multiple cooperative intermolecular interactions.3
The parameter used to experimentally quantify chelate cooperativity in multivalent systems is effective molarity (EM).4 EM is the ratio of the intramolecular association constant to the corresponding intermolecular association constant and provides a quantitative measure of the complementarity of receptor and ligand in non-covalent complexes. Values of EM for intramolecular covalent reactions depend strongly on conformational flexibility and ring strain. However, non-covalent systems are linked relatively loosely, and the overall entropy loss associated with the formation of multivalent complexes is less pronounced than in covalent systems. Williams et al. estimated a value of 3.7 ± 0.9 kJ mol−1 for the free energy cost of restricting an internal rotor in H-bonded complexes by comparing the binding of vancomycin with peptide ligands with different degrees of conformational freedom.5 They also estimated the free energy cost of restricting a rotor in a hydrocarbon chain as 1.6–3.6 kJ mol−1 through analysis of the thermodynamics of hydrocarbon phase transitions.6 Andrews et al. suggested that the free energy cost for restricting a single rotor is 2.9 kJ mol−1, based on a statistical analysis of binding constants for a large number of drug–receptor interactions.7 Based on the association constants of synthetic supramolecular complexes, Schneider et al. proposed a value of 0.5–1.3 kJ mol−1 for the free energy cost of restricting a single rotor.8 The free energy cost for freezing a rotor in non-covalent complexes is significantly lower than the 5–6 kJ mol−1 found for covalent systems.9 The reason is that the rotors are not completely frozen in the non-covalent systems, so the free energy penalty depends on the amount of residual conformational flexibility present in the complex.
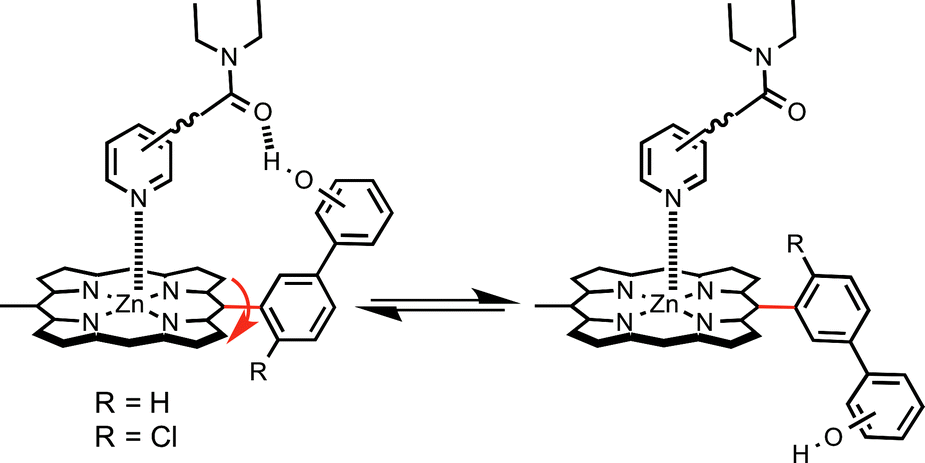
In order to study the effect of receptor flexibility on the strength of intermolecular interactions, we have designed a series of chlorine-substituted zinc porphyrins where it is possible to isolate different atropisomers caused by restricted rotation around the meso phenyl bond labelled in red in Fig. 1.10 The introduction of substituents in the ortho positions of the meso phenyl groups of zinc tetraphenylporphyrins increases the barrier to rotation around the porphyrin–phenyl bond from around 60 kJ mol−1 to 110–130 kJ mol−1.11 The recognition properties of zinc porphyrins with ortho chlorine substituents on the phenyl rings, which are conformationally locked, can be compared with the corresponding porphyrins with no chlorine substituents, where the atropisomers are free to interconvert. Here we study the binding of these two classes of zinc porphyrin with pyridine ligands, which are equipped with amide groups that can make H-bonds with the phenol groups on the porphyrin periphery (Fig. 1). In principle, the effects of conformational restriction on the change in free energy of binding can be predicted using Boltzmann's law, eqn (1).
| |  | (1) |
where
W1 is the number of free states, and
W2 is the number of bound states.
 |
| | Fig. 1 Equilibrium between two atropisomers of a zinc porphyrin–pyridine complex. When R = H, rotation around the meso phenyl bond labelled in red is slow on the NMR timescale but fast on the laboratory timescale. When R = Cl, rotation is also slow on the laboratory timescale, so different atropisomers of tetraphenyl porphyrins can be isolated. | |
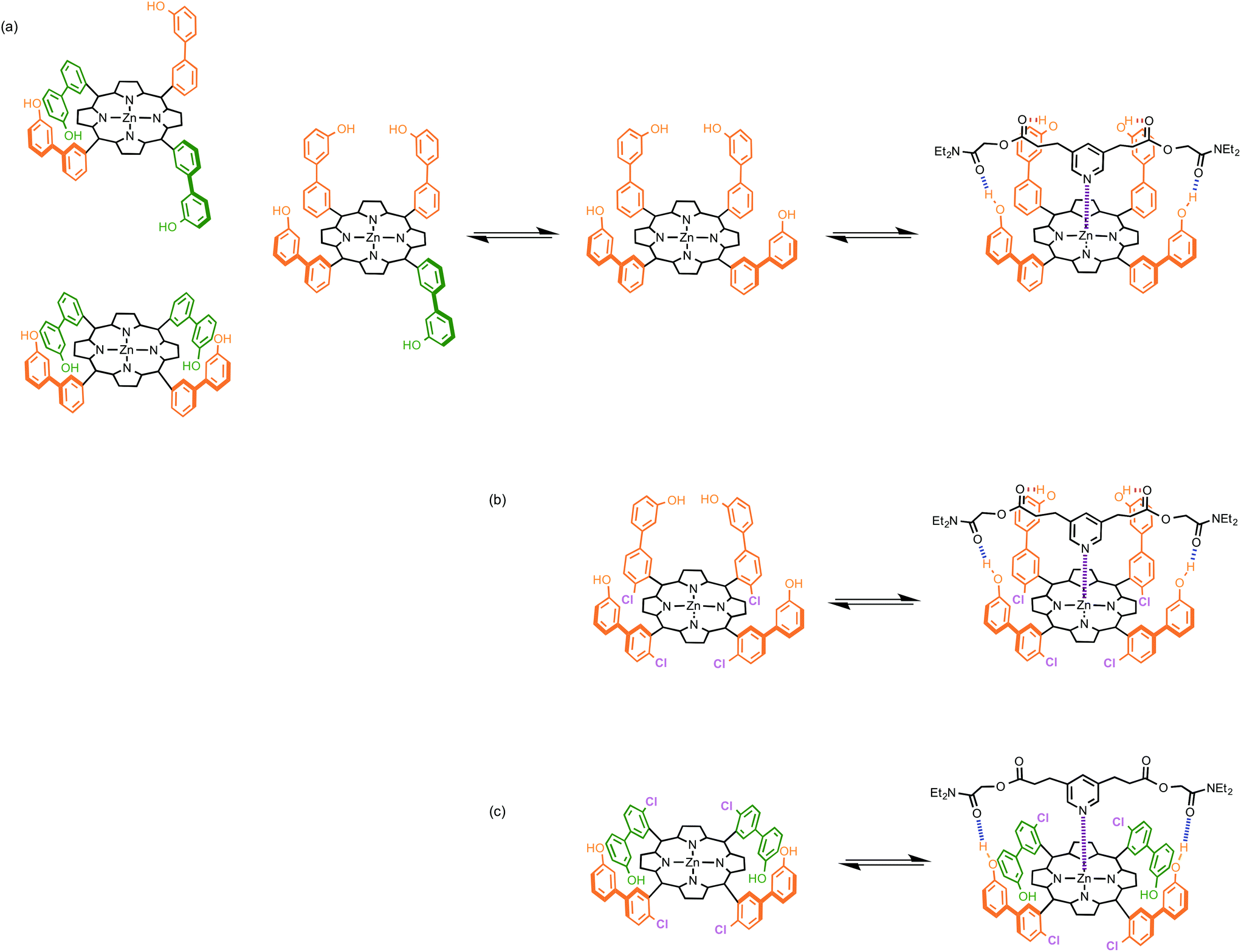
Upon binding with a ligand, the number of conformational states accessible to a porphyrin receptor decreases, because not all conformations can make all of the possible H-bonding interactions with the ligand. The porphyrins used in this work exist as a mixture of four atropisomers: αααα, αααβ, ααββ, αβαβ, and the statistical ratio of populations is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2:1, due to the degeneracy of some of these states (Fig. 2(a)). Note that pyridine binding to zinc porphyrins is in fast exchange between free and bound states, so the two faces of the porphyrin are effectively equilibrated, and pairs of atropisomers are equivalent, e.g. αααα and ββββ. Thus, there are eight states in total for the free porphyrin (W1 = 8). If the porphyrin binds to a ligand that makes four strong H-bonding interactions in addition to the zinc–pyridine coordination bond, then the αααα atropisomer will be the major species in the bound state (W2 = 1). At room temperature, the result would be a free energy penalty of 5 kJ mol−1, due to conformational restriction of the receptor (W1 = 8 and W2 = 1 in eqn (1)).
:4:2:1, due to the degeneracy of some of these states (Fig. 2(a)). Note that pyridine binding to zinc porphyrins is in fast exchange between free and bound states, so the two faces of the porphyrin are effectively equilibrated, and pairs of atropisomers are equivalent, e.g. αααα and ββββ. Thus, there are eight states in total for the free porphyrin (W1 = 8). If the porphyrin binds to a ligand that makes four strong H-bonding interactions in addition to the zinc–pyridine coordination bond, then the αααα atropisomer will be the major species in the bound state (W2 = 1). At room temperature, the result would be a free energy penalty of 5 kJ mol−1, due to conformational restriction of the receptor (W1 = 8 and W2 = 1 in eqn (1)).
 |
| | Fig. 2 Influence of atropisomerism on the formation of a zinc porphyrin–pyridine complex. (a) If the atropisomers interconvert rapidly, the porphyrin populates eight different free states (there are degenerate forms of the four structures illustrated), but formation of four intramolecular H-bonds in the bound state is only possible for the αααα atropisomer. (b) The chlorine substituents allow isolation of the αααα atropisomer, so that there is only one free state, and the side arms are preorganized to form the complex with four H-bonds. (c) For the ααββ atropisomer, only two of the four possible H-bonds can be formed, because the green side arms are preorganised in the wrong geometry. | |
For the chloroporphyrins, the conformation is locked, and the four different atropisomers can be isolated. Thus it is possible to directly measure the binding of a ligand to the αααα atropisomer (Fig. 2(b)). In this case, the number of conformational states of the receptor does not change (W1 = W2 = 1), so the expectation is that the porphyrin-ligand complex in Fig. 2(b) would be eight times more stable than the porphyrin-ligand complex in Fig. 2(a), due to conformational entropy. Similarly, there is no change in conformational entropy for any of the conformationally locked atropisomers: αααβ (W1 = W2 = 4), ααββ (W1 = W2 = 2), and αβαβ (W1 = W2 = 1).
However, not all four H-bonds can be formed for the other three atropisomers, because the side arms are preorganised in the wrong geometry. For example, the ααββ atropisomer can only make two H-bonds (Fig. 2(c)), so conformational restriction leads to an enthalpic penalty corresponding to breaking two ester-phenol H-bonds. In toluene, each ester-phenol H-bond contributes 3 kJ mol−1 to the overall free energy of binding.12 The loss of H-bonding interactions would therefore cancel out the entropic benefit of conformational restriction, so the expectation is that the porphyrin-ligand complex in Fig. 2(c) would have a similar stability to the porphyrin-ligand complex in Fig. 2(a). A similar argument applies to the αβαβ atropisomer, which can only make two H-bonds in the bound state. The αααβ atropisomer can make three H-bonds in the bound state, so the entropic benefit of conformational restriction outweighs the loss of H-bonding interactions, and the conformationally locked αααβ porphyrin-ligand complex is expected to be slightly more stable (2 kJ mol−1) than the porphyrin-ligand complex in Fig. 2(a).
If the ligand can only make one or two H-bonds with the porphyrin phenol groups, then all atropisomers of a conformationally flexible receptor are equally likely in the bound state. There will be no change in the atropisomer distribution on complexation, and there will be no effect on the observed association constant (W1 = W2 = 8). Thus the conformationally restricted porphyrins are expected to bind these ligands with the same affinity as the conformationally flexible porphyrins. However, the experimental data reported here show the situation is more complicated than these idealised theoretical considerations predict.
Approach
In order to measure the effects of conformational restriction, we used chemical double mutant cycles (DMC) to quantify the influence of the chlorine substituents on the free energy contribution due intramolecular H-bonds in a series of zinc porphyrin–pyridine ligand complexes (Fig. 3). A DMC allows dissection of the free energy contribution due to a specific functional group interaction to the overall stability of a supramolecular complex by comparing the properties of four closely related complexes.13 In the DMC in Fig. 3, complex A is a conformationally locked porphyrin-ligand complex held together by a coordination bond and a carbonyl-phenol H-bond. Complex B is similar, but has no H-bonds, so the difference between the free energy changes for formation of complexes A and B provides a measure of the free energy contribution of the H-bond in complex A. The difference between the free energy changes for formation of complexes C and D measures the same H-bond in a system that is conformationally flexible, because the porphyrin is missing the chlorine substituents. Thus, the difference between the A to B mutation and the C to D mutation allows us to dissect out the influence of conformational restriction on the free energy contribution due to the intramolecular carbonyl-phenol H-bond (eqn (2)). Strictly, this H-bond is an intermolecular interaction. However, the zinc–pyridine coordination bond is the dominant interaction, which is fully bound, so we refer to intra-complex H-bond as an intramolecular interaction.| | | ΔΔG° = ΔG°A − ΔG°B − ΔG°C + ΔG°D | (2) |
 |
| | Fig. 3 Double mutant cycle (DMC) constructed to measure the influence of conformational restriction on the free energy contribution due to the intramolecular phenol-carbonyl H-bond in complex A. | |
The DMC cancels out all changes in secondary interactions in a pairwise manner (assuming that free energy contributions are additive). For example, the zinc–pyridine coordination bond may be different for the chloroporphyrins and unsubstituted porphyrins, but the DMC features two chloroporphyrin complexes and two unsubstituted porphyrin complexes. The complex B to complex D mutation measures the change in the zinc–pyridine interaction due to the chlorine substituents, and this difference is subtracted from the complex A to complex C mutation in the DMC.
Results and discussion
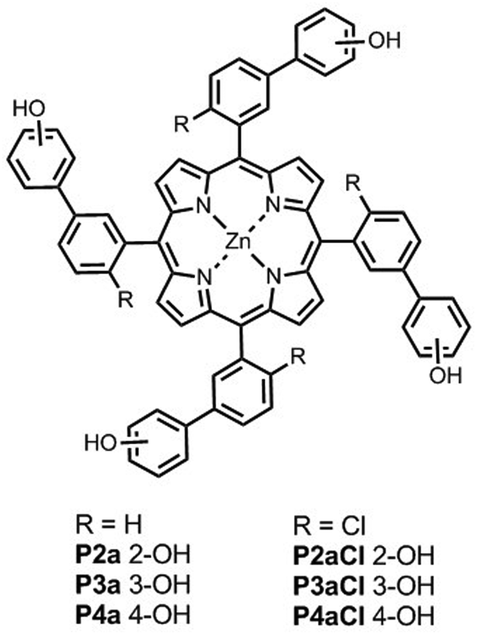
Fig. 4 shows the structures of the conformationally locked chloroporphyrin receptors P2aCl–P4aCl and the corresponding unsubstituted porphyrin receptors P2a–P4a used in this work. Fig. 5 shows the structures of ligands equipped with ester linkers and terminal amide H-bond acceptors (Le), the corresponding ligands with only the ester linkers (Lb), the control ligands with no H-bond acceptors (Lc), and ligand Q, which was used as a competitive ligand in fluorescence displacement experiments to characterise very high affinity complexes.
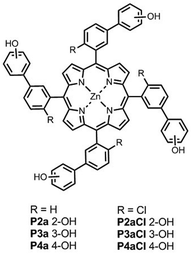 |
| | Fig. 4 Porphyrin receptors P2a–P4a have atropisomers that rapidly interconvert, and P2aCl–P4aCl each have four different atropisomers that can be isolated. | |
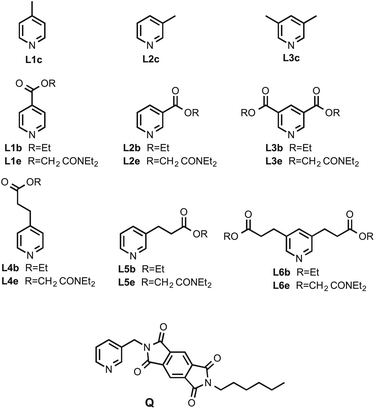 |
| | Fig. 5 Structures of pyridine ligands with ester and amide H-bond acceptors (L1e–L6e), ligands with only ester H-bond acceptors (L1b–L6b), control ligands with no H-bond acceptors (L1c–L3c) and fluorescence displacement assay ligand Q. | |
Synthesis
All ligands in the Lc series are commercially available. Syntheses of porphyrin receptors P2a–P4a, the ligand series Lb and Le and ligand Q were published previously.14 The chloroporphyrins P2aCl–P4aCl were prepared using the route shown in Scheme 1. Suzuki coupling of 2-chloro-5-bromobenzaldehyde with 2-, 3-, or 4-methoxyphenylboronic acid gave the corresponding biphenyl aldehydes, 1, 2, and 3. The Lindsey method was used to convert the aldehydes to the corresponding free base porphyrins 4–6.15 Deprotection of the methoxy groups with boron tribromide gave the tetrahydroxyporphyrins 7–9, and metallation using zinc acetate gave the corresponding zinc porphyrin receptors, P2aCl–P4aCl.
 |
| | Scheme 1 | |
Preparative HPLC and TLC were used to separate the atropisomers of P2aCl–P4aCl (see Experimental section for details). In total, ten of the twelve possible conformationally locked porphyrin receptors were isolated (the ααββ atropisomer of P2aCl and the αααα atropisomer of P4aCl were not formed in sufficient quantities to be isolated). 1H NMR spectra and differences in polarity were used to distinguish between atropisomers. For example, Fig. 6 illustrates the NMR data for the four atropisomers of P3aCl: the signals due to the pyrrole protons appear as one singlet for the αααα and αβαβ atropisomers, a multiplet for the αααβ atropisomer, and two singlets for the ααββ atropisomer. Atropisomers αααα and αβαβ, which have similar NMR spectra, were distinguished based on the Rf on silica TLC plates eluting with a mixture of toluene and diethyl ether: the Rf decreases in the order of polarity (αααα < αααβ < ααββ < αβαβ).10
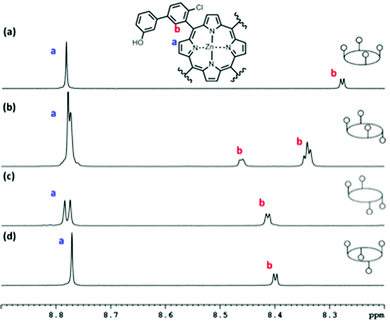 |
| | Fig. 6 Aromatic region of the 1HNMR spectra of the four atropisomers of P3aCl in deuterated methanol at 298 K: (a) αααα, (b) αααβ, (c) ααββ and (d) αβαβ. | |
Binding studies
The association constants for formation of the 120 complexes between the ten chloroporphyrins and the twelve ligands were measured using UV/Vis absorption titrations and fluorescence titrations in both toluene and TCE. For the most strongly bound complexes, a fluorescence displacement titration was required to measure the association constant. In these cases, the pyridine ligands were titrated into a 1:1 mixture of the porphyrin and ligand Q (see Fig. 5), which quenches the porphyrin fluorescence when it is bound but not when it is free. The association constants for the porphyrin·Q complexes were measured by UV/Vis absorption titrations and are recorded in the Experimental section.
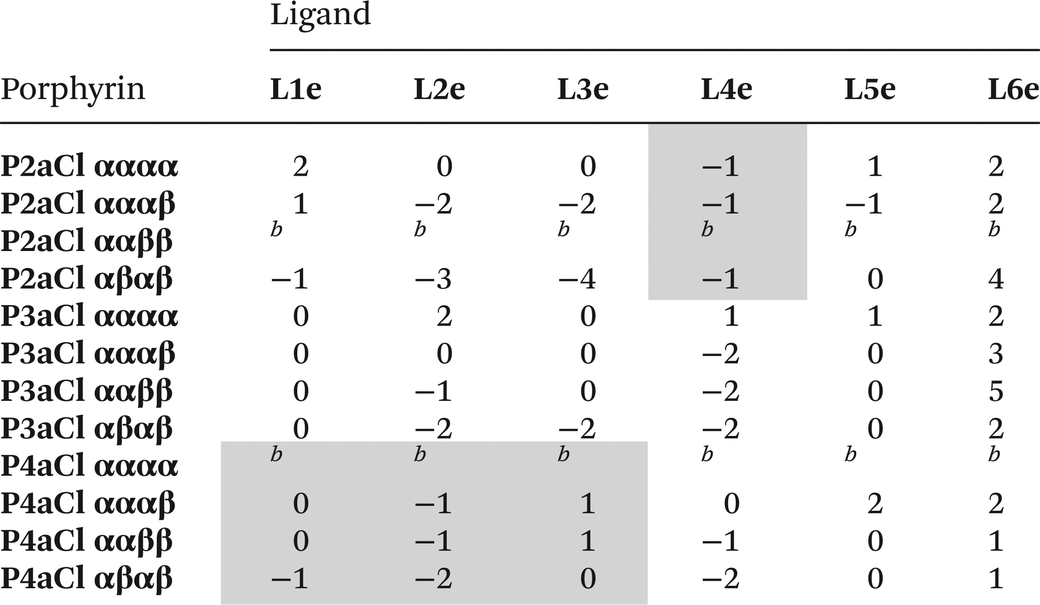
All the titration data fit well to a 1:1 binding isotherm, apart from the complexes of the ααββ and αβαβ atropisomers of P4aCl with L3e in toluene. For these systems, there are additional competing equilibria, which complicate analysis of the data (see ESI†). The association constants and the corresponding data for the unsubstituted porphyrin complexes, which were reported previously, are listed in Tables 1–3.12b The values of the association constants span over five orders of magnitude, 102–107 M−1. The stabilities of the complexes of the chloroporphyrins are generally higher than the corresponding stabilities of the unsubstituted porphyrin complexes. This is true for the complexes that can make H-bonds (Fig. 7(a)) and for the complexes that cannot (Fig. 7(b)). The data in Tables 1–3 suggest that the zinc–nitrogen coordination bond is enhanced by an order of magnitude by the chlorine substituents, but Fig. 7(c) shows that this effect (effectively the Cl⋯L interaction) is removed by the DMC.
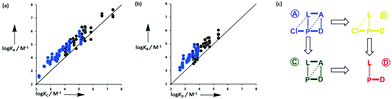 |
| | Fig. 7 Comparison of association constants (logK/M−1) measured in toluene (black) and in TCE (blue) for (a) DMC complexes A and C and (b) DMC complexes B and D. (c) Schematic representation of the chemical double mutant cycle used to dissect the influence of conformational restriction by the chlorine substituent on the magnitude of the intramolecular H-bonding interaction between H-bond acceptor A and H-bond donor D in complexes formed between a zinc porphyrin (P) and a pyridine ligand (L). | |
Table 1 Association constants (K/M−1) for the formation of 1:1 complexes between the P2 porphyrins and pyridine ligands measured at 298 K (with percentage errors in brackets)
| Solvent |
Toluene |
TCE |
| Porphyrin |
P2a
|
P2aCl
αααα
|
P2aCl
αααβ
|
P2aCl
ααββ
|
P2aCl
αβαβ
|
P2a
|
P2aCl
αααα
|
P2aCl
αααβ
|
P2aCl
ααββ
|
P2aCl
αβαβ
|
|
The ααββ atropisomer of P2aCl was not isolated.
Measured by automated fluorescence titration.
Measured by manual fluorescence titration.
|
| Ligand |
|
L1c
|
1.8 × 104 |
1.2 × 105 |
1.0 × 105 |
|
6.3 × 104 |
6.9 × 103 |
4.7 × 104 |
5.8 × 104 |
|
4.4 × 104 |
| (8%) |
(20%) |
(40%) |
(7%) |
(10%) |
(10%) |
(1%) |
(2%) |
|
L2c
|
1.0 × 104 |
7.0 × 104 |
5.4 × 104 |
|
4.2 × 104 |
4.0 × 103 |
3.8 × 104 |
2.7 × 104 |
|
2.8 × 104 |
| (4%) |
(20%) |
(9%) |
(3%) |
(10%) |
(10%) |
(2%) |
(4%) |
|
L3c
|
1.2 × 104 |
1.5 × 105 |
8.8 × 104 |
|
6.9 × 104 |
3.7 × 103 |
6.3 × 104 |
4.9 × 104 |
|
3.7 × 104 |
| (9%) |
(20%) |
(5%) |
(8%) |
(20%) |
(20%) |
(9%) |
(1%) |
|
L1b
|
4.2 × 103 |
1.5 × 104 |
9.6 × 103 |
|
1.3 × 104 |
2.2 × 103 |
1.4 × 104 |
1.2 × 104 |
|
1.5 × 104 |
| (20%) |
(8%) |
(4%) |
(4%) |
(10%) |
(5%) |
(2%) |
(6%) |
|
L2b
|
4.9 × 103 |
1.1 × 104 |
5.3 × 103 |
|
5.3 × 103 |
1.8 × 103 |
1.1 × 104 |
7.1 × 103 |
|
6.7 × 103 |
| (20%) |
(40%) |
(7%) |
(1%) |
(7%) |
(10%) |
(2%) |
(7%) |
|
L3b
|
4.0 × 103 |
6.4 × 103 |
3.6 × 103 |
|
3.3 × 103 |
6.6 × 102 |
3.9 × 103 |
2.9 × 103 |
|
2.2 × 103 |
| (40%) |
(4%) |
(10%) |
(1%) |
(20%) |
(20%) |
(1%) |
(2%) |
|
L4b
|
2.0 × 104 |
8.2 × 104 |
9.9 × 104 |
|
6.8 × 104 |
6.2 × 103 |
3.0 × 104 |
2.8 × 104 |
|
3.2 × 104 |
| (20%) |
(30%) |
(4%) |
(5%) |
(20%) |
(20%) |
(20%) |
(6%) |
|
L5b
|
3.1 × 104 |
7.5 × 104 |
1.3 × 105 |
|
1.1 × 105 |
6.1 × 103 |
3.0 × 104 |
3.8 × 104 |
|
4.2 × 104 |
| (60%) |
(10%) |
(20%) |
(6%) |
(30%) |
(30%) |
(1%) |
(10%) |
|
L6b
|
4.9 × 104 |
1.6 × 105 |
2.7 × 105 |
|
2.9 × 105 |
4.6 × 103 |
6.0 × 104 |
8.1 × 104 |
|
8.8 × 104 |
| (40%) |
(20%) |
(4%) |
(10%) |
(50%) |
(10%) |
(9%) |
(4%) |
|
L1e
|
1.2 × 104 |
1.6 × 104 |
2.9 × 104 |
|
5.0 × 104 |
4.1 × 103 |
1.4 × 104 |
1.7 × 104 |
|
3.5 × 104 |
| (20%) |
(20%) |
(3%) |
(2%) |
(2%) |
(10%) |
(7%) |
(2%) |
|
L2e
|
1.4 × 104 |
1.9 × 104 |
6.0 × 104 |
|
1.1 × 105 |
1.3 × 103 |
7.6 × 103 |
1.1 × 104 |
|
1.9 × 104 |
| (7%) |
(4%) |
(5%) |
(20%) |
(3%) |
(20%) |
(5%) |
(9%) |
|
L3e
|
4.0 × 104 |
1.6 × 105 |
1.7 × 105 |
|
2.8 × 105 |
6.7 × 102 |
4.4 × 103 |
5.7 × 103 |
|
1.0 × 104 |
| (8%) |
(20%) |
(8%) |
(20%) |
(1%) |
(9%) |
(10%) |
(2%) |
|
L4e
|
4.7 × 104 |
9.2 × 104 |
1.3 × 105 |
|
1.9 × 105 |
6.3 × 103 |
4.0 × 104 |
3.7 × 104 |
|
4.4 × 104 |
| (1%) |
(2%) |
(10%) |
(6%) |
(3%) |
(10%) |
(9%) |
(3%) |
|
L5e
|
2.2 × 105 |
3.0 × 105 |
1.3 × 106 |
|
1.2 × 106 |
2.2 × 104 |
8.4 × 104 |
1.9 × 105 |
|
1.5 × 105 |
| (10%) |
(9%) |
(10%) |
(30%) |
(9%) |
(20%) |
(4%) |
(20%) |
|
L6e
|
4.2 × 106 |
1.4 × 107 |
2.1 × 107 |
|
1.5 × 107 |
1.1 × 105 |
7.0 × 105 |
1.0 × 106 |
|
4.2 × 105 |
| (20%)c |
(20%)b |
(30%)b |
(30%)b |
(9%) |
(5%) |
(20%) |
(6%) |
Table 2 Association constants (K/M−1) for the formation of 1:1 complexes between the P3 porphyrins and pyridine ligands measured at 298 K (with percentage errors in brackets)
| Solvent |
Toluene |
TCE |
| Porphyrin |
P3a
|
P3aCl
αααα
|
P3aCl
αααβ
|
P3aCl
ααββ
|
P3aCl
αβαβ
|
P3a
|
P3aCl
αααα
|
P3aCl
αααβ
|
P3aCl
ααββ
|
P3aCl
αβαβ
|
|
Measured by manual displacement fluorescence titration.
Measured by manual fluorescence titration.
|
| Ligand |
|
L1c
|
2.6 × 104 |
1.5 × 105 |
1.2 × 105 |
6.4 × 104 |
1.9 × 105 |
5.1 × 103 |
2.9 × 104 |
1.5 × 104 |
1.2 × 104 |
1.6 × 104 |
| (6%) |
(10%) |
(3%) |
(4%) |
(4%) |
(8%) |
(7%) |
(10%) |
(10%) |
(6%) |
|
L2c
|
1.3 × 104 |
9.1 × 104 |
8.3 × 104 |
4.0 × 104 |
1.2 × 105 |
2.9 × 103 |
2.4 × 104 |
1.3 × 104 |
8.6 × 103 |
1.0 × 104 |
| (2%) |
(8%) |
(5%) |
(3%) |
(10%) |
(10%) |
(1%) |
(4%) |
(2%) |
(2%) |
|
L3c
|
1.7 × 104 |
1.3 × 105 |
1.7 × 105 |
6.7 × 104 |
3.1 × 105 |
2.6 × 103 |
3.6 × 104 |
1.2 × 104 |
7.4 × 103 |
9.5 × 103 |
| (6%) |
(30%) |
(4%) |
(3%) |
(20%) |
(20%) |
(6%) |
(2%) |
(1%) |
(3%) |
|
L1b
|
7.3 × 103 |
1.9 × 104 |
2.0 × 104 |
1.2 × 104 |
2.9 × 104 |
2.0 × 103 |
8.2 × 103 |
7.7 × 103 |
6.2 × 103 |
9.5 × 103 |
| (30%) |
(20%) |
(3%) |
(3%) |
(3%) |
(10%) |
(7%) |
(6%) |
(4%) |
(10%) |
|
L2b
|
7.5 × 103 |
1.6 × 104 |
2.3 × 104 |
2.0 × 104 |
2.3 × 104 |
1.7 × 103 |
6.4 × 103 |
5.7 × 103 |
5.2 × 103 |
5.5 × 103 |
| (20%) |
(6%) |
(3%) |
(3%) |
(5%) |
(20%) |
(6%) |
(8%) |
(5%) |
(3%) |
|
L3b
|
6.9 × 103 |
7.5 × 103 |
8.9 × 103 |
8.7 × 103 |
6.4 × 103 |
6.8 × 102 |
3.3 × 103 |
2.4 × 103 |
2.4 × 103 |
1.7 × 103 |
| (9%) |
(7%) |
(3%) |
(5%) |
(2%) |
(7%) |
(10%) |
(4%) |
(1%) |
(3%) |
|
L4b
|
3.9 × 104 |
1.1 × 105 |
1.3 × 105 |
9.1 × 104 |
2.7 × 105 |
6.2 × 103 |
2.2 × 104 |
1.7 × 104 |
1.4 × 104 |
1.9 × 104 |
| (30%) |
(10%) |
(4%) |
(10%) |
(10%) |
(30%) |
(10%) |
(3%) |
(1%) |
(2%) |
|
L5b
|
5.2 × 104 |
6.1 × 104 |
1.2 × 105 |
1.2 × 105 |
1.7 × 105 |
7.4 × 103 |
1.9 × 104 |
1.6 × 104 |
1.5 × 104 |
1.8 × 104 |
| (20%) |
(30%) |
(9%) |
(3%) |
(20%) |
(30%) |
(9%) |
(10%) |
(17%) |
(20%) |
|
L6b
|
1.2 × 105 |
2.6 × 105 |
4.6 × 105 |
5.4 × 105 |
1.1 × 106 |
8.3 × 103 |
2.9 × 104 |
4.6 × 104 |
4.2 × 104 |
5.6 × 104 |
| (30%) |
(10%) |
(6%) |
(10%) |
(9%) |
(50%) |
(1%) |
(1%) |
(2%) |
(10%) |
|
L1e
|
1.1 × 104 |
1.7 × 104 |
2.7 × 104 |
2.6 × 104 |
5.0 × 104 |
2.3 × 103 |
7.7 × 103 |
8.1 × 103 |
6.5 × 103 |
1.1 × 104 |
| (4%) |
(10%) |
(7%) |
(2%) |
(10%) |
(4%) |
(1%) |
(6%) |
(3%) |
(2%) |
|
L2e
|
6.6 × 104 |
4.8 × 104 |
2.3 × 105 |
2.5 × 105 |
4.3 × 105 |
2.9 × 103 |
5.5 × 103 |
1.2 × 104 |
1.5 × 104 |
2.0 × 104 |
| (3%) |
(1%) |
(8%) |
(8%) |
(5%) |
(3%) |
(20%) |
(7%) |
(5%) |
(9%) |
|
L3e
|
2.9 × 105 |
7.8 × 105 |
8.5 × 105 |
8.4 × 105 |
1.8 × 106 |
3.1 × 103 |
1.5 × 104 |
1.1 × 104 |
1.0 × 104 |
1.7 × 104 |
| (30%) |
(14%) |
(3%) |
(1%) |
(20%) |
(3%) |
(30%) |
(4%) |
(10%) |
(10%) |
|
L4e
|
2.8 × 105 |
1.9 × 105 |
7.9 × 105 |
1.1 × 106 |
2.2 × 106 |
1.4 × 104 |
3.6 × 104 |
7.3 × 104 |
6.8 × 104 |
1.1 × 105 |
| (3%) |
(30%) |
(30%) |
(10%) |
(20%) |
(6%) |
(10%) |
(5%) |
(2%) |
(6%) |
|
L5e
|
7.2 × 105 |
2.4 × 105 |
8.6 × 105 |
1.9 × 106 |
1.8 × 106 |
2.1 × 104 |
3.1 × 104 |
3.9 × 104 |
4.2 × 104 |
6.0 × 104 |
| (30%) |
(10%) |
(4%) |
(10%) |
(4%) |
(5%) |
(8%) |
(10%) |
(7%) |
(10%) |
|
L6e
|
2.5 × 107 |
1.5 × 107 |
4.5 × 107 |
1.2 × 107 |
4.1 × 107 |
1.4 × 105 |
2.2 × 105 |
2.5 × 105 |
1.1 × 105 |
3.6 × 105 |
| (40%)b |
(20%)b |
(10%)b |
(7%)a |
(10%)a |
(10%) |
(10%) |
(5%) |
(10%) |
(7%) |
Table 3 Association constants (K/M−1) for the formation of 1:1 complexes between the P4 porphyrins and pyridine ligands measured at 298 K (with percentage errors in brackets)
| Solvent |
Toluene |
TCE |
| Porphyrin |
P4a
|
P4aCl
αααα
|
P4aCl
αααβ
|
P4aCl
ααββ
|
P4aCl
αβαβ
|
P4a
|
P4aCl
αααα
|
P4aCl
αααβ
|
P4aCl
ααββ
|
P4aCl
αβαβ
|
|
The αααα atropisomer of P4aCl was not isolated.
The titration data did not fit to a 1:1 binding isotherm (see ESI)
|
| Ligand |
|
L1c
|
2.6 × 104 |
1.5 × 105 |
1.2 × 105 |
6.4 × 104 |
1.9 × 105 |
5.1 × 103 |
2.9 × 104 |
1.5 × 104 |
1.2 × 104 |
1.6 × 104 |
| (6%) |
(10%) |
(3%) |
(4%) |
(4%) |
(8%) |
(7%) |
(10%) |
(10%) |
(6%) |
|
L2c
|
1.3 × 104 |
9.1 × 104 |
8.3 × 104 |
4.0 × 104 |
1.2 × 105 |
2.9 × 103 |
2.4 × 104 |
1.3 × 104 |
8.6 × 103 |
1.0 × 104 |
| (2%) |
(8%) |
(5%) |
(3%) |
(10%) |
(10%) |
(1%) |
(4%) |
(2%) |
(2%) |
|
L3c
|
1.7 × 104 |
1.3 × 105 |
1.7 × 105 |
6.7 × 104 |
3.1 × 105 |
2.6 × 103 |
3.6 × 104 |
1.2 × 104 |
7.4 × 103 |
9.5 × 103 |
| (6%) |
(30%) |
(4%) |
(3%) |
(20%) |
(20%) |
(6%) |
(2%) |
(1%) |
(3%) |
|
L1b
|
7.3 × 103 |
1.9 × 104 |
2.0 × 104 |
1.2 × 104 |
2.9 × 104 |
2.0 × 103 |
8.2 × 103 |
7.7 × 103 |
6.2 × 103 |
9.5 × 103 |
| (30%) |
(20%) |
(3%) |
(3%) |
(3%) |
(10%) |
(7%) |
(6%) |
(4%) |
(10%) |
|
L2b
|
7.5 × 103 |
1.6 × 104 |
2.3 × 104 |
2.0 × 104 |
2.3 × 104 |
1.7 × 103 |
6.4 × 103 |
5.7 × 103 |
5.2 × 103 |
5.5 × 103 |
| (20%) |
(6%) |
(3%) |
(3%) |
(5%) |
(20%) |
(6%) |
(8%) |
(5%) |
(3%) |
|
L3b
|
6.9 × 103 |
7.5 × 103 |
8.9 × 103 |
8.7 × 103 |
6.4 × 103 |
6.8 × 102 |
3.3 × 103 |
2.4 × 103 |
2.4 × 103 |
1.7 × 103 |
| (9%) |
(7%) |
(3%) |
(5%) |
(2%) |
(7%) |
(10%) |
(4%) |
(1%) |
(3%) |
|
L4b
|
3.9 × 104 |
1.1 × 105 |
1.3 × 105 |
9.1 × 104 |
2.7 × 105 |
6.2 × 103 |
2.2 × 104 |
1.7 × 104 |
1.4 × 104 |
1.9 × 104 |
| (30%) |
(10%) |
(4%) |
(10%) |
(10%) |
(30%) |
(10%) |
(3%) |
(1%) |
(2%) |
|
L5b
|
5.2 × 104 |
6.1 × 104 |
1.2 × 105 |
1.2 × 105 |
1.7 × 105 |
7.4 × 103 |
1.9 × 104 |
1.6 × 104 |
1.5 × 104 |
1.8 × 104 |
| (20%) |
(30%) |
(9%) |
(3%) |
(20%) |
(30%) |
(9%) |
(10%) |
(17%) |
(20%) |
|
L6b
|
1.2 × 105 |
2.6 × 105 |
4.6 × 105 |
5.4 × 105 |
1.1 × 106 |
8.3 × 103 |
2.9 × 104 |
4.6 × 104 |
4.2 × 104 |
5.6 × 104 |
| (30%) |
(10%) |
(6%) |
(10%) |
(9%) |
(50%) |
(1%) |
(1%) |
(2%) |
(10%) |
|
L1e
|
1.1 × 104 |
1.7 × 104 |
2.7 × 104 |
2.6 × 104 |
5.0 × 104 |
2.3 × 103 |
7.7 × 103 |
8.1 × 103 |
6.5 × 103 |
1.1 × 104 |
| (4%) |
(10%) |
(7%) |
(2%) |
(10%) |
(4%) |
(1%) |
(6%) |
(3%) |
(2%) |
|
L2e
|
6.6 × 104 |
4.8 × 104 |
2.3 × 105 |
2.5 × 105 |
4.3 × 105 |
2.9 × 103 |
5.5 × 103 |
1.2 × 104 |
1.5 × 104 |
2.0 × 104 |
| (3%) |
(1%) |
(8%) |
(8%) |
(5%) |
(3%) |
(20%) |
(7%) |
(5%) |
(9%) |
|
L3e
|
2.9 × 105 |
7.8 × 105 |
8.5 × 105 |
8.4 × 105 |
1.8 × 106 |
3.1 × 103 |
1.5 × 104 |
1.1 × 104 |
1.0 × 104 |
1.7 × 104 |
| (30%) |
(14%) |
(3%) |
(1%) |
(20%) |
(3%) |
(30%) |
(4%) |
(10%) |
(10%) |
|
L4e
|
2.8 × 105 |
1.9 × 105 |
7.9 × 105 |
1.1 × 106 |
2.2 × 106 |
1.4 × 104 |
3.6 × 104 |
7.3 × 104 |
6.8 × 104 |
1.1 × 105 |
| (3%) |
(30%) |
(30%) |
(10%) |
(20%) |
(6%) |
(10%) |
(5%) |
(2%) |
(6%) |
|
L5e
|
7.2 × 105 |
2.4 × 105 |
8.6 × 105 |
1.9 × 106 |
1.8 × 106 |
2.1 × 104 |
3.1 × 104 |
3.9 × 104 |
4.2 × 104 |
6.0 × 104 |
| (30%) |
(10%) |
(4%) |
(10%) |
(4%) |
(5%) |
(8%) |
(10%) |
(7%) |
(10%) |
|
L6e
|
2.5 × 107 |
1.5 × 107 |
4.5 × 107 |
1.2 × 107 |
4.1 × 107 |
1.4 × 105 |
2.2 × 105 |
2.5 × 105 |
1.1 × 105 |
3.6 × 105 |
| (40%)b |
(20%)b |
(10%)b |
(7%)a |
(10%)a |
(10%) |
(10%) |
(5%) |
(10%) |
(7%) |
Fig. 8 illustrates the effect of the chlorine substituents on the stabilities of the P3a complexes. In general, the P3aCl complexes are more stable than the P3a complexes, but there are atropisomer specific patterns. The increase in stability is generally highest for the αβαβ atropisomer, whereas some of the complexes of the αααα atropisomer are significantly less stable than the corresponding P3a complex. These results are quite different from the theoretical predictions based on numbers of bound and free states discussed above. The complex of the P3a αααα atropisomer with L6e is the system illustrated in Fig. 2(b). This complex can make four H-bonds and was predicted to be more stable than the P3a complex by a factor of eight, but it is less stable by a factor of two. As explained above, the differences in Fig. 8 include the effect of the chlorine substituents on the strength of zinc–pyridine interaction, and the DMC is required to dissect out this effect.
 |
| | Fig. 8 Differences between the association constants measured for P3aCl and P3a with the Le ligands in toluene (ΔlogK/M−1). Data for the different atropisomers are coloured red (αααα), blue (αααβ), green (ααββ) and yellow (αβαβ). | |
DMC analysis
The influence of the chlorine substituents on the free energy contribution due to intramolecular H-bonding was determined using the data in Tables 1–3 and eqn (2). The results are summarized in Tables 4–7. Fig. 9 shows the two different DMCs used for the analysis of intramolecular H-bonds with the linker esters (Fig. 9(a)) and for H-bonds with the terminal amides of the Le ligands (Fig. 9(b)). The influence of the chlorine substituents on the free energy contribution due to intramolecular H-bonds ranges from −5 to +6 kJ mol−1. For some of the complexes that do not make any intramolecular H-bonds in the P3a complexes, large positive values of ΔΔG° are observed, and the differences are as high as +7 kJ mol−1. This implies that the DMC does not simply measure the effects of conformational restriction, and there must be adverse steric interactions in the complexes of the chloroporphyrins. The effects are largest for the L2b and L3b ligands, where the ligand side chains are close to the porphyrin meso substituents. No steric effects are apparent for ligands L1b and L4b, where the substituents are in the 4-position on the pyridine ligands and are directed away from the porphyrin meso substituents.
 |
| | Fig. 9 Double mutant cycles (DMC) for measurement of the influence of the conformational restriction on free energy contribution due to (a) intramolecular ester H-bonds (red) and (b) intramolecular amide H-bonds (blue) in complex A. | |
Table 4 Influence of the chlorine substituents on the total free energy contribution due to ester-phenol H-bonds in complex A of the DMC in Fig. 9(a) (ΔΔG°/kJ mol−1) measured at 298 K in toluenea
|
Average error over the data set is ±1 kJ mol−1. Entries for complexes that do not make detectable H-bonds for the corresponding unsubstituted porphyrins are given in italics.
These atropisomers were not isolated.
|

|
Table 5 Influence of the chlorine substituents on the total free energy contribution due to ester-phenol H-bonds in complex A of the DMC in Fig. 9(a) (ΔΔG°/kJ mol−1) measured at 298 K in TCEa
|
Average error over the data set is ±1 kJ mol−1. Entries for complexes that do not make detectable H-bonds for the corresponding unsubstituted porphyrins are given in italics.
These atropisomers were not isolated.
|

|
Table 6 Influence of the chlorine substituents on the total free energy contribution due to amide-phenol H-bonds in complex A of the DMC in Fig. 9(b) (ΔΔG°/kJ mol−1) measured at 298 K in toluenea
|
Average error over the data set is ±1 kJ mol−1. Entries for complexes that do not make detectable H-bonds for the corresponding non-chlorinated porphyrins are given in italics.
These atropisomers were not isolated.
The titration data did not fit to a 1:1 binding isotherm (see ESI).
|

|
Table 7 Influence of the chlorine substituents on the total free energy contribution due to amide-phenol H-bonds in complex A of the DMC in Fig. 9(b) (ΔΔG°/kJ mol−1) measured at 298 K in TCEa
|
Average error over the data set is ±1 kJ mol−1. Entries for complexes that do not make detectable H-bonds for the corresponding non-chlorinated porphyrins are given in italics.
These atropisomers were not isolated.
|

|
Fig. 10 compares the influence of the chlorine substituents on free energy contributions of intramolecular H-bonds in TCE with corresponding values in toluene. The trends in two solvents are in good agreement indicating that there are no significant solvent effects in these systems (other than modulating the intrinsic H-bonding properties of the functional group interactions).
 |
| | Fig. 10 Influence of the chlorine substituents on the free energy contribution due to intramolecular H-bonds measured in TCE compared with corresponding values measured in toluene (ΔΔG°/kJ mol−1). | |
Fig. 11 and 12 compare the influence of the chlorine substituents on the free energy contribution due to intramolecular H-bonds for two-armed ligands with the corresponding one-armed ligands. There is a reasonable correlation for the linker ester-phenol H-bonds (Fig. 11), which indicates that free energy contributions from the ligand side arms are approximately additive, validating the DMC approach. For the terminal amide-phenol H-bonds, the picture is more complicated (Fig. 12). For the short arm ligands (L2e and L3e), the free energy contribution due to H-bonding increases when the chlorine substituents are introduced (ΔΔG° < 0), but for the long arm ligands (L5e and L6e), the free energy contribution due to H-bonding decreases when the chlorine substituents are introduced (ΔΔG° > 0). In addition, the results for the long arm ligands deviate significantly from the ΔΔG°(2) = 2ΔΔG°(1) line. There are a group of outliers in Fig. 12 for which ΔΔG° ≈ 0 for the one arm ligand (L5e), but ΔΔG° is a large positive number for the two arm ligand (L6e). The reason is that L6e is the ligand that can make four H-bonds with the unsubstituted porphyrins, and this is geometrically impossible in the conformationally locked complexes, with the exception of the αααα atropisomer (see Fig. 2(c)).
 |
| | Fig. 11 Influence of the chlorine substituents on the free energy contribution due to intramolecular ester-phenol H-bonds for ligands with two identical side arms, ΔΔG°(2), compared with data for the corresponding one-armed ligands, ΔΔG°(1). Ligands with long arms (L5b and L6b) are shown in grey and ligands with short arms (L2b and L3b) in black. The line corresponds to ΔΔG°(2) = 2ΔΔG°(1). | |
 |
| | Fig. 12 Influence of the chlorine substituents on the free energy contribution due to intramolecular amide-phenol H-bonds for ligands with two identical side arms, ΔΔG°(2), compared with data for the corresponding one-armed ligands, ΔΔG°(1). Ligands with long arms (L5e and L6e) are shown in grey and ligands with short arms (L2e and L3e) in black. The line corresponds to ΔΔG°(2) = 2ΔΔG°(1). | |
Conclusions
Atropisomers of a series of zinc tetraphenyl porphyrins were synthesized, and the binding properties with a series of pyridine ligands were measured. Rotation around the porphyrin-meso phenyl bonds is restricted by installing ortho-chlorine substituents on the phenyl groups. The chlorine substituents allowed chromatographic separation of atropisomers, which did not interconvert at room temperature. The binding properties of these porphyrins were compared with the corresponding porphyrins lacking the chlorine substituents, where the atropisomers are free to interconvert. This comparison provides some insights into the effects of conformational restriction on intramolecular H-bonds formed between H-bond donors on the porphyrin periphery and H-bond acceptor side arms on the ligands.
The chloroporphyrins bind all of the pyridine ligands more strongly than the corresponding unsubstituted porphyrins, due to an electronic effect on the Lewis acidity of the zinc. In order to measure the influence of conformational restriction on intramolecular H-bond formation, double mutant cycles (DMC) were used to dissect out the influence of the chlorine substituents on the free energy contributions due to intramolecular H-bonding in these complexes. However, the results differ from the expectations based on a theoretical analysis of the number of free and bound states. For example, the αααα atropisomer has all of the H-bond donors on the porphyrin receptor preorganised to maximize H-bonding interactions with the ligands, but substantial increases in the magnitude of the free energy contribution due to intramolecular H-bonding were not observed. For some atropisomers, the H-bonding groups are preorganised in a conformation that prevents formation of all of the H-bonds that are formed with the more conformationally flexible unsubstituted porphyrin, and for these systems a decrease in the magnitude of the free energy contribution due to intramolecular H-bonding was observed. The chloroporphyrins appear to be more sensitive to the size of ligands with large adverse steric effects observed for some ligand families. The combination of steric effects and conformational restriction makes it difficult to draw general conclusions.
Experimental section
Synthesis
All reagents were obtained from commercial sources and were used directly without further purification. Thin layer chromatography was carried out using silica gel 60F (Merck) with aluminium sheets as the base. The atropisomers of the porphyrins were separated by preparative HPLC (Varian Prostar system or Shimadzu system) or preparative thin layer chromatography on silica plates (Aldrich chemical company).
Compound 1.
Degassed THF (125 ml), toluene (125 ml) and water (5 ml) were added to a mixture of 2-methoxyphenyl boronic acid (7.08 g, 46.4 mmol), Pd(0)(PPh3)4 (0.80 g, 0.68 mmol) and sodium carbonate (6.07 g, 57.3 mmol) protected by a nitrogen atmosphere. 2-Chloro-5-bromobenzaldehyde14 (10.0 g, 46 mmol) was added to this mixture. The reaction mixture was stirred at 105 °C for 36 h. The solution was allowed to cool, and then the solvent was removed on a rotary evaporator. The residue was dissolved in dichloromethane (250 ml) and then was washed with aqueous sodium hydrogen carbonate (10% w/v, 100 ml), brine (100 ml), and dried with sodium sulfate. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with dichloromethane/hexane. The product was isolated as a colourless solid (9.18 g, 70%). 1H NMR (300 MHz, CDCl3): δH = 10.51 (s, 1H), 8.07 (s, 1H), 7.74 (d, 1H, J = 5), 7.51 (d, 1H, J = 5), 7.37 (t, 1H, J = 5), 7.16 (t, 1H, J = 5), 7.10 (d, 1H, J = 1), 6.93 (dd, 1H, J = 5, J = 1), 3.86 (s, 3H); 13C NMR (75 MHz, CDCl3): δC = 189.7, 160.1, 140.4, 140.1, 136.9, 133.5, 132.5, 130.9, 130.1, 127.6, 119.4, 113.6, 112.7, 55.4; HRMS (ESI): calcd for C14H12ClO2 247.0520, found 247.0532; FT-IR (thin film): νmax/cm−1 3016, 2971, 2874, 2759, 1695, 1601, 1498, 1468, 1299, 1258, 1174, 1122, 1051, 821; M.p. = 56–57 °C.
Compound 2.
Degassed THF (125 ml), toluene (125 ml) and water (5 ml) were added to a mixture of 3-methoxyphenyl boronic acid (7.08 g, 46.4 mmol), 2-chloro-5-bromobenzaldehyde16 (10.0 g, 46 mmol), Pd(0)(PPh3)4 (1.62 g, 1.38 mmol) and sodium carbonate (6.07 g, 57.3 mmol) protected by a nitrogen atmosphere. The reaction mixture was stirred at 105 °C for 24 h. The solution was allowed to cool, and then the solvent was removed on a rotary evaporator. The residue was dissolved in dichloromethane (250 ml and then washed with aqueous sodium hydrogen carbonate (10% w/v, 100 ml), brine (100 ml), dried with sodium sulfate. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with dichloromethane/hexane. The product was isolated as a colorless solid (7.34 g, 56%). 1H NMR (300 MHz, CDCl3): δH = 10.51 (s, 1H), 8.13 (s, 1H), 7.70 (d, 1H, J = 8), 7.46 (d, 1H, J = 8), 7.24–7.38 (m, 2H), 6.97–7.05 (m, 2H), 3.81 (s, 3H); 13C NMR (75 MHz, CDCl3): δC = 189.8, 156.3, 138.0, 136.2, 132.1, 130.5, 130.2, 130.1, 129.6, 128.1, 121.0, 111.3, 55.5; HRMS (ESI): calcd for C14H12ClO2 247.0520, found 247.0509; FT-IR (thin film): νmax/cm−1 3071, 2969, 2865, 2761, 1689, 1611, 1586, 1472, 1268, 1255, 1187, 1166, 1065, 817; M.p. = 60–61 °C.
Compound 3.
Degassed THF (125 ml), toluene (125 ml) and water (5 ml) were added to a mixture of 4-methoxyphenyl boronic acid (7.08 g, 46.4 mmol), Pd(0)(PPh3)4 (0.80 g, 0.68 mmol) and sodium carbonate (6.07 g, 57.3 mmol) protected by a nitrogen atmosphere. 2-Chloro-5-bromobenzaldehyde14 (10.0 g, 46.0 mmol) was then added into the mixture and was stirred at 105 °C for 36 h. The solution was allowed to cool, and then the solvent was removed on a rotary evaporator. The residue was dissolved in dichloromethane (250 ml) and then washed with aqueous sodium hydrogen carbonate (10% w/v, 100 ml), brine (100 ml), dried with sodium sulfate. The solvent was removed on a rotary evaporator and the residue was purified on silica eluting dichloromethane/hexane. The product was isolated as a colorless solid (8.16 g, 62%). 1H NMR (300 MHz, CDCl3): δH = 10.52 (s, 1H), 8.10 (s, 1H), 7.71 (d, 1H, J = 8), 7.49 (d, 1H, J = 8), 7.53 (d, 2H, J = 9), 6.99 (t, 2H, J = 9), 3.86 (s, 3H); 13C NMR (75 MHz, CDCl3): δC = 189.8, 159.9, 140.1, 136.1, 133.0, 132.5, 131.0, 130.9, 128.0, 127.0, 114.5, 114.1, 55.4; HRMS (ESI): calcd for C14H11ClO2Na 269.0340, found 269.0358; FT-IR (thin film): νmax/cm−1 3054, 2924, 2870, 2751, 1693, 1606, 1517, 1465, 1257, 1250, 1179, 1116, 1033, 843; M.p. = 67–68 °C.
Compound 4.
Boron trifluoride diethyl etherate (0.50 ml, 4.00 mmol) was added to a degassed mixture of pyrrole (0.70 ml, 10.0 mmol), compound 1 (2.46 g, 10.0 mmol), ethanol (6.50 ml) and dichloromethane (1000 ml) protected by a nitrogen atmosphere. The reaction mixture was stirred for 90 min, then 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) (2.27 g, 10 mmol) was added, and the reaction mixture was stirred for a further 1 h. Triethylamine (5.60 ml, 40.0 mmol) was added, and the reaction mixture was stirred for 15 min. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with dichloromethane. The product was isolated as a purple solid (0.97 g, 33%). 1H NMR (300 MHz, CDCl3): δH = 8.88 (s, 8H), 8.28–8.52 (m, 4H), 7.96–8.04 (m, 8H), 7.56–7.64 (m, 4H), 7.35–7.41 (m, 4H), 7.13–7.19 (m, 4H), 7.03–7.09 (m, 4H), 3.80–3.96 (m, 12H), −2.77 (s, 4H). M.p. ≥ 300 °C.
Compound 5.
Boron trifluoride diethyl etherate (0.50 ml, 4.00 mmol) was added to a degassed mixture of pyrrole (0.70 ml, 10.0 mmol), compound 2 (2.46 g, 10.0 mmol), ethanol (8.90 ml) and dichloromethane (1300 ml) protected by a nitrogen atmosphere. The reaction mixture was stirred for 90 min, then 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ)(3.32 g, 14.6 mmol) was added, and the reaction mixture was stirred for a further 1 h. Triethylamine (8.20 ml, 58.6 mmol) was added, and the reaction mixture was stirred for another 15 min. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with dichloromethane. The product was isolated as a purple solid (1.09 g, 37%). 1H NMR (300 MHz, CDCl3): δH = 8.81 (s, 8H), 8.38–8.50 (m, 4H), 7.89–8.00 (m, 8H), 7.26–7.37 (m, 12H), 6.92 (s, 4H), 3.80–3.87 (m, 12H), −2.57 (s, 2H). M.p. ≥ 300 °C.
Compound 6.
Boron trifluoride diethyl etherate (0.50 ml, 4.00 mmol) was added to a degassed mixture of pyrrole (0.70 ml, 10.0 mmol), compound 3 (2.46 g, 10.0 mmol), ethanol (8.90 ml) and dichloromethane (1320 ml) protected by a nitrogen atmosphere. The reaction mixture was stirred for 90 min, then 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) (3.32 g, 14.6 mmol) was added, and the reaction mixture was stirred for a further 1 h. Triethylamine (7.20 ml, 51.6 mmol) was added, and the reaction mixture was stirred for another 15 min. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with dichloromethane. The product was isolated as a purple solid (0.97 g, 33%). 1H NMR (300 MHz, CDCl3): δH = 8.81 (s, 8H), 8.34–8.58 (m, 4H), 7.87–7.96 (m, 8H), 7.68–7.73 (m, 8H), 6.96–7.00 (m, 8H), 3.82–3.89 (m, 12H), −2.59 (s, 2H). M.p. ≥ 300 °C.
Compound 7.
Boron tribromide (0.96 ml, 10.0 mmol) in dichloromethane (52.0 ml) was added dropwise to compound 4 (0.59 g, 0.50 mmol) in dichloromethane (52.0 ml) protected by a nitrogen atmosphere at 0 °C over a period of 30 min. The solution was allowed to warm to room temperature and stirred for 24 h. The reaction mixture was then cooled to 0 °C. Methanol (13 ml) and triethylamine (13 ml) were added dropwise, and the reaction mixture was allowed to warm to room temperature. The solvent was removed on a rotary evaporator, and the residue was dissolved in ethyl acetate (260 ml), washed with water (4 × 200 ml), brine (200 ml), dried with sodium sulfate. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with a mixture of dichloromethane and ethyl acetate. The product was isolated as a purple solid (0.431 g, 77%). 1H NMR (300 MHz, CDCl3): δH = 9.58–9.67 (m, 4H), 8.90–8.99 (m, 8H), 8.44–8.55 (m, 4H), 8.03–8.21 (m, 8H), 7.22–7.59 (m, 12H), 6.74–6.90 (m, 4H), −2.81 (s, 2H).
Compound 8.
Boron tribromide (0.96 ml, 10.0 mmol) in dichloromethane (52.0 ml) was added dropwise to compound 5 (0.59 g, 0.50 mmol) in dichloromethane (52.0 ml) protected by a nitrogen atmosphere at 0 °C over a period of 30 min. The solution was allowed to warm to room temperature and stirred for 24 h. The reaction mixture was then cooled to 0 °C. Methanol (13 ml) and triethylamine (13 ml) were added dropwise, and the reaction mixture was allowed to warm to room temperature. The solvent was removed on a rotary evaporator, and the residue was dissolved in ethyl acetate (260 ml), washed with water (4 × 200 ml), brine (200 ml), dried with sodium sulfate. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with a mixture of dichloromethane and ethyl acetate. The product was isolated as a purple solid (0.392 g, 70%). 1H NMR (300 MHz, CDCl3): δH = 9.71–9.78 (m, 1H), 8.85 (s, 8H), 8.36–8.55 (m, 4H), 8.06–8.09 (m, 4H), 7.93–8.00 (m, 4H), 7.49–7.57 (m, 4H), 7.13–7.19 (m, 4H), 6.87–6.97 (m, 8H), −2.70 (s, 2H).
Compound 9.
Boron tribromide (0.96 ml, 10.0 mmol) in dichloromethane (52.0 ml) was added dropwise to compound 6 (0.59 g, 0.50 mmol) in dichloromethane (52.0 ml) protected by a nitrogen atmosphere at 0 °C over a period of 30 min. The solution was allowed to warm to room temperature and stirred for 24 h. The reaction mixture was then cooled to 0 °C. Methanol (13 ml) and triethylamine (13 ml) were added dropwise, and the reaction mixture was allowed to warm to room temperature. The solvent was removed on a rotary evaporator, and the residue was dissolved in ethyl acetate (260 ml), washed with water (4 × 200 ml), brine (200 ml), dried with sodium sulfate. The solvent was removed on a rotary evaporator, and the residue was purified on silica eluting with a mixture of dichloromethane and ethyl acetate. The product was isolated as a purple solid (0.364 g, 65%). 1H NMR (300 MHz, CDCl3): δH = 9.63–9.72 (m, 4H), 8.83 (s, 8H), 8.46–8.61 (m, 4H), 8.10–8.13 (m, 4H), 7.94–7.99 (m, 8H), 7.71–7.77 (m, 4H), 6.83–6.86 (m, 8H), −2.77 (s, 2H).
P2aCl Zinc acetate (0.73 g, 4.00 mmol) was added to compound 7 (0.22 g, 0.20 mmol) in dichloromethane (36.0 ml) and methanol (4.00 ml). The reaction mixture was stirred for 6 h protected by a calcium chloride drying tube. The solvent was removed on a rotary evaporator, and the residue was purified on basic alumina eluting with dichloromethane:methanol (95:5). The product was isolated as a purple solid (0.177 g, 75%). 1H NMR (300 MHz, DMSO-d6): δH = 9.69–9.76 (t, 4H, J = 10), 8.79 (s, 8H), 8.30–8.50 (m, 4H), 8.05 (dd, 4H, J = 2, J = 8), 7.96 (t, 4H, J = 8), 7.50–7.59 (m, 4H), 7.14–7.22 (m, 4H), 6.87–6.99 (m, 8H); UV/Vis (CHCl3) λmax/nm (ε/mol−1 cm2) 435 (ε = 1.2 × 105), 557 (ε = 1.1 × 104), 593 (ε = 2.1 × 103); M.p. ≥ 260 (decomp.)°C. P2aCl was further separated into three atropisomers by preparative TLC silica plate using toluene:ether (8:2). The silica plate was developed in the toluene and ether solvent mixture, dried out, and then developed again. The development and drying out processes were repeated multiple times until a good separation was achieved. P2aClα41H NMR (400 MHz, acetone-d6):δH = 8.88 (s, 8H), 8.64 (s, 4H), 8.41 (d, 4H, J = 2), 8.07 (dd, 4H, J = 2, J = 8), 7.94 (d, 4H, J = 8), 7.58 (dd, 4H, J = 2, J = 8), 7.18 (td, 4H, J = 8, J = 2), 7.00 (dd, 4H, J = 1, J = 8), 6.94 (td, 4H, J = 8, J = 1); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 4.1 × 105); MALDI-TOF reflectron MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.0. P2aClα3β1H NMR (400 MHz, acetone-d6): δH = 8.95–8.83 (m, 8H), 8.72–8.63 (m, 4H), 8.62–8.57 (m, 1H), 8.54–8.48 (m, 2H), 8.41 (d, 1H, J = 2), 8.12–8.04 (m, 4H), 7.98–7.85 (m, 4H), 7.66–7.54 (m, 4H), 7.25–7.14 (m, 4H), 7.06–6.91 (m, 8H); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 4.2 × 105); MALDI-TOF reflectron MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.0. P2aClαβαβ1H NMR (400 MHz, acetone-d6): δH = 8.88 (s, 8H), 8.68 (s, 4H), 8.58 (d, 4H, J = 2), 8.08 (dd, 4H, J = 2, J = 8), 7.89 (d, 4H, J = 8), 7.63 (dd, 4H, J = 2, J = 8), 7.21 (td, 4H, J = 8, J = 2), 7.03 (dd, 4H, J = 1, J = 8), 6.97 (td, 4H, J = 8, J = 1); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 5.3 × 105); MALDI-TOF reflectron MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.1.
P3aCl Zinc acetate (1.10 g, 6.00 mmol) was added to compound 8 (0.34 g, 0.30 mmol) in dichloromethane (54.0 ml) and methanol (6.00 ml). The reaction mixture was stirred for 12 h protected by a calcium chloride drying tube. The solvent was removed on a rotary evaporator, and the residue was purified on basic alumina eluting with dichloromethane:methanol (95:5). The product was isolated as a purple solid (0.209 g, 59%). 1H NMR (300 MHz, DMSO-d6): δH = 8.73 (s, 8H), 8.25–8.44 (m, 4H), 7.95–8.10 (m, 8H), 7.20–7.26 (m, 12H), 6.72–6.77 (m, 4H); UV/Vis (CHCl3) λmax/nm (ε/mol−1 cm2) 431 (ε = 7.0 × 105), 557 (ε = 3.1 × 104), 593 (ε = 1.8 × 103); M.p. ≥ 260 (decomp.)°C. P3aCl was first separated by preparative TLC silica plate unsuccessfully and then further separated into four atropisomers by preparative HPLC on a Varian Prostar system. Column: Xbridge Prep C18 5um OBD 19 × 250 mm column; solvent: acetonitrile:water with 0.1% TFA (8:2) isocratic for 30 min. Detection wavelength: 420 nm. P3aClα41H NMR (400 MHz, Methanol-d4): δH = 8.78 (s, 8H), 8.28 (d, 4H, J = 2), 8.01 (dd, 4H, J = 2, J = 8), 7.93 (d, 4H, J = 8), 7.22 (dd, 8H, J = 1, J = 4), 7.19–7.11 (m, 4H), 6.78–6.70 (m, 4H); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 3.4 × 105); MALDI-TOF reflectron MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.3. P3aClα3β1H NMR (400 MHz, Methanol-d4): δH = 8.80–8.75 (m, 8H), 8.48–8.44 (m, 1H), 8.36–8.32, (m, 3H), 8.03–7.95 (m, 4H), 7.93–7.81 (m, 4H), 7.29–7.16 (m, 12H), 6.80–6.71 (m, 4H); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 5.0 × 105); MALDI-TOF reflectron MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.4. P3aClα2β21H NMR (400 MHz, Methanol-d4): δH = 8.78 (s, 4H), 8.77 (s, 4H), 8.41 (d, 4H, J = 2), 8.01 (dd, 4H, J = 2, J = 8), 7.90 (d, 4H, J = 8), 7.33–7.15 (m, 12H), 6.81–6.71 (m, 4H); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 4.7 × 105); MALDI-TOF reflectron MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.5. P3aClαβαβ1H NMR (400 MHz, Methanol-d4): δH = 8.77 (s, 8H), 8.40 (d, 4H, J = 2), 8.00 (dd, 4H, J = 2, J = 8), 7.88 (d, 4H, J = 8), 7.32–7.16 (m, 12H), 6.81–6.71 (m, 4H); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 3.0 × 105); MALDI-MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.7.
P4aCl Zinc acetate (0.73 g, 4.00 mmol) was added to compound 9 (0.22 g, 0.20 mmol) in dichloromethane (25.0 ml) and methanol (1.25 ml). The reaction mixture was stirred for 24 h protected by a calcium chloride drying tube. The solvent was removed on a rotary evaporator, and the residue was purified on basic alumina eluting with dichloromethane:methanol (95:5). The product was isolated as a purple solid (0.203 g, 86%). 1H NMR (300 MHz, DMSO-d6): δH = 8.89 (s, 8H), 8.27–8.45 (m, 4H), 7.85–7.91 (m, 8H), 7.66 (d, 8H, J = 8), 6.88 (d, 8H, J = 8), 4.80 (s, 4H); UV/Vis (CHCl3) λmax/nm (ε/mol−1 cm2) 431 (ε = 9.5 × 105), 557 (ε = 1.3 × 104), 593 (ε = 2.1 × 103); M.p. ≥ 260 (decomp.) °C. P4aCl was first separated by preparative HPLC Column: Xbridge Prep C18 5um OBD 19 × 250 mm column; solvent: acetonitrile:water with 0.1%TFA (72:28) isocratic for 25 min. Detection wavelength: 420 nm. Three atropisomers were isolated, however zinc was partially removed from the center of porphyrin during the work up due to the protonation of porphyrin by TFA when removing solvent. All three atropisomers were further purified by preparative TLC silica plate runing with DCM:EtOAc (8:2) to isolate the atropisomers from the free porphyrin. P4aClα3β1H NMR (400 MHz, Acetone-d6): δH = 8.91–8.77 (m, 8H), 8.52 (d, 1H, J = 2), 8.49 (brs, 4H), 8.46 (d, 2H, J = 2), 8.34 (d, 1H, J = 2), 8.06–7.97 (m, 4H), 7.90 (d, 1H, J = 8), 7.87 (d, 2H, J = 8), 7.82 (d, 1H, J = 8), 7.77–7.64(m, 8H), 6.93 (d, 2H, J = 9), 6.91 (d, 4H, J = 9), 6.88 (d, 2H, J = 9); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 4.2 × 105); MALDI-MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.2. P4aClα2β21H NMR (400 MHz, Acetone-d6): δH = 8.83(s, 4H),8.83(s, 4H), 8.52 (s, 4H), 8.43 (d, 4H, J = 2), 8.06 (dd, 4H, J = 2, J = 8), 7.90 (d, 4H, J = 8), 7.73 (d, 8H, J = 9), 6.92 (d, 8H, J = 9); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 3.2 × 105); MALDI-MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.2. P4aClαβαβ1H NMR (400 MHz, Acetone-d6): δH = 8.83 (s, 8H), 8.53 (d, 4H, J = 2), 8.50 (s, 4H), 8.07 (dd, 4H, J = 2, J = 8), 7.89 (d, 4H, J = 8), 7.78 (d, 8H, J = 9), 6.95 (d, 8H, J = 9); UV/Vis (TCE) λmax/nm (ε/mol−1 cm2) 422 (ε = 2.8 × 105); MALDI-MS: calcd for C68H40Cl4N4O4Zn: 1180.1095, found 1180.2.
Automated UV/Vis absorption titrations
UV/Vis titrations were carried out using a BMG FLUOstar Omega plate reader equipped with a UV/Vis detector and equilibrated at 298 K. A 5 ml solution of porphyrin was prepared at known concentration (1–5 μM) in spectroscopic grade solvent. A 10 ml solution of ligand was prepared at known concentration (8–40000 μM) using spectroscopic grade solvent. 150 μl of the porphyrin solution was added to a well of a Hellma quartz microplate, and the absorbance at five wavelengths was recorded. Aliquots of the ligand solution (3, 6 or 10 μl) were successively added to the well, and the absorbance was recorded after each addition. Changes in absorbance were fit to a 1:1 binding isotherm in Microsoft Excel to obtain the association constant. Each titration was repeated at least three times, and the experimental error is quoted as twice the standard deviation at a precision of one significant figure.
Automated fluorescence titrations
Fluorescence titrations were carried out at 298 K using the BMG FLUOstar Omega plate reader equilibrated. A 10 ml solution of porphyrin was prepared at known concentration (0.1–1 μM) in spectroscopic grade solvent. A 10 ml solution of ligand was prepared at known concentration (5–63 μM) using spectroscopic grade solvent. 150 μl of the porphyrin solution was added to each of 12 wells of a Hellma quartz microplate. Different volumes of ligand solution (0–150 μl) were added to each well and solvent was added to give a total volume of 300 μl. The excitation wavelength was set at 420 or 430 nm, and the fluorescence emission was measured at four wavelengths (590, 600, 620 and 650 nm) for each well. Changes in fluorescence emission were fit to a 1:1 binding isotherm in Microsoft Excel to obtain the association constant. Each titration was repeated at least three times, and the experimental error is quoted as twice the standard deviation at a precision of one significant figure.
Manual fluorescence titrations
Fluorescence titrations were carried out at 298 K using a Hitachi F-4500 Fluorescence Spectrophotometer. A 10 ml solution of porphyrin at known concentration (0.04–0.05 μM) was prepared in spectroscopic grade solvent. Then 2 ml of this host solution was loaded into a 1 cm path length fluorescence cuvette, and the fluorescence emission spectrum was recorded between500 and 750 nm exciting at 427 nm. A 2 ml stock solution of ligand (0.1–1 μM) was prepared by dissolving the ligand in the host stock solution, so that the concentration of host remained constant throughout the titration. Aliquots of ligand stock solution were added successively to the cuvette, and the emission spectrum was recorded after each addition. Changes in fluorescence emission were fit to a 1:1 binding isotherm in Microsoft Excel to obtain the association constant. Each titration was repeated at least three times, and the experimental error is quoted as twice the standard deviation at a precision of one significant figure.
Fluorescence displacement titrations
Fluorescence displacement titrations were carried out at 298 K using a Hitachi F-4500 fluorescence spectrophotometer. A 20 ml solution of ligand Q at known concentration (about 10 mM) was prepared using spectroscopic grade solvent. A 10 ml solution of porphyrin was prepared at known concentration (about 0.5 μM) by dissolving the porphyrin in the Q stock solution. A 2 ml stock solution of ligand L was prepared at known concentration (about 1 μM) by dissolving L in the porphyrin-Q stock solution, so that the concentration of the porphyrin and Q remained constant throughout the titration. 2 ml of the porphyrin-Q stock solution was loaded into a 1 cm path length fluorescence cuvette, and the fluorescence emission spectrum was recorded between 500 and 750 nm exciting at 427 nm. Aliquots of the L stock solution were added successively to the cuvette, and the emission spectrum was recorded after each addition. Microsoft Excel was used to fit the fluorescence emission intensity at fixed wavelengths to a 1:1 binding isotherm with a linear correction to allow for non-specific effects. This gave the apparent association constant, Kapp, which was used to determine the association constant for formation of the porphyrin-ligand complex using eqn (3).12b| |  | (3) |
Each titration was repeated at least three times, and the experimental error is quoted as twice the standard deviation at a precision of one significant figure (Table 8).
Table 8 Association constants (KQ/M−1) for the formation of zinc porphyrin·Q complexes measured by UV/Vis absorption titrations at 298 K in toluene
| Porphyrin |
K
Q
|
|
P3aCl
αααα
|
3.0 ± 0.3 × 104 |
|
P3aCl
αααβ
|
5.0 ± 0.2 × 104 |
|
P3aCl
ααββ
|
5.5 ± 0.2 × 104 |
|
P3aCl
αβαβ
|
5.2 ± 0.3 × 104 |
Acknowledgements
We thank the EPSRC, the China Scholarship Council, and the University of Sheffield for funding.
Notes and references
-
(a) J. C. Sacchettini, L. G. Baum and C. F. Brewer, Biochemistry, 2001, 40, 3009 CrossRef CAS PubMed;
(b) N. Jayaraman, Chem. Soc. Rev., 2009, 38, 3463 RSC;
(c) V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492 RSC;
(d) J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922 CrossRef CAS PubMed;
(e) D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817 RSC;
(f) M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef;
(g) D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1155 CrossRef CAS PubMed;
(h) P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669 CrossRef CAS PubMed.
-
(a) R. Grunberg, M. Nilges and J. Leckner, Structure, 2006, 14, 683 CrossRef PubMed;
(b) S. A. Bobrovnik, J. Mol. Recognit., 2007, 20, 253 CrossRef CAS PubMed;
(c) H. Adams, E. Chekmeneva, C. A. Hunter, M. C. Misuraca, C. Navarro and S. M. Turega, J. Am. Chem. Soc., 2013, 135, 1853 CrossRef CAS PubMed;
(d) M. C. Misuraca, T. Grecu, Z. Freixa, V. Garavini, C. A. Hunter, P. van Leeuwen, M. D. Segarra-Maset and S. M. Turega, J. Org. Chem., 2011, 76, 2723 CrossRef CAS PubMed;
(e) A. L. Lee, S. A. Kinnear and A. J. Wand, Nat. Struct. Biol., 2000, 7, 72 CrossRef CAS PubMed;
(f) W. Jiang, K. Nowosinski, N. L. Loew, E. V. Dzyuba, F. Klautzsch, A. Schaefer, J. Huuskonen, K. Rissanen and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 1860 CrossRef CAS PubMed;
(g) H. J. Hogben, J. K. Sprafke, M. Hoffmann, M. Pawlicki and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 20962 CrossRef CAS PubMed.
-
(a) V. M. Krishnamurthy, V. Semetey, P. J. Bracher, N. Shen and G. M. Whitesides, J. Am. Chem. Soc., 2007, 129, 1312 CrossRef CAS PubMed;
(b) J. P. Carver, Pure Appl. Chem., 1993, 65, 763 CrossRef CAS.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
- U. Gerhard, M. S. Searle and D. H. Williams, Bioorg. Med. Chem. Lett., 1993, 3, 803 CrossRef CAS.
- M. S. Searle, D. H. Williams and U. Gerhard, J. Am. Chem. Soc., 1992, 114, 10697 CrossRef CAS.
- P. R. Andrews, D. J. Craik and J. L. J. Martin, Med. Chem., 1984, 27, 1648 CrossRef CAS.
-
(a) F. Eblinger and H.-J. Schneider, Angew. Chem., Int. Ed., 1998, 37, 826 CrossRef CAS;
(b) M. A. Hossain and H.-J. Schneider, Chem. – Eur. J., 1999, 5, 1284 CrossRef CAS.
-
(a) M. I. Page and W. P. Jencks, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1678 CrossRef CAS;
(b) C. Galli and L. Mandolini, Eur. J. Org. Chem., 2000, 3117 CrossRef CAS.
- A. S. M. Ressurreicao, M. Pineiro, L. G. Arnaut and A. Gonsalves, J. Porphyrins phthalocyanines, 2007, 11, 50 CrossRef CAS.
-
(a) J. W. Dirks, G. Underwood, J. C. Matheson and D. Gust, J. Org. Chem., 1979, 44, 2551 CrossRef CAS;
(b) C. J. Medforth, R. E. Haddad, C. M. Muzzi, N. R. Dooley, L. Jaquinod, D. C. Shyr, D. J. Nurco, M. M. Olmstead, K. M. Smith, J. G. Ma and J. A. Shelnutt, Inorg. Chem., 2003, 42, 2227 CrossRef CAS PubMed.
-
(a) C. A. Hunter, M. C. Misuraca and S. M. Turega, J. Am. Chem. Soc., 2011, 133, 20416 CrossRef CAS PubMed;
(b) C. A. Hunter, M. C. Misuraca and S. M. Turega, Chem. Sci., 2012, 3, 589 RSC.
-
(a) S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172 RSC;
(b) A. Camara-Campos, D. Musumeci, C. A. Hunter and S. Turega, J. Am. Chem. Soc., 2009, 131, 18518 CrossRef CAS PubMed;
(c) E. Chekmeneva, C. A. Hunter, M. J. Packer and S. M. Turega, J. Am. Chem. Soc., 2008, 130, 17718 CrossRef CAS PubMed;
(d) H. Adams, F. J. Carver, C. A. Hunter, J. C. Morales and E. M. Seward, Angew. Chem., Int. Ed. Engl., 1996, 35, 1542 CrossRef CAS PubMed;
(e) P. J. Carter, G. Winter, A. J. Wilkinson and A. R. Fersht, Cell, 1984, 38, 835 CrossRef CAS.
-
(a) C. A. Hunter, M. C. Misuraca and S. M. Turega, J. Am. Chem. Soc., 2011, 133, 582 CrossRef CAS PubMed;
(b) H. Sun, C. A. Hunter, C. Navarro and S. Turega, J. Am. Chem. Soc., 2013, 135, 13129 CrossRef CAS PubMed.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS.
-
H. Kakinuma, Y. Kobashi, Y. Hashimoto and H. Takahash, Int. Appl. WO Patent, 2007136116 A2, 20070518, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00805k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a