Hydrogen sulfide mediated cascade reaction forming an iminocoumarin: applications in fluorescent probe development and live-cell imaging†
Received
20th April 2015
, Accepted 26th May 2015
First published on 27th May 2015
Abstract
The study on a fluorescent probe that undergoes a H2S mediated cascade reaction to form an iminocoumarin fluorophore is reported. The probe features better water solubility and fast sensing time (t1/2 = 6.1 min and response time = 24 min) as key advances compared to the reported probe that works on a similar mechanism. The sensing mechanism of the probe was demonstrated by mass spectrometric, HPLC titration and FT-IR titration methods. H2S sensing by the probe was characterized by a 31-fold fluorescence enhancement and alimit of detection of 169 nM. Application of the probe was demonstrated by imaging of H2S in live cells.
Introduction
Hydrogen sulfide (H2S) is well known for its pungent smell and noxious nature. The gaseous species is mainly produced from geological and microbial activities. Overexposure to the corrosive and flammable gas can cause eye irritation, inflammation in the respiratory tract,1 loss of consciousness, and sudden cardiac death. H2S is also known to play crucial roles in various diseases such as Alzheimer's disease,2 Down's syndrome,3 diabetes,4 and liver cirrhosis. In mammalian cells, H2S is produced endogenously from cysteine by enzymes, e.g. cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE)5 and 3-mercaptopyruvate sulfurtransferase (MST).6 Recent studies revealed the importance of H2S as a redox signalling molecule7 and made it the third-most important gasotransmitter8 after carbon monoxide (CO) and nitric oxide (NO).9 It can act as a scavenger for reactive oxygen species (ROS)10 by producing sulfate, thiosulfate, sulphite, polysulfane, etc.10 H2S also helps in healing of wounds11 and hippocampal potentiation.12 A state of hibernation can also be achieved by long exposure to the species.13 In recent times, H2S releasing prodrugs are used for treating inflammation14 and cardiovascular diseases.15 These convoluted physiological roles of H2S and its therapeutic applications motivate researchers to monitor its trafficking and production in living cells.
Traditionally, gas-chromatography,16 colorimetric assay,17 and polarographic sensors18 are used for the detection of H2S. However, these methods are less satisfactory for endogenous detection of H2S due to complex sample preparation, annihilation and the volatile nature of the species. Fluorometric methods involving molecular probes are more established for detecting various analytes with excellent selectivity and sensitivity. Generally, a variety of H2S mediated chemical reactions are used for the design of chemodosimeters. For example, precipitation of copper sulphide,19–21 thiolysis of 2,4-dinitrophenyl ether,22 azide to amine reduction,23–31 nitro to amine reduction,32,33 and trapping of H2S by nucleophilic addition34–38 are the established strategies for the development of H2S selective fluorescent probes. Due to the high significance of the azide to amine reduction in H2S sensing, the reaction was further applied for the development of cascade reaction based probes 1–4 (Fig. 1A).39–42
 |
| | Fig. 1 Structures of the reported cascade probes 1–4 (A). Structure and H2S sensing mechanism of the new cascade probe 5 (B). | |
Herein, we report the synthesis of molecule 5 and its development as a fluorescent probe for sensing H2S (Fig. 1B). The design of the present probe significantly contrasts with that of compounds 1–4. In previously reported probes, the fluorophore backbones were pre-existing, while probe 5 was designed to form the fluorophore backbone only upon the sensing process. The azido group was linked to initiate the sensing process via its reduction to the amino group43,44 to form the unstable intermediate 6. Subsequently, the removal of the immolative linker of 6 was expected to facilitate the Pinner cyclization45 to form the iminocoumarin fluorophore 7 and the bi-product 8 (Fig. 1B). Very recently a probe based on a similar concept was reported by Sun and co-workers.46 However their probe suffers from key limitations such as slow response to H2S, use of a high percentage of organic solvent (50% DMF), and a long incubation time (60 min) with live cells (Table S4†). These limitations can be accounted for based on the estimated (using Chemical Properties determination tool of ChemBioDraw program) water solubility (cLog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S = −7.577) and permeability (cLogP = 6.8517) values of the probe. Therefore probe 5 was introduced as a more optimized drug-like molecule by substituting the hydrophobic and bulky benzothiazole moiety with a more polar and smaller cyano group. Molecule 5 was envisaged as a better probe due to its superior cLogS and cLogP values of −5.799 and 4.8887, respectively. In this work, we will demonstrate the formation of the iminocoumarin 7 during the reaction of 5 with Na2S by using HPLC, FT-IR spectroscopy and mass spectral analysis. Subsequently, the proposed drug-like properties will be addressed and application of molecule 5 as a H2S selective fluorescent probe will be established.
S = −7.577) and permeability (cLogP = 6.8517) values of the probe. Therefore probe 5 was introduced as a more optimized drug-like molecule by substituting the hydrophobic and bulky benzothiazole moiety with a more polar and smaller cyano group. Molecule 5 was envisaged as a better probe due to its superior cLogS and cLogP values of −5.799 and 4.8887, respectively. In this work, we will demonstrate the formation of the iminocoumarin 7 during the reaction of 5 with Na2S by using HPLC, FT-IR spectroscopy and mass spectral analysis. Subsequently, the proposed drug-like properties will be addressed and application of molecule 5 as a H2S selective fluorescent probe will be established.
Results and discussion
Synthesis
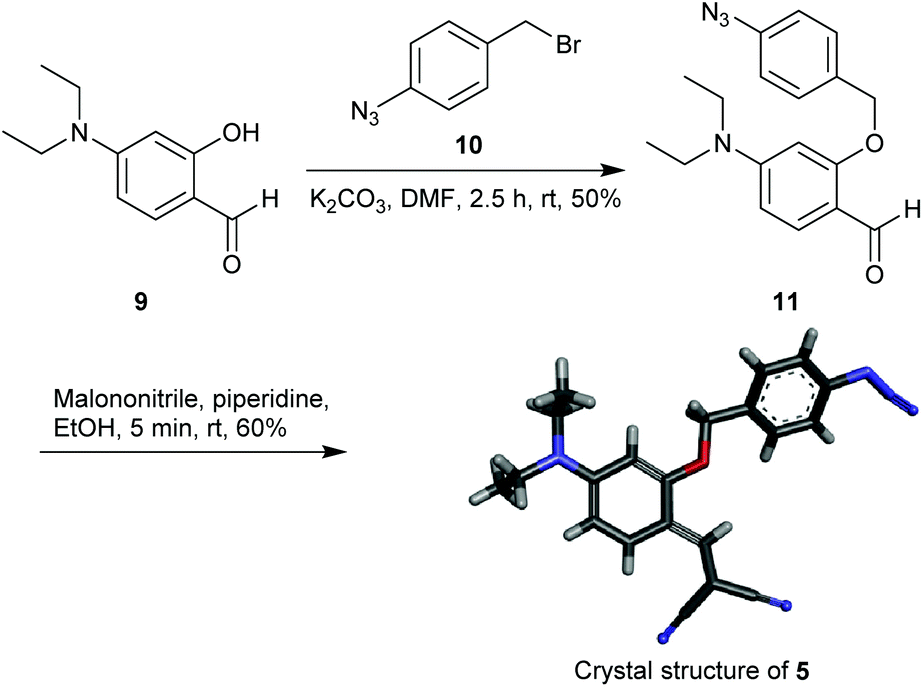
The starting material 4-(diethylamino)salicylaldehyde 9 was purchased from a commercial source and 1-azido-4-(bromomethyl)benzene 10 was synthesized as described in the literature.47 The reaction of 9 with 1 equiv. of 10 in the presence of K2CO3 afforded 11 in 50% yield (Scheme 1). The aldehyde 11 upon treatment with malononitrile provided the Knoevenagel condensation product 5 as a yellow solid (60% yield). The reporter molecule 7 was also synthesized following the methodology reported in the literature (Scheme S1†).45 All new compounds were characterized by NMR, IR-spectroscopy and mass-spectrometry. Compound 5 was additionally characterized by crystal XRD analysis (Scheme 1).
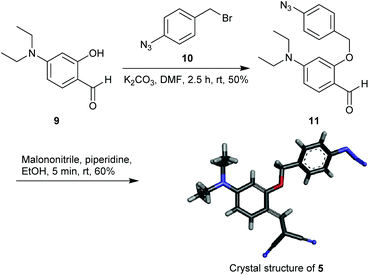 |
| | Scheme 1 Synthesis and crystal structure of probe 5. | |
Photophysical properties
Experimental validation of fluorescence characteristics.
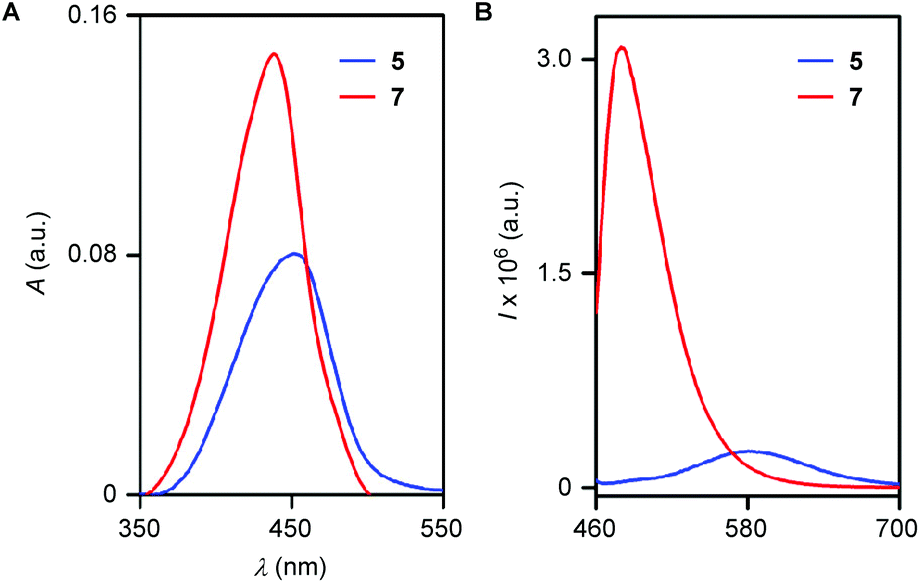
The probe displayed appreciable water solubility and a water (1 mM CTAB)/EtOH (9:1) system was used for photophysical property and sensing studies. UV-vis (Fig. 2A) and fluorescence (Fig. 2B) spectroscopic data of compounds 5 and 7 were also recorded to compare their photophysical properties. The probe 5 (10 μM) displayed a UV-vis band centered at λmax = 453 nm (ε = 30020 M−1 cm−1) while the reporter molecule 7 (10 μM) exhibited a hypsochromic band at λmax = 440 nm (ε = 36150 M−1 cm−1). Upon excitation at 440 nm, these compounds displayed emission bands at λem = 580 nm and λem = 480 nm, respectively. Quantum yield determination for 5 and 7 provided an approximately 43-fold theoretical jump in fluorescence (Φ = 0.00134 for 5 and Φ = 0.05750 for 7).
 |
| | Fig. 2 Absorbance spectra of probe 5 and iminocoumarin 7 (A). Emission spectra of probe 5 and iminocoumarin 7 recorded in water (1 mM CTAB)/EtOH (9:1) at λex = 440 nm (B). | |
HPLC analysis for proving the mechanism.
To confirm the formation of iminocoumarin 7 from probe 5 during H2S mediated reaction, HPLC titrations of 5 with increasing concentration of Na2S were performed. All reactions were carried out in a water (1 mM CTAB)/EtOH (9:1) system and chromatograms were recorded with acetonitrile and water as the eluent in a gradient method (Fig. S5–S8†). Under comparable conditions, compounds 5 and 7 exhibited retention times tR = 9.16 and 4.99 min, respectively (Fig. 3). When reaction mixtures containing 5 and Na2S (5 and 10 equiv.) were analyzed, the peak corresponding to the probe 5 reduced with a simultaneous enhancement of the peak related to 7. When probe 5 was subjected to a reaction with Na2S (2 equiv.) in EtOH for 5 min, and the mixture was analyzed by MALDI mass spectrometry, signals corresponding to various intermediates including 6 (m/z = 384.2296 for [6 + K+])43 and the iminocoumarin 7 (m/z = 242.2710, 264.1002 and 280.1527 for [7 + H+], [7 + Na+] and [7 + K+], respectively) were observed (Fig. S9†).
 |
| | Fig. 3 HPLC chromatograms of 5 (100 μM) upon titration with Na2S (5 and 10 equiv.) in water (1 mM CTAB)/EtOH (9:1), recorded in a solvent system of CH3CN and H2O. | |
FT-IR analysis for proving the mechanism.
To confirm the formation of iminocoumarin 7 from probe 5, FT-IR spectra for 5 (Fig. 4D) and 7 (Fig. 4A) were recorded. Similarly, data were also recorded for the sample containing a mixture of 5 and an increasing equivalent of Na2S (Fig. 4C and B for 0.5 and 1.0 equiv. of Na2S, respectively). These reactions were performed in EtOH for 3 h and spectra were recorded using KBr pellets after complete evaporation of the solvent. FT-IR data indicated significant changes in the 1525–1675 cm−1 region. The signal transmittance around 1557 cm−1 (i.e. for C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 stretching) decreased with the appearance of a new signal around 1590 cm−1 (i.e. for C1′C2′ stretching). Formation of the iminocoumarin ring during sensing was also confirmed by the appearance of a new signal around 1650 cm−1 (i.e. for C3′ N4′ stretching).
C2 stretching) decreased with the appearance of a new signal around 1590 cm−1 (i.e. for C1′C2′ stretching). Formation of the iminocoumarin ring during sensing was also confirmed by the appearance of a new signal around 1650 cm−1 (i.e. for C3′ N4′ stretching).
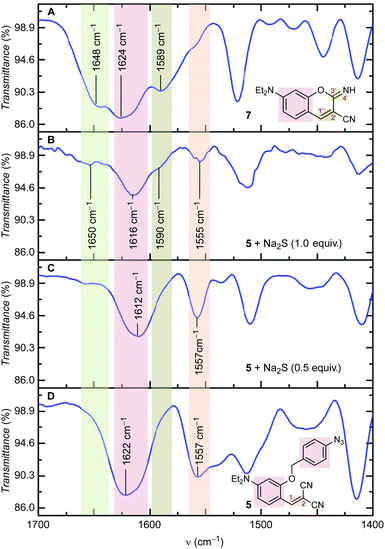 |
| | Fig. 4 FT-IR spectra of 5 (2 mg) upon addition of Na2S (0.5 and 1.0 equiv.) in EtOH. Spectra were recorded using KBr pellets. | |
Determination of the response time.
Based on the aforestated outcome, kinetics of the H2S mediated cascade reaction was studied. The probe 5 (10 μM) was treated with Na2S (150 μM) in water (1 mM CTAB)/EtOH (9:1) and fluorescence spectra (λex = 440 nm) were recorded at various time intervals. Experiments suggested a decrease in the fluorescence intensity at 580 nm and stepwise enhancement of the intensity at 480 nm (Fig. 5A). Subsequently, the disappearance of the probe 5 (i.e. the signal intensity at 580 nm) and formation of 7 (i.e. the signal intensity at 480 nm) were monitored with time (Fig. 5B). The reaction kinetics of reporter release provided the pseudo first order rate constant, k = 0.113 min−1 with t1/2 = 6.1 min and a response time of 24 min (Fig. S10†). Therefore the outcome confirms the better reactivity of the probe 5 over the probe reported by Sun and coworkers.46
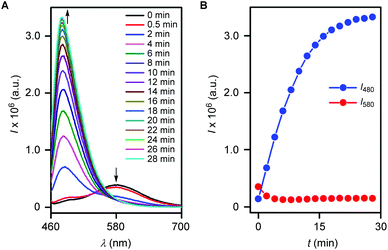 |
| | Fig. 5 Fluorescence spectra of probe 5 (10 μM) upon addition of Na2S (150 μM) at various time intervals in water (1 mM CTAB)/EtOH (9:1) at λex = 440 nm (A). Plots of fluorescence intensities at 480 nm (blue) and 580 nm (red) versus respective time values (B). | |
Kinetic stability and naked-eye detection.
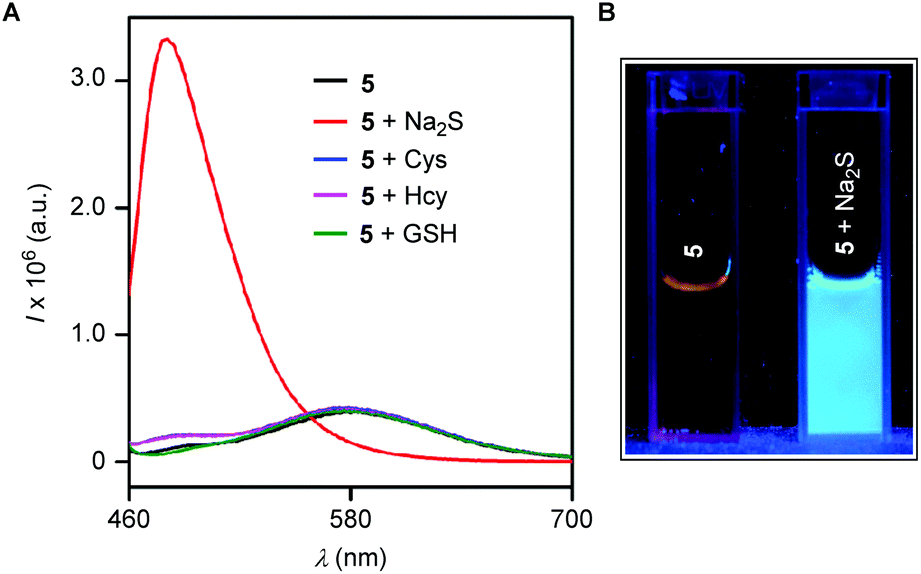
Furthermore, the probe 5 (10 μM) was separately treated with Na2S, Cys, Hcy and GSH (150 μM each) for 30 min and fluorescence spectra were recorded. The fluorescence intensity enhancement at 480 nm was observed only for Na2S, and the probe was inert to other biothiols (Fig. 6A). The reaction of the probe 5 with Na2S was also associated with the change of fluorescence (by placing it under the hand held UV-lamp at λex = 365 nm) from faint orange to a strong blue (Fig. 6B).
 |
| | Fig. 6 Fluorescence spectra of 5 (10 μM) recorded in the absence and in the presence of Na2S, Cys, Hcy, and GSH (150 μM each) at λex = 440 nm (A). Each datum was recorded after 30 minutes of analyte addition in water (1 mM CTAB)/EtOH (9:1). Cuvette images of 5 in the absence and in the presence of Na2S under a hand held UV-lamp (B). | |
Selectivity studies for 5.
Encouraged by these results, the selectivity of probe 5 towards H2S was further evaluated. When 5 (10 μM) was treated separately with ranges of analytes (e.g. GSH, Hcy, Cys, F−, Cl−, Br−, I−, NO2−, NO3−, S2O32−, SO32−, SO42−, SCN−, OH− and H2O2; 150 μM each) for 30 min, no significant fluorescence (at 480 nm with λex = 440 nm) was observed (Fig. 7, blue bars). However, further addition of Na2S to the same cuvettes provided a considerable fluorescence enhancement (Fig. 7, brown bars). The addition of Na2S to 5 provided a 31-fold enhancement in fluorescence intensity (Fig. 6, leftmost brown bar). This selectivity study proved the selectivity of the probe 5 towards H2S, even in the competing environment of other bio-relevant species.
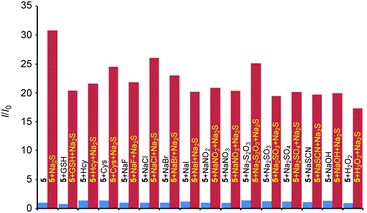 |
| | Fig. 7 Relative fluorescence intensity enhancements [I/I0] at 480 nm for probe 5 (10 μM) towards Na2S (150 μM) in water (1 mM CTAB)/EtOH (9:1). I0 = Fluorescence intensity of the free probe at 480 nm, and I = fluorescence intensity at 480 nm recorded after an analyte addition. Blue bars: intensities observed in the absence and in the presence of various analytes (150 μM); brown bars: intensities observed after the addition of Na2S to a solution containing either free probe 5 or probe 5 in the presence of an analyte. | |
Quantitative response of 5 towards H2S.
Subsequently, the quantitative response of the probe 5 upon detection of H2S was evaluated by fluorescence spectroscopy. Strong enhancement in the fluorescence intensity (at λex = 440 nm) was observed when 5 (10 μM) was treated with the increasing concentration of Na2S (0 to 15 equiv.) for 30 min (Fig. 8). When fluorescence intensities at 480 nm were plotted against the respective Na2S concentration values, an excellent linear correlation was observed up to 5 equiv. of the analyte (Fig. 8, inset). From the linear region of the diagram (concentration range of Na2S = 2–50 μM), the limit of detection, LOD = 169 nM (Table S3†) was calculated by applying the reported formula, LOD = 3σ/m (where σ = standard deviation of the fluorescence intensity of 8 blank measurements, and m = slope of the intensity versus concentration plot). On the basis of these results, applicability of compound 5 as a selective and sensitive probe for the detection of H2S was established.
 |
| | Fig. 8 Fluorescence spectral changes of probe 5 (10 μM) upon addition of different concentrations of Na2S (0–50 μM) in water (1 mM CTAB)/EtOH (9:1). Inset: linear relationship between the fluorescence intensity at 480 nm versus the concentration of Na2S. | |
Application of 5 in live-cell imaging.
Finally, cell permeability of the probe 5 and its ability to detect intracellular H2S were evaluated by live-cell imaging studies. Human cervical cancer cell line (HeLa) was used for cell imaging studies. Very weak fluorescence was observed when HeLa cells were incubated with only probe 5 (10 μM in 1:100 DMSO–DMEM v/v, pH = 7.4) at 37 °C for 30 min (Fig. 9A–C for brightfield, fluorescence and overlay images, respectively). However, further incubation of these cells (pre-incubated with 5) with Na2S (100 μM in 1:100 H2O–DMEM, pH = 7.4) at 37 °C for 30 min resulted in a strong fluorescence inside the cells (Fig. 9D–F for brightfield, fluorescence and overlay images, respectively). The appearance of fluorescence only after the incubation with Na2S confirms the reaction of the probe 5 with H2S present inside these cells (generated from Na2S).
 |
| | Fig. 9 Images of HeLa cells: DIC (A), fluorescence (B), and overlay (C), incubated with probe 5 (10 μM) for 30 min. D–F are the respective DIC, fluorescence and overlay images of HeLa cells pre-incubated with probe 5 followed by incubation with Na2S (100 μM) for 30 min. A concentrated stock solution of probe 5 (5 μL) was added to the cellular media. | |
Conclusion
In short, we synthesised a benzylidenemalononitrile-based fluorescent H2S probe 5 that forms an iminocoumarin derivative 7 as the reporter molecule. Molecule 5 was analyzed first to obtain theoretical estimates of its water solubility (cLogS = −5.799) and permeability (cLogP = 4.8887). Treatment of Na2S with 5 triggered the H2S mediated azide-to-amine reduction, and this further facilitated a cascade reaction sequence leading to the formation of the fluorophore. The mechanism of the reaction was proved by HPLC analysis, mass spectrometry and FT-IR spectroscopy. The probe was capable of sensing H2S in the presence of biological thiols (Cys, Hcy and GSH), ROS, reducing agents and other biological nucleophiles. H2S sensing by the probe provided a fast response (pseudo first order rate constant, k = 0.113 min−1, t1/2 = 6.1 min and a response time of 24 min). The probe also provided a 31-fold fluorescence enhancement and a detection limit of 169 nM. Cell permeability of the probe and its ability to detect intracellular H2S were demonstrated by live-cell imaging studies.
Experimental section
General methods
All the chemicals were purchased from commercial sources and used as received unless stated otherwise. All reactions were conducted under a nitrogen atmosphere, unless stated otherwise. Solvents: petroleum ether and ethyl acetate (EtOAc) were distilled prior to thin layer and column chromatography. Dichloromethane (DCM) was pre-dried over calcium hydride and then distilled under vacuum. Column chromatography was performed on Merck silica gel (100–200 mesh). TLC was carried out with E. Merck silica gel 60-F254 plates.
Physical measurements
The 1H and 13C spectra were recorded on 400 MHz Jeol ECS-400 (or 100 MHz for 13C) spectrometers using either residual solvent signals as an internal reference or from internal tetramethylsilane on the δ scale (DMSO-D6δH, 2.50 ppm, δC 39.52 ppm). The chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. The following abbreviations are used: m (multiplet), s (singlet), br s (broad singlet), d (doublet), t (triplet) and dd (doublet of doublet). High resolution mass spectrometric data were obtained from a MicroMass ESI-TOF MS spectrometer. FT-IR spectra were obtained using a NICOLET 6700 FT-IR spectrophotometer with KBr discs and reported in cm−1. Melting points were measured using VEEGO Melting point apparatus. All melting points were measured in an open glass capillary and values are uncorrected. Absorption spectra were recorded on a Perkin-Elmer, Lambda 45 and SHIMADZU UV-2600 UV-Vis spectrophotometer. Steady state fluorescence experiments were carried out in a microfluorescence cuvette (Hellma, path length 1.0 cm) on a Fluoromax 4 instrument (Horiba Jobin Yvon). Live cell images were recorded in 35 mm (diameter) dishes. The media (DMEM) and PBS buffer were purchased from commercial sources. Fluorescence images were recorded using an Olympus Inverted IX81 equipped with a Hamamatsu Orca R2 microscope. ChemBio Draw Ultra and Image J software were used for drawing structures and for processing cell images respectively.
Synthesis of 2-(4-azidobenzyloxy)-4-(diethylamino)benzaldehyde 11 (C18H20N4O2).
In a 25 mL round bottom flask 1-azido-4-(bromomethyl)benzene4710 (200 mg, 1.34 mmol) was dissolved in 5 mL DMF. Subsequently, 4-(diethylamino)-2-hydroxybenzaldehyde 9 (258 mg, 1.34 mmol) and K2CO3 (278 mg, 2.01 mmol) were added. The reaction mixture was stirred at room temperature for 2.5 h. After completion of the reaction, the reaction mixture was extracted with ethyl acetate (20 mL × 3). The combined organic layer was washed with water (10 mL × 3) and brine (30 mL) and dried over Na2SO4. The solvent was removed under reduced pressure to obtain a brown residue, which was purified by column chromatography over silica gel (eluent: 10% EtOAc in petroleum ether) to furnish the pure 11 (215 mg, 50%) as a colorless liquid. IR (KBr): ν/cm−1 3326, 3210, 2975, 2129, 1728, 1613, 1556, 1513, 1120, 1075; 1H NMR (400 MHz, DMSO-D6): δ 10.06 (s, 1H), 7.53 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 9 Hz, 1H), 7.13 (d, t, J = 8.2, 2.2 Hz, 2H), 6.33 (dd, J = 8.9, 1.9 Hz, 1H), 6.21 (d, J = 2.2 Hz, 1H), 5.24 (s, 2H), 3.40 (q, J = 7.0 Hz, 4H), 1.08 (t, J = 7.0 Hz, 6H); 13C NMR (100 MHz, DMSO-D6): δ 185.72, 163.09, 154.02, 139.42, 134.38, 130.18, 129.70, 119.72, 113.97, 104.96, 94.80, 69.28, 44.62, 12.92; HRMS (ESI): Calculated for C18H21N4O2+ [M + H]+: 325.1644; found: 325.1664.
Synthesis of 2-(2-(4-azidobenzyloxy)-4-(diethylamino)benzylidene)malononitrile 5 (C21H20N6O).
In a 25 mL round bottomed flask 2-(4-azidobenzyloxy)-4-(diethylamino)benzaldehyde 11 (190 mg, 0.58 mmol) was dissolved in ethanol (5 mL). Subsequently, malononitrile (39 mg, 0.58 mmol) and piperidine (248 mg, 2.9 mmol) were added, and the reaction mixture was stirred at room temperature for 5 min. The yellow residue obtained after evaporation of ethanol under reduced pressure was purified by column chromatography over silica gel (eluent: 20% EtOAc in petroleum ether) to furnish the pure 5 (130 mg, 60%) as a yellow solid. M.P.: 139–141 °C; IR (KBr):ν/cm−1 3327, 3210, 2975, 2217, 2131, 1613, 1557, 1514, 1118, 1075; 1H NMR (400 MHz, DMSO-D6):δ 8.03 (d, J = 9.3 Hz, 1H), 7.92 (s, 1H), 7.53 (d, J = 8.3 Hz, 2H), 7.15 (d, J = 8.3 Hz, 2H), 6.54 (dd, J = 9.3, 1.2 Hz, 1H), 6.23 (d, J = 1.3 Hz, 1H), 5.25 (s, 2H), 3.48 (q, J = 6.8 Hz, 4H), 1.09 (t, J = 6.9 Hz, 6H); 13C NMR 100 MHz, DMSO-D6: δ 161.05, 155.04, 150.68, 139.75, 133.70, 130.42, 130.11, 119.84, 117.68, 116.68, 109.20, 106.80, 94.77, 69.79, 66.25, 45.03, 13.03; HRMS (ESI) calculated for C21H21N6O+ [M + H]+: 373.1777; found: 373.1772.
Live-cell imaging
HeLa cells were purchased from the National Centre for Cell Science, Pune (India). HeLa cells were grown in DMEM supplemented with 10% heat inactivated fetal bovine serum (FBS), 100 IU mL−1 penicillin, 100 mg mL−1 streptomycin and 2 mM L-glutamine. Cultures were maintained under a humidified atmosphere with 5% CO2 at 37 °C. The cultured cells were subcultured twice in each week, seeding at a density of about 15 × 103 cells per mL−1. The trypan blue dye exclusion method was used to determine the cell viability. The fluorescence images were recorded using an Olympus Inverted IX81 equipped with a Hamamatsu Orca R2 microscope by exciting at λex = 460–480 nm (by using a GFP filter).
The HeLa cells were incubated with a solution of the probe 5 (10 μM in 1:100 DMSO–DMEM v/v, pH = 7.4) at 37 °C for 30 min. The fluorescence images were acquired after washing with PBS. In this case less significant fluorescence was observed. Another set of HeLa cells was pre-incubated with probe 5 (10 μM in 1:100 DMSO–DMEM v/v, pH = 7.4) at 37 °C for 30 min followed by washing with PBS and incubation with Na2S (100 μM in 1:100 DMSO–DMEM v/v, pH = 7.4) at 37 °C for 30 min. After washing with PBS the fluorescence images showed strong green fluorescence (Fig. 9E).
Acknowledgements
We acknowledge IISER Pune, DST-SERB (Grant no. SR/S1/OC-65/2012) for financial support. We thank Mr Kiran Reddy for solving the crystal data. T. S. thanks UGC for a research fellowship.
Notes and references
- C. L. Evans, Exp. Physiol., 1967, 52, 231–248 CrossRef CAS.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485–1488 CrossRef CAS.
- P. Kamoun, M.-C. Belardinelli, A. Chabli, K. Lallouchi and B. Chadefaux-Vekemans, Am. J. Med. Genet. A, 2003, 116A, 310–311 CrossRef PubMed.
- W. Yang, G. Yang, X. Jia, L. Wu and R. Wang, J. Physiol., 2005, 569, 519–531 CrossRef CAS PubMed.
- D. Boehning and S. H. Snyder, Annu. Rev. Neurosci., 2003, 26, 105–131 CrossRef CAS PubMed.
- M. Stipanuk and I. Ueki, J. Inherit. Metab. Dis., 2011, 34, 17–32 CrossRef CAS PubMed.
- O. Kabil, N. Motl and R. Banerjee, Biochim. Biophys. Acta. Proteins Proteomics, 2014, 1844, 1355–1366 CrossRef CAS PubMed.
- S. Chen, Z.-j. Chen, W. Ren and H.-w. Ai, J. Am. Chem. Soc., 2012, 134, 9589–9592 CrossRef CAS PubMed.
- L. Li, P. Rose and P. K. Moore, Annu. Rev. Pharmacol., 2011, 51, 169–187 CrossRef CAS PubMed.
- Q. Li and J. R. Lancaster Jr., Nitric Oxide, 2013, 35, 21–34 CrossRef CAS PubMed.
- F. Liu, D.-D. Chen, X. Sun, H.-H. Xie, H. Yuan, W. Jia and A. F. Chen, Diabetes, 2014, 63, 1763–1778 CrossRef CAS PubMed.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CAS.
- B. Florian, R. Vintilescu, A. T. Balseanu, A.-M. Buga, O. Grisk, L. C. Walker, C. Kessler and A. Popa-Wagner, Neurosci. Lett., 2008, 438, 180–185 CrossRef CAS PubMed.
- A. Qandil, Int. J. Mol. Sci., 2012, 13, 17244–17274 CrossRef CAS PubMed.
- D. J. Lefer, Br. J. Pharmacol., 2008, 155, 617–619 CrossRef CAS PubMed.
- U. Hannestad, S. Margheri and B. Sörbo, Anal. Biochem., 1989, 178, 394–398 CrossRef CAS.
- L. M. Siegel, Anal. Biochem., 1965, 11, 126–132 CrossRef CAS.
- J. E. Doeller, T. S. Isbell, G. Benavides, J. Koenitzer, H. Patel, R. P. Patel, J. R. Lancaster Jr., V. M. Darley-Usmar and D. W. Kraus, Anal. Biochem., 2005, 341, 40–51 CrossRef CAS PubMed.
- F. Hou, L. Huang, P. Xi, J. Cheng, X. Zhao, G. Xie, Y. Shi, F. Cheng, X. Yao, D. Bai and Z. Zeng, Inorg. Chem., 2012, 51, 2454–2460 CrossRef CAS PubMed.
- K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003–18005 CrossRef CAS PubMed.
- F. Hou, J. Cheng, P. Xi, F. Chen, L. Huang, G. Xie, Y. Shi, H. Liu, D. Bai and Z. Zeng, Dalton Trans., 2012, 41, 5799–5804 RSC.
- X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529–10531 RSC.
- B. Chen, W. Li, C. Lv, M. Zhao, H. Jin, H. Jin, J. Du, L. Zhang and X. Tang, Analyst, 2013, 138, 946–951 RSC.
- S. K. Das, C. S. Lim, S. Y. Yang, J. H. Han and B. R. Cho, Chem. Commun., 2012, 48, 8395–8397 RSC.
- Q. Wan, Y. Song, Z. Li, X. Gao and H. Ma, Chem. Commun., 2013, 49, 502–504 RSC.
- F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 2852–2854 RSC.
- T. Saha, D. Kand and P. Talukdar, Org. Biomol. Chem., 2013, 11, 8166–8170 CAS.
- L. A. Montoya and M. D. Pluth, Chem. Commun., 2012, 48, 4767–4769 RSC.
- G. Zhou, H. Wang, Y. Ma and X. Chen, Tetrahedron, 2013, 69, 867–870 CrossRef CAS PubMed.
- A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS PubMed.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- R. Wang, F. Yu, L. Chen, H. Chen, L. Wang and W. Zhang, Chem. Commun., 2012, 48, 11757–11759 RSC.
- M.-Y. Wu, K. Li, J.-T. Hou, Z. Huang and X.-Q. Yu, Org. Biomol. Chem., 2012, 10, 8342–8347 CAS.
- C. Liu, J. Pan, S. Li, Y. Zhao, L. Y. Wu, C. E. Berkman, A. R. Whorton and M. Xian, Angew. Chem., Int. Ed., 2011, 50, 10327–10329 CrossRef CAS PubMed.
- C. Liu, B. Peng, S. Li, C.-M. Park, A. R. Whorton and M. Xian, Org. Lett., 2012, 14, 2184–2187 CrossRef CAS PubMed.
- Y. Qian, J. Karpus, O. Kabil, S.-Y. Zhang, H.-L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed.
- Y. Qian, L. Zhang, S. Ding, X. Deng, C. He, X. E. Zheng, H.-L. Zhu and J. Zhao, Chem. Sci., 2012, 3, 2920–2923 RSC.
- Z. Xu, L. Xu, J. Zhou, Y. Xu, W. Zhu and X. Qian, Chem. Commun., 2012, 48, 10871–10873 RSC.
- Y. Jiang, Q. Wu and X. Chang, Talanta, 2014, 121, 122–126 CrossRef CAS PubMed.
- L. Zhang, S. Li, M. Hong, Y. Xu, S. Wang, Y. Liu, Y. Qian and J. Zhao, Org. Biomol. Chem., 2014, 12, 5115–5125 CAS.
- S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E.-H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS PubMed.
- Z. Wu, Z. Li, L. Yang, J. Han and S. Han, Chem. Commun., 2012, 48, 10120–10122 RSC.
- B. A. Belinka and A. Hassner, J. Org. Chem., 1979, 44, 4712–4713 CrossRef CAS.
- W. Li, W. Sun, X. Yu, L. Du and M. Li, J. Fluoresc., 2013, 23, 181–186 CrossRef CAS PubMed.
- J. Volmajer, R. Toplak, I. Leban and A. M. L. Marechal, Tetrahedron, 2005, 61, 7012–7021 CrossRef CAS PubMed.
- H. Zhang, Y. Xie, P. Wang, G. Chen, R. Liu, Y.-W. Lam, Y. Hu, Q. Zhu and H. Sun, Talanta, 2015, 135, 149–154 CrossRef CAS PubMed.
- M. Belkheira, D. ElAbed, J.-M. Pons and C. Bressy, Chem. – Eur. J., 2011, 17, 12917–12921 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data (1H-, 13C-NMR and IR spectroscopic, and mass spectrometric), and single crystal X-ray data of 5. CCDC 1059196. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00785b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a