
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate geometry controls the cyclization cascade in multiproduct terpene synthases from Zea mays†
Abith
Vattekkatte
a,
Nathalie
Gatto
a,
Tobias G.
Köllner
b,
Jörg
Degenhardt
c,
Jonathan
Gershenzon
b and
Wilhelm
Boland
*a
aDepartment of Bioorganic Chemistry, Max Planck Institute for Chemical Ecology, Hans-Knöll-Strasse 8, D-07745 Jena, Germany. E-mail: boland@ice.mpg.de
bDepartment of Biochemistry, Max Planck Institute for Chemical Ecology, Hans-Knöll-Strasse 8, D-07745 Jena, Germany
cInstitute for Pharmacy, University of Halle, Hoher Weg 8, D-06120 Halle, Germany
First published on 29th April 2015
Abstract
Multiproduct terpene synthases TPS4-B73 and TPS5-Delprim from maize (Zea mays) catalyze the conversion of farnesyl diphosphate (FDP) and geranyl diphosphate (GDP) into a complex mixture of sesquiterpenes and monoterpenes, respectively. Various isotopic and geometric isomers of natural substrates like (2Z)-[2-2H]- and [2,4,4,9,9,9-2H6]-(GDP) and (2Z,6E)-[2-2H]- and [2,4,4,13,13,13-2H6]-(FDP) were synthesized analogous to presumptive reaction intermediates. On incubation with labeled (2Z) substrates, TPS4 and TPS5 showed much lower kinetic isotope effects than the labeled (2E) substrates. Interestingly, the products arising from the deuterated (2Z)-precursors revealed a distinct preference for cyclic products and exhibited an enhanced turnover on comparison with natural (2E)-substrates. This increase in the efficiency due to (2Z) configuration emphasizes the rate limiting effect of the initial (2E) → (2Z) isomerization step in the reaction cascade of the multiproduct terpene synthases. Apart from turnover advantages, these results suggest that substrate geometry can be used as a tool to optimize the biosynthetic reaction cascade towards valuable cyclic terpenoids.
Introduction
Plants produce a huge variety of secondary metabolites that continue to amaze both plant biologists and natural product chemists.1 The largest group of plant secondary metabolites comprises the terpenes with more than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known compounds.2 These molecules have applications ranging from flavor and fragrance to biological functions such as hormones, attractants for pollinators, or toxins.3 In addition, the terpenoid composition shows major qualitative and quantitative variation among species and also within single species.4 The structural diversity of terpenes is due to terpene synthases, enzymes that convert the prenyl diphosphate substrates like geranyl diphosphate (GDP, C10), farnesyl diphosphate (FDP, C15) and geranylgeranyl diphosphate (GGDP, C20) into monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20), respectively.2b Terpene synthases have been intensively investigated in recent decades and various cDNAs encoding plant terpene synthases responsible for the structural diversity have been characterized.5
000 known compounds.2 These molecules have applications ranging from flavor and fragrance to biological functions such as hormones, attractants for pollinators, or toxins.3 In addition, the terpenoid composition shows major qualitative and quantitative variation among species and also within single species.4 The structural diversity of terpenes is due to terpene synthases, enzymes that convert the prenyl diphosphate substrates like geranyl diphosphate (GDP, C10), farnesyl diphosphate (FDP, C15) and geranylgeranyl diphosphate (GGDP, C20) into monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20), respectively.2b Terpene synthases have been intensively investigated in recent decades and various cDNAs encoding plant terpene synthases responsible for the structural diversity have been characterized.5
Certain terpene synthases are known for their catalytic promiscuity. This catalytic promiscuity is due to a common electrophilic reaction mechanism which is important for deciphering evolution of enzymes and engineering future enzymatic catalysts.6 One of the unique features of terpene synthases is their ability to produce multiple products from a single prenyl diphosphate substrate.7 The δ-selinene synthase and the γ-humulene synthase from Abies grandis hold the present record by producing 52 and 34 different sesquiterpenes.8 Despite their overall sequence diversity, terpene synthases possess several highly conserved amino acid residues.9 An aspartate-rich DDxxD motif located at the entrance of the active site was shown to be involved in the binding of the metal ion-complexed diphosphate ester substrate.10 In the N-terminal part of monoterpene synthases, two arginine residues are present that are believed to influence the isomerization of the initial substrate.11
Two closely related terpene synthase genes encoding multiproduct enzymes, namely TPS4 and TPS5, were recently cloned from Zea mays.12 Both recombinant proteins accepted GDP and FDP as substrates and converted them into two types of cyclic products with cyclohexenyl- and bicyclo[3.1.0]hexyl moieties as common structural features (Scheme 1). Product formation is achieved by initial isomerization of the substrate (E)-GDP or (E)-FDP into the tertiary allylic diphosphates (Scheme 1). After dissociation, the rearranged linaloyl- or nerolidyl-cation cyclizes easily to a cyclohexenyl cation or by formation of a bicyclo[3.1.0]hexyl moiety stabilized in both cases by further deprotonation. Modeling of the TPS4 active site cavity and docking studies with cationic intermediates suggested that discrete steps of the reaction cascade are controlled by two different enzyme pockets.13 We have recently reported about the kinetic isotope effects and enhanced formation of alcohols over olefinic products by deuterated precursors of (2E)-GDP and (2E,6E)-FDP as compared to natural substrates.14
 | ||
| Scheme 1 Isotope sensitive branching strategy. | ||
To reveal further details of the enzyme mechanism, we synthesized geranyl- and farnesyl diphosphates including both geometric isomers of the critical C(2)–C(3) bond (Scheme 1) using deuterium labels as a probe for isotope sensitive branching. We were interested in whether the cyclization of cis-isomers (2Z)-GDP (2a–b) and (2Z,6E) FDP (2c–d) would proceed via the same cascade as observed with their corresponding trans-substrates (1a–d) (Scheme 2). Most studies involving several sesquiterpene synthases using FDP isomers and analogues have been used to compare their kinetic properties and further determine the mechanism of carbocation quenching15 and the initial ionization-isomerization of all-trans-FDP for cis–trans-pathway-specific enzymes.13,16 Here, we describe the effects of substrate's conformation on the initial cyclization and the further course of individual protonation and deprotonation reactions by means of deuterium labeling. In contrast to our previous study,14 both TPS4 and TPS5 cyclize labeled (2Z,6E)-FDP (2c–d) and (2Z)-GDP (2a–b) showed quantitative difference in volatile composition as compared to natural substrates. Interestingly, they exhibited much higher turnover with (2Z) substrates (2a–d) than with their natural (2E) substrates (1a–d) and a reduced ratio of acyclic to cyclic products.
 | ||
| Scheme 2 Monodeuterated and hexadeuterated (2E)-GDP, FDP (1a–d) and (2Z)-GDP, FDP (2a–d). | ||
Results and discussion
To study the rate limiting effects of the initial isomerization step catalyzed by TPS4 and TPS5 and their consequences for the reaction cascade we synthesized labeled substrates (2a–d) with deuterium atoms completely surrounding the C(3) cationic center of the key intermediates (Scheme 1). The deuterium labels serve to investigate the alterations in product distribution that might occur as a consequence of changing the geometry of the C2–C3 double bond, which undergoes isomerization in the reaction sequence. These product alterations depend on the nature of the initial carbocationic intermediates formed whose stability and ease of deprotonation may favor different reaction channels.Synthesis of substrates
[2-2H]- and [2,4,4,9,9,9-2H6]-GDP (2a–b) and [2-2H]- and [2,4,4,13,13,13-2H6]-FDP (2c–d) were prepared by modifying the protocol of Arigoni et al.17 (Scheme 3 and ESI†). For hexadeuterated analogues, [1,1,1,3,3-2H5] ketones 3b and 3d were first prepared by proton-deuterium exchange reaction in MeOD in presence of DBN (1,5-diazabicyclo[4.3.0]non-5-ene).18 Subsequent Peterson olefination of the ketones (3a–d) with [2,2-2H2]-trimethylsilylacetic acid 4 afforded a mixture of carboxylic acids, additionally labeled at C(2). After esterification, the isomers were easily separated by flash chromatography and provided the pure methyl esters (2E)-(5a–d) and (2Z)-(6a–d). Mono- and hexadeuterated alcohols (2Z)-(8a–d) were generated by reduction of the methyl esters with diisopropyl aluminium hydride. Labeled diphosphates were prepared according to Woodside et al.19 and yielded the corresponding trisammonium (2Z)-geranyl diphosphates (2a–b) and trisammonium (2Z,6E)-farnesyl diphosphates (2c–d). The corresponding (2E) substrates (1a–d) were synthesized from (2E)-(5a–d) and have already been described.14 This procedure provides an easy access to various labeled and unlabeled isomers of prenyl diphosphates. | ||
| Scheme 3 Synthesis of deuterated substrates. Reagents and conditions: (a) [2,2-2H2]-trimethylsilylacetic acid, 2 eq. mol. LDA, THF, −78 °C to reflux; (b) Me2SO4, DIPEA; (c) DIBAL-H, CH2Cl2; (d) (i) NBS, Me2S, CH2Cl2; (ii) (Bu4N)3P2O7H, CH3CN; (iii) ion exchange; (iv) cellulose, CH3CN/NH4HCO3. | ||
Enzymatic transformations of labeled GDP and FDP by TPS4 and TPS5
 | ||
| Fig. 1 Comparison of total rate of monoterpene (A) and sesquiterpene (B) formation with incubations of deuterated (E)/(Z)-GDP and FDP with TPS4-B73 and TPS5-Delprim. Horizontal line at 100 represents the relative rate obtained from unlabeled (2E)-GDP (A) and (2E)-FDP (B) respectively. Boxplot: median (horizontal lines in boxes), interquartile range (boxes, 1.5×-interquartile range (whiskers). | ||
Both TPS4 and TPS5 exhibited much higher turnover when incubated with (2Z)-(2a–d) vs. (2E)-(1a–d) substrates (Fig. 1). In Fig. 1, horizontal line at 100 represents the relative rate obtained from unlabeled (2E)-GDP (A) and (2E)-FDP (B) respectively. All labeled (2E)-isomers of C10 and C15 substrates either show a decrease or values around the reference line for both monoterpenes and sesquiterpenes. Whereas in case of labeled (2Z)-isomers of C10 and C15 substrates there was a substantial increase in product formation on comparison with unlabeled reference substrates.
The rate of monoterpene production showed 30% increase after incubation with 2a and 17% with 2b in comparison to their corresponding unlabeled (2E)-analogues. The difference in the rate of volatile formation was even more pronounced in the case of sesquiterpenes (2c–d). When incubated with the monodeuterated 2c the sesquiterpene production increased by ∼200% and with the hexadeuterated 2d the corresponding increase was ∼150% when compared with unlabeled (2E)-FDP. The production of all C10 and C15 cyclic products requires an initial isomerization of the C(2)–C(3) double bond of the original substrate, achieved through the intermediate tertiary allylic phosphates linalyl- and nerolidyl diphosphate, respectively (Scheme 4). The substrates of the (2Z)-series (2a–d) already possess the double bond in the correct configuration allowing the direct cyclization of the emerging carbocationic intermediate after ionization. The increased turnover clearly indicates that the isomerization is the rate limiting step in the reaction cascade with natural substrates. Removal of this rate limiting factor leads to much higher efficiency in terpenoid cyclization by these enzymes.
 | ||
| Scheme 4 General mechanism for generation of acyclic compounds and cyclic compounds by TPS4 and TPS5. | ||
The production of all C10 and C15 cyclic products require an isomerization of the C(2)–C(3) double bond of the original substrate, achieved through the intermediate tertiary allylic phosphates linalyl- and nerolidyl diphosphate, respectively. The substrates of the (2Z)-series possess the double bond already in the correct configuration allowing the direct cyclization of the emerging carbocationic intermediate after diphosphate cleavage. This phenomenon has also been reported for the product formation of some other sesquiterpene synthases like trichodiene synthase from Fusarium sporotrichioides,20 and two terpene synthases from Coprinus cinereus Cop4 and Cop6.16f,21
Three acyclic monoterpene olefins, (E)-β-myrcene, (R)-linalool and (S)-linalool from (2E)-GDP and two acyclic sesquiterpenes, (E)-β-farnesene and (3R)-(E)-nerolidol from (2E,6E)-FDP were also present in the product mixtures of these enzymes. These acyclic terpenes result from deprotonation or water-capture of the first cation formed after cleavage of the diphosphate group. However, a very strong depletion in the amount of acyclic monoterpenes, myrcene (98% reduction) and linalool (65% reduction) was observed after incubation with the 2a (2Z)-GDP (Fig. 2) in comparison to unlabeled (2E)-GDP for both maize cyclases. The rate suppression was even more pronounced with TPS4 after incubation of 2a, resulting in a complete absence of β-myrcene and an 88% reduction of linalool. The same effect was observed after incubation of hexadeuterated (2Z)-GDP 2b with both enzymes: β-myrcene (almost complete elimination) and linalool (48–80% reduction). Similarly, strong decrease in the proportion of the acyclic sesquiterpenes were observed with deuterated (2Z,6E)-FDP (2c, 2d). Thus, incubation with hexadeuterated (2Z,6E)-FDP 2d led to a complete absence of (E)-β-farnesene and nerolidol production with both enzymes. Likewise, monodeuterated (2Z,6E)-FDP 2c lead to a decrease in (E)-β-farnesene formation (97% with TPS5 and complete elimination with TPS4 and nerolidol formation (complete elimination with both enzymes). Nevertheless, the production of minute amounts of acyclic volatiles from (2Z)-substrates can be explained by direct deprotonation from the resulting carbocation or capture of a water molecule. These results strongly support a mechanism by which the substrates of the (2Z)-series (2a–d) are directly cyclized after ionization due to the C(2)–C(3) double bond already being in a suitable configuration for C(6)–C(1) ring closure (Scheme 4).
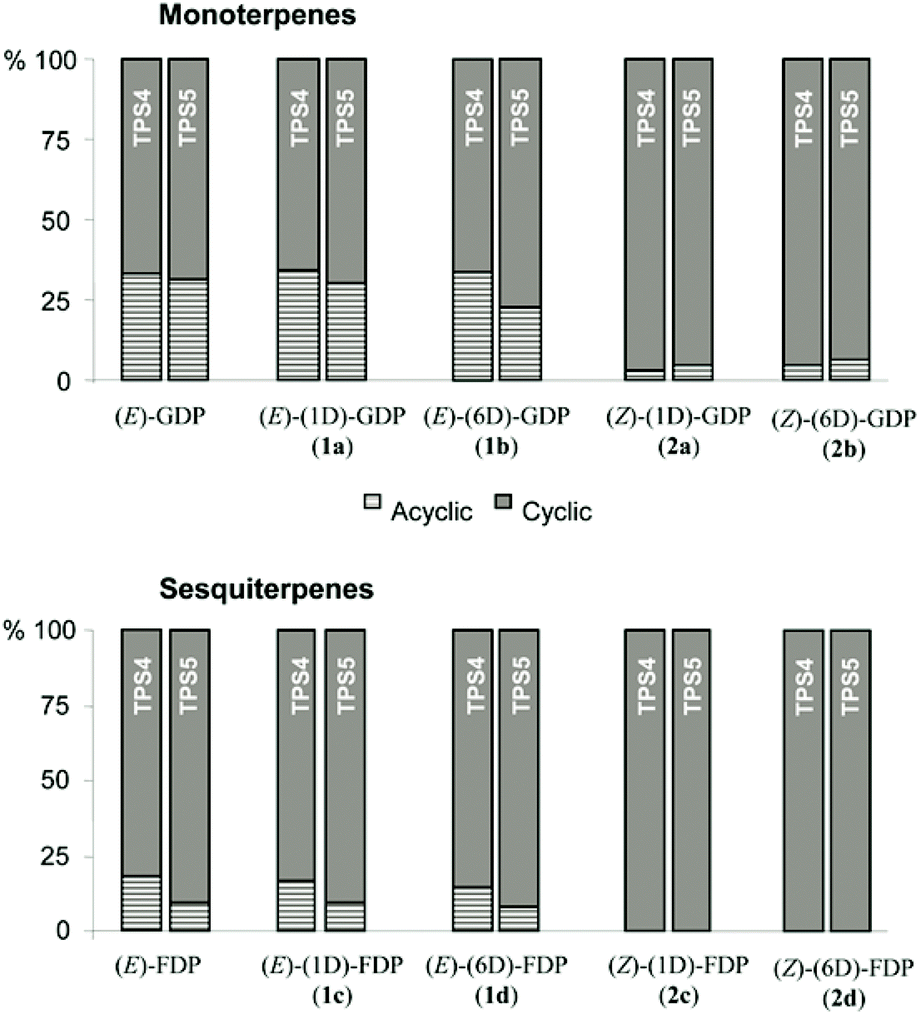 | ||
| Fig. 2 Ratio acyclic/cyclic volatiles released after incubation of labeled substrates with TPS4-B73 and TPS5-Delprim. | ||
Interestingly, both TPS4 and TPS5 showed similar behavior when incubated with same set of isomers, (2Z)-series (2a–d) and (2E)-series (1a–d) (Fig. 2). They maintained a constant ratio between acyclic and cyclic products from the same geometric isomer. This ratio was 1:2 (acyclic/cyclic monoterpenes) for the (2E)-GDP substrates (unlabeled and 1a–b) while an average ratio of 1:22 was observed for the corresponding (2Z)-GDP (2a–b). Similarly, for the unlabeled or deuterated (2E,6E)-FDP, this ratio was 1:5 (acyclic/cyclic sesquiterpenes) for TPS4 and 1:10 for TPS5. These results suggest that labeling with stable isotopes did not influence the kinetics of the first ring closure although the delocalized positive charge is partly surrounded by deuterium. Studies with monoterpene synthases have shown that only terpene synthases that can isomerize the C(2)–C(3) π bond of (2E)-GDP can make cyclic monoterpenes.23 Likewise, only sesquiterpene synthases known to isomerize the C(2)–C(3) π bond of (2E,6E)-FDP have been reported to synthesize cyclic products from (2E)-GDP.24 Thus, when incubated with (2Z)-series labeled GDP and FDP (2a–d) both enzymes showed strong preference for cyclic products with only a weak tendency for isomerization as evidenced by reduced formation of acyclic products (Scheme 4).
| Substrate | TPS4-B73 | TPS5-Delprim | ||
|---|---|---|---|---|
| Relative ratea (%) | k H/kD | Relative ratea (%) | k H/kD | |
| a Relative overall rates compared to those of incubation with unlabeled (E)-GDP or (E,E)-FDP substrates (set at 100). Each experiment was run by the mean of three to six independent replicates. b Apparent total rate isotope effects compared to those of incubation with unlabeled (E)-GDP or (E,E)-FDP substrates. c Apparent total rate isotope effects compared to those of incubation with (2Z)-[2H]-GDP or (2Z,6E)-[2H]-FDP substrates. Note: oxygenated cyclic volatiles not considered. | ||||
| (2E)-[2H]-GDP 1a | 101.09 ± 0.45 | ∼1b | 99.98 ± 0.84 | ∼1b |
| (2E)-[2H6]-GDP 1b | 85.06 ± 0.84 | 1.17b | 75.02 ± 0.78 | 1.33b |
| (2Z)-[2H]-GDP 2a | 132.25 ± 1.18 | — | 135.75 ± 0.63 | — |
| (2Z)-[2H6]-GDP 2b | 117.72 ± 1.67 | 1.12c | 114.38 ± 1.55 | 1.19c |
| (2E,6E)-[2H]-FDP 1c | 106.21 ± 6.95 | ∼1b | 101.30 ± 1.11 | ∼1b |
| (2E,6E)-[2H6]-FDP 2d | 80.95 ± 7.55 | 1.23b | 79.65 ± 1.94 | 1.25b |
| (2Z,6E)-[2H]-FDP 2c | 297.91 ± 2.59 | — | 298.14 ± 1.69 | — |
| (2Z,6E)-[2H6]-FDP 2d | 248.75 ± 2.63 | 1.20c | 215.20 ± 4.58 | 1.38c |
The relative overall rates of reaction with (2Z)-[2H]-GDP 2a and (2Z,6E)-[2H]-FDP 2c were used as reference for the determination of the apparent total rate isotope effects kH/kD for the hexadeuterated (2Z)-substrates (2b, 2d). Thus the relative overall rate of monoterpene formation with (2Z)-[2H6]-GDP 2b decreased for TPS4 (11%) and TPS5 (16%). These rate suppressions correspond to an apparent total rate isotope effect kH/kD of 1.12 and 1.19, respectively. Furthermore TPS4 and TPS5 showed a depletion (17% and 28%, respectively) in the total rates of sesquiterpene volatiles after incubation with (2Z,6E)-[2H6]-FDP 2d, corresponding to an approximated apparent total rate isotope effect kH/kD of 1.20 and 1.38. The observed overall rate reductions after incubation with hexadeuterated GDP (1b, 2b) and FDP (1d, 2d) primarily result from induced primary isotope effects. In contrast the labeling pattern of the monodeuterated GDP (1a, 2c) and FDP (1a, 2c) had no direct influence on the reaction mechanism, since the C–D-bond is not cleaved during the entire cyclization cascade. Moreover, the variations in the product profiles and the overall rates were a result of apparent secondary isotope effects.
 | (1) |
 | ||
| Fig. 3 Product distribution of main monoterpenes from incubations of deuterated GDP (2Z)-[2H]-GDP (2a) and (2Z)-[2H6]-GDP (2b) with TPS4-B73 and TPS5-Delprim from maize (Zea mays). (1) α-Thujene, (2) sabinene, (3) β-myrcene, (4) limonene, (5) α-terpinolene, (6) linalool, (7) α-terpineol. | ||
 | ||
| Fig. 4 Product distribution of main sesquiterpenes from incubations of deuterated FDP (2Z,6E)-[2H]-GDP (2c) and (2Z,6E)-[2H6]-GDP (2d) with TPS4-B73 and TPS5-Delprim from Zea mays. Major sesquiterpene products: (1) 7-epi-sesquithujene, (2) sesquithujene, (3) (Z)-α-bergamotene, (4) (E)-α-bergamotene, (5) sesquisabinene A, (6) sesquisabinene B, (7) (E)-β-farnesene, (8) γ-curcumene, (9) zingiberene, (10) (S)-β-bisabolene, (11) β-curcumene, (12) (E)-γ-bisabolene, (13) 7-epi-sesquithujene hydrate, (14) sesquithujene hydrate, (15) (3R)-(E)-nerolidol. | ||
As mentioned above, the oxygenated cyclic volatiles were not considered in the present data.14 To estimate the weight of this approximation, quantitative kinetic measurements were carried out. Since TPS4-B73 and TPS5-Delprim exhibit similar basic features, only deuterium isotope effects on the catalytic activity of TPS4 were evaluated using the noncompetitive method. Both unlabeled (2E,6E)-FDP and (2E,6E)-[2H6]-FDP 1d substrates were used in two independent enzyme assays. Standard enzyme assays were performed in triplicate with aliquots of the same enzyme extracts under saturated substrate conditions. A decrease of 13% (relative to the reference substrate) of the maximal rate for sesquiterpene formation was observed when (2E,6E)-[2H6]-FDP 1d was incubated with TPS4, while similar Km were obtained for both substrates. Since the kinetic experiments were performed using the same enzyme extract, the total enzyme concentration [ET] was identical for both assays. Therefore, the turnover number (kcat) of the enzyme, usually defined as the ratio of Vmax/[ET], can be approximated to Vmax. The apparent total rate isotope effect kH/kD, determined from the maximal rates, equals 1.15. As discussed before, similar results were obtained when the oxygenated cyclic volatiles were not considered (19% decrease in the volatile production corresponding to a kH/kD = 1.23) and justify the approximation made above.
Monoterpene product distribution
Within a series (2E (1a–b) or 2Z (2a–b)) both cyclases showed negligible changes in the formation of limonene, α-terpinolene and α-terpineol (Fig. 3) independent of the degree of labeling of the substrate used. Thus, minor kinetic isotope effects (KIE) were measured after incubation of (2E)-[2H]-GDP 1a or (2E)-[2H6]-GDP 1b in comparison to the unlabeled (2E)-GDP as well as after incubation of hexadeuterated (2Z)-[2H6]-GDP 2b in comparison to the monodeuterated analog (2Z)-[2H]-GDP 2a. In contrast sabinene and α-thujene showed decreases associated with a corresponding increase in sabinene hydrate after incubation with TPS4 and TPS5. The observed KIEs for the sabinene and α-thujene deprotonation reactions were calculated from eqn (1). In case of substrates differing by only one deuterium atom, the observed KIEs (kH/k1D)E were much weaker (a range of kH/kD = 1.10–1.18). However, the observed KIEs [(kH/k6D)E, (k1D/k6D)E or (k1D/k6D)Z], corresponding to a difference of 5 to 6 deuterium atoms between the substrates, lie in the range of kH/kD = 1.91–5.68. The interesting fact was that the (2Z)-substrates (k1D/k6D)Z showed the lowest KIEs and this effect was negligible when comparison is made with (2E)-substrates.Sesquiterpene product distribution
The same approach was used to evaluate the shift of products after incubation with different FDP substrates (E (1c–d) and Z (2c–d)) (Fig. 4). Despite the blend of volatiles being much more complex than in the case of monoterpenes, isotope effects on sesquiterpene volatiles can be observed after incubation with deuterated substrates. The first set of sesquiterpenes, comprising (S)-β-bisabolene, (E)-γ-bisabolene and zingiberene, were not affected by the isotope sensitive branching experiments and minor changes in product formation were observed after incubation with hexadeuterated substrates. An important decrease in the formation of 7-epi-sesquithujene, sesquithujene, sesquisabinenes A and B and (E)- and (Z)-α-bergamotene isomers were observed from (2E,6E)- or (2Z,6E)-[2H6]-FDP (1d or 2d) (10–64% with TPS4 and 26–61% with TPS5, in comparison with unlabeled (2E,6E)-FDP or (2Z,6E)-[2H]-FDP 2c, respectively). This rate suppression was coupled with a corresponding enhancement in the formation rate of β- and γ-curcumene isomers, sesquithujene hydrate and 7-epi-sesquithujene hydrate (175–400% for TPS4 and 193–255% for TPS5, in comparison with unlabeled (2E,6E)-FDP or (2E,6Z)-[2H]-FDP 2c, respectively). Because of higher complexity involved in biosynthesis of sesquiterpene volatiles as compared to monoterpenes, only the observed KIEs for the deprotonation reactions leading to 7-epi-sesquithujene and sesquithujene were determined. The corresponding KIEs (kH/k1D)E were close to unity, demonstrating that the deuterium label at C(3) had almost no influence on the reaction cascade. The observed KIEs [(kH/k6D)E, (k1D/k6D)E or (k1D/k6D)Z] were in the range of kH/kD = 1.38–4.08 for the terminating deprotonation reaction leading to 7-epi-sesquithujene and kH/kD = 2.17–6.24 for the terminating deprotonation reaction leading to sesquithujene. Again as observed with monoterpenes the lowest KIEs ((k1D/k6D)Z) were calculated between (2Z)-substrates and were much lower as compared with natural isomers. The huge turnover difference and lower KIEs suggest that enzyme is much more efficient when incubated with (2Z)-substrates (2a–d) in spite of isotope effects.β-Secondary KIEs and hyperconjugation
Terpene cyclization cascades catalyzed by cyclases involve the internal additions to remaining double bond, hydride shifts or rearrangements of highly reactive carbocations before their ultimate quenching by deprotonation or nucleophile capture. We had previously proposed12 a reaction mechanism for the formation of mono- and sesquiterpene products by TPS4 and TPS5 consistent with the carbocationic mechanisms described for other sesquiterpene synthases.25 Stabilization of the carbocationic intermediates is partly ensured by interactions with the hydrophobic, aromatic-rich active-site of the enzyme (e.g. π–cation interactions with aromatic residues of the active site).26 Nevertheless, hyperconjugative interactions within carbocations themselves also play an important role in their stability. In molecular orbital terms, hyperconjugation is described as the interaction of the vacant p-type orbital on the cationic center with adjacent C–H or C–C σ-bonds.20 Magnitude of this hyperconjugative effect depends on the number of hydrogen atoms attached to the carbon atom immediately adjacent to the unsaturated system. Because the energy required for breaking a C–D bond is higher than that for a C–H bond, a C–D hyperconjugation stabilizes an adjacent positive charge less than a C–H hyperconjugation. Consequently, reactions in which C–D bonds are broken proceed more slowly than reactions in which C–H bonds are broken. Such hyperconjugative weakening in intermediates due to isotope substitution induces secondary kinetic isotope effects. In the present study, these secondary KIEs combine with the primary KIEs and alter the product distributions after isotopically sensitive branching with both enzymes.To illustrate the effects of hyperconjugation and the secondary KIEs, the case of (2Z)-[2H6]-GDP 2b is depicted in Fig. 5 as a representative example. From (2Z)-[2H6]-GDP 2b (Fig. 5), there is distinct lack of acyclic products due to the lack of an isomerization step. The cyclization cascade is initiated by the formation of the (S)- and (R)-terpinyl carbocations A1 and A2. Deuterium isotope effects on the monoterpene product distribution can rationalized in terms of hyperconjugation The positive charge is positioned far from the area of the deuterated carbons, hence deprotonation or water capture terminating steps leading to (S)-(−)-limonene and α-terpineol or α-terpinolene are not influenced by kinetic isotope effects. The minor KIEs observed for limonene, α-terpinolene and α-terpineol reflect the low destabilizing influence of the labeled carbon center in the two α-terpinyl carbocations. From A1 and A2 the cyclization cascade can proceed with the formation of carbocation B followed by subsequent rearrangement into the tertiary highly unstable carbocations C1 and C2. Here, the positive charge is fully surrounded by deuterium atoms and consequently less stabilized by C–H hyperconjugation than in the corresponding unlabeled compound. This leads to comparatively higher deuterium isotope effects on the formation of sabinene, sabinene hydrate and α-thujene. This leads to decreased deprotonation and higher formation of sabinene hydrate after capture by water molecule.
 | ||
| Fig. 5 Proposed reaction mechanism for the formation of monoterpenes by TPS4 and TPS5 from (2Z)-[2H6]-GDP (2b) and (2E)-[2H6]-GDP (1b). | ||
Similar considerations apply to the formation of sesquiterpenes by maize TPS4 and TPS5. From (2Z,6E)-[2H6]-FDP 2d, few acyclic products are formed and the cyclization cascade is initiated by the formation of (S)- and (R)-bisabolyl cations. These first carbocations can be directly deprotonated to produce (S)-β-bisabolene without noticeable KIEs. These observations are consistent with the product distribution obtained from isotopically sensitive branching experiments. Deuterium isotope effects are less pronounced in the case of the monodeuterated analogue 2cvs. hexadeuterated analogue 2d since the positive charge can at most be surrounded by only one deuterium atom, with the other hydrogen atoms being able to undergo C–H hyperconjugative interactions. Higher KIEs were obtained for the formation of sesquithujene, 7-epi-sesquithujene or sesquisabinenes A and B after incubation with the hexadeuterated (2Z)-substrate (2d) since these products are formed from carbocations that are fully surrounded by deuterium labeled carbons. This results in higher KIEs as the final product formation can only proceed via isotope labeled centers. In case of bisabolyl carbocations, minor KIEs were observed for β or γ-bisabolene formation because the positive charge is not disturbed by the deuterium labeled carbon center. Similarly, slight KIEs were observed for zingiberene isomers because carbocations were not highly destabilized by deuterium substitution. These experiments clearly show that isotope effects follow the same patterns within both the geometric isomers. Thus, (2Z) substrate geometry provides an advantage in the early steps of reaction cascade that accounts for higher turnover and selection towards cyclic products.
A major determinant of product selectivity is the degree of conformational flexibility of the substrate in the active site of a terpene synthase. To explore the structural basis for the proposed reaction mechanism of maize TPS4, we had modeled the protein structure of this enzyme (TPS4) using the data available for 5-epi-aristolochene synthase (TEAS).13 The active site cavity of TPS4 is divided into two pockets by the G2 helix which reaches slightly into the cavity. Previously, four of the amino acids that make up the G2 helix were shown to have a major impact on the product specificity of TPS4.12 Docking of the (E,E)-FDP substrate showed that the olefin moiety of the FDP substrate is located predominantly in one of the two pockets (pocket I). The early steps of the catalytic sequence including dephosphorylation, isomerization, and cyclization, up to the formation of the bisabolyl carbocation, all take place in pocket-I. When the bisabolyl carbocation adopts an alternate conformation, it shifts to pocket II. Then, a variety of additional cyclizations, hydride shifts, and deprotonations occur in pocket II leading to the formation of bicyclic products like 7-epi-sesquithujene. The energy requirement for the conformational change that drives pocket shifting is likely to be very small determined both by the chemical nature of the intermediate and the surrounding amino acids.13 This structural feature of having two pockets, is likely to be advantageous for initial interactions of enzyme with the deuterated (2Z)-GDP and (2Z,6E)-FDP tested in this study. Both of these have already undergone isomerization around the C(2)–C(3) bond, so the only activity left in pocket I is conversion to the corresponding carbocation. After these 2Z substrates cross the small energy barrier to pocket II, they are converted to a larger proportion of cyclic products than the (2E)-substrates. The enzyme likely has better efficiency with (2Z)-substrates because of lower energy requirements, as substrates are directly cyclized after ionization. This could explain not only the reduction in acyclic substrates and smaller kinetic isotope effects, but also the availability of more energy that can be utilized for processes in pocket II.
Recently, isotopically sensitive branching experiments have been used to identify the carbocation cascade reaction leading to the tricyclic sesquiterpene pentalenene which was first predicted using quantum chemical calculations.27 Deuterium isotope effects have also been used to study the reaction kinetics of initial dissociation of pyrophosphate moiety in sesquiterpene cyclization by tobacco epi-aristolochene synthase and monoterpene cyclization by pinene synthases from Salvia officinalis.15c,28 In this study we have utilized isotope sensitive branching experiments to study the effects of alternate substrate geometry around the C(2)–C(3) double bond on volatile production in multiproduct terpene synthase enzymes. The observation of increased turnover of cyclic products with (2Z)-substrate geometry may be useful in direction terpene synthase reaction cascades towards desired cyclic products.
Conclusion
In this work we investigate the effects of alternate substrate stereochemistry on the reaction mechanism of two maize multiproduct synthases that isomerize the C(2)–C(3) π bond of (2E,6E)-FDP via an NDP intermediate. We were interested in the impact of geometry as well as isotope effects on the product distribution of multiproduct enzymes. There was indeed major influence of the (2Z)-isomers on the enzymatic cascade as confirmed by deuterium labeling of products. The product distribution results showed a strong preference for cyclic products as a result of the alternate C(2)–C(3) geometry, indicating the rate limiting effects of the isomerization step in the natural biosynthesis of terpenes. There was also a major impact on efficiency with higher turnover and lower kinetic isotope effects observed with (2Z)-isomers as compared with natural (2E)-substrates. This can rationalized in terms that availability of a preferable conformer and lower energy requirements which increases the efficiency of catalysis. This strong preference for cyclic products and huge turnover can be exploited to direct biosynthesis by certain terpene synthases already known for their catalytic promiscuity.Acknowledgements
We thank Kerstin Ploss for the HRMS measurements, Stephan von Reuss and Stefan Garms for editorial assistance and Grit Winnefeld for administrative support. This work was supported by the Max Planck Society.References
- M. Wink, Phytochemistry, 2003, 64, 3 CrossRef CAS.
- (a) J. S. Dickschat, Nat. Prod. Rep., 2011, 28, 1917 RSC; (b) J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman & Hall, New York, 1992 Search PubMed.
- (a) E. Nambara and A. Marion-Poll, Annu. Rev. Plant Biol., 2005, 56, 165 CrossRef CAS PubMed; (b) E. Pichersky and J. Gershenzon, Curr. Opin. Plant Biol., 2002, 5, 237 CrossRef CAS; (c) J. H. Langenheim, J. Chem. Ecol., 1994, 20, 1223 CrossRef CAS PubMed.
- (a) T. G. Köllner, C. Schnee, J. Gershenzon and J. Degenhardt, Plant Cell, 2004, 16, 1115 CrossRef PubMed; (b) J. Gershenzon and R. B. Croteau, Lipid Metab. Plants, 1993, 339 CAS.
- (a) J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621 CrossRef CAS PubMed; (b) D. J. Schenk, C. M. Starks, K. R. Manna, J. Chappell, J. P. Noel and R. M. Coates, Arch. Biochem. Biophys., 2006, 448, 31 CrossRef CAS PubMed.
- (a) N. Tokuriki and D. S. Tawfik, Science, 2009, 324, 203 CrossRef CAS PubMed; (b) D. W. Christianson, Curr. Opin. Chem. Biol., 2008, 12, 141 CrossRef CAS PubMed.
- E. M. Davis and R. Croteau, in Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Springer-Verlag, Berlin, 2000, pp. 53–95 Search PubMed.
- D. B. Little and R. B. Croteau, Arch. Biochem. Biophys., 2002, 402, 1201 CrossRef.
- J. Bohlmann, G. Meyer-Gauen and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4126 CrossRef CAS.
- C. M. Starks, K. W. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815 CrossRef CAS.
- D. C. Williams, D. J. McGarvey, E. J. Katahira and R. Croteau, Biochemistry, 1998, 37, 12213 CrossRef CAS PubMed.
- T. G. Köllner, C. Schnee, J. Gershenzon and J. Degenhardt, Plant Cell, 2004, 16, 1115 CrossRef PubMed.
- T. G. Köllner, P. E. O'Maille, N. Gatto, W. Boland, J. Gershenzon and J. Degenhardt, Arch. Biochem. Biophys., 2006, 448, 83 CrossRef PubMed.
- N. Gatto, A. Vattekkatte, T. Kollner, J. Degenhardt, J. Gershenzon and W. Boland, Chem. Commun., 2015, 51, 3797 RSC.
- (a) I. Alchanati, J. A. A. Patel, J. Liu, C. R. Benedict, R. D. Stipanovic, A. A. Bell, Y. Cui and C. W. Magill, Phytochemistry, 1998, 47, 961 CrossRef CAS; (b) S. Picaud, P. Mercke, X. F. He, O. Sterner, M. Brodelius, D. E. Cane and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 448, 150 CrossRef CAS PubMed; (c) D. J. Schenk, C. M. Starks, K. R. Manna, J. Chappell, J. P. Noel and R. M. Coates, Arch. Biochem. Biophys., 2006, 448, 31 CrossRef CAS PubMed; (d) S.-H. Kim, K. Heo, Y.-J. Chang, S.-H. Park, S.-K. Rhee and S.-U. Kim, J. Nat. Prod., 2006, 69, 758 CrossRef CAS PubMed.
- (a) C. R. Benedict, J. L. Lu, D. W. Pettigrew, J. G. Liu, R. D. Stipanovic and H. J. Williams, Plant Physiol., 2001, 125, 1754 CrossRef CAS PubMed; (b) D. E. Cane, G. H. Yang, Q. Xue and J. H. Shim, Biochemistry, 1995, 34, 2471 CrossRef CAS; (c) L. S. Vedula, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 12719 CrossRef CAS PubMed; (d) L. S. Vedula, M. J. Rynkiewicz, H.-J. Pyun, R. M. Coates, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 6153 CrossRef CAS PubMed; (e) J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621 CrossRef CAS PubMed; (f) T. G. Köllner, C. Schnee, S. Li, A. Svatos, B. Schneider, J. Gershenzon and J. Degenhardt, J. Biol. Chem., 2008, 283, 20779 CrossRef PubMed.
- D. Arigoni, D. E. Cane, J. H. Shim, R. Croteau and K. a. Wagschal, Phytochemistry, 1993, 32, 623 CrossRef CAS.
- M. Kunert, P. Rahfeld, K. H. Shaker, B. Schneider, A. David, K. Dettner, J. M. Pasteels and W. Boland, ChemBioChem, 2013, 14, 353 CrossRef CAS PubMed.
- A. B. Woodside, Z. Huang and C. D. Poulter, Org. Synth., 1993, 8, 616 Search PubMed.
- D. E. Cane, H. T. Chiu, P. H. Liang and K. S. Anderson, Biochemistry, 1997, 36, 8332 CrossRef CAS PubMed.
- (a) P. E. O'Maille, J. Chappell and J. P. Noel, Arch. Biochem. Biophys., 2006, 448, 73 CrossRef PubMed; (b) T. G. Köllner, J. Gershenzon and J. Degenhardt, Phytochemistry, 2009, 70, 1139 CrossRef PubMed; (c) F. Lopez-Gallego, S. A. Agger, D. Abate-Pella, M. D. Distefano and C. Schmidt-Dannert, ChemBioChem, 2010, 11, 1093 CrossRef CAS PubMed.
- (a) J. A. Faraldos, P. E. O'Maille, N. Dellas, J. P. Noel and R. M. Coates, J. Am. Chem. Soc., 2010, 132, 4281 CrossRef CAS PubMed; (b) J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess, L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377 CrossRef CAS PubMed.
- (a) R. Croteau and D. M. Satterwhite, J. Biol. Chem., 1989, 264, 15309 CAS; (b) W. Schwab, D. C. Williams, E. M. Davis and R. Croteau, Arch. Biochem. Biophys., 2001, 392, 123 CrossRef CAS PubMed.
- (a) C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, J. Biol. Chem., 1998, 273, 2078 CrossRef CAS PubMed; (b) S. Picaud, M. E. Olsson, M. Brodelius and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 452, 17 CrossRef CAS PubMed; (c) J. Bohlmann, J. Crock, R. Jetter and R. a. Croteau, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6756 CrossRef CAS; (d) S. M. Colby, J. E. Crock, P. G. Lemaux and R. B. Croteau, Official Gazette of the United States Patent and Trademark Office Patents, 1254, 2002 Search PubMed.
- D. E. Cane, Compr. Nat. Prod. Chem., 1999, 2, 155 CrossRef CAS.
- C. A. Lesburg, J. M. Caruthers, C. M. Paschall and D. W. Christianson, Curr. Opin. Struct. Biol., 1998, 8, 695 CrossRef CAS.
- L. Zu, M. Xu, M. W. Lodewyk, D. E. Cane, R. J. Peters and D. J. Tantillo, J. Am. Chem. Soc., 2012, 134, 11369 CrossRef CAS PubMed.
- (a) K. C. Wagschal, H. J. Pyun, R. M. Coates and R. Croteau, Arch. Biochem. Biophys., 1994, 308, 477 CrossRef PubMed; (b) J. R. Mathis, K. Back, C. Starks, J. Noel, C. D. Poulter and J. Chappell, Biochemistry, 1997, 36, 8340 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Product distributions of main monoterpenes and sesquiterpenes from incubations of deuterated GDP and FDP, respectively, with TPS4 and TPS5. Synthetic procedure and 1H, 13C NMR spectra of compounds 3b, 3d, 4, 5a–d, 6a–d, 7a–d, 8a–d, 1-a–d and 2a–d and 31P NMR spectra of compounds 1-a–d and 2a–d. IR spectra of compounds 3b, 3d, 4, 5a–d, 6a–d, 1-a–d and 2a–d. See DOI: 10.1039/c5ob00711a |
| This journal is © The Royal Society of Chemistry 2015 |