
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and properties of unsymmetrical azatrioxa[8]circulenes†
Malene
Plesner
,
Thomas
Hensel
,
Bjarne E.
Nielsen
,
Fadhil S.
Kamounah
,
Theis
Brock-Nannestad
,
Christian B.
Nielsen
,
Christian G.
Tortzen
,
Ole
Hammerich
and
Michael
Pittelkow
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: pittel@kiku.dk
First published on 30th April 2015
Abstract
Insights to the subtle reactivity patterns of hydroxy-substituted carbazoles allows the precise synthesis of unsymmetrical azatrioxa[8]circulenes by the reaction of N-benzyl-2,7-di-tert-butyl-3,6-dihydroxycarbazole with two different 1,4-benzoquinones in the presence of an oxidant (chloranil) and a Lewis acid (BF3OEt2). The unique synthetic control obtained originates from the selectivity obtained upon reacting N-benzyl-2,7-di-tert-butyl-3,6-dihydroxycarbazole with an electron-rich benzoquinone to give first the C–C bond formation and then subsequently the dibenzofuran formation with high regioselectivity. Herein the first synthesis of unsymmetrical antiaromatic azatrioxa[8]circulenes and the full characterization using NMR spectroscopy, optical spectroscopy, electrochemistry, computational techniques and single crystal X-ray crystallography is reported. The controlled stepwise condensation of N-benzyl-2,7-di-tert-butyl-3,6-dihydroxycarbazole with two different 1,4-benzoquinones gives selectively the unsymmetrical azatrioxa[8]circulenes.
Introduction
Classical aromatic compounds such as benzene obey Hückel's 4n + 2 π-electron rule, while anti-aromatic compounds have 4n π-electrons.1,2 The idea of extending the π-conjugated framework of polycyclic aromatic- and heteroaromatic molecules has fascinated chemists for more than a century.3 An interest in the exploration of fundamental properties inspired early studies of structure–property relationships, while more recent work has focused on using π-conjugated compounds as the active materials in optical and electronic devices.3 Recent work by Haley4 on the preparation of indeno[1,2-b]fluorenes, by Osuka,5 Sessler6 and Latos-Grażyński7 on the preparation of porphyrioids, and by Bunz9 on the preparation of hetero-acenes have paved the way to studies of the properties of large polycyclic aromatic-antiaromatic conjugates.19Planarized conjugated cyclooctatetraenes (COTs) are among the classic antiaromatic moieties, and as part of heterocyclic [8]circulenes COTs have attracted the interest of Komatsu,8 Osuka,10 Nenajdenko,11 Wong,12 Christensen,13 Erdtman and Högberg,14 and us.15 We have previously used DTF based nuclear independent chemical shift (NICS) calculations on the diazadioxa[8]circulenes (1), azatrioxa[8]circulenes (2) and tetraoxa[8]circulenes (3) to address the question of the aromaticity/antiaromaticity of the central planarized COTs, and found that the COT indeed possess anti-aromatic character.16 Another feature that we have explored with heterocyclic [8]circulenes is their (blue) fluorescent properties. We have found that the azatrioxa[8]circulenes and the diazadioxa[8]circulenes fluoresce in the blue region making them attractive for application in light emitting devices.17 Aggregation of π-conjugated systems tends to shift the fluorescence from the blue to the green region, and we have found it useful to functionalize the [8]circulenes with tert-butyl groups to modulate aggregation behaviour.18
The previously described synthetic methodologies developed for heterocyclic [8]circulenes limit the range of compounds available to symmetrical structures. Preparing unsymmetrical π-extended heterocyclic[8]circulenes may open the possibility of systematically studying the interplay between aromatic and antiaromatic moieties in large π-conjugated frameworks. In this paper we showcase a new synthetic protocol towards this goal, and we report on the spectroscopic, computational and electrochemical properties. The work described in this paper focuses on utilizing the intrinsic reactivity of hydroxyl-substituted carbazoles for the construction of azatrioxa[8]circulenes. We have found that we can modulate the reactivity of the hydroxyl-substituted carbazoles by careful choice of benzoquinone reaction partners, and thus opening the pathway for construction of complex heterocyclic [8]circulenes.
Results and discussion
The synthetic strategy is built upon the observation that 1,4-benzoquinones tetramerize by treatment with acid to yield tetraoxa[8]circulenes 3via a 3,6-dihydroxydibenzofuran intermediate.17 Recently we applied a similar logic and condensed a 3,6-dihydroxycarbazole with two equivalents of suitable substituted 1,4-benzoquinones in the presence of a Lewis acid to give azatrioxa[8]circulenes (2, Scheme 1, middle) in high yields.15a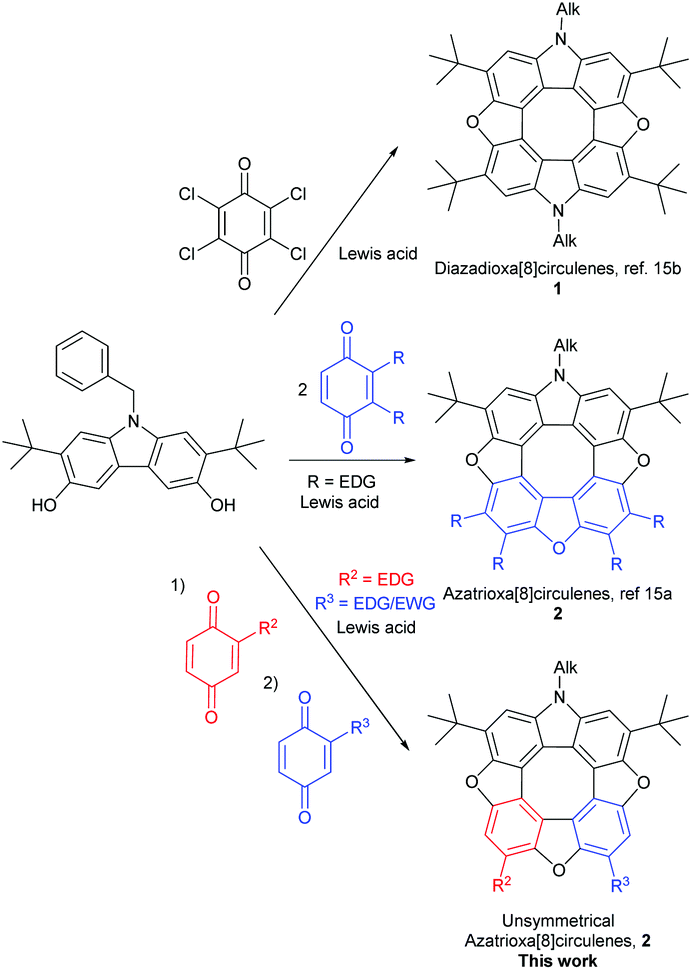
We found that the 3,6-dihydroxycarbazole may be dimerized under oxidative conditions to yield diazadioxa[8]circulenes 1 by simply changing from a 1,4-benzoquinone substituted with electron donating substituents to 1,4-benzoquinones substituted with electron withdrawing substituents (1, Scheme 1, top).15b It is important that the 1,4-benzoquinones are substituted in the 2- and 3-positions and that the 3,6-dihydroxycarbazole is substituted in the 2- and 7-positions, as otherwise the reaction gives uncontrolled polymerisation.
When N-benzyl-2,7-di-tert-butyl-3,6-dihydroxycarbazole (4) is treated with only one equivalent of a 1,4-quinone that does not cause it to dimerize (the benzoquinone should not have an electron-withdrawing substituent) to the diazadioxa[8]circulene (1) the reaction mixture contains mainly the product where one C–C bond has been formed (5, Scheme 2, top).
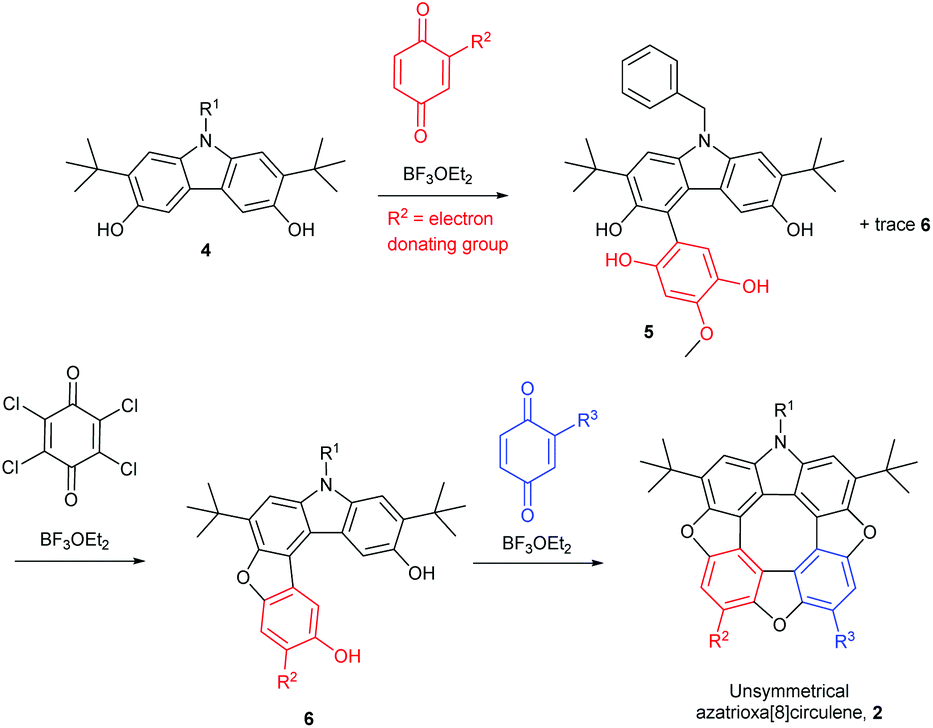 | ||
| Scheme 2 Pathway to unsymmetrical azatrioxa[8]circulene. R2 of the first 1,4-benzoquinone must be an electron donating group, otherwise the diazadioxa[8]circulene (1) is the main product. | ||
Addition of one equivalent of an oxidant such as chloranil to the tetrahydroxy compound 5 in the presence of BF3OEt2 yields the ring-closed furan 6. Subsequent addition of a second 1,4-quinone to this material gives the unsymmetrical azatrioxa[8]circulene 2 in a process where two new C–C bonds and two furan rings are formed (Scheme 2, bottom).
We have found that when using either 2-tert-butyl-1,4-benzoquinone or 2-methoxy-1,4-benzoquinone only one regioisomer of the tetrahydroxy compound 5 was formed, and as a consequence only one regioisomer of the dibenzofuran 6 is formed after oxidation. Thus, the R2 group is situated in the position indicated in Scheme 2 (red). When treating the tert-butyl derivative 5 generated in situ with either 2-methoxy-1,4-benzoquinone (43%), 2-thiododecyl-1,4-benzoquinone (35%, 2-tert-butyl-1,4-benzoquinone (13%) or 1,4-naphthoquinone (31%) we isolate the unsymmetrical azatrioxa[8]circulenes (2, Fig. 1) in good yields.
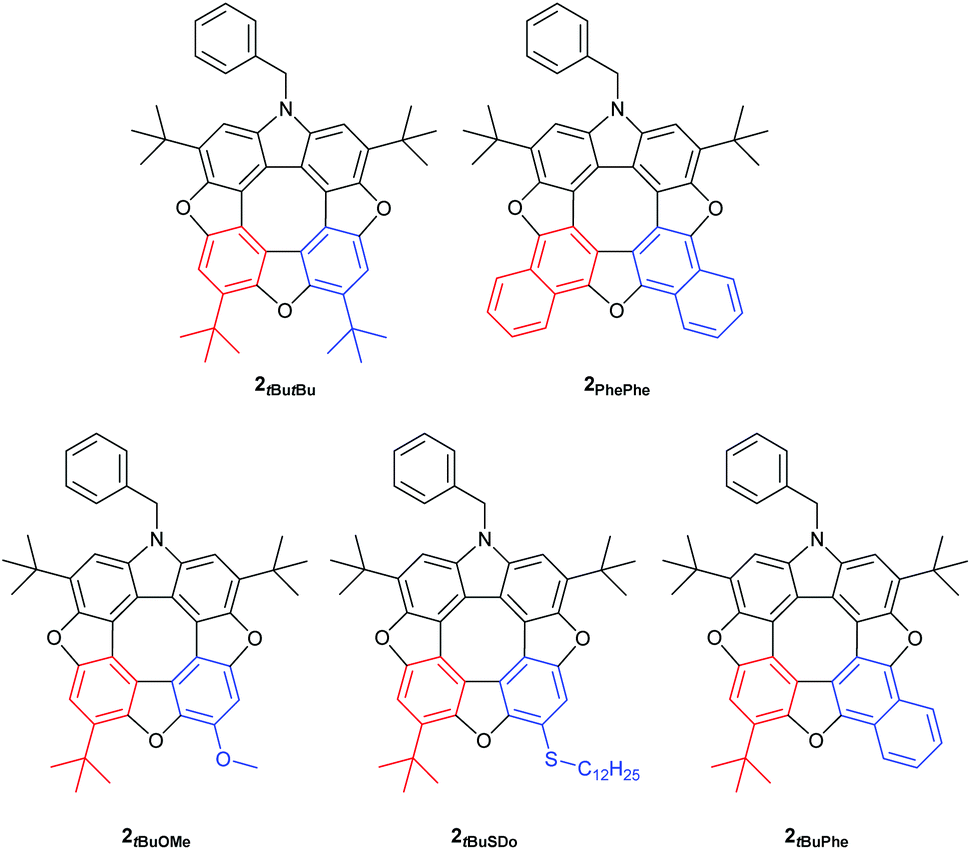 | ||
| Fig. 1 Azatrioxa[8]circulenes (2) described in this paper. | ||
In all three cases where unsymmetrical azatrioxa[8]circulenes are made, one regioisomer is formed as the main product. This is a remarkable control of regiochemistry in a reaction protocol where three C–C bonds and three C–O bonds are formed while three molecules of water are eliminated. When starting by reacting the N-benzyl-2,7-di-tert-butyl-3,6-dihydroxycarbazole (4) with 2-methoxy-1,4-benzoquinone followed by 2-tert-butyl-1,4-benzoquinone it gave exactly the same azatrioxa[8]circulene product as when the reaction was performed with the reverse order, as seen by 1H-NMR analysis of the isolated pure product. We have also found that if the sequence is started by reacting the N-benzyl-2,7-di-tert-butyl-3,6-dihydroxycarbazole (4) with 1,4-naphthoquinone, and then with 2-tert-butyl-1,4-benzoquinone, then we obtained a mixture of the two azatrioxa[8]circulene regioisomers with respect to the tert-butyl group originating from the 2-tert-butyl-1,4-benzoquinone.
The unsymmetrical azatrioxa[8]circulenes were all crystalline compounds, however we were not able to grow crystals with large enough dimensions to be suitable for single crystal X-ray crystallography. We were able to grow crystals of the C2-symmetrical azatrioxa[8]circulene (2tButBu, Fig. 2). The single crystal X-ray structure confirms outcome of the azatrioxa[8]circulenes synthesis and also the regiochemical outcome of the reaction forming the azatrioxa[8]circulene with respect to the tert-butyl groups originating from the 2-tert-butyl-benzoquinones. The azatrioxa[8]circulene is planar with respect to the π-conjugated system and due to the four tert-butyl substituents no intermolecular π–π-stacking is observed.
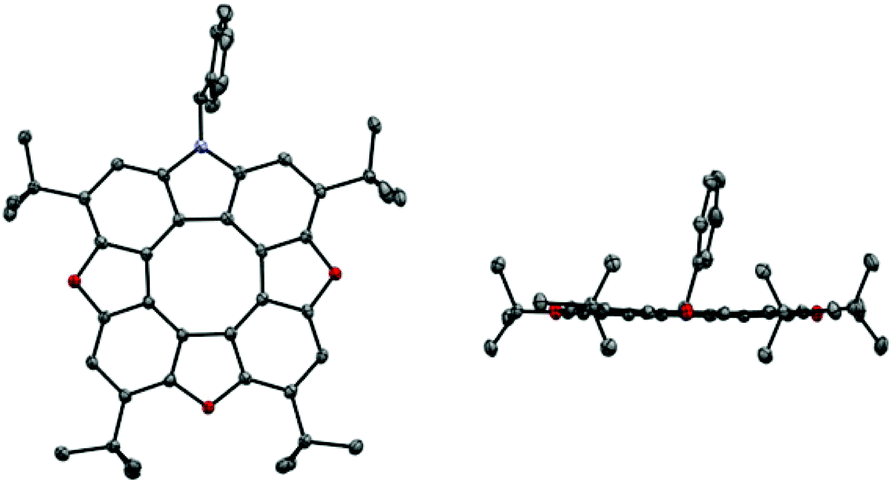 | ||
| Fig. 2 Single crystal X-ray structure of azatrioxa[8]circulene (2tButBu). Hydrogens are omitted for clarity. Left: top view. Right: side view. | ||
We have previously performed NICS calculations on planar heterocyclic [8]circulenes (1–3) in order to address the question of the antiaromaticity of the central planarized COTs.15,16 NICS(0) and NICS(1)zz values for 2tBuOMe were determined at the B3LYP/6-311+G(d,p) level of theory using geometries obtained from B3LYP/6-31G(d) confirming the antiaromatic nature of the COT (Fig. 3).
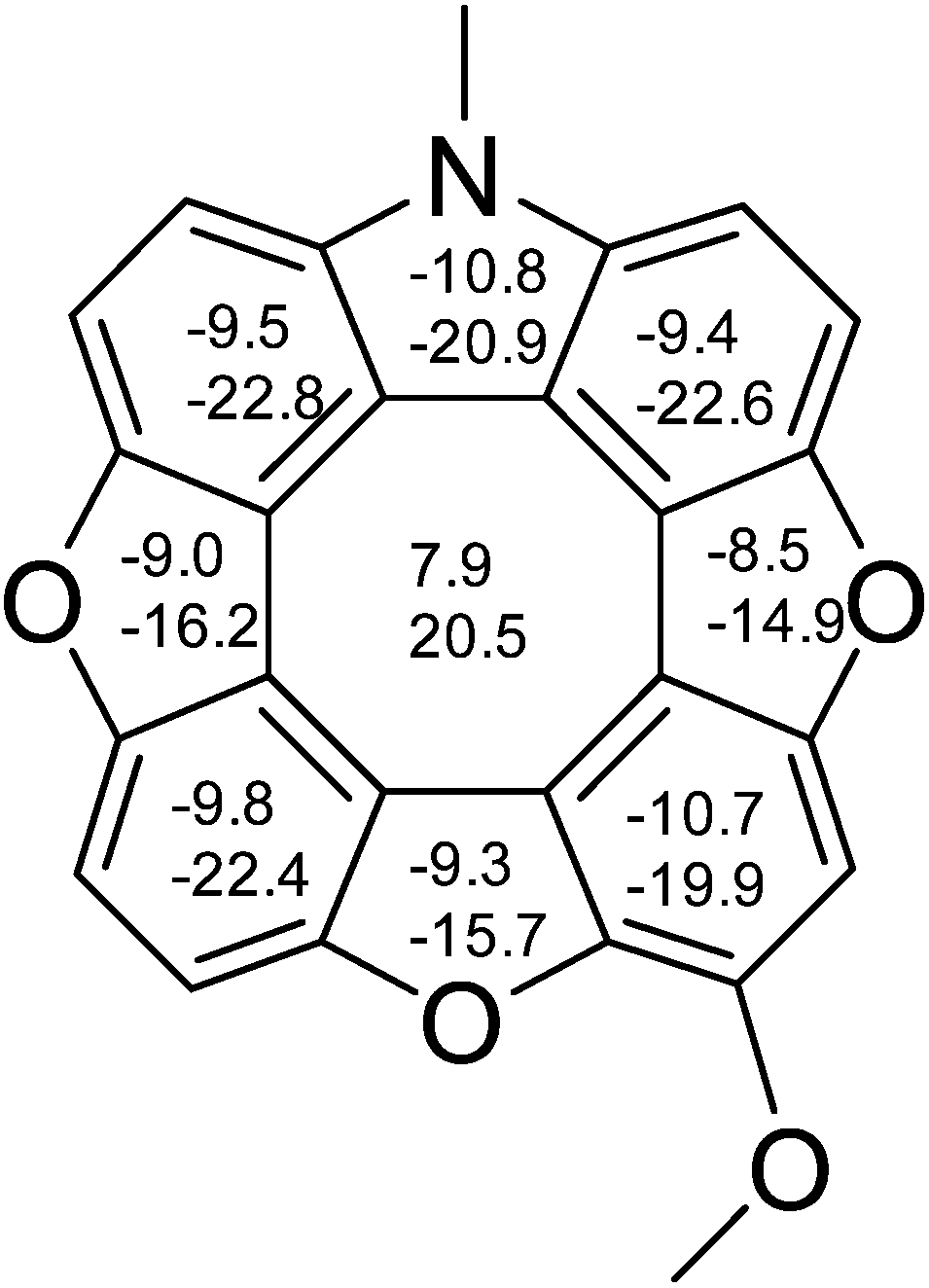 | ||
| Fig. 3 NICS(0) and NICS(1)zz (lower numbers) values calculated for the methoxy substituted azatrioxa[8]circulene (2). | ||
The UV/Vis and fluorescence spectra of the series of new azatrioxa[8]circulene 2 were recorded to assess whether these derivatives would be suitable for applications as the fluorescent component in blue OLEDs (Fig. 4). Three common electronic transitions are centered at ∼260 nm (∼4.8 eV), ∼360 nm (∼3.4 eV) and at ∼425 nm (∼2.9 eV) with varying intensities.
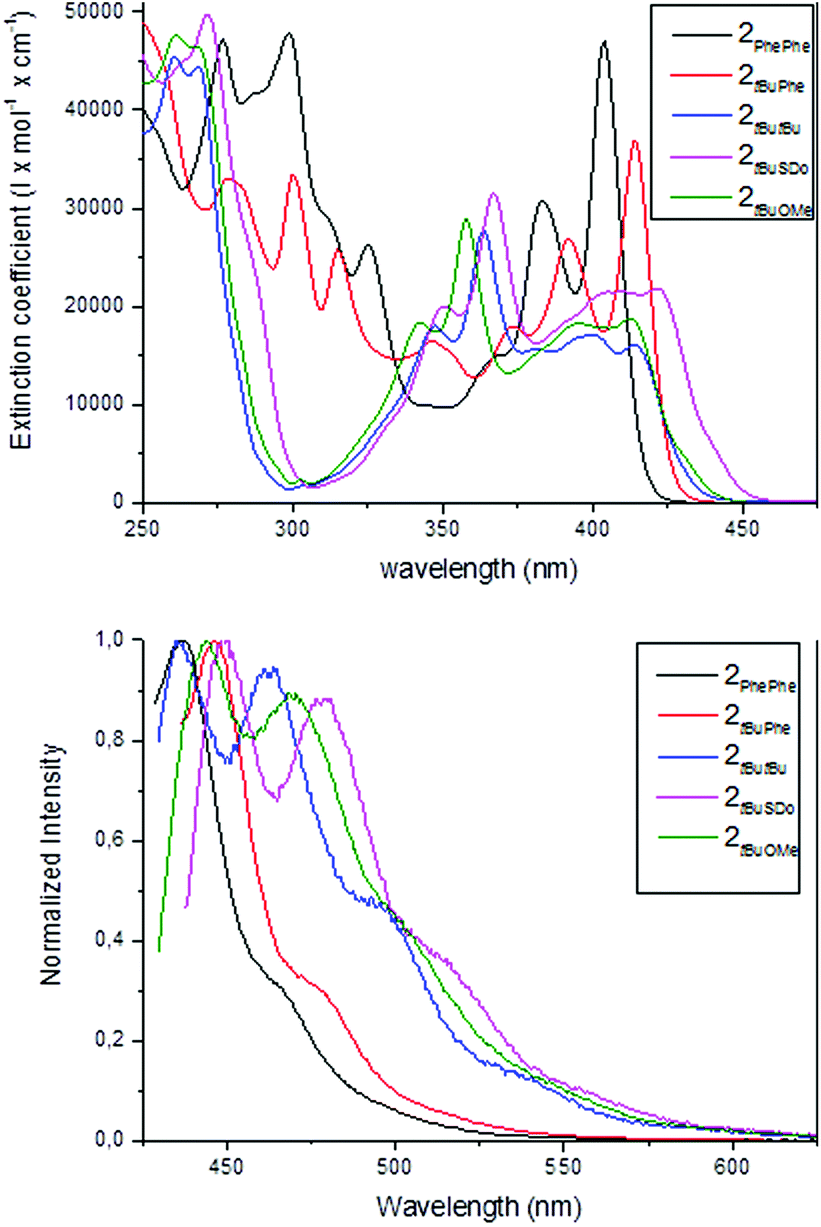 | ||
| Fig. 4 UV/Vis (top) and normalized fluorescence spectra (bottom) of the series of azatrioxa[8]circulenes (CH2Cl2). | ||
The oscillator strength and Franck–Condon factors for each transition are not identical when comparing the transitions at the low energy transition. The transition at ∼425 nm is weak in intensity in the spectrum of the three azatrioxa[8]circulenes without a naphthalene units (extinction coefficient of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 M−1 cm−1) compared to the spectra of the two azatrioxa[8]circulenes with either one or two naphthalene moieties (extinction coefficients of ∼35000–45000 M−1 cm−1). The two C2 symmetrical azatrioxa[8]circulenes (2PhePhe and 2tButBu) have a slightly blue-shifted fluorescence as compared to the three unsymmetrical azatrioxa[8]circulenes. Again the two azatrioxa[8]circulenes containing naphthalene units (2PhePhe and 2tBuPhe) give fluorescence spectra with comparable features, but the spectrum of the compound containing two naphthalenes is blue-shifted by ca. 20 nm. The fluorescence spectra of the three unsymmetrical azatrioxa[8]circulenes with identical π-systems (2tBuOMe, 2tBuSDo and 2tButBu) have similar features, with the high-energy bands of the 2tBuOMe (10 nm) and 2tBuSDo (15 nm) slightly redshifted as compared to 2tButBu. We also determined the fluorescence quantum yields for the series, and the quantum yields increase significantly with the increasing area of the π-conjugated system (Table 1).
000–20000 M−1 cm−1) compared to the spectra of the two azatrioxa[8]circulenes with either one or two naphthalene moieties (extinction coefficients of ∼35000–45000 M−1 cm−1). The two C2 symmetrical azatrioxa[8]circulenes (2PhePhe and 2tButBu) have a slightly blue-shifted fluorescence as compared to the three unsymmetrical azatrioxa[8]circulenes. Again the two azatrioxa[8]circulenes containing naphthalene units (2PhePhe and 2tBuPhe) give fluorescence spectra with comparable features, but the spectrum of the compound containing two naphthalenes is blue-shifted by ca. 20 nm. The fluorescence spectra of the three unsymmetrical azatrioxa[8]circulenes with identical π-systems (2tBuOMe, 2tBuSDo and 2tButBu) have similar features, with the high-energy bands of the 2tBuOMe (10 nm) and 2tBuSDo (15 nm) slightly redshifted as compared to 2tButBu. We also determined the fluorescence quantum yields for the series, and the quantum yields increase significantly with the increasing area of the π-conjugated system (Table 1).
| Compound | Quantum yielda | Oxidation potentialb |
|---|---|---|
| a Quantum yields determined in CH2Cl2. b Formal potentials determined by cyclic voltammetry as the average of Eoxp and Eredp for the first oxidation wave. Solvent: dichloromethane, supporting electrolyte: (nBu)4PF6 (0.1 M), voltage sweep rate: 0.1 V s−1, working electrode: glassy carbon (d = 3 mm). | ||
| 2PhePhe | 0.89 | 0.67 V |
| 2tButBu | 0.31 | 0.67 V |
| 2tBuOMe | 0.17 | 0.62 V |
| 2tBuSDo | 0.28 | 0.66 V |
| 2tBuPhe | 0.67 | 0.65 V |
The optical data suggests that the π-extended azatrioxa[8]circulenes may be suitable candidates as the fluorescent component in light emitting devices, as this derivative fluoresce in the blue region and it does not aggregate in solution. Analysis of the crystal structure of 2PhePhe15a suggests that this derivative aggregates in the solid state, so despite having the highest quantum yield of fluorescence it may be more desirable to utilize the 2tBuPhe derivative in light emitting devices.
The electrochemical oxidation of the five compounds was studied by cyclic voltammetry in dichloromethane with (nBu)4PF6 (0.1 M) as the supporting electrolyte. Reversible one-electron oxidations corresponding to the formation of persistent radical cations were observed for 2tBuOMe, 2tBuSDo, 2tBuPhe and 2tButBu (Fig. S14†). In the case of 2PhePhe, the oxidation process appeared as a double peak composed of two closely spaced electron transfer processes (Fig. 5). The voltammogram gradually developed into that for a simple one-electron transfer upon dilution. In contrast, the double peak behaviour became increasingly pronounced with decreasing temperature (Fig. S15†). Similar observations were recently reported for indenofluorene-extended tetrathiafulvalenes and were shown to originate from the formation of radical cation-neutral and radical cation-radical cation associates.20 It is likely that similar associates result from the oxidation of 2PhePhe that in contrast to the four other members of the series has one dibenzofuran subunit not carrying a space filling tert-butyl group, but rather an extended π-system resulting from the presence of two additional benzene rings. The values of the formal potentials for the one-electron oxidations are summarized in Table 1 and it is seen that the minor structural changes that distinguish the five azatrioxa[8]circulenes are reflected by oxidation potentials that are very similar. A second oxidation wave corresponding to the subsequent formation of the dication was observed close to the solvent wall for all compounds (Fig. S16†). For 2tBuSDo the dication was observed to react with residual water; deprotonation resulted in the formation of the corresponding S-oxide that was further oxidized to a persistent radical cation at the potential of the second wave (Fig. S17†).
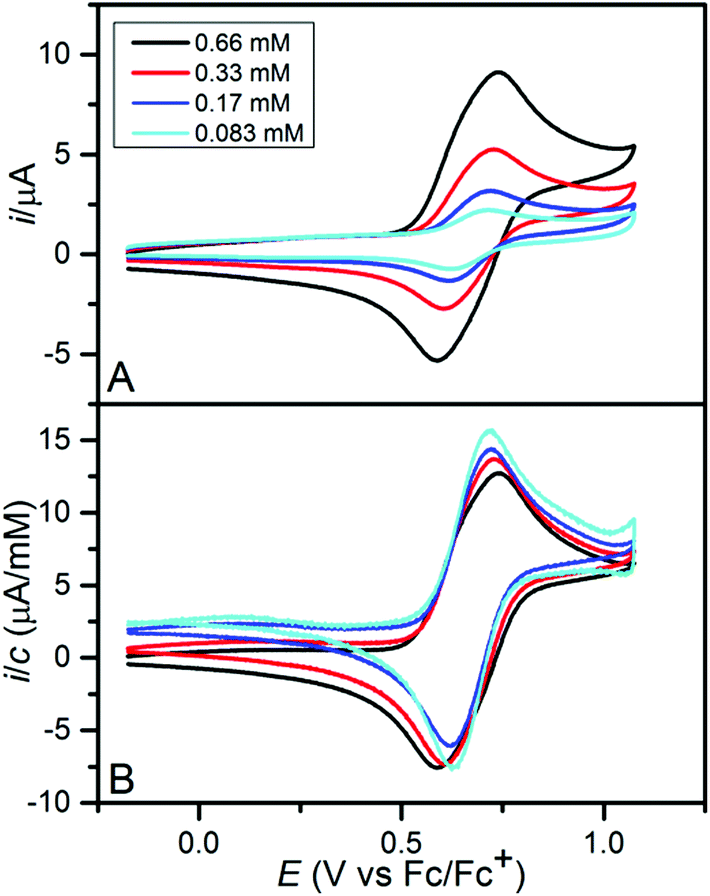 | ||
| Fig. 5 The influence of the substrate concentration (see the insert) on the shape of the cyclic voltammogram for 2PhePhe. (A) the raw data and (B) the same data after background subtraction and normalization with respect to the substrate concentration. The working electrode was a circular glassy carbon disk (d = 3 mm); the voltage sweep rate was 0.1 V s−1. | ||
Conclusion
In summary, we have developed a novel synthetic method for the preparation of a series of unsymmetrical azatrioxa[8]circulenes and have demonstrated the feasibility of functionalising fully conjugated, formally antiaromatic azatrioxa[8]circulenes. Through X-ray crystallography and DFT calculations we have visualised the planar COT core and confirmed the structure of 2 by comparison to the other known molecules that contain this moiety. Examination of the optoelectronic properties of the series of azatrioxa[8]circulenes unambiguously confirms that the optical properties of this antiaromatic framework can modulated by careful design.Experimental section
All chemicals and solvents, unless otherwise stated, were purchased from commercial suppliers and used as received. Analytical thin layer chromatography (TLC) was performed on SiO2 plates and visualized under UV-light (254 or 360 nm). Dry column vacuum chromatography was carried out using SiO2 (60 Å, 15–40 μm). NMR spectra were recorded on a 500 MHz spectrometer. Temperature dependent NMR spectra were recorded on a 300 MHz spectrometer. 1H NMR and 13C NMR spectra were recorded at 500 MHz and 125 MHz, respectively, on a 500 spectrometer using residual non-deuterated solvent as the internal standard. All chemical shifts (δ) are quoted in ppm and all coupling constants (J) are expressed in Hertz (Hz). The following abbreviations are used for convenience in reporting the multiplicity for NMR resonances: s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. Samples were prepared using CDCl3 or DMSO-d6. Assignment of all 1H and 13C resonances was achieved using standard 2D NMR techniques as 1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC. Elemental analysis was conducted at the Department of Chemistry, University of Copenhagen. MALDI-TOF MS was recorded using dithranol as matrix. The MALDI HRMS was recorded using dithranol as matrix, and calibration was performed externally, using sodium trifluoroacetate cluster ions. UV-Vis and fluorescence spectra were measured in CH2Cl2. Cyclic voltammetry was carried out at 5×10−4 mol L−1. (nBu)4NPF6 (1 M) was used as supporting electrolyte. The working electrode was a circular glassy carbon disk (d = 3 mm). A Pt-wire served as the counter electrode and the reference electrode was Ag/Ag+. The potential scale was subsequently calibrated by recording the potential of the ferrocene/ferrocenium redox couple. The voltage sweep rate was 0.1 V s−1 and all experiments were carried out at room temperature unless otherwise stated. The geometry optimization was performed by the DFT B3LYP/6-31G(d) level using the GAUSSIAN 03 package. Based on the calculated coordinates of the ring critical points of the (3, +1) type the NICS (0) and NICS(1) indices are calculated by the B3LYP/6-311+G(d,p) level with the gauge-independent atomic orbital (GIAO) approach. Negative NICS values in the center of the ring corresponds to the presence of the “aromatic” diatropic ring currents, whereas positive NICS values at the same points denote the “antiaromatic” paratropic ring currents.2-Methoxy-1,4-benzoquinone
In a 100 ml round bottomed flask was placed silica for flash column chromatography (6 g) and the flask was equipped with a magnet and sealed with a rubber septum. Cerium ammonium nitrate (2.75 g: 5.02 mmol) dissolved in water (3 ml) was added drop wise whilst the silica was stirred using magnetic stirring. The silica was stirred for an additional 5 min. CH2Cl2 (25 ml) was added followed by the addition of 2-methoxy-1,4-hydroquinone (280 mg: 2.00 mmol) suspended in CH2Cl2 (5 ml). The reaction mixture was stirred for 30 minutes and filtered through a sintered glass funnel. The filter cake was washed with CH2Cl2 (3 × 50 ml) and the solvent was removed from the combined filtrates yielding 2-methoxy-1,4-benzoquinone, fine yellow powder.Yield: 96% (266 mg: 1.92 mmol) melting point: 47–49 °C TLC: Rf-value = 0.26 (CH2Cl2) 1H-NMR (500 MHz, CDCl3): δ = 6.71 (m, 2H), 5.95 (s, 1H), 3.83 (s, 3H) ppm. 13C-NMR (126 MHz, CDCl3): δ = 187.6 (Cq), 181.9 (Cq), 158.8 (Cq), 137.4 (CH), 134.6 (CH), 107.9 (CH), 56.4 (–OCH3) ppm. Elemental analysis: C: 60.90%, H: 4.10%, N: 0% (theoretical – C: 60.87%, H: 4.38% N: 0%). GC-MS: calcd for C7H6O3 [M]+m/z 138.0, found 138.1.
Yield: 82.5% (773 mg: 1.92 mmol) melting point: 205–209 °C TLC: Rf-value = 0.35 (MeCN/toluene, 1:8) 1H-NMR (500 MHz, DMSO): δ = 8.87 (s, 2H, 2 × ROH), 7.28 (t, J = 7.4 Hz, 2H), 7.24 (s, 2H), 7.23 (s, 2H), 7.22–7.17 (m, 3H), 5.46 (s, 2H, R2NCH2Ph), 1.40 (s, 18H, 2 × RC(CH3)3) ppm. 13C-NMR (126 MHz, DMSO): δ = 149.2 (Cq), 138.7 (Cq), 134.8 (Cq), 134.6 (CH), 128.5 (Cq), 127.1 (CH), 126.9 (CH), 119.7 (CH), 106.7 (CH), 105.6 (CH), 45.7 (CH2), 35.0 (Cq), 29.7 (CH3) ppm. Elemental analysis: C: 81.00%, H: 7.76%, N: 3.41% (theoretical – C: 80.76%, H: 7.78% N: 3.49%), MALDI HRMS: calcd for C27H31NO2 [M]+m/z 401.23548, found 401.23464.
Yield: 49% (309 mg: 0.48 mmol) melting point: 304–308 °C TLC: Rf-value = 0.57 (toluene/n-heptane, 1:4) 1H-NMR (500 MHz, CDCl3): δ = 8.71–8.63 (m, 4H), 7.77–7.70 (m, 4H), 7.49 (s, 2H), 7.36–7.29 (m, 5H), 5.81 (s, 2H), 1.78 (s, 18H) ppm. 13C-NMR (126 MHz, CDCl3): δ = 150.5 (Cq), 148.9 (Cq), 148.8 (Cq), 137.9 (Cq), 137.6 (Cq), 134.1 (Cq), 129.0 (CH), 127.7 (CH), 126.9 (CH), 126.2 (CH), 126.0 (CH), 122.2 (CH), 121.9 (CH), 121.0 (Cq), 120.8 (Cq), 117.4 (Cq), 114.1 (Cq), 113.1 (Cq), 112.6 (Cq), 104.9 (CH), 47.8 (CH2), 35.5 (C(CH3)3), 30.7 (R-CH3) ppm. MALDI HRMS: calcd for C47H35NO3 [M]+m/z 661.26169, found 661.26095.
Yield: 13% (45 mg: 0.06 mmol) melting point: 245–248 °C TLC: Rf-value = 0.69 (toluene/n-heptane, 1:4) 1H-NMR (500 MHz, CDCl3): δ = 7.73 (s, 2H), 7.39 (s, 2H), 7.36–7.27 (m, 5H), 5.75 (s, 2H), 1.77 (s, 18H), 1.69 (s, 18H) ppm. 13C-NMR (126 MHz, CDCl3): δ = 152.9 (Cq), 151.3 (Cq), 151.1 (Cq), 138.0 (Cq), 137.3 (Cq), 134.8 (Cq), 134.3 (Cq), 129.0 (CH), 127.7 (CH), 126.9 (CH), 117.1 (Cq), 116.8 (Cq), 115.1 (Cq), 112.7 (Cq), 107.4 (CH), 105.5 (CH), 47.8 (CH2), 35.4 (Cq), 35.3 (Cq), 30.5 (CH3) 30.4 (CH3) ppm. MALDI HRMS: calcd for C47H47NO3 [M]+m/z 673.35559, found 673.35398.
Yield: 31% (60 mg: 0.09 mmol) melting point: 300–303 °C TLC: Rf -value = 0.66 (toluene/n-heptane, 1:4) 1H-NMR (500 MHz, CDCl3): δ = 8.71–8.66 (m, 2H, ArH), 7.79 (s, 1H), 7.76 (dt, J = 6.3, 3.5 Hz, 2H), 7.47 (s, 1H), 7.46 (s, 1H), 7.36–7.28 (m, 5H), 5.79 (s, 2H), 1.83 (s, 9H), 1.78 (s, 9H), 1.71 (s, 9H) ppm. 13C-NMR (126 MHz, CDCl3): δ = 153.2 (Cq), 151.3 (Cq), 151.0 (Cq), 150.5 (Cq), 149.2 (Cq), 148.8 (Cq), 138.0 (Cq), 137.6 (Cq), 137.3 (Cq), 135.0 (Cq), 134.3 (Cq), 134.1 (Cq), 129.0 (CH), 127.7 (CH), 126.9 (CH), 126.4 (CH), 126.3 (CH), 122.2 (CH), 122.2 (CH), 121.4 (Cq), 121.1 (Cq), 117.7 (Cq), 117.4 (Cq), 116.8 (Cq), 115.0 (Cq), 113.3 (Cq), 113.2 (Cq), 112.7 (Cq), 112.7 (Cq), 106.8 (CH), 105.1 (2 × CH), 47.8 (CH2), 35.6 (Cq), 35.5 (Cq), 35.4 (Cq), 30.7 (CH3), 30.6 (CH3), 30.4 (CH3) ppm. MALDI HRMS: calcd for C47H41NO3 [M]+m/z 667.30864, found 667.30724.
Yield: 43% (139 mg: 0.21 mmol) melting point: 326–329 °C TLC: Rf -value = 0.66 (toluene/n-heptane, 1:4) 1H-NMR (500 MHz, CDCl3): δ = 7.75 (s, 1H), 7.39 (s, 1H), 7.36 (s, 1H), 7.34 (s, 1H), 7.33–7.27 (m, 5H), 5.74 (s, 2H), 4.25 (s, 3H, O-CH3), 1.76 (s, 9H), 1.68 (s, 9H) ppm. 13C-NMR (126 MHz, CDCl3): δ = 153.3 (Cq), 153.0 (Cq), 151.8 (Cq), 151.1 (Cq), 150.8 (Cq), 145.8 (Cq), 142.7 (Cq), 138.0 (Cq), 137.5 (Cq), 137.3 (Cq), 135.3 (Cq), 134.2 (Cq), 134.2 (Cq), 129.0 (CH), 127.6 (CH), 126.8 (CH), 117.6 (Cq), 117.2 (Cq), 117.0 (Cq), 116.6 (Cq), 115.3 (Cq), 112.7 (Cq), 112.3 (Cq), 110.0 (Cq), 107.8 (CH), 105.5 (CH), 104.4 (CH), 94.9 (CH), 57.2 (O–CH3), 47.7 (CH2), 35.5 (Cq), 35.4 (2 × Cq), 30.5 (CH3), 30.5 (CH3), 30.4 (CH3) ppm. MALDI HRMS: calcd for C44H41NO4 [M]+m/z 647.30356, found 647.30208.
Acknowledgements
We acknowledge financial support from the Lundbeck Foundation for a Young Group Leader Fellowship (MP), the Villum foundation for a diffractometer for determining the single crystal X-ray structures and the Danish high technology foundation. We thank Dr Sophie R. Beeren for insightful discussions.Notes and references
- R. Breslow, Acc. Chem. Res., 1973, 6, 393–398 CrossRef CAS.
- Carbon-Rich Compounds, ed. M. M. Haley and R. R. Tykwinski, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) Functional Organic Materials, ed. T. J. J. Muller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) Organic Light Emitting Devices: Synthesis Properties and Applications, ed. K. Müllen and U. Scherf, Wiley-VCH, Weinheim, 2006 Search PubMed.
- D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 11103–11106 CrossRef CAS PubMed.
- S. Mori and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8030–8031 CrossRef CAS PubMed.
- M. Ishida, S.-J. Kin, C. Preihs, K. Ohkubo, J. M. Lim, B. S. Lee, J. S. Park, V. M. Lynch, V. V. Roznyatovskiy, T. Sarba, P. K. Panda, C.-H. Lee, S. Fukuzumi, D. Kim and J. L. Sessler, Nat. Chem., 2012, 5, 15–20 CrossRef PubMed.
- M. Stephien, L. Latos-Grażyński and L. Szterenberg, J. Org. Chem., 2007, 72, 2259–2270 CrossRef PubMed.
- A. Matsuura and K. Komatasu, J. Am. Chem. Soc., 2001, 123, 1768–1769 CrossRef CAS.
- S. Miao, S. M. Brombosz, P. v. R. Schleyer, L. I. Wu, S. Barlow, S. R. Marden, K. I. Hardcastle and U. H. F. Bunz, J. Am. Chem. Soc., 2008, 130, 7339–7344 CrossRef CAS PubMed.
- Y. Nakamura, N. Aratani, H. Shinokubo, A. Takagi, T. Kawai, T. Matsumoto, Z. S. Yoon, D. Y. Kim, T. K. Ahn, D. Kim, A. Muranaka, N. Kobayashi and A. Osuka, J. Am. Chem. Soc., 2006, 128, 4119–4127 CrossRef CAS PubMed.
- K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367–7370 CrossRef CAS PubMed.
- X. Xiong, C.-L. Deng, B. F. Minaev, G. V. Baryshnikov, X.-S. Peng and H. N. C. Wong, Chem. – Asian J., 2014, 10, 969–975 CrossRef PubMed.
- J. Eskildsen, T. Reenberg and J. B. Christensen, Eur. J. Org. Chem., 2000, 1637–1640 CrossRef CAS.
- (a) H. G. H. Erdtman, Proc. R. Soc. London., Ser A, 1933, 143, 228–241 CrossRef CAS; (b) H. G. H. Erdtman and H.-E. Högberg, Chem. Commun., 1968, 773–774 RSC; (c) H. Erdtman and H.-E. Högberg, Tetrahedron Lett., 1970, 11, 3389–3392 CrossRef; (d) H.-E. Högberg, Acta Chem. Scand., 1973, 27, 2559–2566 CrossRef PubMed.
- (a) T. Hensel, D. Trpcevski, C. Lind, R. Grosjean, P. Hammershøj, C. B. Nielsen, T. Brock-Nannestad, B. E. Nielsen, M. Schau-Magnussen, B. F. Minaev, G. V. Baryshnikov and M. Pittelkow, Chem. – Eur. J., 2013, 19, 17097–17102 CrossRef CAS PubMed; (b) C. B. Nielsen, T. Brock-Nannestad, P. Hammershøj, T. K. Reenberg, M. Schau-Magnussen, D. Trpcevski, T. Hensel, R. Salcedo, G. V. Baryshnikov, B. F. Minaev and M. Pittelkow, Chem. – Eur. J., 2013, 19, 3898–3904 CrossRef CAS PubMed.
- (a) G. V. Baryshnikov, R. R. Valiev, N. N. Karaush and B. F. Minaev, Phys. Chem. Chem. Phys., 2014, 16, 15367–15374 RSC; (b) G. V. Baryshnikov, B. F. Minaev, M. Pittelkow, C. B. Nielsen and R. Salcedo, J. Mol. Model., 2013, 19, 847–850 CrossRef CAS PubMed.
- C. B. Nielsen, T. Brock-Nannestad, T. K. Reenberg, P. Hammershøj, J. B. Christensen, J. W. Stouwdam and M. Pittelkow, Chem. – Eur. J., 2010, 16, 13030–13034 CrossRef CAS PubMed.
- T. Brock-Nannestad, C. B. Nielsen, M. Schau-Magnussen, P. Hammershøj, T. K. Reenberg, A. B. Petersen, D. Trpcevski and M. Pittelkow, Eur. J. Org. Chem., 2011, 6320–6325 CrossRef CAS PubMed.
- H.-E. Högberg, Acta Chem. Scand., 1972, 26, 2752–2758 CrossRef PubMed.
- M. A. Christensen, C. R. Parker, T. J. Sørensen, S. de Graaf, T. J. Morsing, T. Brock-Nannestad, J. Bendix, M. M. Haley, P. Rapta, A. Danilov, S. Kubatkin, O. Hammerich and M. B. Nielsen, J. Mater. Chem. C, 2014, 2, 10428–10438 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for new compounds, general experimental procedures, a detailed NMR assignment and further electrochemical assignments. CCDC 1032471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00676g |
| This journal is © The Royal Society of Chemistry 2015 |