
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous introduction of trifluoromethyl and λ6-pentafluorosulfanyl substituents using F5S–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) C–CF3 as a dienophile†
C–CF3 as a dienophile†
Blazej
Duda
* and
Dieter
Lentz
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstr. 34-36, 14195 Berlin, Germany. E-mail: blazejduda@zedat.fu-berlin.de; dieter.lentz@fu-berlin.de
First published on 15th April 2015
Abstract
F5S–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CF3 can be easily prepared in high yields in two steps from 3,3,3-trifluoropropyne. It is a powerful, versatile dienophile in Diels–Alder reactions. Reactions at room temperature provide the corresponding products in up to quantitative yields allowing the introduction of the pentafluorosulfanyl group and trifluoromethyl group at the 1,2 position.
C–CF3 can be easily prepared in high yields in two steps from 3,3,3-trifluoropropyne. It is a powerful, versatile dienophile in Diels–Alder reactions. Reactions at room temperature provide the corresponding products in up to quantitative yields allowing the introduction of the pentafluorosulfanyl group and trifluoromethyl group at the 1,2 position.
Since the discovery of the Diels–Alder reaction (DA) in 1928,1 its value in organic chemistry as a perfect synthetic protocol towards the formation of six-membered rings has been continuously growing.2 The use of this methodology has also been applied in modern medicinal, agrochemical and materials science.2 It is well known that introducing fluorinated groups into organic molecules remarkably alters their physical, chemical and biological properties.3 As the CF3 group is one of the most important substituents there are several reviews concerning its introduction including radical, nucleophilic and electrophilic reactions.4 The incorporation of the trifluoromethyl group via DA reactions produces new six-membered hetero- and carbocyclic compounds.5 In this matter, CF3-substituted alkynes are used instead of the corresponding dienes.
So far there exist no comparable protocols for the introduction of the pentafluorosulfanyl group, which possesses unique features, including high stability (chemical and thermal) and lipophilicity of SF5-substituted molecules.6 Because it is regarded as a larger version of the CF3 group, the SF5 group is very often referred to as a “super CF3” group. In comparison with trifluoromethyl, the chemistry of SF5 is still largely unexplored probably because of the limited availability of SF5-containing reagents.6 Therefore, new methods to introduce this group into organic molecules are challenging and highly required.
Despite the significance of organofluorine chemistry, there are no reports of using a reagent that can introduce these two particularly interesting groups simultaneously. Thus, a suitable protocol that will give rise to substituted organic molecules with unique physical, biological and chemical properties is undoubtedly very important. We envisioned that 3,3,3-trifluoromethyl-1-pentafluorosulfanyl-1-propyne,7 F5S–CC–CF3 (2) would be the ideal reagent for this purpose (Scheme 1).
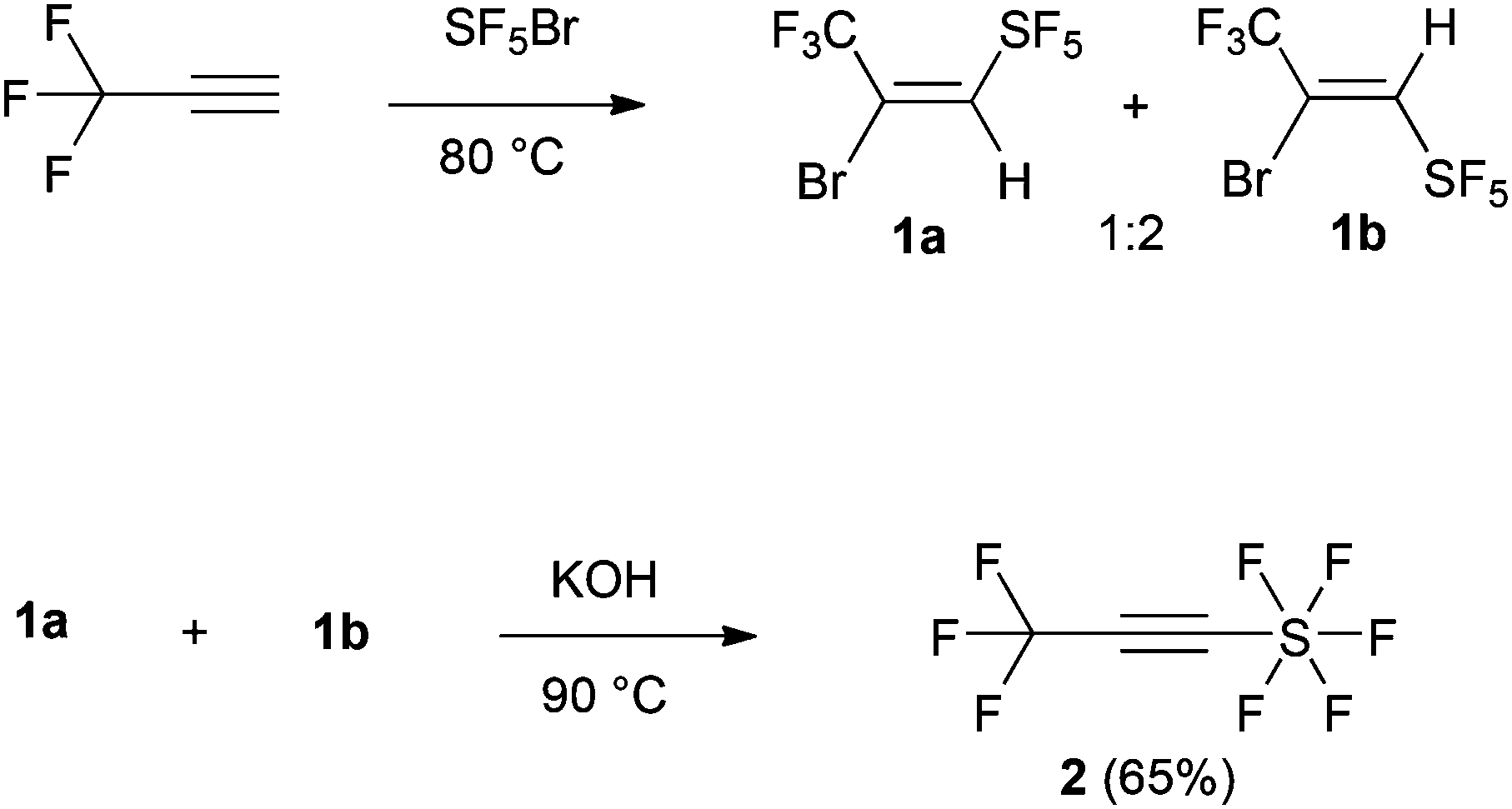 | ||
| Scheme 1 Synthesis of the alkyne 2. | ||
The synthesis of the dienophile (2) follows the literature protocol and involved the addition of SF5Br to terminal CF3-alkyne (1) to form the alkenes 1a–b as a mixture of E/Z isomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) (Scheme 1) in 80% yield.8 Carrying out the reaction in dry DCM or n-hexane at −40 °C and −10 °C in the presence of catalytic BEt3 as described by Dolbier et al.9 for other systems gave lower yields (47–50%, 1:1 ratio of E/Z isomers). A mixture of 1a–b was subsequently treated with KOH resulting in the target alkyne (2) in 65% yield.7
:2) (Scheme 1) in 80% yield.8 Carrying out the reaction in dry DCM or n-hexane at −40 °C and −10 °C in the presence of catalytic BEt3 as described by Dolbier et al.9 for other systems gave lower yields (47–50%, 1:1 ratio of E/Z isomers). A mixture of 1a–b was subsequently treated with KOH resulting in the target alkyne (2) in 65% yield.7
Diphenylbenzoisofuran (3a) was chosen as a model substrate for the DA reaction. The progress of the reaction in dry DCM at room temperature was controlled by 19F NMR spectroscopy. Within 1 h we observed 100% conversion of 2 towards the target product 4a (Scheme 2). Lowering the reaction temperature to −60 °C results in a 50% conversion after 1 h. Screening of the reaction conditions, by using different solvents, revealed that this cycloaddition was not affected by the polarity of solvents. For instance, performing such a process in DCM and n-pentane gave comparable isolated yields within the same reaction time. Based on the 19F NMR spectroscopic analysis no side-reactions occurred even when performing this process at elevated temperatures. The crude mixture was purified by flash column chromatography using DCM/n-pentane (1:2 v/v) as the eluent giving 4a in almost quantitative yield. Compound 4a was easily identified by 19F NMR spectroscopy. The resonance of the trifluoromethyl group splits into a quintet (5JFF(eq) = 12 Hz) due to coupling with the four cis fluorine nuclei of the SF5 group. As expected we observed a characteristic inversion of the axial and equatorial chemical shifts of the SF5 group.10 With the reaction conditions established, 2 was then treated with selected dienes (Scheme 2), such as furan (3b), 2,5-dimethylfuran (3c), cyclopentadiene (3d), pentamethylcyclopentadiene (3e), 6,6-dimethylfulvane (3f), cyclohexa-1,3-diene (3g), and cyclohepta-1,3,5-triene, which is in equilibrium with 3h. Apart from 4f and 4h conversions and yields are excellent although due to steric hindrance longer reaction times can be required.
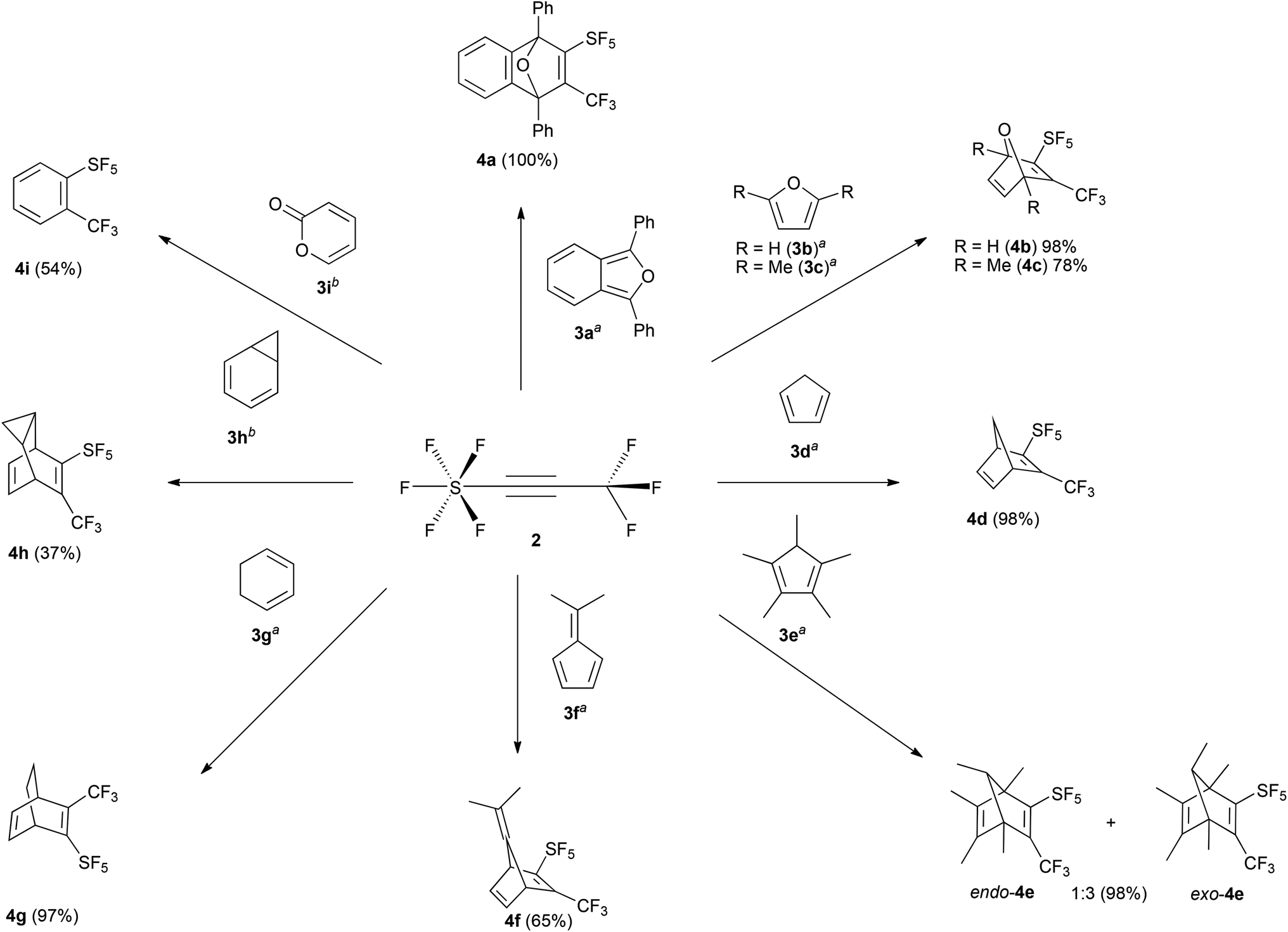 | ||
| Scheme 2 Diels–Alder reaction of 2 with selected dienes. Reaction conditions: (a) 2 (1 mmol), 3a–g (1.2 mmol), DCM (or n-pentane) 5 mL, RT, 3 h; (b) 2 (1 mmol), 3h–i (1.2 mmol), PhMe 5 mL, 110 °C, 24 h. | ||
The 1H NMR spectrum of 4d exhibits a doublet at δ 2.09 (2JHH = 7 Hz) and a doublet of quintets at 2.37 (2JHH = 7 Hz, 5JHF(eq) = 1 Hz) (see the ESI†) for the chemically inequivalent protons of the –CH2 group. The regiochemistry of the endo and exo-4e was determined by 1H NMR spectroscopy. The bridged C–H proton shows characteristic splittings: for exo-4e the resonance is split into a quartet with a coupling constant of 3JHH = 6 Hz; whereas for endo-4e we observed a quartet of quintets (3JHH = 6 Hz, 5JHF(eq) = 1 Hz) due to additional coupling to the four equatorial fluorine nuclei of the SF5 group.
The ratio of endo/exo-4e depends on the reaction temperature; at ambient temperature the isomers are formed in a 1:3 ratio whereas at −40 °C a 1:1 ratio is obtained. Thus we can conclude that endo-4e is kinetically favored due to less steric hindrance in the transition state.
Cyclopentadienones and 2H-pyran-2-ones (3i) are ideal precursors for the synthesis of substituted aromatic compounds by Diels–Alder reaction with an alkyne in one step. The latter reacted with 2 in toluene at 110 °C affording a benzene ring with only CF3 and SF5 substituents 4i in 54% isolated yield (Scheme 2). The former has been used by us and others in reactions to construct fluoranthene derivatives with adjacent substituents as precursors of substituted corannulenes.11 The acenaphthaquinone derivative (5) reacts with pentan-3-one in the presence of KOH yielding the cyclopentadienone derivative (6) (see Scheme 3). Compound 6 is usually not isolated and treated directly with a dienophile, for example 2 in Ac2O at 80 °C for 24 h furnishing the target fluoranthene 7 in 75% yield containing both SF5 and CF3 groups in the ortho position (Scheme 3). The structures of compounds 6 and 7 were elucidated by single-crystal X-ray diffraction (Fig. 1).‡ Compound 6 forms perfect planar molecules with columnar stacks along the crystallographic c axis with the shortest intermolecular C–C distance of 3.28 Å. The cyclopentadienone 6 is not stable in the solution and undergoes rapid dimerization (see the 1H NMR spectrum in the ESI†). Compound 7 showed a characteristic Cs-fold12 configuration of the methyl groups.
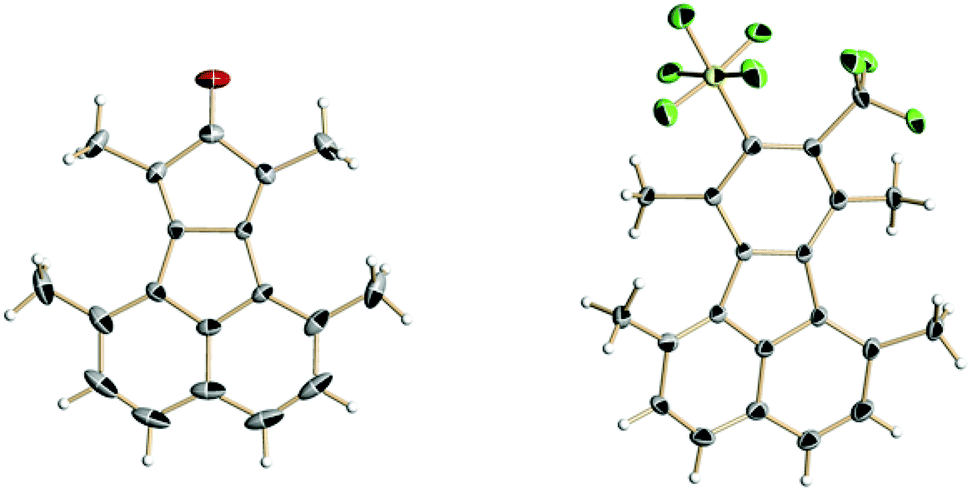 | ||
| Fig. 1 Single-crystal X-ray structures (thermal ellipsoids at 50% probability) of cyclopentadienone 6 (left) and fluoranthene 7 (right). | ||
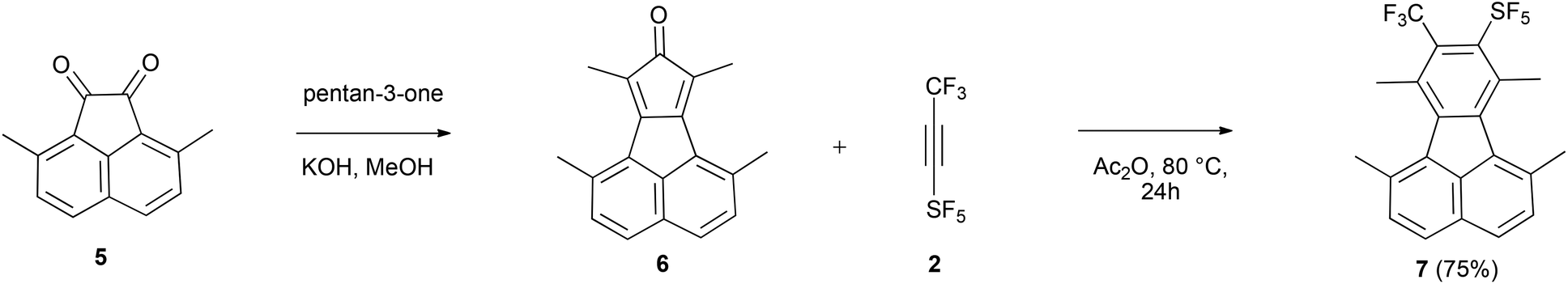 | ||
| Scheme 3 Synthesis of fluoranthene 7. | ||
In conclusion, we have reported a protocol for the synthesis of compounds with SF5 and CF3 in the 1,2-position via Diels–Alder reaction by employing a corresponding alkyne. The reaction occurs at room temperature or below within a few hours and in the absence of any side-reactions. Further studies using SF5 substituted alkynes as synthones for a variety of novel organic compounds with interesting new physical, biological and chemical properties are currently underway in our group.
Acknowledgements
Support from the Deutsche Forschungsgemeinschaft GRK 1582 “Fluorine as a Key Element” is gratefully acknowledged.Notes and references
- O. Diels and K. Alder, Justus Liebiegs Ann. Chem., 1928, 460, 98 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS; (b) J.-A. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822 CrossRef CAS PubMed.
- (a) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed; (b) R. D. Chambers, Fluorine in Organic Chemistry, John Wiley & Sons, 2005 Search PubMed; (c) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, John Wiley & Sons, 2009 Search PubMed; (d) I. Ojima, J. Org. Chem., 2013, 78, 6358 CrossRef CAS PubMed; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (f) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- For the recent reviews see: (a) S. Barata-Vallejo, M. R. Torviso, B. Lantaño, S. M. Bonesi and A. Postigo, J. Fluorine Chem., 2014, 161, 134 CrossRef CAS PubMed; (b) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (c) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2012, 473, 470 CrossRef PubMed; (e) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (f) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed.
- (a) G. Pawlowski and M. Hanack, Synth. Commun., 1981, 11, 351 CrossRef CAS; (b) M. Kuwabara, K. Fukunishi, M. Nomura and H. Yamanaka, J. Fluorine Chem., 1988, 41, 227 CrossRef CAS; (c) S. N. Tverdomed, G.-V. Roeschenthaler, N. Kalinovich, E. Lork, A. V. Dogadina and B. I. Ionin, J. Fluorine Chem., 2008, 129, 478 CrossRef CAS PubMed; (d) B. Duda, S. N. Tverdomed and G.-V. Röschenthaler, J. Fluorine Chem., 2013, 152, 29 CrossRef CAS PubMed; (e) A. A. Kislukhin, C. J. Higginson, V. P. Hong and M. G. Finn, J. Am. Chem. Soc., 2012, 134, 6491 CrossRef CAS PubMed; (f) M. Toshiya, I. Takashi, T. Konno and Y. Hiroki, Chem. Lett., 2000, 666 Search PubMed; (g) R. T. Conlin and Y. W. Kwak, Organometallics, 1984, 3, 918 CrossRef CAS; (h) P. G. Gassman and J. B. Hall, J. Am. Chem. Soc., 1984, 106, 4267 CrossRef CAS; (i) J. Stapersma, I. Rood and G. W. Klumpp, Tetrahedron, 1982, 38, 2201 CrossRef CAS.
- (a) P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130–1190 CrossRef CAS PubMed; (b) S. Altomonte and M. Zanda, J. Fluorine Chem., 2012, 143, 57 CrossRef CAS PubMed; (c) J. T. Welch in Fluorine in Pharmaceutical and Chemical Biology, ed. V. Gouverneur and K. Müller, 2012 Search PubMed; (d) D. Lentz and K. Seppelt in Chemistry of Hypervalent Compounds, ed. K. Akiba, 1999 Search PubMed; (e) W. A. Sheppard, J. Am. Chem. Soc., 1962, 84, 3064 CrossRef; (f) R. D. Bowdem, P. J. Comina, M. P. Greenhall, B. M. Kariuki, A. Loveday and D. Philip, Tetrahedron, 2000, 56, 3399 CrossRef; (g) R. W. Winter, R. A. Dodean and G. A. Gard in Fluorine-Containing Synthones, ACS Symposium Series 911, ed. V. A. Soloshonok, 2005 Search PubMed; (h) J. T. Welch and D. S. Lim, Bioorg. Med. Chem., 2007, 15, 6659 CrossRef CAS PubMed; (i) P. Wipf, T. Mo, S. Geib, D. Caridha, G. Dow, L. Gerena, N. Roncal and E. Milner, Org. Biomol. Chem., 2009, 7, 4163 RSC; (j) B. Stump, C. Eberle, W. B. Schweizer, M. Kaiser, R. Brun, R. L. Kraut-Siegel, D. Lentz and F. Diederich, ChemBioChem, 2009, 10, 79 CrossRef CAS PubMed; (k) P. Kirsch and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4216 CrossRef CAS; (l) D. S. Lim, J. S. Choi, C. S. Pak and J. T. Welch, J. Pestic. Sci., 2007, 32, 255 CrossRef CAS PubMed; (m) M. V. Ponomarenko, N. Kalinovich, Y. A. Serguchev, M. Bremer and G.-V. Röschenthaler, J. Fluorine Chem., 2012, 135, 68 CrossRef CAS PubMed.
- A. D. Berry, R. A. De Marco and W. B. Fox, J. Am. Chem. Soc., 1979, 101, 737 CrossRef CAS.
- Q. C. Wang, H. F. White and G. L. Gard, J. Fluorine Chem., 1979, 13, 455 CrossRef CAS.
- S. Ait-Mohand and W. R. Dolbier, Org. Lett., 2002, 4, 3013 CrossRef PubMed.
- (a) R. Winter, R. J. Terjeson and G. L. Gard, J. Fluorine Chem., 1998, 89, 105 CrossRef CAS; (b) J. A. M. Canich, M. M. Ludvig, W. W. Paudler, G. L. Gard and J. M. Shreeve, Inorg. Chem., 1985, 24, 3668 CrossRef CAS.
- (a) B. Schmidt, S. Seki, B. Topolinski, K. Ohkubo, S. Fukuzami, H. Sakurai and D. Lentz, Angew. Chem., Int. Ed., 2012, 51, 11385 CrossRef CAS PubMed; (b) B. Schmidt, B. Topolinski, M. Yamada, S. Higashibayashi, M. Shionoya, H. Sakurai and D. Lentz, Chem. – Eur. J., 2013, 19, 13872 CrossRef CAS PubMed; (c) E. L. Elliott, A. Orita, D. Hasegawa, P. Gantzel, J. Otera and J. S. Siegel, Org. Biomol. Chem., 2005, 3, 581 RSC; (d) A. Sygula, S. D. Karlen, R. Sygula and P. W. Rabideau, Org. Lett., 2002, 4, 3135 CrossRef CAS PubMed; (e) T. J. Seiders, E. L. Elliott, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 1999, 121, 7804 CrossRef CAS; (f) A. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1998, 120, 12666 CrossRef CAS.
- N. Rahanyan, A. Linden, K. K. Baldridge and J. S. Siegel, Org. Biomol. Chem., 2009, 7, 2082 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental details and copy of 1H, 13C and 19F NMR spectra. CCDC 1020179 and 1020180. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00610d |
| ‡ Crystal data for 6: C19H16O, M = 260.32, orthorhombic, a = 11.088(2) Å, b = 18.565(4) Å, c = 13.258(3) Å, V = 3591(2) Å3, T = 138(2) K, space group C2221, Z = 8, μ(Mo Kα) = 0.077 mm−1, 15171 reflections measured, 4184 independent reflections (Rint = 0.0231), The final R1 value was 0.0476 (I > 2σ(I)). The final wR(F2) value was 0.1150 (I > 2σ(I)). The final R1 value was 0.0565 (all data). The final wR(F2) value was 0.1215 (all data). The goodness of fit on F2 was 1.119. Crystal data for 7: C21H16F8S, M = 452.40, monoclinic, a = 16.519(6) Å, b = 12.951(5) Å, c = 17.522(6) Å, β = 106.652(7)°, V = 2729.2(10) Å3, T = 133(2) K, space group C2/c, Z = 8, μ(Mo Kα) = 0.266 mm−1, 28 |
| This journal is © The Royal Society of Chemistry 2015 |