
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective chemical modification of DNA with alkoxy- and benzyloxyamines†
Lorina
Gjonaj
and
Gerard
Roelfes
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: j.g.roelfes@rug.nl; Web: http://roelfesgroup.nl
First published on 29th April 2015
Abstract
A new method for the selective chemical modification of DNA at cytosine nucleobases using alkoxy- and benzyloxyamines is presented. It is shown that in particular benzyloxyamines are effective DNA modifying agents, giving rise to almost exclusive formation of the mono addition products. By using a bifunctional derivative, that is, p-azidobenzyloxyamine hydrochloride, an azide moiety, which is a convenient handle for further functionalization, could be introduced into the DNA. The azido modified DNA was then further reacted in a copper(I)-monophos catalysed 1,3-dipolar cycloaddition. These results illustrate the potential of the presented method for application in site and chemo-selective modification of DNA.
Introduction
The unique chemical and physical properties of DNA have paved the way for widespread application of this molecule far beyond its natural role as carrier of genetic information. The selective recognition of complementary nucleobases, resulting in Watson–Crick base pairing, has proven to be a very powerful tool for the assembly of complex DNA structures.1 For many applications, however, additional chemical functionalities need to be introduced into the DNA. Functionalized DNAs have found applications in diverse fields such as nanotechnology, synthesis and catalysis and chemical biology, for example as DNA sensors, for electrochemical analysis,2 DNA labelling,3 DNA-based catalysis,4 hybrid materials and drug delivery systems5 and many more.The most common method used for the preparation of functionalized DNAs involves solid phase synthesis, which is convenient for obtaining small quantities of short oligonucleotides containing novel functionalities.6 Another popular approach for the preparation of functionalized DNA involves the enzymatic incorporation of modified nucleotide triphosphates (dNTPs) by DNA polymerases.7
Both these approaches have in common that they require multistep chemical synthesis of the necessary nucleotide building blocks, which is laborious and time consuming. Ideally, it would be possible to introduce a novel chemical functionality selectively in the DNA in a post-(bio)-synthetic step. The fact that DNA is built up from only 4 building blocks that present very similar functional groups makes this a highly challenging task.
One example of such an approach is the sequence specific modification of DNA by use of DNA methyltransferase.8 While this method has enormous potential, it again relies on synthetic nucleotides, that are, S-adenosyl methionine derivatives.9
A limited number of methods for the selective chemical modification of DNA have been developed. This includes bio-orthogonal labelling of 5-hydroxymethylcytosine,10 selective DNA G-quadruplex alkylating agents,11 reaction of cytosine with bisulfite followed by transamination,12 selective alkylation of exocyclic amines with rhodium and copper carbenoid complexes,13 and using cisplatin as a covalent anchor for catalytically active metal complexes.4a,14
Alkoxyamines are frequently used in bioconjugation reactions, also to DNA, albeit that this generally involves reactions with an introduced bio-orthogonal aldehyde moiety, resulting in oxime formation.15 However, hydroxyl and methoxyamines have long been known to modify DNA selectively. This is, for example, the foundation for their use as mutagenic agents and in Maxam–Gilbert sequencing.16
Both hydroxyl- and methoxyamine react specifically and hundred times more efficiently with exposed cytosine residues compared to the other three nucleobases.17 The main products of this reaction are the result of nucleophilic attack on either C4 or C6, which are the most electrophilic positions. At high concentrations of alkoxyamine, commonly the C4 substitution product A and the double addition product B are obtained as the main products (Scheme 1). The efficiency and the selectivity of the reaction are dependent on the pH, temperature and concentration.18 Here we report that alkoxyamines, and in particular benzyloxyamines, are excellent reagents for the selective mono-functionalization of cytosine in DNA. Moreover, we show that by using a bifunctional benzyloxyamine derivative containing an azide moiety, further functionalization of DNA can be achieved using azide–alkyne click chemistry.
 | ||
| Scheme 1 Reaction of hydroxylamine and methoxyamine with cytosine. | ||
Results and discussion
Synthesis of alkoxyamines
The alkoxyamines hydrochloride salts 1–7 were used in this study (Fig. 1). 1, 5 and 6 were available from commercial sources, 3 and 4 were prepared following literature procedures.19 | ||
| Fig. 1 Alkoxyamines used in this study. | ||
2 was prepared from decyl bromide (8) by reaction with N-hydroxyphtalamide in the presence of K2CO3 giving rise to 9 (Scheme 2). This was followed by reaction with hydrazine hydrate, resulting in the free alkoxyamine 10. Treatment of 10 with HCl [6 M] provided 1-decylhydroxylamine hydrochloride salt 2.
 | ||
| Scheme 2 Synthesis of 2. (i) N-Hydroxyphtalamide, K2CO3, DMF, overnight, 83%; (ii) hydrazine hydrate, DCM, overnight, 69%; (iii) 10 M HCl, quantitative. | ||
Azide modified benzyloxyamine 7 was prepared using the synthetic route outlined in Scheme 3. p-Azido-toluene (12) was prepared by reacting p-toluidine (11) with NaNO2/HCl and NaN3. The subsequent bromination reaction was performed using NBS and AIBN, which did not result in full conversion, even after 3 days. Treatment of 13 with N-hydroxyphtalamide in presence of a base yielded 14, which possessed the aminoxy functionality. Deprotection of 14 with hydrazine hydrate furnished the free alkoxyamine, which was transformed immediately to the corresponding hydrochloride salt 7.
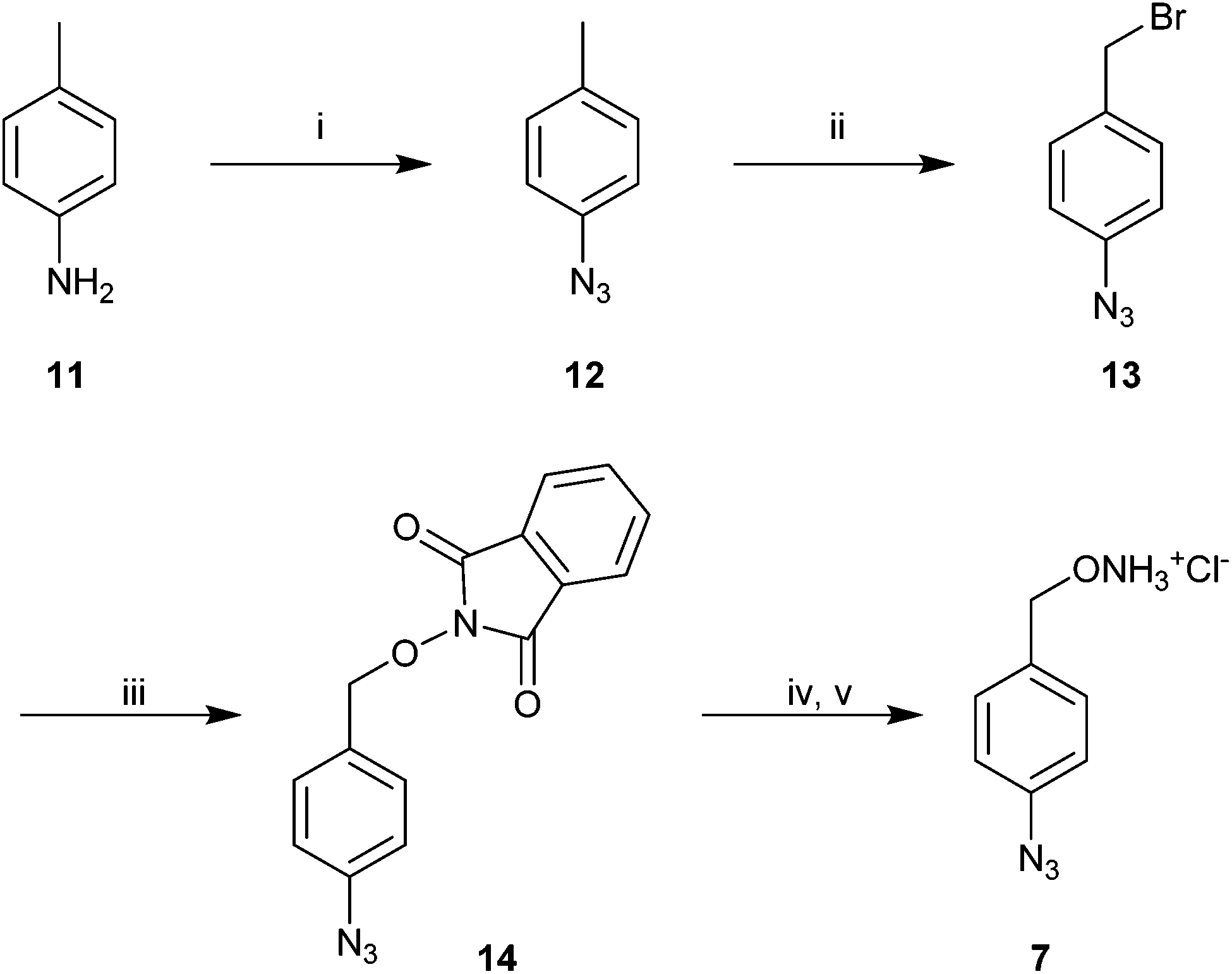 | ||
| Scheme 3 Synthesis of azido modified benzyloxyamine 7. (i) NaNO2, HCl, NaN3, H2O, 85%; (ii) NBS, AIBN, benzene, 3 days, 69%; (iii) N-hydroxyphtalamide, K2CO3, DMF, overnight, 76%; (iv) hydrazine hydrate, DCM, overnight, 56%; v. Conc. HCl, quantitative. | ||
DNA modification with alkoxyamines
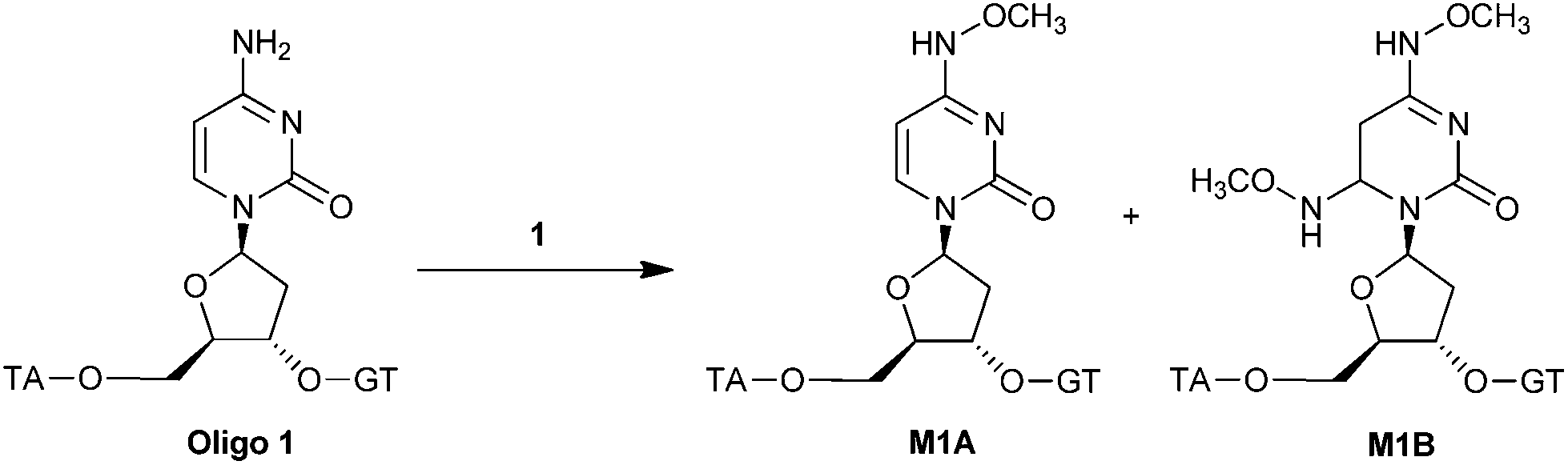
The optimal reaction conditions for DNA modification were investigated in the reaction of O-methoxyamine·HCl (1) with a single stranded DNA pentamer with sequence dTACGT (O1), containing only one cytosine. This reaction gives rise to 2 possible products: the mono-substituted adduct A and double substituted product B. A first screening of conditions revealed that the shortest reaction time and optimal selectivity was achieved at pH = 4 and a reaction temperature of 50 °C (Table S1†). Next, the dependence on the concentration of 1 was determined (Table 1). The highest ratio A/B, i.e. A/B = 12, was obtained at a final concentration of 1 of 0.5 M (entry 3), but with relatively low conversions after 8 h. A longer reaction time resulted in almost full conversion, albeit accompanied by a small decrease in selectivity (entry 4). Lowering the concentration of 1 further resulted in a decrease of both the ratio A/B and the reaction rate. Therefore it was concluded that the optimal conditions to achieve both high conversion and high selectivity were pH = 4.0, 50 °C and final concentration of 0.5 M of the methoxyamine (Table 1, entry 4). The products were purified using reversed phase HPLC and their identity was confirmed by MALDI-TOF (A m/z = 1507 and B m/z = 1554, respectively).|
|
||||
|---|---|---|---|---|
| Entry | 1 (M) | Conversionb (%) | Timec (h) | Ratio A/Bd |
| a All experiments were carried out in the presence of oligo 1 [100 μM], methoxyamine 1 (pH = 4 and 50 °C). b Determined by RP-HPLC. c Time after which the samples were taken. d Determined with RP-HPLC. | ||||
| 1 | 1.25 | 94 | 24 | 3 |
| 2 | 0.75 | 97 | 24 | 3 |
| 3 | 0.5 | 40 | 8 | 12 |
| 4 | 0.5 | 95 | 24 | 9 |
| 5 | 0.25 | 100 | 48 | 6 |
| 6 | 0.1 | 80 | 48 | 4.5 |
Having established the optimal conditions for modifying DNA with 1, the scope of the reaction was explored using a variety of other alkoxyamines. First the alkoxyamines 2–4 were investigated. 2 contains a long alkyl chain and the DNA adduct potentially can be used for the synthesis of DNA surfactants, which have found applications in nanotechnology.20 After optimization it was found that at room temperature (r.t) and at 0.45 M methoxyamine concentration gave the best result with a ratio A/B of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table S2†).
:1 (Table S2†).
Amino modified DNAs are useful scaffolds for derivatization.3 The goal here was to modify the DNA with an amino functionality using amino alkoxyamines. The DNA functionalization with aminoxy aminoalkyl dihydrochlorides, 3 and 4, was investigated using the optimized conditions found for 1. Unfortunately, in both cases the reaction times were significantly longer and many unidentified side products were obtained. Even when the final concentration of the modifying agents was increased to 1.0 M, full conversion was not obtained after 5 days. The corresponding mono-substituted products were observed, but due to the fact that the reaction results in complex product mixtures, this reaction was not pursued further.
DNA modification with benzyloxyamines
In light of the results described above, it was decided to focus on aryloxy- and benzyloxyamines. Phenyloxyamine hydrochloride 5 gave no reaction, even after several days. This suggests that this reagent is not sufficiently nucleophilic. In contrast, excellent results were achieved using benzyloxyamine hydrochloride 6. After 48 h, the reaction had almost reached completion (97% conversion) and the desired mono-substituted product A was obtained as the major product with an A/B ratio of 12:1 (Table 2, entry 2). Increasing the final concentration of the alkoxyamine, resulted in a lower ratio compared to entry 2. The product was purified by reversed phase-HPLC, giving rise to 12% isolated yield.
|
|
||||
|---|---|---|---|---|
| Entry | Reagent | Conversionb (%) | Timec (d) | Ratio A/Bd |
| a All experiments were carried out with oligo 1 [100 μM] and a final concentration of alkoxyamine reagent of 0.5 M, at pH = 4 and 50 °C, unless noted otherwise. b Determined by RP-HPLC. c Time after which the samples were taken. d Ratio mono/double substituted products, determined by RP-HPLC. e Reaction with oligo 2 [100 μM]. f Final concentration of compound 7 0.25 M. g Disubstituted product B was not detected. | ||||
| 1 | 5 | — | 3 | — |
| 2 | 6 | 97 | 2 | 12 |
| 3 | 6 | 90 | 5 | Only Ag |
| 4 | 7 | 98 | 3 | Only Ag |

The product M6A was subjected to an MS/MS analysis to verify that indeed the cytosine had been modified selectively (Fig. S2†). Assignment of the peaks resulting from the small fragments of the product confirmed the presence of unmodified A, G and T nucleobases; no peaks corresponding to any of these bases carrying the benzyloxyamino functionality were found. However, unmodified cytosine fragments were not observed after fragmentation. Instead, a fragment with m/z = 216.1 was found which corresponds to the benzyloxyamino modified cytosine nucleobase. Moreover, analysis of the larger fragments confirmed that A, T and G were not modified (ESI†). Additionally, the assignment of fragments at m/z 1110.6 and 1279 supports modification at C.
Given the success of modifying oligo 1 with benzyloxyamine hydrochloride a longer sequence of DNA was tested. The reaction with oligo 2 (dAATACGTG) was investigated using the optimized conditions. The reaction was found to be highly selective, i.e. the double substituted product could not be detected, but slow (Table 2, entry 3). After 5 days, 90% conversion was obtained, as determined by HPLC. The product was purified by preparative RP-HPLC and its identity was confirmed by MALDI-TOF (m/z = 2534).
DNA modification with a bifunctional benzyloxyamine
Having established the efficient and selective modification of DNA with benzyloxyamine, we focused on using the bifunctional benzyloxyamine derivative 7, carrying an azide moiety that can be used for further functionalization. The reaction was followed by RP-HPLC and analysed by MALDI-TOF. Full conversion was obtained after 3 days at 50 °C. The modification process was highly selective towards the mono-substituted product M7A, i.e. formation of the disubstituted product M7B was not observed. M7A was purified by size exclusion chromatography (75–80% yield).The azide functionality can be reacted further using either the Huisgen 1,3-dipolar cycloaddition.21 Thus, DNA labelled with an azide moiety is an attractive building block for introducing fluorophores, carbohydrates and ligation to other biomolecules.22 The copper catalysed azide–alkyne cycloaddition procedure that was applied involves a recently reported copper-monophos catalyst, which has been shown to significantly accelerate the reaction.23 This procedure is compatible with peptides and enzymes and it can be performed in aqueous media. The reaction was explored using two different alkynes (Scheme 4), phenylacetylene 15 and the fluorescent alkyne 16, a derivate of dansyl chloride.
 | ||
| Scheme 4 Click reaction of M7A. Conditions: CuSO4:monophos:sodium ascorbate (1:1.1:10) (5 mol% copper), excess of alkyne. | ||
Copper(II) sulphate was used as the copper source, which was reduced to Cu(I) in situ by sodium ascorbate. The mixture of copper and ligand was added to the solution of the alkyne and azide, under nitrogen atmosphere. The click reactions were followed by reversed phase. Full conversion was achieved within 2 hours in both reactions. The DNA functionalized products, M8A and M9A, were obtained after preparative HPLC in isolated yields of 14–15% over 2 steps.
Conclusions
A new method for the selective chemical modification of DNA at cytosine nucleobases using alkoxy- and benzyloxyamines was developed. The most effective modifying agents were found to be benzyloxyamines, which proved to give almost exclusively formation of the mono addition products. By using a bifunctional derivative, that is, p-azidobenzyloxyamine hydrochloride, an azide moiety, which is a convenient handle for further functionalization, could be introduced into the DNA. The azido modified DNA was then further reacted in a copper(I)-monophos catalysed 1,3-dipolar cycloaddition. This illustrates the potential of the presented method for application to site and chemo-selective modification of DNA.Experimental
General remarks
Reagents were purchased from Aldrich or Fluka and they were used as received. 1H-NMR and 13C-NMR spectra were recorded on a Varian AMX400 (400 and 100 MHz) in CDCl3. Reversed phase-HPLC (RP-HPLC) analysis were performed on a Shimadzu LC-10AD VP, Waters Xterra MS C18 column (3.0 × 150 mm, particle size 3.5 μm) using a gradient of CH3CN/TEAA (triethylammonium acetate) buffer 50 mM pH = 7 at 40 °C For purification purposes Waters Xterra Prep MS C18 column (7.8 × 150 mm, particle size 10 μm) at flow 1.0 mL min−1 was used. Gradient A (general gradient unless noted otherwise): CH3CN/TEAA buffer 50 mM pH = 7; gradient: 05/95 0 to 10 min, to 10/90 at 15 min, to 20/80 at 20 min to 30/70 at 30 min, to 50/50 at 50 min to 70/30 at 65 to 05/95 at 95 min for 15 min. Flow of 0.5 mL min−1 for analytical runs and flow of 1.0 mL min−1 for the preparative runs. MALDI-TOF measurements were done on a Voyager-DE Pro apparatus (Matrix: 20 μL of a solution of 2,4,6-trihydroxyacetophenone 0.5 M in EtOH + 10 μL of a solution of ammonium citrate dibasic 0.1 M in double distilled (dd) H2O + 2 μL sample solution in dd H2O). HRMS spectra were recorded on a LTQ XL Orbitrap from Thermo Fisher Scientific. Oligo 1 (dTACTG) and oligo 2 (dAATACGTG) were obtained from BioTez Berlin. Oligonucleotide concentrations were determined using Nanodrop ND-100 from Thermo Fisher Scientific. Column chromatography was performed on silica gel.Synthesis of alkoxy amines
:ethyl acetate, 7:3) to give 10 as a colorless oil (0.83 g, 4.8 mmol. 69% yield). 1H NMR (400 MHz, CDCl3): δ 5.34 (br s, 2H –ONH2), 3.65 (t, J = 6.7 Hz, 2H), 1.57 (m, 2H), 1.18–1.39 (s, 14H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 76.3, 32.0, 29.7, 29.6, 29.6, 29.4, 28.4, 26.1, 22.7, 14.5. HRMS (positive mode) (m/z) calculated mass for C10H24NO [M + H]+ 174.18524, found mass 174.18518.
The product was immediately converted to the corresponding hydrochloride salt by treatment with of HCl [6 M]. After filtration, the white solid was lyophilized.
:HCl (1:1) at 0 °C. Sodium nitrite (6.3 g, 92.8 mmol) dissolved in 26 mL of cold H2O was added drop wise, followed by drop wise addition of sodium azide (6.1 g, 92.8 mmol) in 58 mL H2O. After 1 h CHCl3 (100 mL) was added. The aqueous phase was extracted twice with CHCl3 (100 mL). The combined organic layers were washed with H2O (2 × 30 mL) and dried over MgSO4, concentrated under pressure to give 12 as a brown oil (11.1 g, 79.1 mmol, 85%). 1H NMR (400 MHz, CDCl3): δ 7.17 (d, J = 8.0 Hz, 2H), 6.94 (d, J = 8.0 Hz, 2H), 2.35 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 137.1, 134.86, 130.3, 118.7, 20.5.
The product was immediately converted to the corresponding hydrochloride salt by treatment with of HCl [6 M]. After filtration, the yellow solid was lyophilized.
DNA modifications
The oligonucleotides were dissolved in ddH2O (final concentrations of 200 μM). The concentrations of the oligonucleotides were determined from their UV/Vis absorptions at 260 nm (Nanodrop) using the calculated extinction coefficients.
Purity of the samples was evaluated with analytical reversed phase HPLC (gradient A) and the identity was confirmed with MALDI-TOF:
M1A: Retention time 33.3 min. MALDI-TOF (m/z) found 1507 (calculated 1506).
M1B: Retention time 34.2 min. MALDI-TOF (m/z) found 1554 (calculated 1553).
M2A: Retention time (gradient C) = 35.2 min MALDI-TOF (m/z) found 1632; calculated 1632.
M2B: Retention time (gradient C) = 40.3 min MALDI-TOF (m/z) found 1808; calculated 1805.
M3A: MALDI-TOF (m/z) 1537; calculated 1535.
M4A: MALDI-TOF (m/z) 1567; calculated 1563.
M6A: Retention time 40.2 min. MALDI-TOF (m/z) 1585; calculated 1582 (yield 12%).
M6B: Retention time 50.2 min. MALDI-TOF (m/z) 1706; calculated 1705 (yield 2%).
Retention time = 44.0 min MALDI-TOF (m/z) 2534; calculated 2538 (yield 10%).
M7A: Retention time 43.5 min MALDI-TOF (m/z) 1624; calculated 1623.
General procedure for click reaction
Before use, all the solutions were flushed with nitrogen for 10–15 min. The azido-modified M7A was dissolved in 600 μL of ddH2O.The catalyst solution was prepared by dissolving CuSO4·5H2O (2.5 mg, 0.01 mmol) and sodium ascorbate (19.8 mg, 0.1 mmol) in 1.5 mL of ddH2O, followed by addition of monophos (4.1 mg) in 500 μL of DMSO. This solution was allowed to stir for 10–15 min and then used in the click reaction.
Acknowledgements
The authors thank the European Research Council (ERC starting grant no. 280010) and the Ubbo Emmius Foundation for financial support. G.R. gratefully acknowledges support from the Ministry of Education, Culture and Science (Gravitation program 024.001.035). Dr W. Szymanski is thanked for kindly providing the dansyl derivative 16.Notes and references
- (a) T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. N. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138 RSC; (b) P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef CAS PubMed; (c) N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed.
- M. Hocek and M. Fojta, Chem. Soc. Rev., 2011, 40, 5802 RSC.
- S. H. Weisbrod and A. Marx, Chem. Commun., 2008, 5675 RSC.
- (a) N. Sancho Oltra and G. Roelfes, Chem. Commun., 2008, 6039 RSC; (b) L. Gjonaj and G. Roelfes, ChemCatChem, 2013, 5, 1718 CrossRef CAS.
- M. Kwak and A. Herrmann, Chem. Soc. Rev., 2011, 40, 5745 RSC.
- (a) S. L. Beaucage and R. P. Iyer, Tetrahedron, 1993, 49, 1925 CrossRef CAS; (b) C. B. Reese, Org. Biomol. Chem., 2005, 3, 2851 RSC; (c) A. H. El-Sagheer and T. Brown, Acc. Chem. Res., 2012, 45, 1258 CrossRef CAS PubMed.
- (a) M. Hocek and M. Fojta, Org. Biomol. Chem., 2008, 6, 2233 RSC; (b) R. Kranaster and A. Marx, ChemBioChem, 2010, 11, 2077 CrossRef CAS PubMed.
- S. Klimaśauskas and E. Weinhold, Trends Biotechnol., 2007, 25, 99 CrossRef PubMed.
- (a) G. Pljevaljcic, F. Schmidt and E. Weinhold, ChemBioChem, 2004, 5, 265 CrossRef CAS PubMed; (b) Z. Liutkevičiūte, E. Kriukiene, I. Grigaityte, V. Masevičius and S. Klimašauskas, Angew. Chem., Int. Ed., 2011, 123, 2138 CrossRef.
- C. Song and C. He, Acc. Chem. Res., 2011, 44, 709 CrossRef CAS PubMed.
- M. Di Antonio, F. Doria, S. N. Richter, C. Bertipaglia, M. Mella, C. Sissi, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2009, 131, 13132 CrossRef CAS PubMed.
- G. M. Blackburn, M. J. Gait, D. Loakes and D. M. Williams, in Nucleic acids in Chemistry and Biology, RSC, Cambridge, 3rd edn, 2006, ch. 8, pp. 302–303 Search PubMed.
- (a) K. Tishinov, K. Schmidt, D. Häussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000 CrossRef CAS PubMed; (b) K. Tishiniov, N. Fei and D. Gillingham, Chem. Sci., 2013, 4, 4401 RSC.
- (a) P. de Hoog, C. Boldron, P. Gamez, K. Sliedregt-Bol, I. Roland, M. Pitié, R. Kiss, B. Meunier and J. Reedijk, J. Med. Chem., 2007, 50, 3148 CrossRef CAS PubMed; (b) P. de Hoog, P. M. Pitié, G. Amedei, P. Gamez, B. Meunier, R. Kiss and J. Reedijk, J. Biol. Inorg. Chem., 2008, 13, 575 CrossRef CAS PubMed; (c) Ş. Özalp-Yaman, P. de Hoog, G. Amedei, P. M. Pitié, P. Gamez, J. Dewelle, T. Mijatovic, B. Meunier, R. Kiss and J. Reedijk, Chem. – Eur. J., 2008, 14, 3418 CrossRef PubMed.
- (a) M. Villien, S. Deroo, E. Gicquel, E. Defrancq, C. Moucheron, A. Kirsch-DeMesmaeker and P. Dumy, Tetrahedron, 2007, 63, 11299 CrossRef CAS; (b) N. Tuccitto, N. Giamblanco, S. Ghosh, V. Spampinato, P. Labbé, P. Dumy, S. Quici, G. Marletta, E. Defrancq and A. Liccardello, Langmuir, 2011, 27, 8595 CrossRef CAS PubMed; (c) P. Crisalli, A. R. Hernández and E. T. Kool, Bioconjugate Chem., 2012, 23, 1969 CrossRef CAS PubMed.
- A. M. Maxam and W. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 560 CrossRef CAS.
- B. H. Johnston, Methods Enzymol., 1992, 212, 180 CAS.
- (a) C. Nagata and O. Mårtensson, J. Theor. Biol., 1968, 19, 133 CrossRef CAS PubMed; (b) E. I. Budowsky, E. D. Sverdlov and T. N. Spasokukotskaya, Biochim. Biophys. Acta, 1972, 287, 195 CrossRef CAS PubMed.
- M. C. Pankaskie and S. A. Stolz, Synth. Commun., 1989, 19, 339 CrossRef CAS.
- (a) M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932 CrossRef CAS PubMed; (b) Y. H. Roh, J. B. Lee, P. Kiatwuthinon, M. R. Hartman, J. J. Cha, S. H. Um, D. A. Miller and D. Luo, Small, 2011, 7, 74 CrossRef CAS PubMed; (c) F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899 CrossRef CAS.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984, p. 1 Search PubMed.
- A. H. El-Sagheer and T. D. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC.
- L. S. Campbell-Verduyn, L. Mirfeizi, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Chem. Commun., 2009, 2139 RSC.
- M. T. Klapötke, B. Krumm, H. Piotrowski, K. Polborn and G. Holl, Chem. – Eur. J., 2003, 9, 687 CrossRef PubMed.
- W. Trommer and M. Hendrick, Synthesis, 1973, 484 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00595g |
| This journal is © The Royal Society of Chemistry 2015 |