
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHIV-1 drug discovery: targeting folded RNA structures with branched peptides
Jessica E.
Wynn
and
Webster L.
Santos
*
Department of Chemistry and Virginia Tech Center for Drug Discovery, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: santosw@vt.edu; Fax: +1540 231 3255; Tel: +1 540 231 5041
First published on 28th April 2015
Abstract
Human immunodeficiency virus type 1 (HIV-1) is an RNA virus that is prone to high rates of mutation. While the disease is managed with current antiretroviral therapies, drugs with a new mode of action are needed. A strategy towards this goal is aimed at targeting the native three-dimensional fold of conserved RNA structures. This perspective highlights medium-sized peptides and peptidomimetics used to target two conserved RNA structures of HIV-1. In particular, branched peptides have the capacity to bind in a multivalent fashion, utilizing a large surface area to achieve the necessary affinity and selectivity toward the target RNA.
Introduction
Human immunodeficiency virus (HIV) is a retrovirus that has been isolated and studied by scientists for over three decades.1 It is responsible for the development of acquired immunodeficiency syndrome (AIDS), a condition that ultimately destroys the immune system of the persons infected, eventually leading to death. From its discovery in the early 1980s, it has taken the lives of approximately 25 million people, and over 33 million are currently infected.1b,2 Its nature as a retrovirus, as well as its high rate of replication and mutation, has kept HIV/AIDS in the forefront as a continued worldwide epidemic that requires diligent efforts in discovering alternate methods of treatment.Upon discovery of HIV, initial research focused on understanding the mechanism of infection (Fig. 1).3 HIV infects CD4+ T cells, as well as other lymphatic cells, by fusing largely to the cells’ receptors on the cell membrane through gp120 and gp41 proteins.4 As HIV is a retrovirus, it must first reverse transcribe its RNA to DNA via reverse transcriptase, and once this is accomplished, the enzyme, integrase, incorporates the DNA into the host cell's genome.5 The DNA is then transcribed back to RNA, and upon entry into the cytoplasm the genetic information is translated into proteins.4a HIV-1 protease cleaves the proteins and with use of export factors such as Crm-1, eIF-5A and Ran-GTP, the newly translated viral proteins are reencapsulated and released to repeat the viral cycle.6
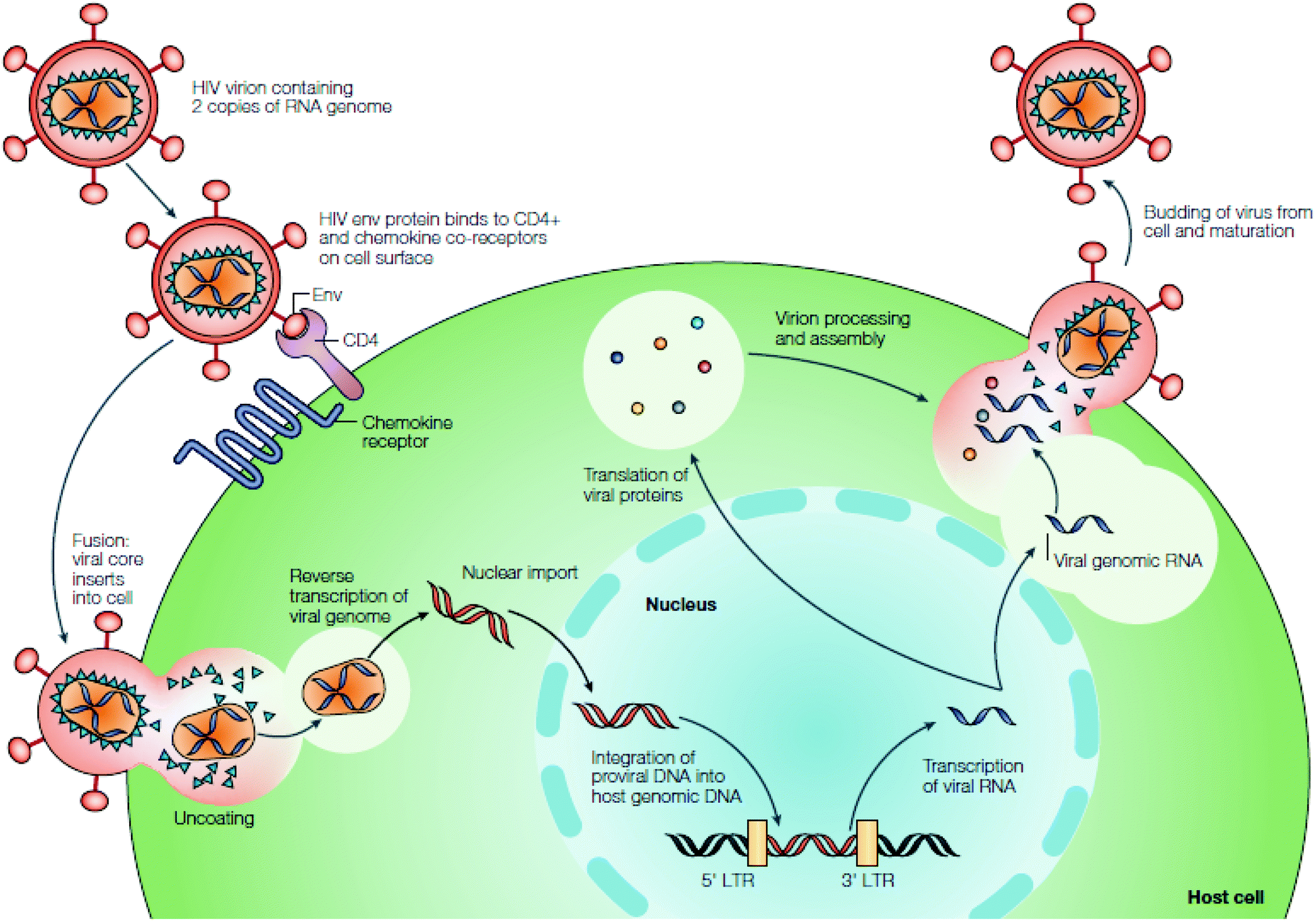 | ||
| Fig. 1 HIV replication cycle. Reprinted from ref. 3 with permission from Nature. | ||
Due to the rapid spread of HIV, many different anti-retroviral therapy (ART) drugs have been designed to combat CD4 cell infection and reduce viral loads. By the mid 1990s, nucleoside/nucleotide reverse transcriptase inhibitors were approved by the FDA for treatment of HIV, and a few years later non-nucleoside reverse transcriptase inhibitors were introduced. The intent of both inhibitors is to prevent HIV from replicating by preventing the viral RNA from being transcribed to DNA, one by terminating replication through insertion of a faulty nucleoside/nucleotide, and the other by binding HIV-1 reverse transcriptase, respectively.7 In the early 2000s, protease and fusion inhibitors were introduced as new ART drug classes. Protease inhibitors function by blocking the action of HIV protease, preventing the cleavage and subsequent packaging of viral proteins,7a,8 while fusion inhibitors prevent HIV from entering CD4 cells by mimicking a portion of the gp41 protein, inhibiting the formation of the helical bundle necessary to allow the fusion of HIV with the host cell membrane.9 In 2007, integrase inhibitors were approved by the FDA; these prevent the viral DNA from becoming incorporated into the cell's genome by binding to the enzyme.7a CCR5 receptor antagonists were also approved as a drug class that binds to CCR5, a co-receptor on the surface of CD4 cells that interacts with HIV to allow for binding of the virus to the cell.9
The number and variety of drugs developed to treat HIV testify to the immense difficulty in treating the retrovirus. This is due to a number of factors, including genetic recombination from the two genomic copies in each viral capsid via reverse transcriptase,10 the high mutation rate of reverse transcriptase, and fast replication of the virus.3,11 Combinations of ART drugs taken together, known as Highly Active Anti-retroviral Therapy (HAART), are employed as a means of combating drug resistance,3 but issues such as adverse side effects and maintaining strict dosing regimens remain problematic to treating HIV. Therefore, new drugs that target HIV-1 through new modes of action are needed as next generation therapeutics.
Recognition of RNA as a therapeutic target
RNA serves many critical biological functions, from transfer of genetic information to regulating roles in the cell, such as transcription, translation, catalysis, as well as splicing.12 A unique facet of RNA is its exquisite three-dimensional architecture derived from secondary structural elements such as hairpin loops, bulges, stems, turns, and pseudoknots, which minimize the energy of the structure. Also, the structure of RNA differs from DNA in that the major groove of A-form RNA is deeper and narrower than the B-form of DNA and the minor groove is shallower. This tertiary structure allows for binding interactions that could impart selectivity towards certain ligand constructs; thus, both the inherent functions and structure of RNA make it an ideal therapeutic target.13 However, with the exception of antibiotics that work by binding to portions of rRNA,14 and RNA gene suppression by antisense technology,15 there has been limited success in developing small-molecule ligands that selectively target RNA.16 This is due to several factors, including the large targetable surface area of most RNAs, the high binding affinity of endogenous ligands towards the RNA, permeability and stability of siRNA ligands, and conformational dynamics that make binding to as well as crystallizing a particular structure of RNA in solution difficult.15,17In order to surmount these challenges, in silico studies have been employed to virtually screen ligands against various RNA motifs, and certain ligand-RNA dynamics have been studied using nuclear magnetic resonance and molecular dynamics studies.16b,18 Another method is to perform a high-throughput screen using chemical libraries, in which a large number of diverse ligands can be screened against various RNAs with rapid turnover of results.19 Several RNAs have been well-studied as therapeutic targets, including viral RNAs such as the HIV-1 dimerization initiation site (DIS)20 and the HCV internal ribosome entry site (IRES),21 as well as expanded nucleotide repeats r(CCUG) involved with the development of myotonic dystrophy type 2.22 Herein, we focus on the review of medium-sized peptides and peptidomimetics used in targeting two conserved RNA structures of HIV-1: the transactivation response element (TAR) and rev response element (RRE) RNA, as well as the utilization of branched peptide scaffolds in therapeutics and our contribution in this field.
HIV-1 TAR RNA as a therapeutic target
HIV-1 TAR RNA has been widely investigated due to its critical role in HIV-1 replication. TAR RNA is a highly conserved 59 base pair sequence located at the 5′ end of transcribed HIV-1 RNA. The secondary structure reveals a double-stranded stem that contains a hexanucleotide loop as well as a three nucleotide bulge UCU (Fig. 2), through which the arginine rich motif (ARM) of the transcriptional activator protein Tat binds; this leads to further binding of cofactors cyclin T1-ckd1 and cyclin-dependent kinase 9 (CDK9), and the resulting complex promotes efficient transcription elongation of the RNA from the long terminal repeat.23 Without the formation of a Tat–TAR complex, the rate of viral transcription is minimal, leading to the production of fragmented transcripts.24 Therefore, disrupting the Tat–TAR interaction is a utilized strategy to interrupt viral replication, and has been pursued through the use of a wide variety of ligands such as intercalators,25 aminoglycosides,26 small molecules,27 siRNA,28 and nucleic acids.29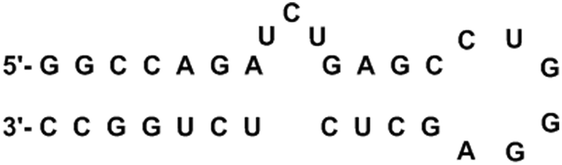 | ||
| Fig. 2 Secondary structure of TAR RNA. | ||
Peptides and peptidomimetics have also been used as medium-sized molecules to disrupt Tat–TAR interactions. A Tat-derived linear analog, RKKRRQRRK, was shown to compete with Tat for TAR, inhibiting the virus at the post-transcriptional level.30 Concomitantly in the 1990s, Hamy and co-workers employed the use of peptoids and D-amino acids in a combinatorial library to generate inhibitors containing unique secondary structures. One of the first peptidomimetics developed was a hybrid peptoid/peptide CGP64222, which was shown to inhibit formation of the Tat–TAR complex at nanomolar concentrations by inducing a conformational change of the RNA upon binding (Fig. 3).31 This compound was also shown to block viral entry through interaction with a CXC-chemokine receptor 4 co-receptor, making CGP64222 a dual-acting HIV-1 inhibitor.32 In 2000, Friedler and co-workers synthesized cyclic peptides with an arginine-rich scaffold to generate ARM mimetics, with one compound Tat11 shown to inhibit nuclear import and disrupt Tat-RNA binding.33 Cyclic peptides were also investigated by inducing a β-hairpin turn through use of a D-Pro-L-Pro motif.34 Several of these cyclic peptides bound TAR in the low nanomolar regime, with one compound L50 (cyclic L-ProRVRTRGKRRI-D-Pro) displaying a Kd of 1 nM and an IC50 of 250 nM, inhibiting both reverse transcription and Tat-dependent transcription.35
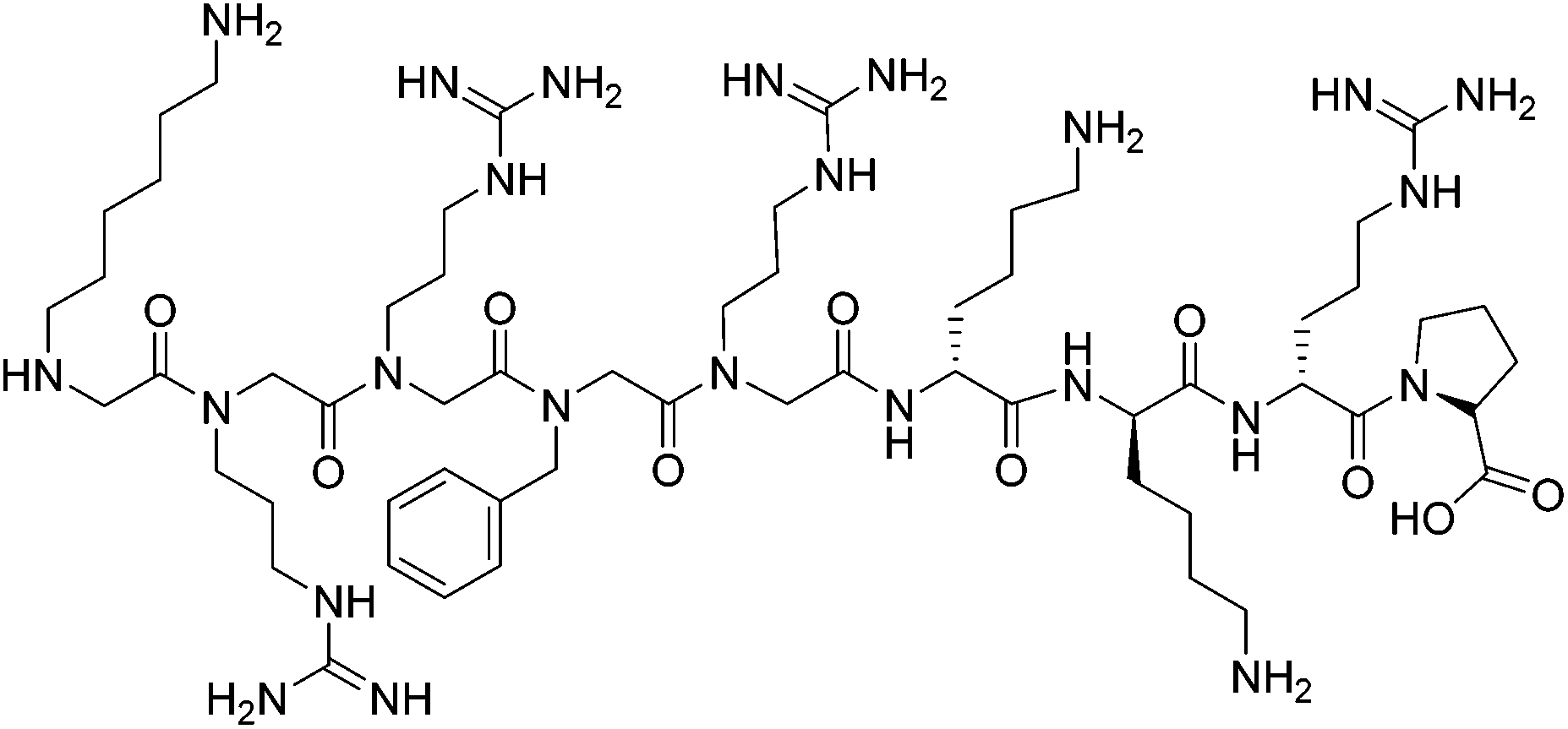 | ||
| Fig. 3 Peptoid/peptide hybrid CGP64222. | ||
HIV-1 RRE RNA as a therapeutic target
The continued struggle to combat HIV has led to great interest in also examining the rev response element (RRE) RNA as a potential drug target. RRE is a highly conserved region in the HIV-1 genome, consisting of approximately 351 nucleotides in the env gene.36 RRE interacts with a Rev protein, also encoded in the env region, to allow for transport of singly spliced and unspliced mRNAs from the nucleus into the cytoplasm with complexation of nuclear export factors such as Ran-GTP, eLF-5A and Crm-1.37 Unspliced mRNAs are required for translation of gag and pol genes in order to both encode structural proteins for packaging as well as to serve as the genome for new viruses.37,38 Thus, disruption of the Rev–RRE interaction would serve to inhibit the replication of HIV-1, making RRE a suitable drug target. RRE contains a high affinity binding site, stem IIB, where a Rev dimer initially binds, generating cooperative binding that extends to stem IA (Fig. 4). Frankel and co-workers proposed models showing that the hydrophobic portion of Rev bound with itself to form a V-shaped dimer, while the ARM segment interacted with the RRE RNA.39 Recently, this model was expanded upon with the report of the RRE–Rev complex in solution, revealing an “A” shape structure of RRE, with Rev dimers cooperatively binding on the “leg” portions that correspond to stems IIB and IA (Fig. 5).40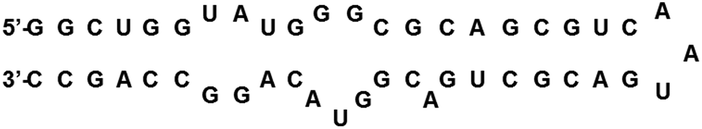 | ||
| Fig. 4 Secondary structure of RRE IIB. | ||
 | ||
| Fig. 5 Right: secondary structure of full-length RRE RNA with domain locations; left: SAXS topological structure, with the high affinity binding site IIB in green and oligomerization site between IIB and IA (cyan) legs. Reprinted from ref. 40 with permission from Elsevier. | ||
Similar ligands have been designed to disrupt Rev–RRE binding, including aminoglycosides,41 small molecules,42 peptide nucleic acids,43 nucleobase amino acids,44 metallopeptide complexes,45 and bifunctional compounds such as aminoglycoside-acridine41b,46 and aminoglycoside–PNA conjugates.47 Also, developments have been made to covalently link drug-RNA targets; for example, hv was utilized to covalently photocrosslink a diazinine-containing Met analogue with RRE RNA.48 However, there has been a greater focus in the use of α-helical peptidomimetics to target RRE RNA. Using a highly specific RRE-binding peptide R6QR7, Kaplin and co-workers induced alpha helicity in linear peptide analogs through macrolactam constraints generated from the amide bond formation at Lys and Asp residues, generating a hit compound Ac-RRRERQRKRRR-OH with a Kd of 45 nM and a 26-fold selectivity for RRE.49 Yu and co-workers also pursued the development of peptidomimetics by first designing N-methylated peptides modeled from calmodulin (Table 1, 1). A compound containing two N-methylated lysines (K*), LKK*LLKLLK*KLLKLKG, had a Kd of 9.1 nM and showed a five-fold selectivity for RRE against a mixture of tRNAs as well as TAR RNA.50 Further modifications of calmodulin included the introduction of acridine (K**) at the epsilon amine of lysine as an intercalator, and mono- and bis-acridinylated peptides were examined as RRE binders. It was found that the position of the acridinyl lysine affected the binding affinity of the peptides, with mono acridinylated peptides 3, 4, and 5 demonstrating better binding affinities towards RNA but with little selectivity. Sequences containing two acridinyl lysines (6–8) showed improvement of binding affinity, with 6 and 8 displaying increased selectivity for RRE and TAR over tRNAmix by almost 10 and 20-fold, respectively. The addition of another acridinyl lysine 8 slightly raised binding affinity but selectivity was lost. Through an alanine scan using 1 as a lead peptide, sequences 10 and 11 were found to improve binding affinities towards RRE, with each showing selectivity against TAR RNA.21a Yu and co-workers also developed constrained peptides from 1 in which the 5th and 12th Leu residues were replaced with Cys and then cross-linked by various maleimido derivatives.21b Unfortunately, none of the peptides showed increased binding affinity towards RRE in relation to 1, with Kds ranging from 46 to 90 nM. Kds in the picomolar range were achieved through the covalent crosslinking of the peptides through intermolecular disulfide bonds; however, these peptides did not display selectivity for RRE or TAR RNA.
| Peptide | Sequence (position(s) of K*, K**)a,b | α-Helicityc (%) | K d vs. RRE [nM] | K d vs. TARd [nM] | K d vs. tRNAmix [nM] |
|---|---|---|---|---|---|
| a K* = Nε,Nε-dimethyl Lys. b K** = Nε-acridinyl-Lys. c In 10 mM H3PO4/50% 2,2,2-trifluoroethanol (TFE) in 10 mM H3PO4, pH 7.4. d Discrimination ratios (Kd against other RNA/Kd against RRE) are given in parenthesis. | |||||
| 1 | LKKLLKLLKKLLKLKG | 26/57 | 22 (2.5) | 62 (2.8) | 55 (2.5) |
| 2 | LKK*LLKLLK*KLLKLKG | 8/52 | 9.1 | 53 (5.8) | 42 (4.6) |
| 3 | LK**KLLKLLKKLLKLKG | 14/52 | 3.2 (4.4) | 2.5 (5.6) | 14 |
| 4 | LKKLLKLLK**KLLKLKG | 36/62 | 4.3 (3.0) | 1.3 (10) | 13 |
| 5 | LKKLLKLLKK**LLKLKG | 25/63 | 6.8 (2.1) | 1.5 (9.3) | 14 |
| 6 | LK**KLLKLLK**KLLKLKG | 35/57 | 0.61 (8.0) | 0.55 (8.9) | 4.9 |
| 7 | LK**KLLKLLKK**LLKLKG | 15/48 | 0.72 (8.5) | 0.64 (9.5) | 6.1 |
| 8 | LKKLLKLLK**K**LLKLKG | 12/54 | 0.92 (7.5) | 0.37 (18) | 6.9 |
| 9 | LK**KLLKLLK**K**LLKLKG | 15/49 | 0.25 (0.92) | 0.20 (1.1) | 0.23 |
| 10 | WKKLLKLLKKLLKLAG | 48/65 | 2.5 ± 0.3 | 2.0 ± 0.3 | nd |
| 11 | LKKLLKWLKKLLKLAG | 19/71 | 1.2 ± 0.07 | 21 ± 1 | nd |
Branched peptides as medicinal scaffolds
Branched peptides have been used as a scaffold for a variety of applications, including vaccine development, metal chelation, tumor targeting, as well as transfection agents.51 Their prominence in drug therapy stems from their modulation of biological activity through multivalent binding, as well as improved resistance to proteolysis in vivo compared to linear counterparts.51,52 This resistance is thought to occur in part due to the deep channel in the active site of metallopeptidases such as neurolysin; only small peptides have access to this channel, allowing for more bulky peptides to escape rapid proteolysis.53 The concept of utilizing branched peptides surfaced in 1988; Tam developed the multiple antigen peptide (MAP) system where multiple lysines served as a core matrix on which peptide antigens could be attached via a triglyceryl linker.54 This MAP design has also been exploited as a drug delivery system, with conjugation of polyhedral boron and dihydroxyboryl-phenylalanine to branched poly-lysine-alanine systems for boron neutron capture therapy,55 and the use of branched cell permeable peptides (CPPs) or branched histidine-lysine peptides to improve the efficiency in internalization and gene delivery through transduction or transfection of cells.56 Recently, histidine-rich branched peptides (GH)2K and (HH)2K have also been utilized as a potential therapeutic for Alzheimer's Disease through the chelation of Cu2+ and Zn2+ ions, which have been shown to encourage the formation of beta-amyloid (Aβ) plaques.57Branched peptide libraries targeting folded RNA structures
As discussed earlier, the genetic diversity and high rate of mutation of HIV-1 have made attempts to eradicate the virus unsuccessful, so the conserved properties of both TAR and RRE RNA make them suitable targets to inhibit the virus through a new mode of action. In developing an RNA binder, several features are desired: the inhibitor (i) must be cell permeable to reach the target in the nucleus, (ii) must be selective against a variety of RNAs including tRNAs, and (iii) must have the potency required to disrupt RNA:cognate protein binding. Synthesis of the binder should be efficient, and the reaction should be one in which structural diversity can be readily introduced. Due to the structural complexity of RNA, the binder may benefit from multivalent interactions that could aid in both selectivity and binding affinity towards accessible pockets of the RNA. Considering these factors, medium-sized (∼1000 to 2500 Da) branched peptides were proposed as a scaffold for targeting RNA.58 Amino acids have a wide variety of functional groups that can interact with RNA via non-canonical modes of binding, such as with electrostatic interactions (Lys, Arg), π–π interactions (Phe, Trp), hydrophobic interactions (Val, Leu) and hydrogen bonding (Ser, Tyr). Also, imposed architecture and function of other reactive groups due to the wide availability and synthesis of unnatural amino acids can be achieved. First developed by Merrifield in 1963,59 the synthesis of peptides using solid phase is straightforward and large numbers of sequences can be generated using a split and pool method to generate libraries of peptides in an expedient manner, with subsequent on-bead screening against the desired RNA target.60 Branching the peptide allows the potential for increased surface area interaction with the RNA, and the structural diversity present in a combinatorial library should afford sequences that are biased toward specific tertiary structures amidst an ensemble of RNA conformations. Further, branching in peptides typically results in improved metabolic stability making them amenable to therapeutic development.First-generation 3.3.3 branched peptide library targeting HIV-1 TAR RNA
Our first-generation branched peptide library was developed to target HIV-1 TAR RNA.61 A combinatorial library of 4096 peptides linked to TentaGel resins via a photocleavable linker was synthesized (Fig. 6). The goal was to interrogate all possible binding modes that maximize RNA binding; hence, monomers with the capability to interact via electrostatic, π–π, and hydrogen bonding were utilized. Equally important is the ability to distinguish non-selective binders from false positives. For these, we opted to block promiscuous binders with BSA and competitor RNA. After high throughput screening with fluorescently labelled RNA, branched peptide hits containing an Arg–Arg motif were identified and characterized. In this proof-of-principle assay, the presence of positively charged moieties was not a surprise due to the negatively charged phosphates on RNA as well as the fact that the native ligand Tat interacts with TAR via an ARM (vide supra). Interestingly, large scale synthesis of FITC-labelled peptides for fluorescence polarization and dot blot assays resulted in an acid-mediated deletion of the N-terminal residue. Luckily, use of an aminohexanoic acid (Ahx) spacer between the N-terminus and FITC group eliminated this autocleavage and improved overall yields.62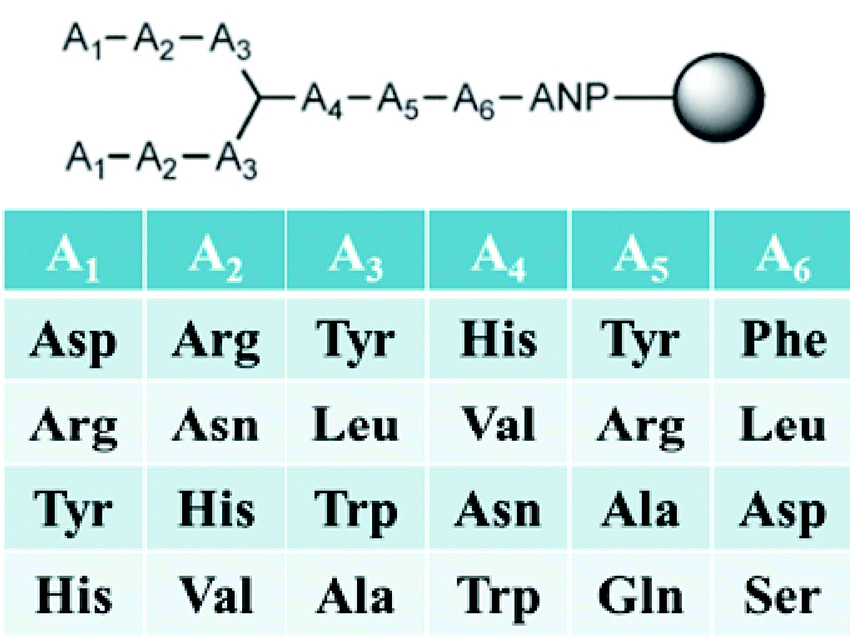 | ||
| Fig. 6 3.3.3 Branched peptide library. | ||
Results indicated binding affinities towards TAR RNA in the low micromolar range, with the best binder FL4 [(RRW)2*HAL] having a Kd of 600 nM, comparable to native Tat–TAR binding (Kd = 780 nM). It was also discovered that the lack of arginine groups at the N-terminus for several hits led to poor aqueous solubility, thus suggesting the need for basic residues within the peptides. To probe the effect of branching on RNA binding affinity, a linear version of T4-1 (RRWGHAL) was synthesized and revealed a 125-fold decrease in binding affinity, demonstrating the key role of branching in the peptide. As electrostatic interactions can, in principle, account for a substantial amount of binding energy, multiple basic residues that arise from the screening assay may generate false positives. To our delight, several peptides containing Arg–Arg motifs at the N-terminus as well as peptides containing a larger number of positive charges relative to FL4 also had lower binding affinities. This suggested that the decrease in binding affinity seen for T4-1 was not simply due to the loss of electrostatic interactions, thus supporting the role of branching design and sequence in increasing the binding affinity of FL4 towards the RNA.
The selectivity of branched peptides towards the native TAR structure was also probed using competition assays. In the presence of excess competitor tRNA, the binding affinity of FL4 to 32P-labeled TAR RNA was shifted, indicating partial selectivity of the compound towards TAR. Further titrations against several mutant versions of TAR including point mutation TAR 24C>U, bulgeless TAR, and tetraloop TAR, showed a decrease in binding affinity for the tetraloop and bulgeless mutants, indicating that FL4 interacted with the bulge and apical loop moieties on TAR RNA (Fig. 7). Hill analyses showed noncooperative binding for both the native structure TAR and TAR 24C>U, supporting the multivalent interaction of FL4 towards the RNA. In the case of the point mutation TAR 24C>U, the binding affinity was similar to the native structure. This indicated that the compound may not interact specifically with the C24 nucleobase; however, since this mutation should not alter the native structure of TAR this result was not completely unexpected.63
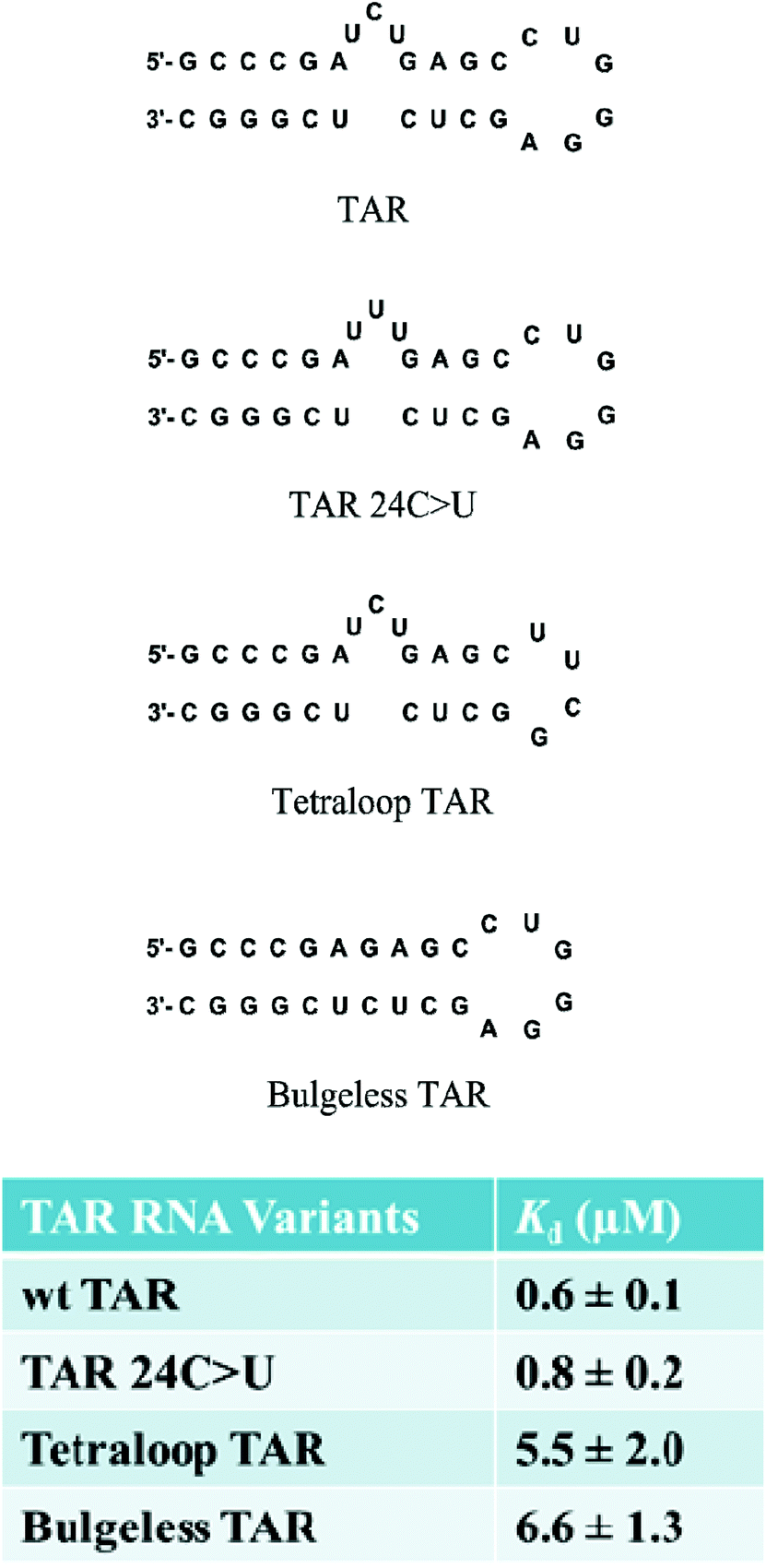 | ||
| Fig. 7 Structures of TAR RNA variants and dissociation constants for FL4. | ||
Lastly, the cellular uptake and cytotoxicity of the branched peptides were examined. We predicted these compounds to be cell permeable due to their medium molecular weight (500 < x < 1500 Da) as well as the presence of arginine moieties in the peptides.56c,64 It was demonstrated that medium-sized branched peptides were internalized into the cytoplasm and nucleus of HeLa cells. MTT assays also revealed relative cell viabilities of greater than 70% for all compounds at a concentration of 1 μM.
With proof-of-principle studies in hand—i.e., the demonstration of the pivotal role of branching in peptides, cell permeability as a result of their ‘medium’ molecular weight, and good binding affinity, an improved peptide library was envisioned along with the possibility of targeting other structured RNAs.
Second-generation 3.3.4 branched peptide library targeting HIV-1 RRE RNA
Due to increased interest in another conserved structure of HIV-1 RNA, we focused on targeting RRE RNA.65 In this library, two unnatural amino acids containing boronic acid moieties were incorporated: a phenylalanine derivative, FBPA and a benzoyl lysine derivative, KBBA (Fig. 8). Installation of boronic acids in biomolecules present a unique mode of interaction due to their ability to form reversible covalent bonds with Lewis bases. Further, we envisioned the boronic acid moiety as a potential surrogate for positive charge. Peptides containing boronic acids have been shown to form reversible covalent bonds with sugars such as alizarin and glucose, and have been employed to inhibit various proteases.66 In fact, in 2003 a peptidyl proteasome inhibitor, Bortezomib (Velcade), was approved by the FDA to treat multiple myeloma,67 and another boron-containing drug, Tavaborole (AN2690), recently received FDA approval in 2014 for treatment of onychomycosis.68 Both of these drugs work by binding hydroxyl groups to form reversible boronate adducts: Bortezomib via the Thr-OH in the 26S proteasome and Tavaborole via the 2′ and 3′-oxygen atoms of the terminal adenosine in leucyl-tRNA synthetase. Inspired by this work, the use of boronic acid to form reversible covalent bonds with the 2′-OH of RNA was employed. In principle, a likely increase in selectivity for the RNA target over DNA as well as improvement of binding affinity via a non-canonical mode of binding is envisioned (Fig. 9).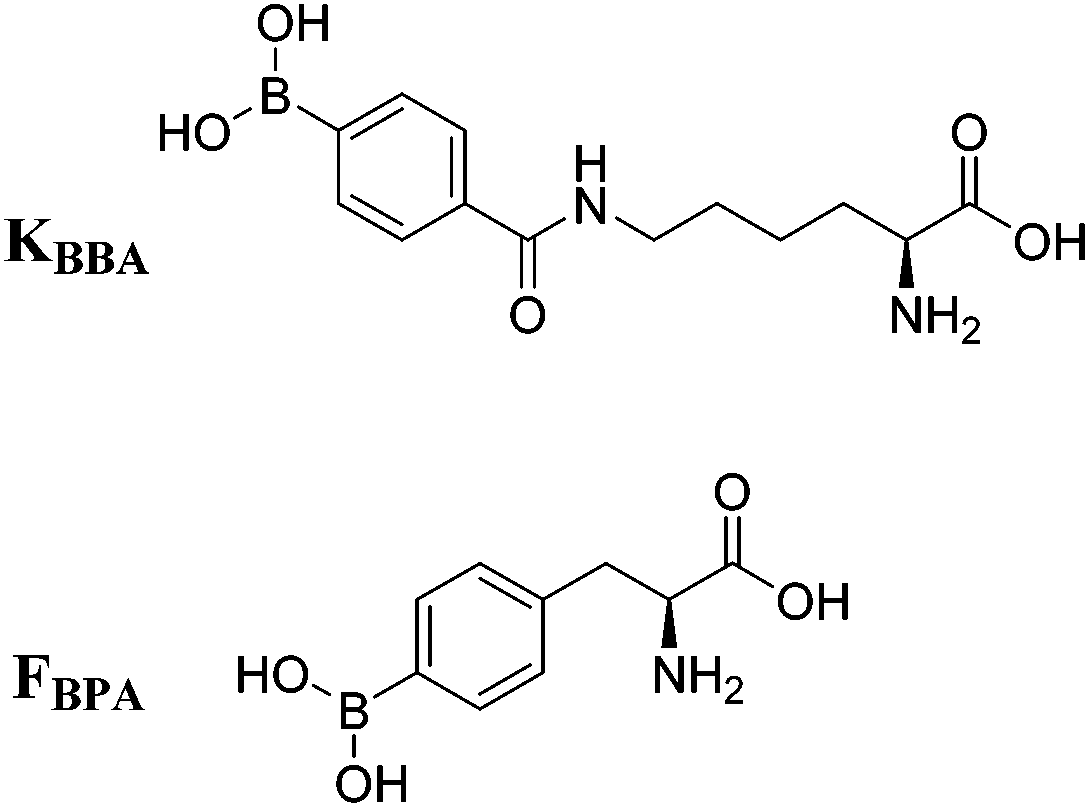 | ||
| Fig. 8 Structures of boronic acid monomers KBBA and FBPA. | ||
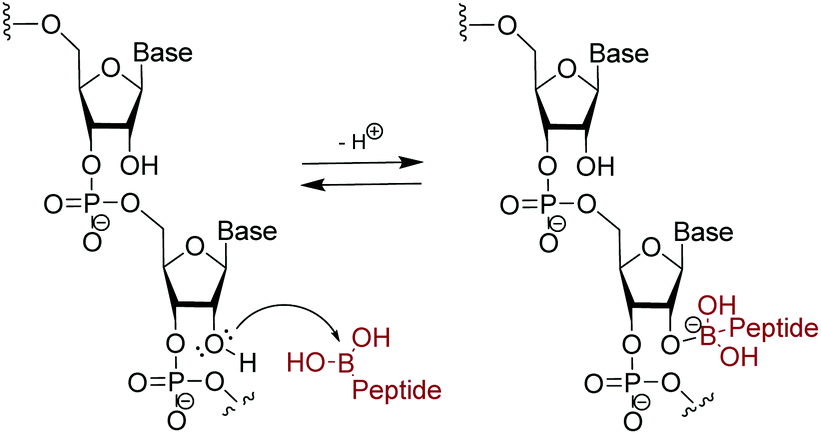 | ||
| Fig. 9 Suggested reversible covalent bond formation between boronic acid on branched peptide and 2′-OH on RNA. | ||
In this second generation of branched peptides, the library was expanded to 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 656 possible sequences, and a tyrosine was included at position A7 as a spectroscopic handle for quantification of the peptides (Fig. 10). Once again, amino acids were selected at each position to engage different interactions with the RNA as described above, and KBBA/FBPA was chosen at each variable position to explore the binding potential of boronic acid both with a shorter or longer alkyl tether. Of the eleven hits sequenced using MALDI MS/MS,69BPBA1-BPBA3 had dissociation constants in the low micromolar range and seven contained one or both of the boronic acid moieties. These boronic acids were preferred at the A1–A3 N-terminus region of the sequences, while the longer-chain boronic acid KBBA was preferred over FBPA. Lysine was a preferred residue in the majority of positions. However, the number of lysines present had no correlation to an increase or decrease in binding affinity suggesting again that while electrostatic interactions were important for peptide:RNA interactions, the binding affinity was not solely dependent on this type of interaction. Also, it was shown that the hydrophobic side chain residues were least preferred, indicating these interactions were not beneficial for binding interactions with the RNA.
656 possible sequences, and a tyrosine was included at position A7 as a spectroscopic handle for quantification of the peptides (Fig. 10). Once again, amino acids were selected at each position to engage different interactions with the RNA as described above, and KBBA/FBPA was chosen at each variable position to explore the binding potential of boronic acid both with a shorter or longer alkyl tether. Of the eleven hits sequenced using MALDI MS/MS,69BPBA1-BPBA3 had dissociation constants in the low micromolar range and seven contained one or both of the boronic acid moieties. These boronic acids were preferred at the A1–A3 N-terminus region of the sequences, while the longer-chain boronic acid KBBA was preferred over FBPA. Lysine was a preferred residue in the majority of positions. However, the number of lysines present had no correlation to an increase or decrease in binding affinity suggesting again that while electrostatic interactions were important for peptide:RNA interactions, the binding affinity was not solely dependent on this type of interaction. Also, it was shown that the hydrophobic side chain residues were least preferred, indicating these interactions were not beneficial for binding interactions with the RNA.
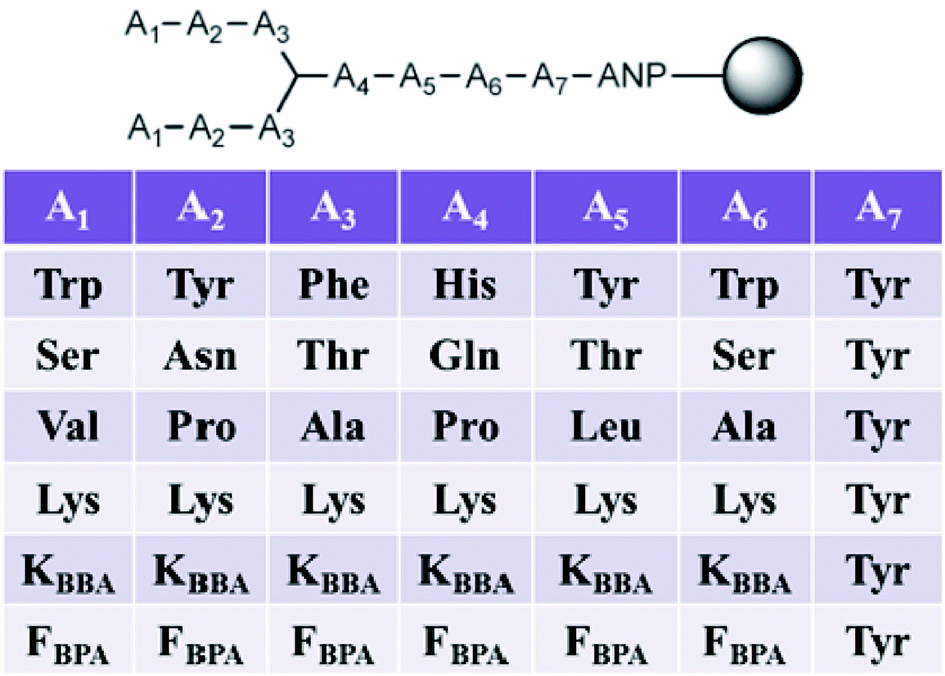 | ||
| Fig. 10 3.3.4 Branched peptide boronic acid (BPBA) library. | ||
The role of boronic acid in binding was explored using BPBA1 and BPBA3.65a When the boronic acid moiety was removed from KBBA to afford BPBA1.1, a six-fold decrease of binding affinity towards RRE IIB was observed (Fig. 11). In contrast, when an electron withdrawing fluorine atom was installed ortho to the boronic acid group (BPBA1.2), the binding affinity improved to 0.8 μM. This result supports the hypothesis that increasing the Lewis acidity of boron can lead to better complexation with the RNA. Further, when multiple boronic acid residues were removed in BPBA 3.1, binding was undetectable, indicating that these boronic acids play a pivotal role in recognizing and binding RRE IIB. These results support the use of boronic acid as a unique mode of binding for peptide:RNA interactions, and show the capacity for tuning the Lewis acidity of these boronic acids to increase their affinity towards RNA.
 | ||
| Fig. 11 Left: structures of modified boronic acid monomers KBZ and KFBA; right: dissociation constants of the highest affinity binders from the 3.3.4 BPBA library (BPBA1-3) and their analogues (1.1, 1.2, and 3.1). NB = no binding. | ||
Biophysical characterizations of branched peptide BPBA1 revealed several observations. First, branching in peptides is critical to binding with RRE RNA. For example, removal of the WKK N-terminus linked either at the (i) α- or (ii) ε-nitrogen of the lysine branch or (iii) removal of the C-terminus branch showed a marked decrease in binding affinity. Second, electrostatic interactions play an important contribution to binding affinity, but the location within the branched peptide is far more critical. Indeed, linear peptides where the ε-nitrogen N-terminal branch was translocated either to N- or C-terminus revealed Kd values that were 5-fold weaker than parent BPBA1. Further, a sequence scrambled branched peptide had a >75 fold increase in Kd. These results indicated that both branching and sequence were important in contributing towards the binding affinity of BPBA1 towards RRE IIB.
Investigations on the selectivity of BPBA1 towards other variants of RRE IIB RNA demonstrated significant improvement when compared to the first generation library. In summary, all variants (stem and loop deletions/mutations) showed an increase in Kd values compared to the native structure, and a variant in which all loops and bulges were deleted resulted in a ∼50-fold decrease in binding affinity (Kd of 91.7 μM), indicating that optimal binding of BPBA1 was achieved with the native tertiary structure of RRE IIB wild type RNA. Competition studies with tRNAmix and a DNA analogue of RRE IIB RNA also showed over 30 fold preference for the target RNA. These studies highlight the importance of three dimensional architecture and functional group exposure such as hydroxyl groups to binding affinity and selectivity. Ribonuclease protection assays revealed specific nucleotide contacts of BPBA1 with RRE IIB RNA (Fig. 12). For example, RNase VI showed protection along the upper stem portion, where the native protein Rev binds,37a,70 whereas RNase A showed protection in the internal loops of RRE IIB, specifically U7 and U36. These data indicate that BPBA1 spans a large portion of the RRE IIB RNA, with multiple contact points along the RNA.
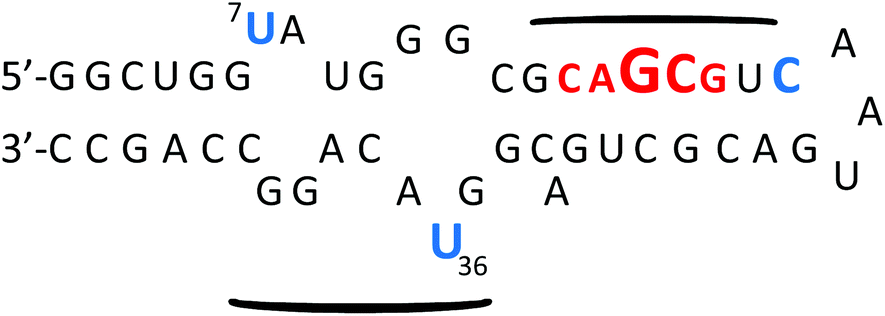 | ||
| Fig. 12 Summary of RNase protection of RRE IIB in the presence of increasing concentrations of BPBA1. Font size reflects level of protection from RNase A (blue) and RNase VI (red). Curved lines indicate regions of peptide:RNA contact. | ||
Conclusions
Drug discovery using RNA as a therapeutic target remains a challenging, herculean task. Our approach in using branched peptides provides an alternative strategy to sequence selective recognition of RNA; that is, conserved, highly structured RNA can be targeted based on the three-dimensional arrangement of nucleic acid bases. Indeed, HIV-1 TAR and RRE RNAs fall into this category and have been a focus of many studies by researchers. An attractive facet of this chemical biology approach is that the RNA:peptide interaction can be aided using virtually any exotic amino acid functional group; in our case, we focused on boronic acids in order to capitalize on their Lewis acidic properties. Thus far, we have demonstrated that selective tuning of this Lewis acidity can increase or decrease binding affinity to RRE RNA. To effectively inhibit RNA:protein interactions where the binding constants are in the low nanomolar range, RNA inhibitors need to be tight, selective binders. Bias towards a specific RNA target can be generated by conformational restriction of the peptide that induces a preorganized scaffold and protrudes key functional groups to interact favourably with RNA. In our investigations, branched peptide boronic acids possess Kds near the 1 μM range and improvements are clearly needed. The ability of medium molecular weight branched peptides to utilize a large surface area for binding, and thus create additional opportunities of selective engagement with the RNA, suggests a viable strategy towards this goal and perhaps the targeting of other RNA structures as well.Acknowledgements
We thank NIGMS of the National Institutes of Health for financial support (GM093834).Notes and references
- (a) R. C. Gallo, Retrovirology, 2006, 3, 72 CrossRef PubMed; (b) M. A. Wainberg and K.-T. Jeang, BMC Med., 2008, 6, 31 CrossRef PubMed.
- UNAIDS, Regional HIV and AIDS statistics, 2001 and 2009, http://www.unaids.org/documents/20101123_globalreport_slides_chapter2_em.pdf.
- A. Rambaut, D. Posada, K. A. Crandall and E. C. Holmes, Nat. Rev. Genet., 2004, 5, 52–61 CrossRef CAS PubMed.
- (a) Y.-H. Zheng, N. Lovsin and B. M. Peterlin, Immunol. Lett., 2005, 97, 225–234 CrossRef CAS PubMed; (b) D. C. Chan and P. S. Kim, Cell, 1998, 93, 681–684 CrossRef CAS PubMed.
- C. S. Adamson and E. O. Freed, Antiviral Res., 2010, 85, 119–141 CrossRef CAS PubMed.
- V. W. Pollard and M. H. Malim, Annu. Rev. Microbiol., 1998, 52, 491–532 CrossRef CAS PubMed.
- (a) C. Armbruster, Anti-Infect. Agents Med. Chem., 2008, 7, 201–214 CrossRef CAS; (b) M.-P. de Bethune, Antiviral Res., 2010, 85, 75–90 CrossRef CAS PubMed.
- A. M. J. Wensing, N. M. van Maarseveen and M. Nijhuis, Antiviral Res., 2010, 85, 59–74 CrossRef CAS PubMed.
- (a) M. A. Lobritz, A. N. Ratcliff and E. J. Arts, Viruses, 2010, 2, 1069–1105 CrossRef CAS PubMed; (b) J. C. Tilton and R. W. Doms, Antiviral Res., 2010, 85, 91–100 CrossRef CAS PubMed.
- D. L. Robertson, B. H. Hahn and P. M. Sharp, J. Mol. Evol., 1995, 40, 249–259 CrossRef CAS PubMed.
- (a) A. S. Perelson and R. M. Ribeiro, Stat. Med., 2008, 27, 4647–4657 CrossRef PubMed; (b) R. A. Weiss, Science, 1993, 260, 1273–1279 CAS.
- D. E. Draper, Annu. Rev. Biochem., 1995, 64, 593–620 CrossRef CAS PubMed.
- (a) R. Schroeder, A. Barta and K. Semrad, Nat. Rev. Mol. Cell Biol., 2004, 5, 908–919 CrossRef CAS PubMed; (b) G. J. R. Zaman, P. J. A. Michiels and C. A. A. van Boeckel, Drug Discovery Today, 2003, 8, 297–306 CrossRef CAS PubMed.
- (a) T. Hermann, Curr. Opin. Struct. Biol., 2005, 15, 355–366 CrossRef CAS PubMed; (b) A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Nature, 2000, 407, 340–348 CrossRef CAS PubMed; (c) Y. Tor, Biochimie, 2006, 88, 1045–1051 CrossRef CAS PubMed; (d) D. E. Brodersen, W. M. Clemons, Jr., A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Cell, 2000, 103, 1143–1154 CrossRef CAS PubMed; (e) D. N. Wilson, J. M. Harms, K. H. Nierhaus, F. Schluenzen and P. Fucini, Biol. Chem., 2005, 386, 1239–1252 CrossRef CAS PubMed; (f) K. L. Leach, S. M. Swaney, J. R. Colca, W. G. McDonald, J. R. Blinn, L. M. Thomasco, R. C. Gadwood, D. Shinabarger, L. Xiong and A. S. Mankin, Mol. Cell, 2007, 26, 393–402 CrossRef CAS PubMed.
- B. L. Davidson and P. B. McCray, Jr., Nat. Rev. Genet., 2011, 12, 329–340 CrossRef CAS PubMed.
- (a) J. R. Thomas and P. J. Hergenrother, Chem. Rev., 2008, 108, 1171–1224 CrossRef CAS PubMed; (b) M. M. Lee, A. Pushechnikov and M. D. Disney, ACS Chem. Biol., 2009, 4, 345–355 CrossRef CAS PubMed.
- J. Lu, B. M. Kadakkuzha, L. Zhao, M. Fan, X. Qi and T. Xia, Biochemistry, 2011, 50, 5042–5057 CrossRef CAS PubMed.
- (a) S. J. Seedhouse, L. P. Labuda and M. D. Disney, Bioorg. Med. Chem. Lett., 2010, 20, 1338–1343 CrossRef CAS PubMed; (b) A. C. Stelzer, A. T. Frank, J. D. Kratz, M. D. Swanson, M. J. Gonzalez-Hernandez, J. Lee, I. Andricioaei, D. M. Markovitz and H. M. Al-Hashimi, Nat. Chem. Biol., 2011, 7, 553–559 CrossRef CAS PubMed; (c) A. V. Filikov, V. Mohan, T. A. Vickers, R. H. Griffey, P. D. Cook, R. A. Abagyan and T. L. James, J. Comput. Aided Mol. Des., 2000, 14, 593–610 CrossRef CAS PubMed.
- (a) G. Galicia-Vazquez, L. Lindqvist, X. Wang, I. Harvey, J. Liu and J. Pelletier, Anal. Biochem., 2009, 384, 180–188 CrossRef CAS PubMed; (b) L. Yen, M. Magnier, R. Weissleder, B. R. Stockwell and R. C. Mulligan, RNA, 2006, 12, 797–806 CrossRef CAS PubMed.
- (a) J.-C. Paillart, M. Shehu-Xhilaga, R. Marquet and J. Mak, Nat. Rev. Microbiol., 2004, 2, 461–472 CrossRef CAS PubMed; (b) E. Skripkin, J.-C. Paillart, R. Marquet, B. Ehresmann and C. Ehresmann, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4945–4949 CrossRef CAS PubMed; (c) E. Ennifar, P. Walter, B. Ehresmann, C. Ehresmann and P. Dumas, Nat. Struct. Mol. Biol., 2001, 8, 1064–1068 CrossRef CAS PubMed; (d) E. Ennifar, M. Yusupov, P. Walter, R. Marquet, B. Ehresmann, C. Ehresmann and P. Dumas, Structure, 1999, 7, 1439–1449 CrossRef CAS PubMed.
- (a) S. J. Lee, S. Hyun, J. S. Kieft and J. Yu, J. Am. Chem. Soc., 2009, 131, 2224–2230 CrossRef CAS PubMed; (b) S. Hyun, J. Na, S. J. Lee, S. Park and J. Yu, ChemBioChem, 2010, 11, 767–770 CrossRef CAS PubMed.
- (a) L. Guan and M. D. Disney, ACS Chem. Biol., 2011, 7, 73–86 CrossRef PubMed; (b) K. Sobczak, G. Michlewski, M. de Mezer, E. Kierzek, J. Krol, M. Olejniczak, R. Kierzek and W. J. Krzyzosiak, J. Biol. Chem., 2010, 285, 12755–12764 CrossRef CAS PubMed; (c) S. G. Rzuczek, Y. Gao, Z.-Z. Tang, C. A. Thornton, T. Kodadek and M. D. Disney, ACS Chem. Biol., 2013, 8, 2312–2321 CrossRef CAS PubMed; (d) J. L. Childs-Disney, J. Hoskins, S. G. Rzuczek, C. A. Thornton and M. D. Disney, ACS Chem. Biol., 2012, 7, 856–862 CrossRef CAS PubMed.
- (a) F. Aboul-ela, J. Karn and G. Varani, J. Mol. Biol., 1995, 253, 313–332 CrossRef CAS PubMed; (b) T. M. Rana and K.-T. Jeang, Arch. Biochem. Biophys., 1999, 365, 175–185 CrossRef CAS PubMed.
- N. J. Keen, M. J. Gait and J. Karn, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2505–2510 CrossRef CAS.
- (a) L. S. Ratmeyer, R. Vinayak, G. Zon and W. D. Wilson, J. Med. Chem., 2002, 35, 966–968 CrossRef; (b) V. Peytou, R. Condom, N. Patino, R. Guedj, A.-M. Aubertin, N. Gelus, C. Bailly, R. Terreux and D. Cabrol-Bass, J. Med. Chem., 1999, 42, 4042–4053 CrossRef CAS PubMed.
- (a) P. W. Huber, M. Cui, A. W. Czarnik and H.-Y. Mei, Biochemistry, 1998, 37, 5549–5557 CrossRef PubMed; (b) A. Litovchick, A. G. Evdokimov and A. Lapidot, FEBS Lett., 1999, 445, 73–79 CrossRef CAS PubMed; (c) A. G. Evdokimov and A. Lapidot, Biochemistry, 2000, 39, 2838–2852 CrossRef.
- (a) N. T. Seongwoo Hwang, K. Kibler, H. Cao, A. Ali, Y.-H. Ping, K.-T. Jeang and T. M. Rana, J. Biol. Chem., 2003, 278, 39092–39103 CrossRef PubMed; (b) B. Davis, M. Afshar, G. Varani, A. I. H. Murchie, J. Karn, G. Lentzen, M. Drysdale, J. Bower, A. J. Potter, I. D. Starkey, T. Swarbrick and F. Aboul-ela, J. Mol. Biol., 2004, 336, 343–356 CrossRef CAS PubMed; (c) A. I. H. Murchie, B. Davis, C. Isel, M. Afshar, M. J. Drysdale, J. Bower, A. J. Potter, I. D. Starkey, T. M. Swarbrick, S. Mirza, C. D. Prescott, P. Vaglio, F. Aboul-ela and J. Karn, J. Mol. Biol., 2004, 336, 625–638 CrossRef CAS PubMed.
- J. J. Turner, M. Fabani, A. A. Arzumanov, G. Ivanova and M. J. Gait, BBA-Biomembr., 2006, 1758, 290–300 CrossRef CAS PubMed.
- (a) J. J. Turner, G. D. Ivanova, B. Verbeure, D. Williams, A. A. Arzumanov, S. Abes, B. Lebleu and M. J. Gait, Nucleic Acids Res., 2005, 33, 6837–6849 CrossRef CAS PubMed; (b) A. P. Walsh, V. K. Rajwanshi, R. Kumar, J. Wengel and M. J. Gait, Biochemistry, 2001, 40, 14645–14654 CrossRef; (c) N. Kaushik, P. K. Pandey, F. Kashanchi, L. Deng and V. N. Pandey, Biochemistry, 2000, 39, 11532–11539 CrossRef; (d) M. Watrin, F. Von Pelchrzim, E. Dausse, R. Schroeder and J.-J. Toulme, Biochemistry, 2009, 48, 6278–6284 CrossRef CAS PubMed; (e) A. Arzumanov, M. J. Gait, C. Di Primo and J.-J. Toulme, Biochemistry, 2002, 41, 12186–12192 CrossRef.
- I. Choudhury, J. Wang, A. B. Rabson, S. Stein, S. Pooyan, S. Stein and M. J. Leibowitz, J. Acquir. Immune Defic. Syndr. Hum. Retrovirol., 1998, 17, 104–111 CrossRef CAS PubMed.
- F. Hamy, E. R. Felder, G. Heizmann, J. Lazdins, F. Aboul-ela, G. Varani, J. Karn and T. Klimkait, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3548–3553 CrossRef CAS.
- D. Daelemans, D. Schols, M. Witvrouw, C. Pannecouque, S. Hatse, S. van Dooren, F. Hamy, T. Klimkait, E. de Clercq and A.-M. VanDamme, Mol. Pharmacol., 2000, 57, 116–124 CAS.
- F. D. Friedler A, N. W. Luedtke, Y. Tor, A. Loyter and C. Gilon, J. Biol. Chem., 2000, 275, 23783–23789 CrossRef PubMed.
- A. Davidson, T. C. Leeper, Z. Athanassiou, K. Patora-Komisarska, J. Karn, J. A. Robinson and G. Varani, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11931–11936 CrossRef CAS PubMed.
- M. S. Lalonde, M. A. Lobritz, A. Ratcliff, M. Chamanian, Z. Athanassiou, M. Tyagi, J. Wong, J. A. Robinson, J. Karn, G. Varani and E. J. Arts, PLoS Pathog., 2011, 7, e1002038 CAS.
- (a) D. A. Mann, I. Mikaelian, R. W. Zemmel, S. M. Green, A. D. Lowe, T. Kimura, M. Singh, P. J. G. Butler, M. J. Gait and J. Karn, J. Mol. Biol., 1994, 241, 193–207 CrossRef CAS PubMed; (b) D. I. Van Ryk and S. Venkatesan, J. Biol. Chem., 1999, 274, 17452–17463 CrossRef CAS PubMed.
- (a) J. Kjems, M. Brown, D. D. Chang and P. A. Sharp, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 683–687 CrossRef CAS PubMed; (b) V. W. Pollard and M. H. Malim, Annu. Rev. Microbiol., 1998, 52, 491–532 CrossRef CAS PubMed.
- M. H. Malim, L. S. Tiley, D. F. McCarn, J. R. Rusche, J. Hauber and B. R. Cullen, Cell, 1990, 60, 675–683 CrossRef CAS PubMed.
- M. D. Daugherty, D. S. Booth, B. Jayaraman, Y. Cheng and A. D. Frankel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12481–12486 CrossRef CAS PubMed.
- X. Fang, J. Wang, I. P. O'Carroll, M. Mitchell, X. Zuo, Y. Wang, P. Yu, Y. Liu, J. W. Rausch, M. A. Dyba, J. Kjems, C. D. Schwieters, S. Seifert, R. E. Winans, N. R. Watts, S. J. Stahl, P. T. Wingfield, R. A. Byrd, S. F. J. Le Grice, A. Rein and Y.-X. Wang, Cell, 2013, 155, 594–605 CrossRef CAS PubMed.
- (a) N. W. Luedtke, T. J. Baker, M. Goodman and Y. Tor, J. Am. Chem. Soc., 2000, 122, 12035–12036 CrossRef CAS; (b) N. W. Luedtke, Q. Liu and Y. Tor, Biochemistry, 2003, 42, 11391–11403 CrossRef CAS PubMed.
- (a) K. L. M. Fernandez-Saiz, C. T. Rigl, A. Kumar, K. G. Ragunathan, A. W. McConnaughie, D. W. Boykin, H.-J. Schneider and W. D. Wilson, Bioorg. Med. Chem., 1997, 5, 1157–1172 CrossRef; (b) M. Zhao, L. Ratmeyer, R. G. Peloquin, S. Yao, A. Kumar, J. Spychala, D. W. Boykin and W. D. Wilson, Bioorg. Med. Chem., 1995, 3, 785–794 CrossRef CAS PubMed; (c) E. S. DeJong, C. E. Chang, M. K. Gilson and J. P. Marino, Biochemistry, 2003, 42, 8035–8046 CrossRef CAS PubMed; (d) K. Nakatani, S. Sando and I. Saito, J. Am. Chem. Soc., 2000, 122, 2172–2177 CrossRef CAS.
- (a) I. Kumagai, T. Takahashi, K. Hamasaki, A. Ueno and H. Mihara, Bioorg. Med. Chem. Lett., 2000, 10, 377–379 CrossRef CAS PubMed; (b) I. Kumagai, T. Takahashi, K. Hamasaki, A. Ueno and H. Mihara, Bioorg. Med. Chem. Lett., 2001, 11, 1169–1172 CrossRef CAS PubMed.
- T. Takahashi, K. Hamasaki, A. Ueno and H. Mihara, Bioorg. Med. Chem., 2001, 9, 991–1000 CrossRef CAS PubMed.
- Y. Jin and J. A. Cowan, J. Am. Chem. Soc., 2005, 128, 410–411 CrossRef PubMed.
- S. R. Kirk, N. W. Luedtke and Y. Tor, J. Am. Chem. Soc., 2000, 122, 980–981 CrossRef CAS.
- (a) D.-R. Ahn and J. Yu, Bioorg. Med. Chem., 2005, 13, 1177–1183 CrossRef CAS PubMed; (b) S. Hyun, K. H. Lee and J. Yu, Bioorg. Med. Chem. Lett., 2006, 16, 4757–4759 CrossRef CAS PubMed.
- H. Nakashima, M. Hashimoto, Y. Sadakane, T. Tomohiro and Y. Hatanaka, J. Am. Chem. Soc., 2006, 128, 15092–15093 CrossRef CAS PubMed.
- N. L. Mills, M. D. Daugherty, A. D. Frankel and R. K. Guy, J. Am. Chem. Soc., 2006, 128, 3496–3497 CrossRef CAS PubMed.
- S. Hyun, H. J. Kim, N. J. Lee, K. H. Lee, Y. Lee, D. R. Ahn, K. Kim, S. Jeong and J. Yu, J. Am. Chem. Soc., 2007, 129, 4514–4515 CrossRef CAS PubMed.
- A. Pini, C. Falciani and L. Bracci, Curr. Protein Pept. Sci., 2008, 9, 468–477 CrossRef CAS PubMed.
- (a) L. Bracci, C. Falciani, B. Lelli, L. Lozzi, Y. Runci, A. Pini, M. G. De Montis, A. Tagliamonte and P. Neri, J. Biol. Chem., 2003, 278, 46590–46595 CrossRef CAS PubMed; (b) C. Falciani, M. Fabbrini, A. Pini, L. Lozzi, B. Lelli, S. Pileri, J. Brunetti, S. Bindi, S. Scali and L. Bracci, Mol. Cancer Ther., 2007, 6, 2441–2448 CrossRef CAS PubMed.
- (a) C. K. Brown, K. Madauss, W. Lian, M. R. Beck, W. D. Tolbert and D. W. Rodgers, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3127–3132 CrossRef CAS PubMed; (b) R. Natesh, S. L. U. Schwager, E. D. Sturrock and K. R. Acharya, Nature, 2003, 421, 551–554 CrossRef CAS PubMed.
- J. P. Tam, Proc. Natl. Acad. Sci. U. S. A, 1988, 85, 5409–5413 CrossRef CAS.
- (a) F. Hudecz, Anticancer Drugs, 1995, 6, 171–193 CrossRef CAS PubMed; (b) G. Mezo, F. Hudecz, M. Szekeike, J. Kajtar, G. Sarmay, J. Gergely, Z. Nagy and J. A. Clegg, J. Bioact. Compat. Polym., 1996, 11, 263–285 Search PubMed.
- (a) K. S. Kawamura, M. Sung, E. Bolewska-Pedyczak and J. Gariepy, Biochemistry, 2006, 45, 1116–1127 CrossRef CAS PubMed; (b) C. Rudolph, C. Plank, J. Lausier, U. Schillinger, R. H. Mueller and J. Rosenecker, J. Biol. Chem., 2003, 278, 11411–11418 CrossRef CAS PubMed; (c) S. H. Park, J. Doh, S. I. Park, J. Y. Lim, S. M. Kim, J. I. Youn, H. T. Jin, S. H. Seo, M. Y. Song, S. Y. Sung, M. Kim, S. J. Hwang, J. M. Choi, S. K. Lee, H. Y. Lee, C. L. Lim, Y. J. Chung, D. Yang, H. N. Kim, Z. H. Lee, K. Y. Choi, S. S. Jeun and Y. C. Sung, Gene Ther., 2010, 17, 1052–1061 CrossRef CAS PubMed; (d) Q. Leng and A. J. Mixson, Nucleic Acids Res., 2005, 33, e40/41–e40/49 CrossRef PubMed; (e) Q. Leng, P. Scaria, J. Zhu, N. Ambulos, P. Campbell and A. J. Mixson, J. Gene Med., 2005, 7, 977–986 CrossRef CAS PubMed.
- A. Lakatos, B. Gyurcsik, N. V. Nagy, Z. Csendes, E. Weber, L. Fueloep and T. Kiss, Dalton Trans., 2012, 41, 1713–1726 RSC.
- D. I. Bryson, W. Zhang, W. K. Ray and W. L. Santos, Mol. BioSyst., 2009, 5, 1070–1073 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- (a) K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, Nature, 1991, 354, 82–84 CrossRef CAS PubMed; (b) A. Furka, F. Sebestyen, M. Asgedom and G. Dibo, Int. J. Pept. Protein Res., 1991, 37, 487–493 CrossRef CAS PubMed.
- (a) D. I. Bryson, W. Zhang, W. K. Ray and W. L. Santos, Mol. BioSyst., 2009, 5, 1070–1073 RSC; (b) D. I. Bryson, W. Zhang, P. M. McLendon, T. M. Reineke and W. L. Santos, ACS Chem. Biol., 2012, 7, 210–217 CrossRef CAS PubMed.
- M. Jullian, A. Hernandez, A. Maurras, K. Puget, M. Amblard, J. Martinez and G. Subra, Tetrahedron Lett., 2009, 50, 260–263 CrossRef CAS.
- D. Wang, J. Iera, H. Baker, P. Hogan, R. Ptak, L. Yang, T. Hartman, R. W. Buckheit, Jr., A. Desjardins, A. Yang, P. Legault, V. Yedavalli, K.-T. Jeang and D. H. Appella, Bioorg. Med. Chem. Lett., 2009, 19, 6893–6897 CrossRef CAS PubMed.
- (a) A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed; (b) V. P. Torchilin, Adv. Drug Delivery Rev., 2008, 60, 548–558 CrossRef CAS PubMed.
- (a) W. Zhang, D. I. Bryson, J. B. Crumpton, J. Wynn and W. L. Santos, Chem. Commun., 2013, 49, 2436–2438 RSC; (b) W. Zhang, D. I. Bryson, J. B. Crumpton, J. Wynn and W. L. Santos, Org. Biomol. Chem., 2013, 11, 6263–6271 RSC.
- (a) G. Springsteen and B. Wang, Chem. Commun., 2001, 1608–1609 RSC; (b) D. H. Kinder and J. A. Katzenellenbogen, J. Med. Chem., 1985, 28, 1917–1925 CrossRef CAS PubMed; (c) E. S. Priestley, I. De Lucca, B. Ghavimi, S. Erickson-Viitanen and C. P. Decicco, Bioorg. Med. Chem. Lett., 2002, 12, 3199–3202 CrossRef CAS PubMed.
- (a) R. C. Kane, P. F. Bross, A. T. Farrell and R. Pazdur, Oncologist, 2003, 8, 508–513 CrossRef PubMed; (b) S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed.
- F. L. Rock, W. Mao, A. Yaremchuk, M. Tukalo, T. Crepin, H. Zhou, Y.-K. Zhang, V. Hernandez, T. Akama, S. J. Baker, J. J. Plattner, L. Shapiro, S. A. Martinis, S. J. Benkovic, S. Cusack and M. R. K. Alley, Science, 2007, 316, 1759–1761 CrossRef CAS PubMed.
- J. B. Crumpton, W. Zhang and W. L. Santos, Anal. Chem., 2011, 83, 3548–3554 CrossRef CAS PubMed.
- J. Kjems, B. J. Calnan, A. D. Frankel and P. A. Sharp, EMBO J., 1992, 11, 1119–1129 CAS.
| This journal is © The Royal Society of Chemistry 2015 |