
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A practical deca-gram scale ring expansion of (R)-(−)-carvone to (R)-(+)-3-methyl-6-isopropenyl-cyclohept-3-enone-1†
Leandro de C.
Alves
a,
André L.
Desiderá
a,
Kleber T.
de Oliveira
a,
Sean
Newton
b,
Steven V.
Ley
*b and
Timothy J.
Brocksom
*a
aDepartamento de Química, Universidade Federal de São Carlos, P.O. Box 676, 13565-905, São Carlos – SP, Brazil. E-mail: brocksom@terra.com.br; Web: http://www.lqbo.ufscar.br/ Fax: +55(16)3351-8250
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: svl1000@cam.ac.uk; Web: http://www.leygroup.ch.cam.ac.uk/
First published on 8th May 2015
Abstract
A route to enantiopure (R)-(+)-3-methyl-6-isopropenyl-cyclohept-3-enone-1, an intermediate for terpenoids, has been developed and includes a highly chemo- and regioselective Tiffeneau–Demjanov reaction. Starting from readily available (R)-(−)-carvone, this robust sequence is available on a deca-gram scale and uses flow chemistry for the initial epoxidation reaction. The stereochemistry of the addition of two nucleophiles to the carbonyl group of (R)-(−)-carvone has been determined by X-ray diffraction studies and chemical correlation.
Introduction
The synthesis of bioactive terpenoids and alkaloids containing an all carbon seven-membered ring is of current interest.1,2 Examples of such natural product syntheses include longifolene,3 and the guaiane sesquiterpenes thapsigargins,4 the englerins,5 echinopines6 and nanolobatolide (a sesqui- plus 3 C).7 The higher terpenoid examples include phorbol (prostratin),8 ingenol,9 guanacastapenes,10 icetexanes,11 and the mero-terpenoids perovskone,12 frondosin13 and the cortistatins.14 Among the alkaloids, colchicine,15 diterpenoid alkaloids,16 nominine16a,17 and the daphniphyllums18 can be cited as being part of this class of compounds (Fig. 1).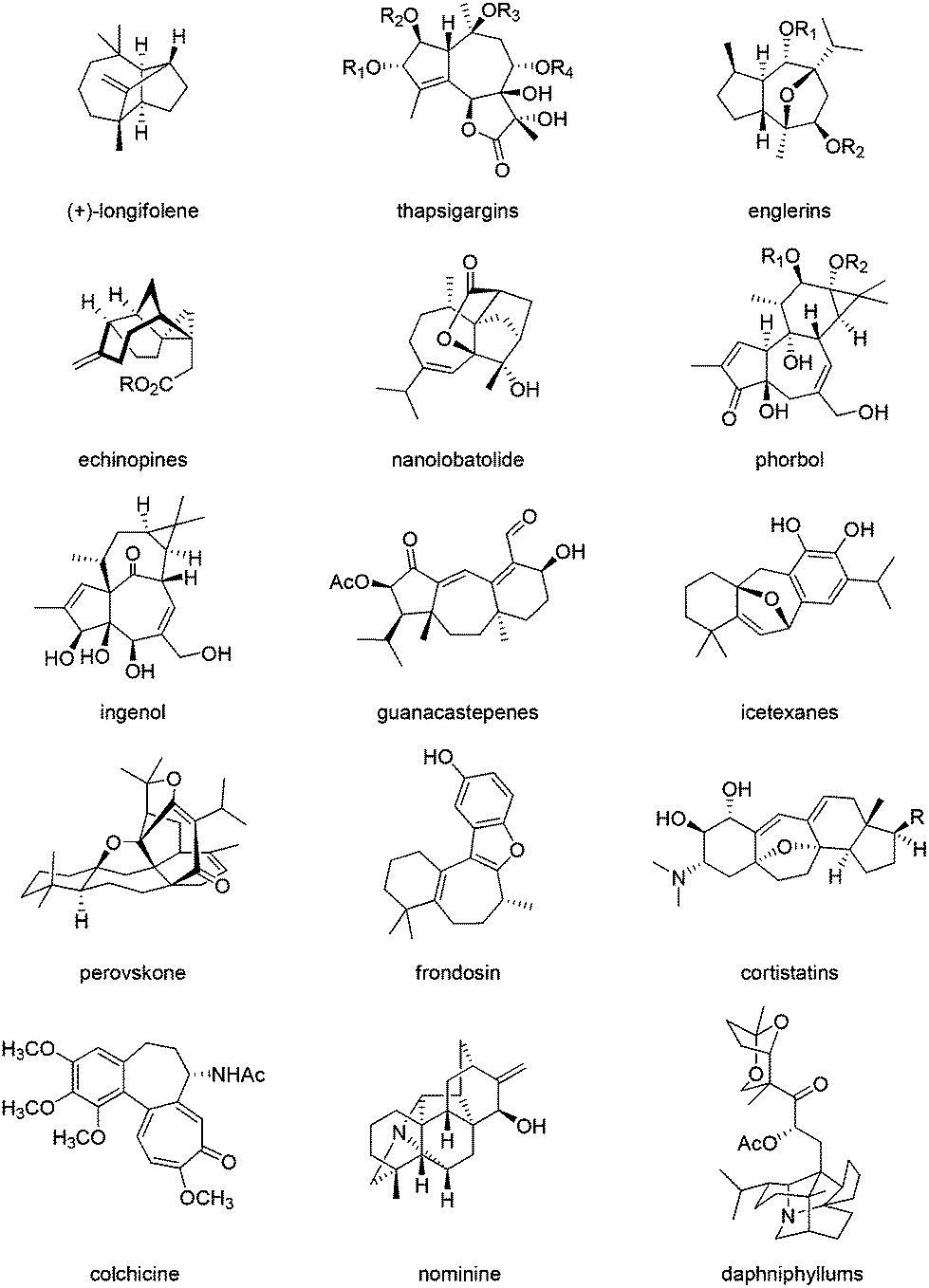 | ||
| Fig. 1 Examples of natural product synthetic targets containing an all carbon cycloheptane moiety. | ||
The synthesis of all carbon seven-membered rings can be effected by cyclization reactions19 and ring-closing metathesis,20 (4 + 3)21 or (5 + 2)22 or (3 + 2 + 2)23 cycloadditions, and 6 + 1 or 5 + 2 ring expansions.24 The terpenoid natural products11a,25 frequently present the seven membered-ring in fusion with a five-membered ring (sesquiterpenoids), the diterpenoids having a further six-membered ring (Scheme 1).
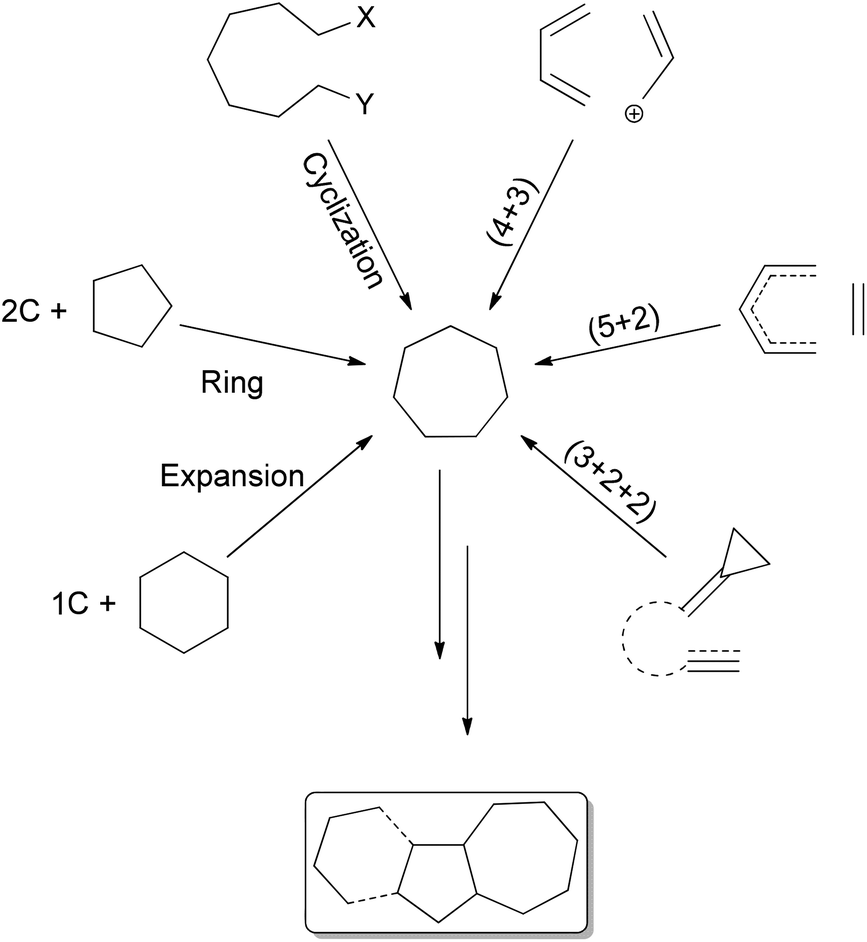 | ||
| Scheme 1 Synthetic methods to obtain cycloheptane rings, with target terpenoid carbon skeletons. | ||
The perhydroazulene sesquiterpenes26,27 and diterpenes28,29 generally possess methyl and isopropyl group substitutions in a 1,4-relationship, thus displaying a carbon skeleton similarity to naturally occurring para-menthane monoterpenes, and embedding a 1,4-methyl, isopropyl-cycloheptane residue (Fig. 2).
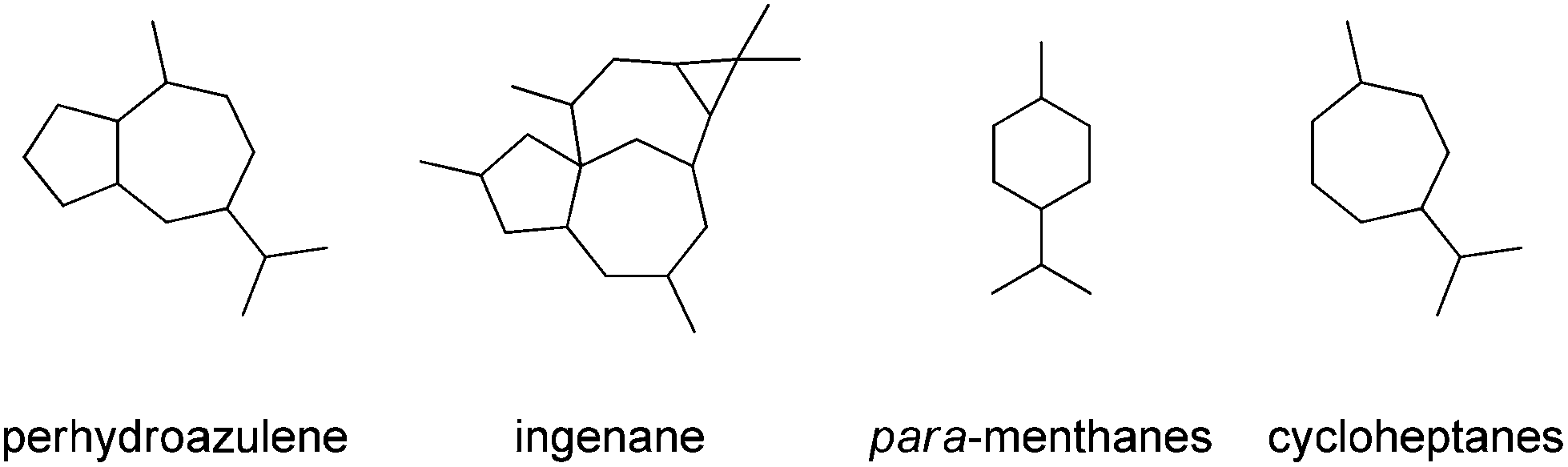 | ||
| Fig. 2 Structural relationships amongst perhydroazulene sesquiterpenes and diterpenes, para-menthane monoterpenes and cycloheptanes. | ||
Synthetic approaches to the perhydroazulene structures from para-menthanes have involved the contraction of the six-membered ring to cyclopentanoids followed by heptanyl-annulation,30,31 or much less frequently expansion to cycloheptanoids and pentanyl-annulation.32 This last strategy has been studied in our laboratory for the synthesis of cycloheptanoids,33 and thus access to perhydroazulene terpenoids and alkaloids.34 The ring expansions occur by two different methods: cyclopropanation of a suitable para-menthene-1 to an overbred bicyclic system followed by cleavage of the common C–C bond.35 The second sequence requires an appropriate nucleophile addition to a para-menthanone-2 followed by a regioselective rearrangement, as presented in Scheme 2.
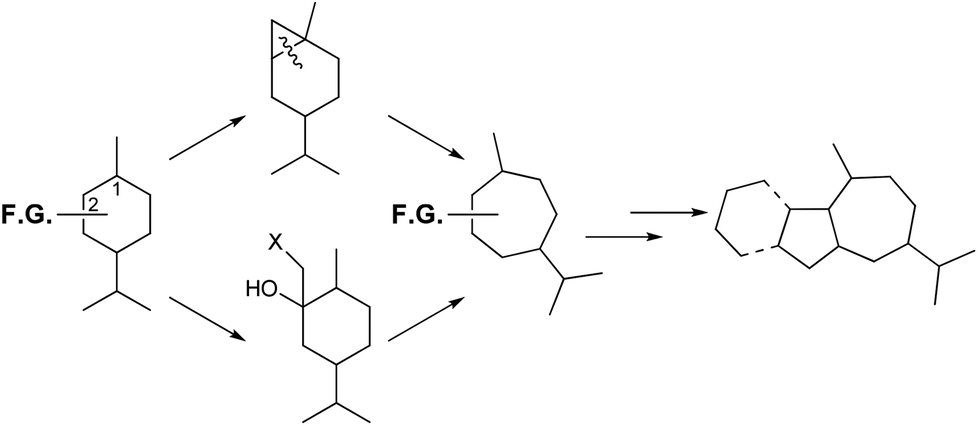 | ||
| Scheme 2 Ring expansion strategies to obtain 1,4-methyl, isopropyl-cycloheptanes, and thus terpenoids. | ||
Recently, we developed a synthetic route to transform (R)-(−)-carvone (1) into (R)-(+)-3-methyl-6-isopropenyl-cyclohept-3-enone-1 (2), based upon a completely chemo- and regioselective Tiffeneau–Demjanov ring expansion reaction (Scheme 3).36 The addition of TMS-cyanide to (R)-(−)-carvone (1) furnishes the TMS-cyanohydrins 3a/3b, as a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 diastereoisomeric mixture. Reduction of 3a/3b with LiAlH4 provides the required amino-alcohols 4a/4b for the Tiffeneau–Demjanov rearrangement, which leads to the non-conjugated cycloheptenone (R)-(+)-2.
:10 diastereoisomeric mixture. Reduction of 3a/3b with LiAlH4 provides the required amino-alcohols 4a/4b for the Tiffeneau–Demjanov rearrangement, which leads to the non-conjugated cycloheptenone (R)-(+)-2.
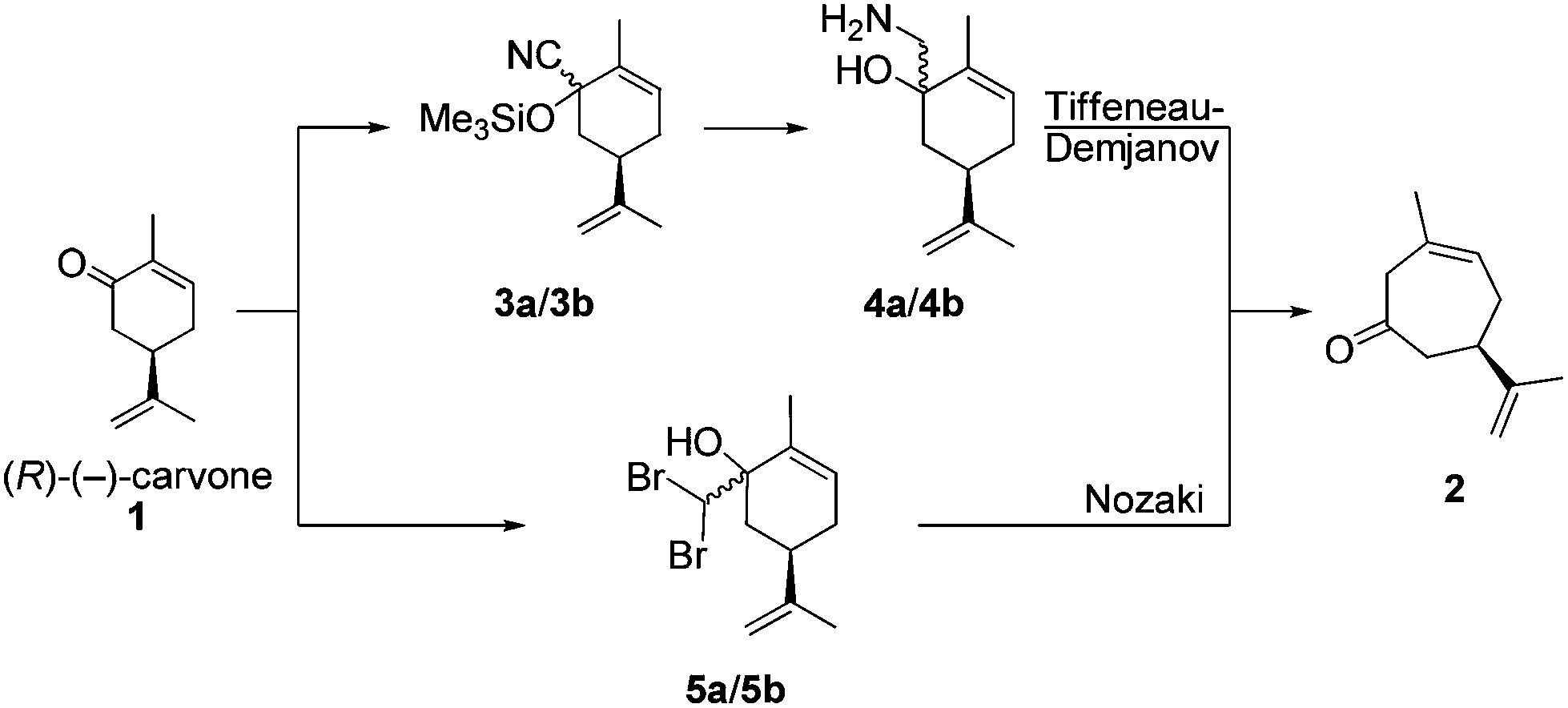 | ||
| Scheme 3 Ring expansions of carvone by the Tiffeneau–Demjanov rearrangement and Nozaki reaction. | ||
We were pleasantly surprised to discover the complete chemo- and regioselectivity in this rearrangement, as can be seen in Scheme 4, where the epoxide 6 and the regio-isomeric cycloheptenone 7 were not observed. Similarly, the Nozaki ring expansion with addition of the dibromomethyl carbanion to carvone, and subsequent rearrangement of 5 are also highly effective (Scheme 3).36
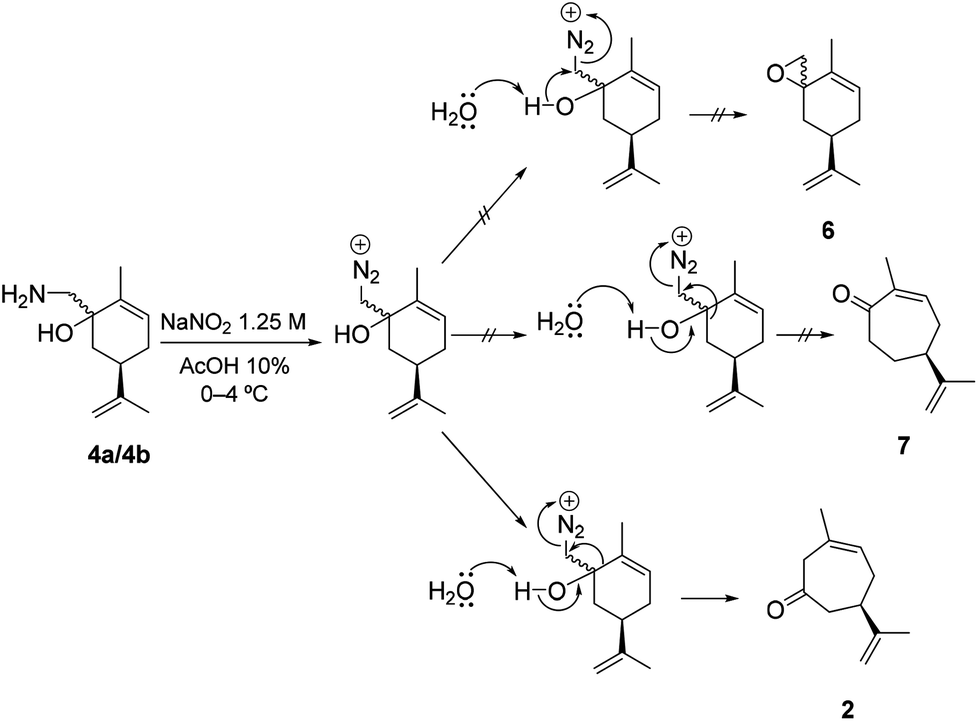 | ||
| Scheme 4 Chemo- and regioselectivity in the Tiffeneau–Demjanov rearrangement. | ||
Encouraged by the total chemo- and regioselectivity observed in the Tiffeneau–Demjanov rearrangement, we have now examined an attractive alternative by Corey–Chaykovsky epoxidation of (R)-(−)-carvone (1) and N-nucleophile ring opening to the same intermediate amino-alcohols 4a/4b obtained in Scheme 3. We now demonstrate significant improvements over the previous synthetic routes, which allow a deca-gram scale-up, in less bench time, with very simple purifications, thus reducing substantially problems of synthesis logistics. This route also avoids the practical problems of using KCN and LiAlH4, or the preparation and use of the CHBr2 carbanion, on a large scale. We also present a structural assignment of 4a by X-ray diffraction and a chemical correlation, and thus the stereochemistry of 1,2 nucleophilic additions to the carbonyl group of (R)-(−)-carvone (1).
Results and discussion
Our initial experiments with the Corey–Chaykovsky epoxidation of (R)-(−)-carvone (1),37 using the original NaH procedure for generating the sulfur ylide, were not reproducible on a large scale, besides presenting some practical difficulties with the manipulation of large quantities of NaH. This led us to try the modification using methyl lithium or n-butyl lithium hexane solutions with DMSO,38 and in these conditions, the epoxidation of (R)-(−)-carvone (1) led to a 90:10 ratio (by GC and 1H-NMR) of 6a and 6b in 90–95% yields (Scheme 5), and this mixture was used as such in the following reaction.
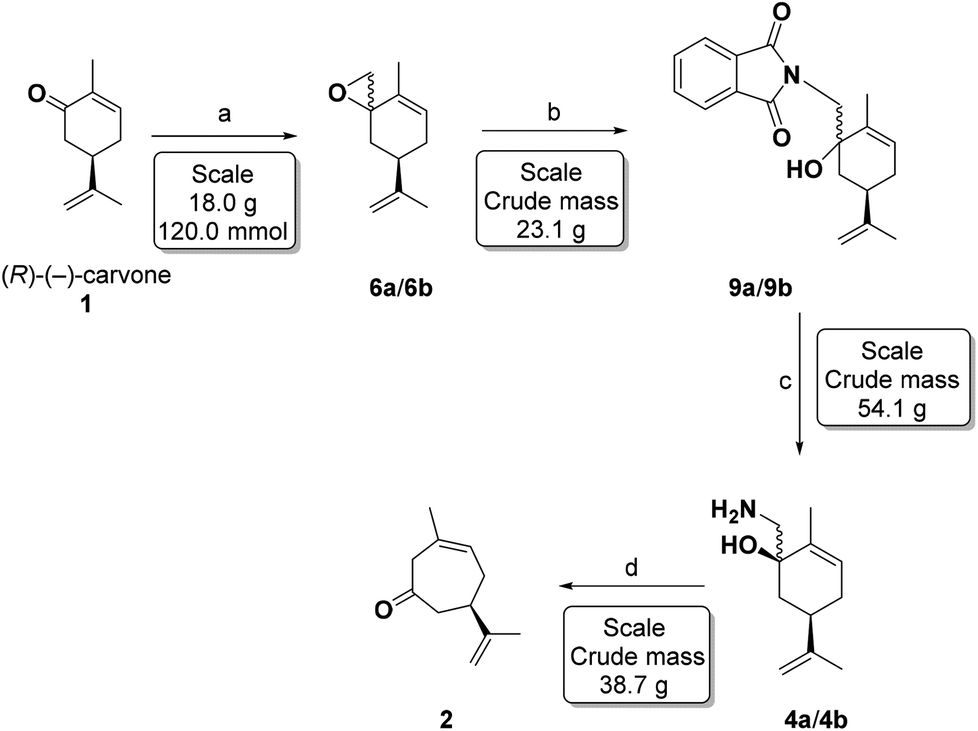 | ||
| Scheme 5 Reagents and conditions: (a) Me3S+I−, n-BuLi, DMSO-THF, 90–95%, (b) K phthalimide, phthalimide, DMF, 155–160 °C, 3 h, (c) NH2NH2·H2O, EtOH, 80–85 °C, 2 h, 64% for 2 steps (d) NaNO2 1.25 M, AcOH 10% (v/v), 0–4 °C, 4 h, 35–71%. | ||
To test the quality of our commercial Me3S+I−, we prepared this reagent freshly by reaction of Me2S with MeI,39 and after recrystallisation the Me3S+I− turned out to be slightly more efficient in our reaction. We conclude, however, that the commercial product is perfectly adequate for our purposes, not justifying the time spent on its preparation and purification.
The base used to form the ylide and reaction conditions were also studied, and the results are shown in Table 1. A solution of the alkyl-lithium was added to DMSO at room temperature, a biphasic mixture was formed, the dimsyl-Li (heavier phase) was transferred, via cannula, to a previously prepared solution of Me3S+I− in THF/DMSO at −10 °C. The (R)-(−)-carvone solution in THF was added, and after 3 h a crude oil containing the mixture of epoxides 6a/6b was obtained, to be used as such in the following reaction.
| Entry | Scale gram (mmol) | Base (eq.) | Me3S+I− (eq.) | Conversiona (%) | Crude mass obtainedb (g) |
|---|---|---|---|---|---|
|
a Conversion based on 1H-NMR signal ratios between H-2 of (R)-(−)-carvone and H-2 of 6a/6b.
b Based on the crude product mass after aqueous work-up; 90:10 ratio in all entries; increased base and Me3SI with scale was used to maintain high conversions.
|
|||||
| 1 | 0.15 (1.0) | MeLi 1.56 M in Et2O (1.2) | 1.2 | 87 | 0.065 |
| 2 | 0.45 (3.0) | MeLi 1.56 M in Et2O (1.2) | 1.2 | 78 | 0.48 |
| 3 | 0.15 (1.0) | n-BuLi 1.50 M in hexanes (1.2) | 1.2 | 80 | 0.17 |
| 5 | 0.45 (3.0) | MeLi 1.56 M in Et2O (1.5) | 1.5 | 100 | 0.52 |
| 6 | 1.50 (10) | n-BuLi 2.50 M in hexanes (1.5) | 1.5 | 100 | 1.41 |
| 7 | 3.02 (20) | n-BuLi 2.50 M in hexanes (2.0) | 2.0 | 95 | 3.38 |
| 8 | 9.00 (60) | n-BuLi 2.50 M in hexanes (2.0) | 2.0 | 95 | 15.03 |
| 9 | 18.02 (120) | n-BuLi 2.50 M in hexanes (2.0) | 2.0 | 95 | 23.13 |
The epoxides 6a/6b were formed on a 23 gram scale from 18 g (120 mmol) of carvone under the conditions shown in Table 1 (entry 9), and used as such in the subsequent reaction.
However, a significant improvement was found while conducting this reaction under continuous flow conditions.40 We used a Vapourtec E-series41,42 flow equipment, with 3 peristaltic pumps (A, B and C), a 10 mL coil reactor and 1.0 mm i.d. PTFE tubing. The equipment schematics with the best results are shown in Scheme 6.
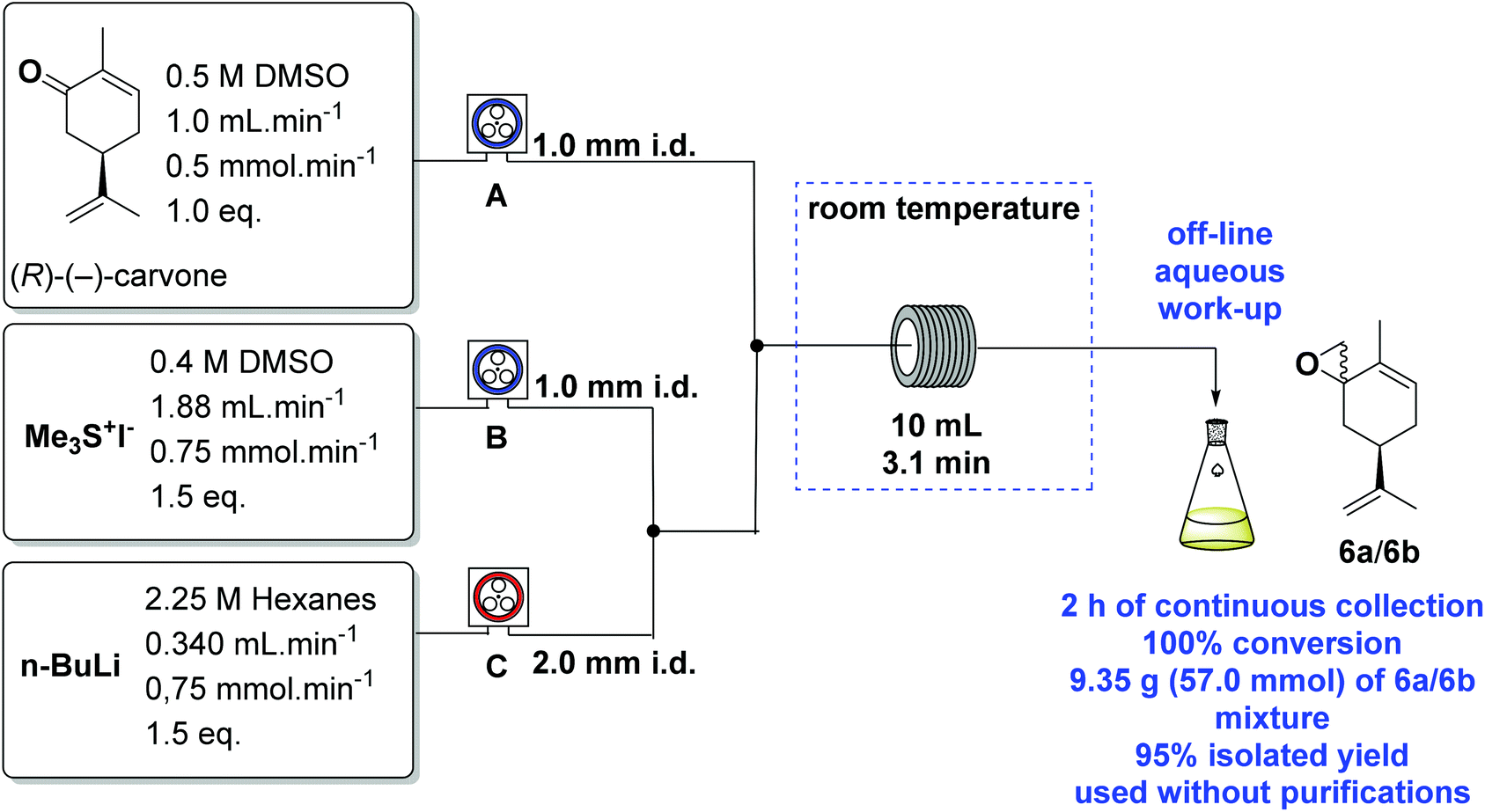 | ||
| Scheme 6 Corey–Chaykovsky epoxidation of (R)-(−)-carvone under continuous flow conditions. | ||
n-BuLi in hexanes was pumped through C (we used a red end crimped fluoropolymer in this pump, compatible with organometallic solutions) and then mixed with a solution of Me3S+I− in DMSO pumped by B. The ylide then meets the (R)-(−)-carvone stream from A, and the reaction takes place in the 10 mL reactor coil kept at room temperature. After the continuous off-line aqueous work-up the desired epoxides 6a/6b were obtained. DMSO (99%+, Alfa-Aesar) was used directly from the bottle, without any further treatment in the flow experiments.
In the initial experiments, our attempts to optimise the procedure were accompanied by blockages within 15 min of processing. The blockages came from the precipitation of the Me3S+I− and/or LiOH from n-BuLi hydrolysis. This problem was solved using a wider bore T-piece and tubing (2.0 mm i.d.) as shown in Scheme 6.
After the standard parameters had been optimised, the preparation was continuously run for over 2 hours, to generate the epoxides 6a/6b (9.4 g, 57 mmol) in 95% isolated yield, from 9.0 g (60 mmol) of (R)-(−)-carvone, 18.4 g (90 mmol) of Me3S+I− and 40 mL (90 mmol) of n-BuLi 2.25 M in hexanes. The crude colourless oil was used in the next step without any need of further purification.
In the batch process using NaH as the base, we obtained the epoxides 6a/6b in 87% yield, whereas the batch process using n-BuLi as the base and a continuous flow process led to 95% yield without the need for purification, and ready for the next synthetic step. The continuous flow procedure was also better due to the ease of operation at much larger scales with minimum manual interactions, resulting in an increase in bench time and simplicity.
Initially the ring opening of the epoxides 6a/6b (Scheme 5) was studied with the obvious nucleophiles ammonia43 and sodium amide,44 but with limited success. For example, we used a commercial ammonia solution in H2O (25–30%, Fisher Scientific) and prepared other ammonia solutions by bubbling ammonia gas, at room temperature, into the desired solvent for 2 h. The solutions were titrated with a 0.12 M aqueous HCl standard solution containing bromocresol green.
We obtained ammonia solutions in MeOH (8.0 N), isopropanol (4.0 M), dimethoxyethane (2.9 M). These ammonia solutions were then reacted in screw cap sealed pressure tubes with the epoxide mixture 6a/6b, and led to the formation of the desired amino-alcohols 4a/4b, and unidentified by-products. The corresponding diols were also formed when we used aqueous ammonia solutions, due to the presence of water (in a 26:10 ratio) (Table 2). The separation of the amino-alcohols on a multi-gram scale we deemed to be impracticable.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Scale grams (mmol) | Solvent (eq. NH3) | Conditions | 4a/4ba (%) | 8 (%) | By-productsa (%) |
| a Conversion based on 1H-NMR signal ratios of H-2 of 4a/4b, 8 and by-products. | ||||||
| 1 | 2 (12.2) | H2O/THF (10) | 90–100 °C, 6 h | 26 | 10 | 64 |
| 2 | 0.2 (1.2) | MeOH (10) | 50 °C, 1 h | 2 | — | 8 |
| 3 | 0.2 (1.2) | MeOH (10) | 70 °C, 1 h | 11 | — | 32 |
| 4 | 0.2 (1.2) | MeOH (10) | 90 °C, 2.5 h | 33 | — | 67 |
| 5 | 0.16 (1.0) | MeOH (10) | 130 °C, 15 min, MW | 29 | — | 61 |
| 6 | 0.16 (1.0) | Isopropanol (10) | 90 °C, 2 h | 8 | — | 10 |
| 7 | 0.32 (2.0) | DME (10) | 90 °C, 1.5 h | — | — | — |
| 8 | 0.32 (2.0) | DME (10) | 130 °C, 15 min, MW | — | — | — |
| 9 | 0.16 (1.0) | Dioxane (10) | 130 °C, 25 min, MW | — | — | — |
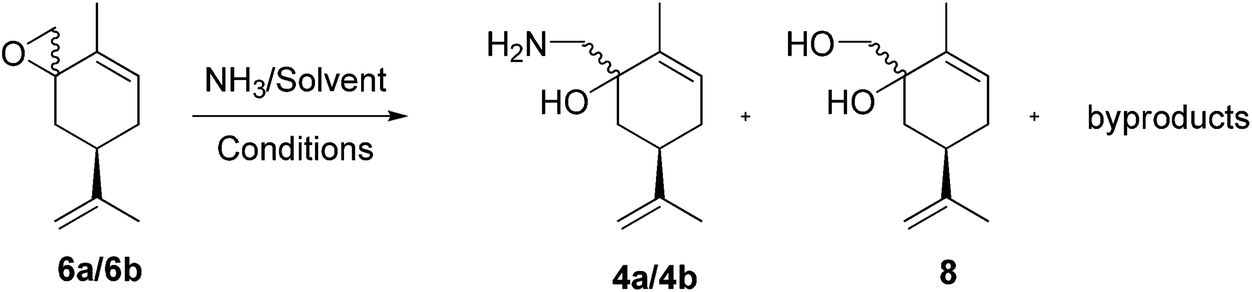
On the other hand, sodium amide led to complex mixtures as shown by TLC analysis, probably due to its reactivity as a base.
Ring opening of the epoxides 6a/6b was easily accomplished by potassium phthalimide and phthalimide in dimethylacetamide (DMA) or DMF at 155–160 °C, according to the classic Gabriel procedure.45Table 3 summarizes our epoxide ring opening results under conventional or microwave (MW) heating, with DMA presenting better results.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Scale grams (mmol) | Conditions |
6a/6ba (%) |
9a/9ba (%) |
10a (%) |
| a Conversion based on the 1H-NMR signal ratios of H-11 of 6a/6b, 9a/9b and 10. b Performed simultaneously in two flasks containing the same quantities (8.21 g, 50.0 mmol) of epoxides 6a/6b. | |||||
| 1 | 0.49 (3.0) | t-BuOH, 160 °C, 2 h | 100 | — | — |
| 2 | 0.49 (3.0) | EtOAc, 160 °C, 2 h | 100 | — | — |
| 3 | 0.49 (3.0) | MeCN, 160 °C, 5 h | Degradation | ||
| 4 | 0.82 (5.0) | DMF, 80 °C, 2 h | 100 | — | — |
| 5 | 0.82 (5.0) | DMF, 110 °C, 2 h | 100 | — | — |
| 6 | 0.82 (5.0) | DMF, 140 °C, 7 h | — | 70 | 30 |
| 7 | 0.16 (1.0) | DMF, 160 °C, 1.5 h | — | 80 | 20 |
| 8 | 0.16 (1.0) | DMF, 100 °C, 7.5 min (MW) | 97 | 2 | 1 |
| 9 | 0.16 (1.0) | DMF, 125 °C, 7.5 min (MW) | 88 | 7 | 5 |
| 10 | 0.16 (1.0) | DMF, 150 °C, 7.5 min (MW) | — | 77 | 23 |
| 11 | 0.16 (1.0) | DMA, 150 °C, 7.5 min (MW) | — | 86 | 14 |
| 12 | 0.16 (1.0) | DMA, 150 °C, 5.0 min (MW) | — | 86 | 14 |
| 13 | 0.16 (1.0) | DMA, 150 °C, 2.5 min (MW) | — | 85 | 15 |
| 14 | 1.64 (10.0) | DMA, 160 °C, 1 h (MW) | — | 84 | 16 |
| 15 | 16.40 (100.0)b | DMA, 160 °C, 6 h | — | 87 | 13 |
| 16 | 23.13 (120.0) | DMF, 160 °C, 3 h | — | 78 | 22 |
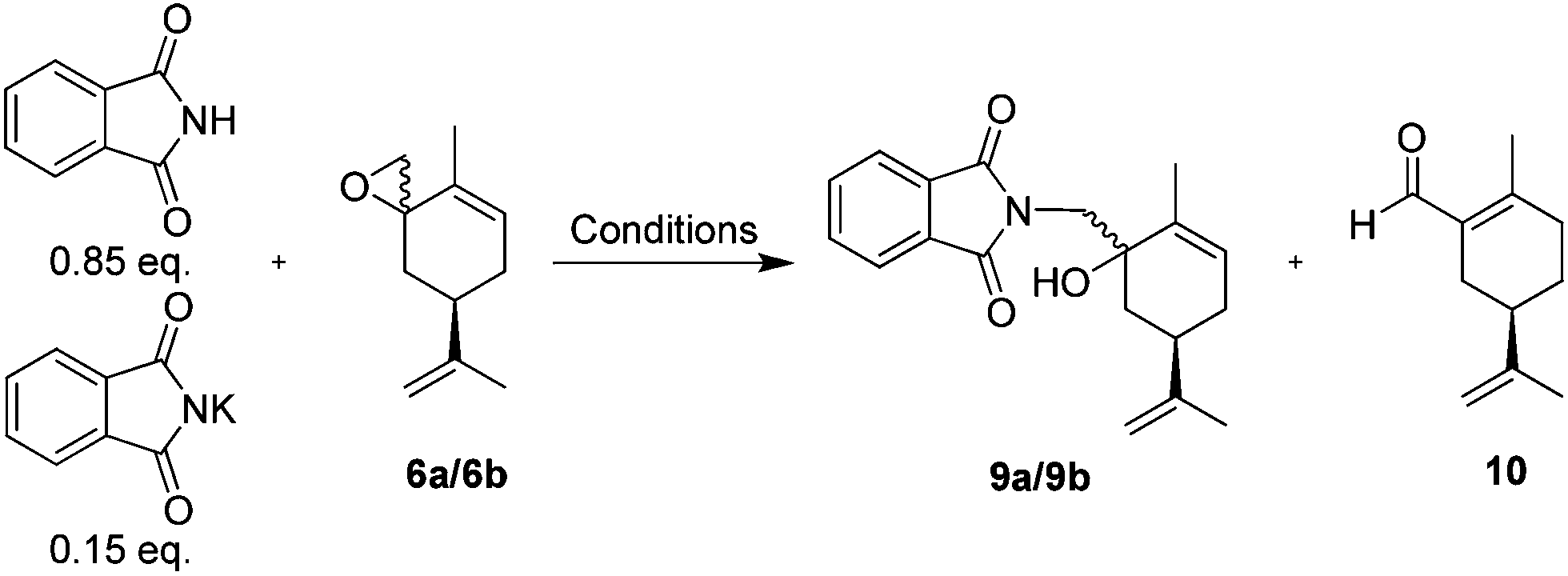
Microwave heating at 150–160 °C in DMA gave the best results, but due to the volume limitation of the microwave tubes (∼14 mL), the large scale reactions (2 flasks with 8.2 g, 50 mmol each and 23.1 g, 120 mmol; Table 3 entries 15 and 16) were performed with conventional heating. The crude product obtained as a brown oil was used directly in the next step.
Hydrazinolysis of the phthalimido-alcohols 9a/9b produced the same amino-alcohols 4a/4b obtained in the previous sequence (Scheme 3). After reacting with hydrazine monohydrate (1.5 equivalents) in refluxing ethanol for 3 hours, the starting material 9a/9b was completely consumed. The crude reaction product (13.9 g, 77 mmol) was characterized as the amino-alcohols 4a/4b with an 85:15 ratio as determined by 1H-NMR. Recrystallization from hot hexanes afforded 12.2 g (73.7 mmol) of amino-alcohol 4a in 64% yield for the last 2 steps.
The phthalimido-alcohol 9a/9b reduction described by Ganem et al.46 was also examined using NaBH4 in isopropanol:H2O at room temperature for 24 h. A yellow oil was obtained containing the amino-alcohols 4a/4b together with a complex mixture of by-products. As the product 4a/4b crystallizes along with the by-product phthalide, and required large amounts of sodium borohydride, this procedure was not investigated further.
The Tiffeneau–Demjanov47,48 rearrangement was performed by treatment of a solution of 4a/4b (38.7 g, 214 mmol, divided in 3 flasks with 12.9 g each), in 10% (v/v) aqueous AcOH with an aqueous NaNO2 solution (1.25 M, 103 mL, 8.83 g, 128 mmol, 1.8 equiv.) for each flask. The temperature was maintained at 0–4 °C (ice-water bath) for 4 hours. Aqueous work-up then produced a brown oil which underwent immediate chromatographic purification affording the non-conjugated cycloheptenone 2 (15.9 g, 96 mmol) in 35% yield ([α]25D = +44.3 (c 1.15, CHCl3): lit.36 [α]25D = +30.0 (c 0.26 CHCl3). Under these conditions we did not observe the formation of the conjugated cycloheptenone.
The assignment of the stereochemistry of 3a/3b and thus 6a/6b was made by chemical correlation with 4a, from the mono-crystal X-ray diffraction studies (Fig. 3) and nOe irradiation (see ESI†) of the major amino-alcohol 4a, obtained by reduction of the TMS–cyanohydrin mixture 3a/3b. Other nucleophilic addition reactions to the carbonyl group of carvone have been studied previously,49 and the stereochemistry of major approaches has been shown to be preferentially anti- to the isopropenyl group.50 We have now confirmed that this is the correct stereochemistry for the major isomers 3a and 6a. The structure of 4a is shown in Fig. 3, and the chemical correlation of the amino-alcohols 4a/4b obtained in both sequences, establishes the same 90:10 diastereomeric preference of addition of cyanide and the sulfonium ylide nucleophiles.
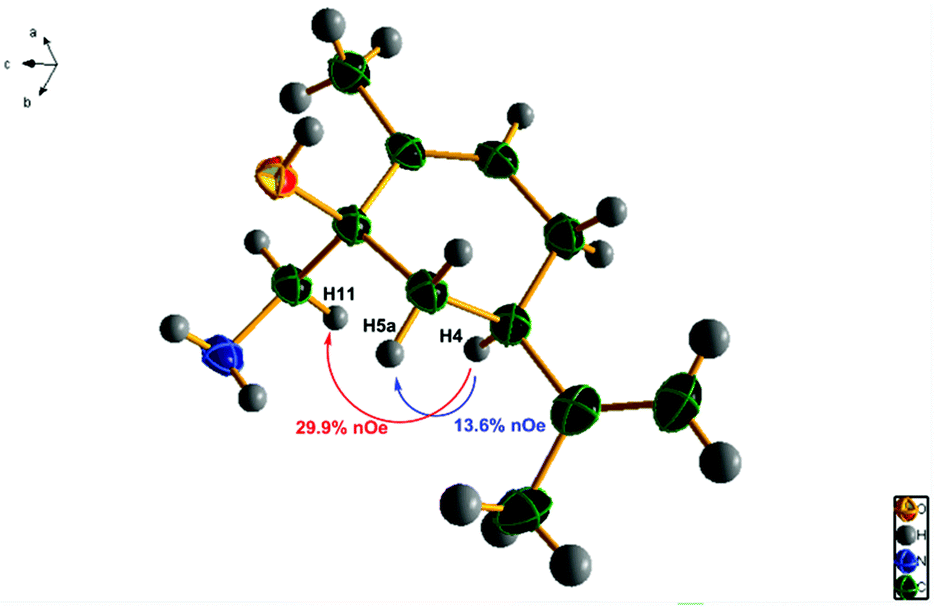 | ||
| Fig. 3 Structure of the major amino-alcohol 4a, obtained by X-ray diffraction studies, indicating observed nOe interactions. (Ellipsoids shown at 40% probability level). | ||
Conclusions
In summary, we have developed a very efficient synthetic route to the appropriately functionalized (R)-(+)-3-methyl-6-isopropenyl-3-cycloheptenone-1 (2), a useful enantiopure intermediate for bioactive terpenoid synthesis. Using this methodology, cycloheptenone 2 has been prepared on a deca-gram scale with minimal chromatographic separation needed. Starting from 18.0 g of (R)-(−)-carvone (1), we have obtained 8.1 g (41% overall yield) of the cycloheptenone 2.Experimental
General protocol for preparation of 6a/6b in flow
The continuous flow preparation of the epoxides 6a/6b was carried out using a three-stream reactor assembly. The Vapourtec E-Series machine was charged with a 0.5 M solution of (R)-(−)-carvone in DMSO (pump A) at the rate of 1.0 mL min−1, a 0.4 M solution of Me3S+I− in DMSO (pump B) at the rate of 1.88 mL min−1 and a solution of n-BuLi (2.25 M in hexanes) pumped directly from the bottle through C at the rate of 0.340 mL min−1. The DMSO was used directly without any purification. The desired flow rates were set and all pumps were started. DMSO and hexane were pumped for 5 min. Pump C was timed to switch to pumping n-BuLi for 5 min before switching pumps A and B simultaneously to (R)-(−)-carvone and Me3S+I− at the rates as determined above. The streams of pumps B and C were mixed through a T-piece generating the sulfur ylide which was mixed with a stream of (R)-(−)-carvone from pump A. A PTFE tubing of 2.00 mm i.d. was used between pump C and the second T-piece. The resulting stream was driven to a 10 mL coil reactor at room temperature with a residence time of 3.1 min at these flow rates. The quenching was achieved by continuously collecting the output in a conical flask with cold water for 2 h.:5); ratio6a/6b: 90:10 (1H NMR and GC); [α]25D = +24.3 (c 1.42, CHCl3); 1H NMR (CDCl3, 400 MHz) major isomer 6a: δ 5.82–5.70 (1H, m), 4.74 (1H, br s), 4.72 (1H, br s), 2.93 (1H, dd, J = 4.9, 1.4 Hz), 2.67 (1H, d, J = 5.0 Hz), 2.44–2.56 (1H, m), 2.16–2.26 (1H, m), 2.06–2.12 (1H, m), 1.98–2.05 (1H, m), 1.73 (3H, br s), 1.50 (3H, br s), 1.45–1.56 (1H, m); 13C NMR (CDCl3, 100 MHz) major isomer 6a: δ 148.4, 133.0, 128.7, 109.5, 59.0, 53.3, 41.6, 36.8, 31.4, 20.6, 15.6; IR (neat, cm−1): 2971, 2919, 1645, 1450, 1436, 888; LRMS: m/z 164, 149, 135, 121, 107, 93, 91, 77, 55, 41; HRMS (ESI+): m/z calc. for C11H17O [M + H]+ 165.1279, found 165.1278; GC: 9.975 min = 6a, 9.817 min = 6b.
Representative procedure for the epoxide opening with ammonia
A screw-cap pressure tube was charged with the epoxides 6a/6b and ammonia solution. The pressure tubes were stoppered and heated. The mixtures were cooled down to room temperature, and the pressure tubes were opened and gently heated in order to remove the residual ammonia.:50); [α]25D = −0.71 (c 0.11, CHCl3); m.p. 103.6–104.5 °C; 1H NMR (CDCl3, 400 MHz): δ 5.66–5.74 (1H, m), 4.74–4.77 (2H, m), 3.70 (1H, d, J = 10.7 Hz), 3.54 (1H, d, J = 10.7 Hz), 2.29–2.40 (1H, m), 2.12–2.20 (1H, m), 1.90–1.96 (1H, m), 1.82–1.89 (1H, m), 1.73–1.79 (6H, m), 1.55–1.64 (1H, m), 1.49–2.45 (2H, m, after D2O exchange this resonance disappears); 13C NMR (CDCl3, 100 MHz): δ 149.1, 134.2, 128.7, 109.3, 72.8, 68.8, 39.2, 37.1, 31.4, 21.0, 18.0; IR (neat, cm−1): 3305, 2946, 2911, 2854, 1645, 1445, 1359, 1011, 889; LRMS: m/z 182, 164, 151, 123, 109, 93, 91, 67, 55, 41; HRMS (ESI+): m/z calc. for C11H18O2Na [M + Na]+ 205.1205, found 205.1201; GC: 12.983 min = 8.
Representative procedure for the epoxide opening with sodium amide
A screw cap pressure tube was charged with the epoxides 6a/6b, THF and sodium amide. Ammonia was condensed from the cylinder with a cold-finger condenser and added to a screw cap tube maintained at −78 °C with magnetic stirring. The pressure tube was closed and allowed to warm to room temperature, and left stirring for 12 h. After this time, the tube was opened and gently heated in order to remove the residual ammonia. The amino-alcohols 4a/4b were not observed by TLC.Procedure for the epoxide opening with phthalimide/phthalimide K
To a round-bottomed flask were added sequentially phthalimide, potassium phthalimide, the epoxides 6a/6b and DMF. The suspension was heated at 160 °C for 3 h and after cooling down to room temperature the reaction mixture was diluted with EtOAc, water and brine. The crude product was used in the next step without any further purification.:20); ratio9a/9b: 85:15 (1H NMR); [α]25D = −34.4 (c 1.40, CHCl3); 1H NMR (CDCl3, 400 MHz) major isomer 9a: δ 7.81–7.90 (2H, m), 7.68–7.75 (2H, m), 5.58 (1H, br s), 4.76 (1H, br s), 4.70 (1H, br s), 3.98 (1H, d, J = 14.6 Hz), 3.84 (1H, d, J = 14.6 Hz), 3.18 (1H, s, after D2O exchange this resonance disappears), 2.52–2.65 (1H, m), 2.07–2.17 (1H, m), 1.92–2.02 (1H, m), 1.85 (3H, br s), 1.77–1.87 (1H, m), 1.70 (3H, br s), 1.44–1.53 (1H, m); 13C NMR (CDCl3, 100 MHz) major isomer 9a: δ 169.5, 148.7, 135.7, 134.3, 131.9, 126.0, 123.6, 109.5, 75.0, 44.4, 39.0, 38.1, 31.1, 20.5, 17.2; IR (neat, cm−1): 3486, 2940, 2920, 1772, 1705, 890, 716; LRMS:m/z 311, 293, 268, 252, 238, 196, 178, 161, 151, 133, 123, 109, 91, 77, 67, 41; HRMS (ESI+): m/z calc. for C19H22NO3 [M + H]+ 312.1600, found 312.1594; GC: 26.925 min = 9a, 27.025 min = 9b.
:10); [α]25D = +113 (c 1.16, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 10.13 (1H, s), 4.73 (1H, br s), 4.69 (1H, br s), 2.44–2.65 (1H, m), 2.26–2.37 (2H, m), 2.13 (3H, s), 2.01–2.10 (1H, m), 1.87–1.95 (1H, m), 1.78–1.86 (1H, m), 1.73 (3H, br s), 1.38–1.51 (1H, m); 13C NMR (CDCl3, 100 MHz): δ 191.0, 155.7, 149.0, 133.3, 109.2, 40.3, 34.8, 27.6, 26.9, 20.9, 18.1; IR (neat, cm−1): 2933, 2865, 1666, 1643, 1438, 1232, 888; LRMS: m/z 164, 149, 123, 121, 95, 93, 68, 53, 41; HRMS: m/z calc. mass for C11H17O [M + H]+ 165.1279, found 165.1273; GC: 11.825 min = 10.
Procedure for hydrazinolysis of phthalimido-alcohols
To a suspension of the crude products 9a/9b in EtOH was added NH2NH2·H2O. The reaction system was heated at 80–85 °C for 3 h, cooled down to room temperature, and the white solid formed (phthalyl hydrazide) was filtered off in a sintered funnel and washed with EtOH. The ethanolic filtrate afforded after evaporation in vacuo a yellowish oil mixed with a solid characterized as 4a/4b. The crude mixture of amino-alcohols 4a/4b can be used directly in the next step.:50); ratio4a/4b: 85:15 (1H NMR and GC); [α]25D = −118 (c 1.03, CHCl3) {lit.36 [α]25D = −92.2 (c 2.0 CHCl3)}; m.p. 100.9–101.5 °C {lit.36 m.p. 99.2–99.7 °C}; 1H NMR (CDCl3, 600 MHz): δ 5.51 (1H, br s), 4.72 (1H, br s), 4.71 (1H br s), 2.78 (1H, d, J = 13.0 Hz), 2.72 (1H, d, J = 13.0), 2.21–2.29 (1H, m), 2.04–2.11 (1H, m) 1.88–1.96 (2H, m), 1.72 (3H, br s), 1.70 (3H, br s), 1.45–1.54 (1H, m,), 0.5–3.5 (3H, m, after D2O exchange this resonance disappears). 13C NMR (CDCl3, 150 MHz): δ 149.1, 137.2, 125.2, 109.2, 73.0, 46.6, 39.5, 38.3, 31.3, 20.6, 17.3; IR (film, cm−1): 3372, 3309, 3082, 2955, 2914, 1645, 1596, 940, 891; LRMS: m/z 181, 164, 151, 123, 109, 91, 81, 67, 55, 41; HRMS (ESI+): m/z calc. for C11H20NO [M + H]+ 182.1545, found 182.1541; GC: 12.958 min = 4a, 13.017 min = 4b.
Procedure for Tiffeneau–Demjanov rearrangement
A solution of the amino-alcohols 4a/4b in 10% (v/v) aqueous AcOH at 0 °C was treated with a 1.25 M aqueous solution of NaNO2. The reaction mixture was stirred for 4 h at 0 °C. The aqueous phase was extracted with EtOAc and the combined organic extracts were washed with 10% (m/v) solution of NaHCO3, brine, water and dried over Mg2SO4. The solvent was removed in vacuo and the residue was immediately purified by flash column chromatography to afford 2.:10); [α]25D = +44.3 (c 1.15, CHCl3) {lit.36 [α]25D = +30.0 (c 0.26 CHCl3)}; 1H NMR (CDCl3, 400 MHz): δ 5.51–5.59 (1H, m), 4.75 (1H, br s), 4.72 (1H, br s), 3.30 (1H, d, J = 14.8 Hz), 2.99 (1H, d, J = 14.8 Hz), 2.70–2.80 (1H, m), 2.60 (1H, br s), 2.58 (1H, br s), 2.16–2.35 (2H, m), 1.77 (3H, br s), 1.72 (3H, br s); 13C NMR (CDCl3, 100 MHz): δ 208.0, 148.3, 130.4, 124.5, 110.2, 49.0, 48.3, 43.3, 33.1, 26.1, 20.5; IR (neat, cm−1): 2969, 2913, 1704, 890; LRMS: m/z 164, 149, 136, 122, 107, 93, 80, 68, 53, 41; HRMS: m/z calc. for C11H17O [M + H]+ 165.1279, found 165.1278; GC: 10.242 min = 2.
Acknowledgements
The authors wish to thank the following agencies for financial support: FAPESP (2014/12001-4, 2011/13993-2 and 2013/06532-4), CNPq and CAPES in Brazil, and the EPSRC (UK) for funds award number EP/FO69685/1 (to S.N. and S.V.L). We also thank Dr John Davies, the X-Ray Crystallography Facilities at the Department of Chemistry, University of Cambridge, and Firmenich S.A., São Paulo, for a generous donation of (R)-(−)-carvone. We acknowledge the contribution of the referees for their excellent and constructive suggestions.Notes and references
- K. T. de Oliveira, B. M. Servilha, L. de C. Alves, A. L. Desiderá and T. J. Brocksom, Stud. Nat. Prod. Chem., 2014, 42, 421–463 CrossRef CAS.
- T. V. Nguyen, J. M. Hartmann and D. Enders, Synthesis, 2013, 845–873 CAS.
- (a) E. J. Corey, M. Ohno, P. A. Vatakencherry and R. B. Mitra, J. Am. Chem. Soc., 1961, 83, 1251–1253 CrossRef CAS; (b) E. J. Corey, M. Ohno, R. B. Mitra and P. A. Vatakencherry, Am. Chem. Soc., 1964, 86, 478–485 CrossRef CAS.
- (a) S. V. Ley, A. Antonello, E. P. Balskus, D. T. Booth, S. B. Christensen, E. Cleator, H. Gold, K. Högenauer, U. Hünger, R. M. Myers, S. F. Oliver, O. Simic, M. D. Smith, H. Søhoel and A. J. A. Woolford, Proc. Nat. Acad. Sci. U. S. A., 2004, 101, 12073–12078 CrossRef CAS PubMed; (b) S. P. Andrews, M. Ball, F. Wierschem, E. Cleator, S. Oliver, K. Högenauer, O. Simic, A. Antonello, U. Hünger, M. D. Smith and S. V. Ley, Chem. – Eur. J., 2007, 13, 5688–5712 CrossRef CAS PubMed.
- (a) R. H. Pouwer, J.-A. Richard, C.-C. Tseng and D. Y.-K. Chen, Chem. – Asian J., 2012, 7, 22–35 CrossRef CAS PubMed; (b) K. Molawi, N. Delpont and A. M. Echavarren, Angew. Chem., Int. Ed., 2010, 49, 3517–3519 CrossRef CAS PubMed.
- (a) D. Y.-K. Chen, Synlett, 2011, 2459–2481 CrossRef CAS; (b) W. Xu, S. Wu, L. Zhou and G. Liang, Org. Lett., 2013, 15, 1978–1981 CrossRef CAS PubMed.
- (a) H. M. Cheng, W. Tian, P. A. Peixoto, B. Dhudshia and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2011, 50, 4165–4168 CrossRef CAS PubMed; (b) L. Chang, H. Jiang, J. Fu, B. Liu, C.-c. Li and Z. Yang, J. Org. Chem., 2012, 77, 3609–3614 CrossRef CAS PubMed.
- (a) P. A. Wender, K. D. Rice and M. E. Schnute, J. Am. Chem. Soc., 1997, 119, 7897–7898 CrossRef CAS; (b) P. A. Wender, H. Kogen, H. Y. Lee, J. D. Munger, R. S. Wilhelm and P. D. Williams, J. Am. Chem. Soc., 1989, 111, 8957–8958 CrossRef CAS; (c) See also, P. A. Wender, Tetrahedron, 2013, 69, 7529–7550 CrossRef CAS PubMed.
- (a) L. Jørgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878–882 CrossRef PubMed; (b) G. Grue-Sørensen, X. Liang, K. Månsson, P. Vedsø, M. D. Sørensen, A. Soor, M. Stahlhut, M. Bertelsen, K. M. Engell and T. Högberg, Bioorg. Med. Chem. Lett., 2014, 24, 54–60 CrossRef PubMed; (c) S. J. McKerrall, L. Jørgensen, C. A. Kuttruff, F. Ungeheuer and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5799–5810 CrossRef CAS PubMed.
- (a) A. K. Miller, C. C. Hughes, J. J. Kennedy-Smith, S. N. Gradl and D. Trauner, J. Am. Chem. Soc., 2006, 128, 17057–17062 CrossRef CAS PubMed; (b) C. C. Hughes, J. J. Kennedy-Smith and D. Trauner, Org. Lett., 2003, 5, 4113–4115 CrossRef CAS PubMed; (c) A. Srikrishna and D. H. Dethe, Org. Lett., 2004, 6, 165–168 CrossRef CAS PubMed; (d) C. A. McGowan, A.-K. Schmieder, L. Roberts and M. F. Greaney, Org. Biomol. Chem., 2007, 5, 1522–1524 RSC.
- (a) J. R. Hanson, Nat. Prod. Rep., 2013, 30, 1346–1356 RSC; (b) D. L. Chen, X. Y. Liu, H. Cheng and F. P. Wang, Chin. Chem. Lett., 2011, 22, 774–776 CrossRef CAS; (c) F. de J. Cortez, D. Lapointe, A. M. Hamlin, E. M. Simmons and R. Sarpong, Tetrahedron, 2013, 69, 5665–5676 CrossRef CAS PubMed; (d) F. de J. Cortez and R. Sarpong, Org. Lett., 2010, 12, 1428–1431 CrossRef PubMed.
- (a) G. Majetich and Y. Zhang, J. Am. Chem. Soc., 1994, 116, 4979–4980 CrossRef CAS; (b) G. Majetich, Y. Zhang, X. Tian, J. E. Britton, Y. Li and R. Phillips, Tetrahedron, 2011, 67, 10129–10146 CrossRef CAS.
- (a) M. Reiter, S. Torssell, S. Lee and D. W. C. MacMillan, Chem. Sci., 2010, 1, 37–42 RSC; (b) D. R. Laplace, B. Verbraeken, K. V. Hecke and J. M. Winne, Chem. – Eur. J., 2014, 20, 253–262 CrossRef CAS PubMed.
- (a) A. R. Hardin-Narayan, E. M. Simmons and R. Sarpong, Eur. J. Org. Chem., 2010, 3553–3567 CrossRef CAS; (b) C. F. Nising and S. Bräse, Angew. Chem., Int. Ed., 2008, 47, 9389–9391 CrossRef CAS PubMed; (c) M. Dai and S. J. Danishefsky, Tetrahedron Lett., 2008, 49, 6610–6612 CrossRef CAS PubMed; (d) K. C. Nicolaou, X.-S. Peng, Y.-P. Sun, D. Polet, B. Zou, C. S. Lim and D. Y.-K. Chen, J. Am. Chem. Soc., 2009, 131, 10587–10597 CrossRef CAS PubMed; (e) E. M. Simmons, A. R. Hardin-Narayan, X. Guo and R. Sarpong, Tetrahedron, 2010, 66, 4696–4700 CrossRef CAS PubMed.
- T. Graening and H.-G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 3230–3256 CrossRef CAS PubMed.
- (a) K. M. Peese and D. Y. Gin, J. Am. Chem. Soc., 2006, 128, 8734–8735 CrossRef CAS PubMed; (b) S. S. Bhar and M. M. V. Ramana, Stud. Nat. Prod. Chem., 2008, 35, 399–422 CAS.
- H. Muratake and M. Natsume, Angew. Chem., Int. Ed., 2004, 43, 4646–4649 CrossRef CAS PubMed.
- (a) C. H. Heathcock, M. M. Hansen, R. B. Ruggeri and J. C. Kath, J. Org. Chem., 1992, 57, 2544–2553 CrossRef CAS; (b) Z. Lu, Y. Li, J. Deng and A. Li, Nat. Chem., 2013, 5, 679–684 CrossRef CAS PubMed; (c) H. Wu, X. Zhang, L. Ding, S. Chen, J. Yang and X. Xu, Planta Med., 2013, 79, 1589–1598 CrossRef CAS PubMed.
- C. Thebtaranonth and Y. Thebtaranonth, Cyclization Reactions, CRC Press Inc., Boca Raton, FL, 1994 Search PubMed.
- (a) Y.-D. Zhang, Y.-F. Tang, T.-P. Luo, J. Shen, J.-H. Chen and Z. Yang, Org. Lett., 2006, 8, 107–110 CrossRef CAS PubMed; (b) J. A. Enquist and B. M. Stoltz, Nature, 2008, 453, 1228–1231 CrossRef CAS PubMed; (c) J. A. Enquist, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2011, 17, 9957–9969 CrossRef CAS PubMed; (d) T. Magauer, J. Mulzer and K. Tiefenbacher, Org. Lett., 2009, 11, 5306–5309 CrossRef CAS PubMed; (e) M. S. Dowling and C. D. Vanderwal, J. Am. Chem. Soc., 2009, 131, 15090–15091 CrossRef CAS PubMed; (f) B. Shi, N. A. Hawryluk and B. B. Snider, J. Org. Chem., 2003, 68, 1030–1042 CrossRef CAS PubMed; (g) D. S. Tan, G. B. Dudley and S. J. Danishefsky, Angew. Chem., Int. Ed., 2002, 41, 2185–2188 CrossRef CAS; (h) S. Lin, G. B. Dudley, D. S. Tan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2002, 41, 2188–2191 CrossRef CAS; (i) G. S. Weatherhead, G. A. Cortez, R. R. Schrock and A. H. Hoveyda, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5805–5809 CrossRef CAS PubMed.
- (a) M. Harmata, Tetrahedron, 1997, 53, 6235–6280 CrossRef CAS; (b) M. Harmata, Chem. Commun., 2010, 46, 8886–8903 RSC; (c) E. H. Krenske, K. N. Houk and M. Harmata, Org. Lett., 2010, 12, 444–447 CrossRef CAS PubMed; (d) Y. Lian, L. C. Miller, S. Born, R. Sarpong and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 12422–12425 CrossRef CAS PubMed.
- P. A. Wender, H. Takahashi and B. Witulski, J. Am. Chem. Soc., 1995, 117, 4720–4721 CrossRef CAS.
- (a) P. A. Evans, T. Baikstis and P. A. Inglesby, Tetrahedron, 2013, 69, 7826–7830 CrossRef CAS; (b) L. Saya, G. Bhargava, M. A. Navarro, M. Gulías, F. López, I. Fernández, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2010, 49, 9886–9890 CrossRef CAS PubMed; (c) N. Etkin, T. L. Dzwiniel, K. E. Schweibert and J. M. Stryker, J. Am. Chem. Soc., 1998, 120, 9702–9703 CrossRef CAS.
- E. J. Kantorowski and M. J. Kurth, Tetrahedron, 2000, 56, 4317–4353 CrossRef CAS.
- B. M. Fraga, Nat. Prod. Rep., 2013, 30, 1226–1264 RSC.
- D. A. Foley and A. R. Maguire, Tetrahedron, 2010, 66, 1131–1175 CrossRef CAS.
- (a) T.-L. Ho, Carbocycle Construction in Terpene Synthesis, VCH, New York, NY, 1988 Search PubMed; (b) T.-L. Ho, Enantioselective Synthesis: Natural Products From Chiral Terpenes, Wiley, New York, NY, 1992 Search PubMed.
- A. Mendoza, Y. Ishihara and P. S. Baran, Nat. Chem., 2012, 4, 21–25 CrossRef CAS PubMed.
- (a) I. Ojima, M. Tzamarioudaki, Z. Li and R. J. Donovan, Chem. Rev., 1996, 96, 635–662 CrossRef CAS PubMed; (b) C. P. Dell, Contemp. Org. Synth., 1997, 4, 87–117 RSC; (c) K. I. Booker-Milburn and A. Sharpe, Contemp. Org. Synth., 1996, 3, 473–498 RSC; (d) R. C. Hartley and S. T. Caldwell, J. Chem. Soc., Perkin Trans. 1, 2000, 477–501 RSC.
- T. Harayama, Y. Shinkai, Y. Hashimoto, H. Fukushi and Y. Inubushi, Tetrahedron Lett., 1983, 24, 5241–5244 CrossRef CAS.
- A. R. Angeles, D. C. Dorn, C. A. Kou, M. A. S. Moore and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 1451–1454 CrossRef CAS PubMed.
- G. Kreiselmeier and B. Föhlisch, Tetrahedron Lett., 2000, 41, 1375–1379 CrossRef CAS.
- T. J. Brocksom, E. T. C. Pesquero and F. T. Lopes, Synth. Commun., 1990, 20, 1181–1191 CrossRef CAS.
- (a) J. Zukerman-Schpector, I. Caracelli, C. C. Carvalho, M. L. de Faria, F. C. Silva, L. G. de O Matias and T. J. Brocksom, J. Braz. Chem. Soc., 2001, 12, 154–158 CrossRef CAS; (b) T. J. Brocksom, U. Brocksom and D. Frederico, Tetrahedron Lett., 2004, 45, 9289–9291 CrossRef CAS; (c) T. J. Brocksom, A. G. Corrêa, R. M. Naves, F. Silva Jr., V. Catani, M. A. Ceschi, J. Zukerman-Schpector, A. P. Toloi, M. L. Ferreira and U. Brocksom, in Organic Synthesis: Theory and Applications, ed. T. Hüdlický, Elsevier Science Ltd, 2001, vol. 5, pp. 39–87 Search PubMed.
- (a) M. L. de Faria, R. de A. Magalhães, F. C. Silva, L. G. de O. Matias, M. A. Ceschi, U. Brocksom and T. J. Brocksom, Tetrahedron: Asymmetry, 2000, 11, 4093–4103 CrossRef; (b) T. J. Brocksom, U. Brocksom and F. P. Barbosa, J. Braz. Chem. Soc., 2006, 17, 792–796 CrossRef CAS . The term overbred was introduced in R. W. Hoffmann, Elements of Synthesis Planning, Springer, Berlin, p. 106, 2009, to describe synthesis by way of an intermediate with more rings than in the final product.
- T. J. Brocksom, U. Brocksom, D. P. Sousa and D. Frederico, Tetrahedron: Asymmetry, 2005, 16, 3628–3632 CrossRef CAS . The exclusive regioselectivity of our Tiffeneau–Demjanov rearrangement is attributed to the preferential migratory aptitude of sp2 carbon atoms over sp3 carbon atoms.
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 3782–3783 CrossRef CAS; (b) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1345–1353 CrossRef CAS; (c) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1354–1364 Search PubMed; (d) E. J. Corey and M. Chaykovsky, Org. Synth. Coll., 1973, 5, 755–757 Search PubMed.
- (a) B. Zhou, X. Li, H. Feng and Y. Li, Tetrahedron, 2010, 66, 5396–5401 CrossRef CAS; (b) M. E. Kuehne and F. Xu, J. Org. Chem., 1993, 58, 7490–7497 CrossRef CAS; (c) S. J. Danishefsky, J. J. Masters, W. B. Young, J. T. Link, L. B. Snyder, T. V. Magee, D. K. Jung, R. C. A. Isaacs, W. G. Bornmann, C. A. Alaimo, C. A. Coburn and M. J. di Grandi, J. Am. Chem. Soc., 1996, 118, 2843–2859 CrossRef CAS.
- H. J. Emeléus and H. G. Heal, J. Chem. Soc., 1946, 1126–1131 RSC.
- Selected publications featuring flow chemistry: (a) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS; (b) J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42 CrossRef CAS PubMed; (c) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC; (d) I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519–552 CrossRef CAS; (e) S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC; (f) J.-i. Yoshida, Chem. Rec., 2010, 10, 332–341 CrossRef CAS PubMed; (g) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC; (h) S. Newton, C. F. Carter, C. M. Pearson, L. de C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915–4920 CrossRef CAS PubMed; (i) X. Y. Mak, P. Laurino and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5, 19 Search PubMed; (j) L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634–1637 CrossRef CAS PubMed.
- http://www.vapourtec.co.uk/products/eseriessystem (accessed December 2014).
- Recent publications featuring organometallics in flow: (a) P. R. D. Murray, D. L. Browne, J. C. Pastre, C. Butters, D. Guthrie and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1192–1208 CrossRef CAS; (b) J. A. Newby, L. Huck, D. W. Blaylock, P. M. Witt, S. V. Ley and D. L. Browne, Chem. – Eur. J., 2014, 20, 263–271 CrossRef CAS PubMed; (c) W. Shu and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 5355–5358 CrossRef CAS PubMed; (d) D. Webb and T. F. Jamison, Org. Lett., 2011, 14, 568–571 CrossRef PubMed.
- (a) W. F. Newhall, J. Org. Chem., 1959, 24, 1673–1676 CrossRef CAS; (b) M. W. Bedore, N. Zaborenko, K. F. Jensen and T. F. Jamison, Org. Process Res. Dev., 2010, 14, 432–440 CrossRef CAS; (c) M. Nagel, H.-J. Hansen and G. Fráter, Synlett, 2002, 280–284 CrossRef CAS.
- M. A. McKinney and P. P. Patel, J. Org. Chem., 1973, 38, 4059–4064 CrossRef CAS.
- (a) J. C. Sheehan and V. A. Bolhofer, J. Am. Chem. Soc., 1950, 72, 2786–2788 CrossRef; (b) M. S. Gibson and R. W. Bradshaw, Angew. Chem., Int. Ed. Engl., 1968, 7, 919–930 CrossRef CAS.
- J. O. Osby, M. G. Martin and B. Ganem, Tetrahedron Lett., 1984, 25, 2093–2096 CrossRef CAS.
- (a) M. Tiffeneau, M. P. Weill and B. Tehoubar, Compt. Rend., 1937, 54–56 CAS; (b) P. A. S. Smith and D. R. Baer, Org. React., 1960, 11, 157–188 CAS.
- D. Fattori, S. Henry and P. Vogel, Tetrahedron, 1993, 49, 1649–1664 CrossRef CAS.
- (a) P. Klahn, A. Duschek, C. Liébert and S. F. Kirsch, Org. Lett., 2012, 14, 1250–1253 CrossRef CAS PubMed; (b) H.-J. Liu and B.-Y. Zhu, Can. J. Chem., 1991, 69, 2008–2013 CrossRef CAS; (c) Y. Liu, K. Lu, M. Dai, K. Wang, W. Wu, J. Chen, J. Quan and Z. Yang, Org. Lett., 2007, 9, 805–808 CrossRef CAS PubMed; (d) J. Le Nôtre, A. C. Martinez, P. H. Dixneuf and C. Bruneau, Tetrahedron, 2003, 59, 9425–9432 CrossRef.
- (a) B. M. Trost, J. Florez and D. J. Jebaratnam, J. Am. Chem. Soc., 1987, 109, 613–615 CrossRef CAS; (b) Y.-D. Wu, K. N. Houk and B. M. Trost, J. Am. Chem. Soc., 1987, 109, 5560–5561 CrossRef CAS; (c) B. M. Trost and D. J. Jebaratnam, Tetrahedron Lett., 1987, 28, 1611–1614 CrossRef CAS; (d) E. Toromanoff, Tetrahedron, 1980, 36, 2809–2931 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical characterization of compounds, NMR spectra, experimental details and X-ray details. CCDC 1051801 CIF format for structure 4a has been deposited at the Cambridge Crystallographic Data Centre. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00525f |
| This journal is © The Royal Society of Chemistry 2015 |