
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inhibition of a multiproduct terpene synthase from Medicago truncatula by 3-bromoprenyl diphosphates†
Abith
Vattekkatte
a,
Nathalie
Gatto
a,
Eva
Schulze
b,
Wolfgang
Brandt
b and
Wilhelm
Boland
*a
aDepartment of Bioorganic Chemistry, Max Planck Institute for Chemical Ecology, Hans-Knöll-Strasse 8, D-07745 Jena, Germany. E-mail: boland@ice.mpg.de
bDepartment of Bioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Weinberg 3, 06120 Halle (Saale), Germany
First published on 18th March 2015
Abstract
The multiproduct sesquiterpene synthase MtTPS5 from Medicago truncatula catalyzes the conversion of farnesyl diphosphate (FDP) into a complex mixture of 27 terpenoids. 3-Bromo substrate analogues of geranyl diphosphate (3-BrGDP) and farnesyl diphosphate (3-BrFDP) were evaluated as substrates of MTPS5 enzyme. Kinetic studies demonstrated that these compounds were highly potent competitive inhibitors of the MtTPS5 enzyme with fast binding and slow reversibility. Since there is a lack of knowledge about the crystal structure of multiproduct terpene synthases, these molecules might be ideal candidates for obtaining a co-crystal structure with multiproduct terpene synthases. Due to the structural and mechanistic similarity between various terpene synthases we expect these 3-bromo isoprenoids to be ideal probes for crystal structure studies.
Terpenes constitute the largest and most diverse class of plant natural products with more than 55
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members.1 They serve many biological functions such as hormones, structural components of membranes, attractants for pollinators, toxins, feeding or as oviposition deterrents to insects.2–5 Despite their enormous structural variety, all terpenes are essentially derived from simple linear precursors such as geranyl diphosphate (GDP), farnesyl diphosphate (FDP), and geranylgeranyl diphosphate (GGDP). The cyclization of GDP to monoterpenes, FDP to sesquiterpenes and GGDP to diterpenes is accomplished by enzymes known as terpene synthases. Structurally, sesquiterpenes are one of the most diverse classes of terpenes isolated from plants, fungi, bacteria, and marine invertebrates.6 All sesquiterpenoids known to date are based on 300 basic hydrocarbon skeletons; these skeletons are generated by cyclization of FDP by sesquiterpene synthases.7
000 members.1 They serve many biological functions such as hormones, structural components of membranes, attractants for pollinators, toxins, feeding or as oviposition deterrents to insects.2–5 Despite their enormous structural variety, all terpenes are essentially derived from simple linear precursors such as geranyl diphosphate (GDP), farnesyl diphosphate (FDP), and geranylgeranyl diphosphate (GGDP). The cyclization of GDP to monoterpenes, FDP to sesquiterpenes and GGDP to diterpenes is accomplished by enzymes known as terpene synthases. Structurally, sesquiterpenes are one of the most diverse classes of terpenes isolated from plants, fungi, bacteria, and marine invertebrates.6 All sesquiterpenoids known to date are based on 300 basic hydrocarbon skeletons; these skeletons are generated by cyclization of FDP by sesquiterpene synthases.7
Sesquiterpene synthases are capable of catalyzing the formation of some of the most complex carbon–carbon bond forming reactions found in nature.8 Despite of their promiscuous nature, active site of these synthases strictly control the reaction pathway by directing the way isoprenoid chain folds, shielding the cation from early nucleophilic attack, and guiding carbocation cascade to its final products by quenching. Even in sesquiterpene synthases, while some generate a single product, others produce complex bouquets of acyclic and cyclic products from a single precursor.9 The δ-selinene synthase and the γ-humulene synthase from Abies grandis hold the current record for producing 52 and 34 different sesquiterpenes, respectively.10 The lack of crystal structures of multiproduct terpene synthases, has led to great interest in the three-dimensional contour of the active site which retains such control over the reaction cascade. This knowledge is pivotal as it forms the mechanistic basis for the varying selectivity resulting in different conformations of the reactive cationic intermediates. Earlier we had reported that a multiproduct terpene synthase MtTPS5 (Fig. 1) from Medicago truncatula produces 27 products from FDP.11,12
 | ||
| Fig. 1 GC chromatogram of sesquiterpenoids formed on incubation of FDP with MtTPS5 from M. truncatula. | ||
We had also previously proposed the complex mechanistic pathway controlled by the MtTPS5 using a combination of techniques including labelling experiments.11 In brief, the reaction cascade is initiated by the formation of a highly reactive farnesyl carbocation by the disassociation of diphosphate moiety. The C1 to C11 ring closure affords the humulyl cation, which generates terpenoids such as α-humulene and β-caryophyllene (Scheme 1). Most of the products require the initial C1 to C10 closure generating the germacren-11-yl cation which is further cyclized to products like germacrene D and germacrenyl based alcohols. The other key cationic intermediates are (2Z,6E)-germacren-1-yl cation and cadinan-7-yl cation, leading to about 80% of products. These intermediates are generated by isomerization of FDP to nerolidyl diphosphate (NDP). Even alteration of a single amino acid has a dramatic effect on the product profile; alteration of tyrosine to phenylalanine in MtTPS5 prevents the formation of a key intermediate via protonation of germacrene D. The tight control of the enzyme leads to optically pure products resulting from cyclization steps and hydride shifts. Hence, it is of vital importance to understand the electrostatic interactions within active site. One possibility is to link the structure with complex catalytic cascade by using alternate substrates.13
 | ||
| Scheme 1 Simplified cyclization pathways of FDP with MtTPS5. | ||
Substrate analogues have been used successfully to probe the specificity of the enzymes and obtain a substrate-bound crystal structure of enzymes.14–16 Natural biosynthesis allows these substrates to carry only methyl substituents, but synthetic approaches can provide various analogues of the natural substrates. Especially, substitution with sulphur has been used quite successfully for co-crystallization with different terpene synthases.17,18 In a recent example from Heaps et al.,14 chlorinated substrates have been shown to be alternative co-substrates for farnesyl diphosphate synthase and also acting as inhibitors when both substrates were chlorinated. Fluorinated substrates have been successfully used to study the cyclization mechanism of several terpene synthases, in particular the aristolochene synthases.19 Thus, non-natural substrates have shown great potential as substrates or inhibitors to probe mechanisms and can be further utilized for the investigation of multiproduct terpene synthases.
Based on the information obtained from mechanistic studies of MtTPS5,11 we have designed a set of functional analogues of prenyl diphosphates for this enzyme that could destabilize the developing allylic carbocation. The GDP analogue (3-BrGDP) and FDP analogue (3-BrFDP) (Fig. 2) were considered because of their geometrical similarity and virtually identical van der Waals radius and volume (ca. 2.0 Å,20cf. also Fig. 6) with the methyl group at C3 position of natural substrates. The highly electronegative bromine atom can strongly influence the stability of the neighboring carbocationic species but imposes no additional steric effect in comparison with the corresponding natural substrates. These analogues could either provide novel sesquiterpenes that could be further utilized to investigate mechanistic aspects of the MtTPS5, or they could act as potent inhibitors of MtTPS5. In case of inhibition it can be used to provide an active site resolved crystal structure like in the case of aristolochene synthase with farnesyl-S-thiolodiphosphate.21 There is a lack of definitive understanding about the structure of multiproduct terpene synthases due to the absence of their crystal structures. These easy to synthesize inhibitors with identical sterical features could prove to be valuable tool for understanding the complex cyclization sequences in the active sites of these terpene cyclases.
 | ||
| Fig. 2 GDP analogue (3-BrGDP) and FDP analogue (3-BrFDP). | ||
Results and discussion
Synthesis
In order to investigate the potential of modified prenyl diphosphates, we have synthesized 3-bromo analogs of GDP (3-BrGDP) and FDP (3-BrFDP) as shown in Scheme 2. Alkylation of the THP ether of propargyl alcohol with 1-bromo-3-methylbut-2-ene (1a) and (E)-1-bromo-3,7-dimethylocta-2,6-diene (1b) provided the alkynes (2a,b) that were deprotected (MeOH, PPTS) to the propargyl alcohols (3a) and (3b). Reduction of the alcohols (3a) and (3b) with Red-Al, followed by reaction with N-bromosuccinimide gave the 3-bromo (E)-allylic alcohols (4a) and (4b). These alcohols were converted to 3-BrGDP (5a) and 3-BrFDP (5b) by sequential treatment with N-chlorosuccinimide and tris-(tetrabutyl-ammonium) hydrogen pyrophosphate in acetonitrile to yield the corresponding 3-bromoprenyl diphosphates. | ||
| Scheme 2 Synthesis of 3-BrGDP (5a) and 3-BrFDP (5b). | ||
Enzymatic characterizations of substrates
Both 3-BrGDP (5a) and 3-BrFDP (5b) were then assayed as inhibitors of the MtTPS5. With immediate mixing of enzyme, substrate, and inhibitor (no preincubation of enzyme and inhibitor), 3-BrGDP (5a) and 3-BrFDP (5b) were found to inhibit the MtTPS5 (Fig. 3). After preincubation for 1 h, standard assays containing protein in assay buffer with inhibitor and with natural substrate were performed (Fig. 3). The Ki, which was measured against MtTPS5 with 3-BrFDP (5b), was determined to be 1.54 ± 0.21 μM and Ki with BrGDP (5a) was determined to be 1.25 ± 0.23 μM. Additional preincubations with the enzyme for 1, 2, 4 and 12 h prior to substrate addition at t = 0, showed no increase in potency of inhibition, demonstrating a fast onset of the inhibition of the enzyme.
 | ||
| Fig. 3 Double reciprocal plots of initial rates versus the concentration of substrate for MtTPS5 catalyzed turnover of FDP in the presence of 3-BrGDP (5a) and 3-BrFDP (5b) are shown on panels A and B respectively. | ||
 | ||
| Fig. 4 Reversibility of Inhibition of MtTPS5 by 3-BrFDP (5b). | ||
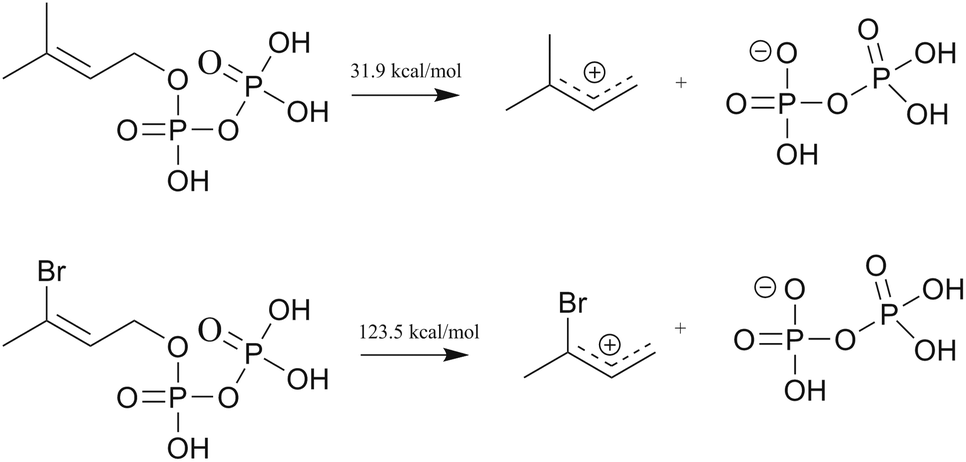 | ||
| Fig. 5 Ab initio energy calculations for cleavage of DMAPP and 3-BrDMAPP. | ||
To investigate the influence of the 3-bromo substitution on the destabilization of the intermediate allyl cation, reaction energies calculations were carried out using TURBOMOLE24 using DFT (B3-LYP25,26) with the def-TZVPP basis set.27 These calculations were performed for the corresponding cleavage of dimethylallyl diphosphate (DMAPP) and compared with 3-bromoallyl diphosphate (3-BrDMAPP) (Fig. 5). Whereas the cleavage of DMAPP requires only 31.9 kcal mol−1, the corresponding 3-bromo analogue requires much more energy (123.5 kcal mol−1) and is clearly not energetically favorable. In the case of longer isoprenyl chains like GDP and FDP, the resulting carbocations are stabilized by specific cyclizations guided by the enzymatic active site. However, in case of 3-bromo analogues the huge endothermic energy required for the formation of the intermediate allyl cation prevents its formation, and hence, subsequent energy gain by cyclization is not possible. However, 3-Br GDP and 3-Br FDP are able to bind to the enzyme because there is no steric difference on comparison with the natural substrates. Since the dissociation of the diphosphate moiety is the first step, common to all prenyl diphosphate based terpenoid synthases and cyclases, we expect this simple analogues as a broadly applicable inhibitors. Preliminary experiments with a terpenoid synthase from insects isoprenyl diphosphate synthase 1 (PcIDS1)28 from Phaedon cochleariae showed no catalytic formation of GDP with the incubation of substrates DMADP and IDP.
Molecular mechanics simulations
In the case of the MtTPS5 enzyme, the three-dimensional contour of the active site forms the structural basis of varying selectivity that allows different conformations of the reactive cationic intermediates. Since the crystal structure for MtTPS5 is not yet known, we earlier compared the structure with the 5-epi-aristolochene synthase (TEAS) from Nicotiana tabacum that transforms FDP to 5-epi-aristolochene through the stable intermediate germacrene A which is also an intermediate of the MtTPS5 catalyzed reaction sequence.7 Starting from the TEAS crystal structure, Starks et al.29 suspected that the hydroxyl group of tyrosine in the interaction with the two aspartate participates as a proton donor in the activation of germacrene A and forms a catalytic triad.29 A comparison of the TEAS amino acid sequence shows that all three amino acids were conserved in MtTPS5 as well. This sequence similarity is can be used to our advantage to model the active site interactions leading to products.Recently, the co-crystal structure of aristolochene synthase (PDB code: 4KUX) from Aspergillus terreus with a bound FSDP, a stable FDP analogue was reported. Despite differences in sequence alignment, the discussed triad the 84DDXXE motif essential for the diphosphate recognition corresponds with a similar motif 306DDXXD in MtTPS5. Thus, there is more likely to be comparable binding of the substrate or inhibitor is more likely. This is further supported when analyzing the active site (Fig. 6) of 4KUX with bound FSDP. The influence of the bromine substitution was analyzed first by replacement of the sulfur in FSDP with oxygen to form FDP and further modifying the ligand to 3-BrFDP (5b). The active site with ligands was energy minimized within an area of 7 Å around the ligand using the YASARA package.30 There were no changes in key structural features in case of interaction between the catalytic triad and C3 methyl group placed in the hydrophobic area created by F147 and partly by L80 (Fig. 4). The sequence alignment with MtTPS5 shows that both F147 and L80 are superposed with F374 and L302, respectively, Thus, almost identical hydrophobic interactions especially of the 3-Me or 3-Br moiety in MtTPS5 can be expected. The folding pattern of both substrates in the active site might be identical and the dissociation of the diphosphate moiety should lead to initiation of formation of cyclic products. To check if the bromine is able to exhibit special stabilizing interactions, distances to spatially neighboring atoms were measured. Except of the mentioned interactions with the side chains of F147 and L80 there is only one carboxyl oxygen atoms of D84 in a distance of 3.9 Å which is, however, too large for significant stabilizing interactions. This distance is larger than 3.4 Å, the sums of the respective van der Waals radii, and also the C–Br–O angle (50°) is not appropriate to lead to an ideal scenario for the formation of an attractive Br–O halogen bond.31 Thus, we can conclude that natural FDP and 3-BrFDP (5b) bind to the active site in identical ways, and due to higher endothermic energy requirements the cleavage to a carbocation intermediate is not possible leading to absence of products.
 | ||
| Fig. 6 Docking arrangement of FDP (A) and 3-BrFDP (5b) (B) in the active site of the X-ray structure of aristolochene synthase (4KUX). The van der Waals radii are shown by a cloud around CH3 and Br groups. | ||
Conclusion
The 3-bromo substituted GDP (3-BrGDP) and FDP (3-BrFDP) were evaluated as substrates or inhibitors for the MTPS5 enzyme. The brominated analogues were not accepted as natural substrates for MtTPS5 and no product formation was observed. Our kinetic analyses demonstrated that these compounds were highly potent, acting as linear competitive inhibitors of the MtTPS5 enzyme, with fast binding and slow reversibility. Since there is a lack of knowledge about the crystal structure of multiproduct terpene synthases, these molecules would be ideal candidates for obtaining a co-crystal structure with e.g. the MtTPS5 from M. truncatula. They would not only support the co-crystallization process, but also give information on the conformation of the substrate bound in the crystal structure which is imperative in determining the precise nature of mechanism catalyzed by the enzyme. Due to the structural similarity between various terpene synthases, and similarity in behavior in active site, we expect these 3-bromo isoprenoids as ideal probes for crystal structure studies. The bromo substituent provides additional advantage to determine the absolute configuration based on the anomalous dispersion effect.32,33Experimental
General methods
Reactions were performed under Ar. Solvents were dried according to standard procedures. 1H, 13C and 31P NMR were recorded at 400 MHz. Chemical shifts of 1H, 13C and 31P NMR are given in ppm (δ) based on solvent peaks. CDCl3: 7.27 (1H NMR) and 77.4 ppm (13C NMR). D2O/ND4OD: 4.79 (1H NMR); 13C NMR and 31P NMR were referenced to external standard 3-(trimethylsilyl)-propionic acid-d4 sodium salt (TSP; 3% in D2O) and phosphoric acid (H3PO4, 10% in D2O), respectively. IR: Bruker Equinox 55 FTIR spectrophotometer.Protein expression
Strains of E. coli (BL21-CodonPlus(DE3)) with recombinant vectors of MtTPS512 and N-terminal His8-tag were grown to A600 = 0.5 at 37 °C in LB-medium with kanamycin (50 μg ml−1). After induction with isopropyl β-D-1-thiogalactopyranoside (IPTG), cultures were shaken overnight at 16 °C. Cells were harvested by centrifugation and the pellet was resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) and incubated with lysozyme. After disruption of the cells by sonication, cell debris were removed by centrifugation. The supernatant was passed over a column of Ni2+-NTA-Agarose (QIAGEN, Germany), equilibrated with lysis buffer. After being washed twice with washing buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole), the protein was eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). The purified protein was desalted into a TRIS-buffer (50 mM TRIS, pH 7.5, 10 mM NaCl, 10% glycerol) by passing through a NAP 25 column (Amersham Biosciences, Sweden), diluted to reach a concentration of 0.2 and 1 mg ml−1 and stored at −20 °C.Assay for terpene synthase activity
Standard assays contained 600 nM purified protein in assay buffer (25 mM HEPES, pH 7.5, 10% glycerol, 10 mM MgCl2, 1 mM DTT) with 50 μM substrate in 1 mL final volume. The reaction mixture was covered with 100 μL of pentane (1 ng μL−1 of dodecane as an internal standard) to trap the reaction products. After being incubated for 90 min at 30 °C, the reaction was stopped by vortexing for 20 s. The whole mixture was frozen in liquid nitrogen, and the pentane layer was removed after thawing and analyzed by GC-MS.Gas chromatography
GC-MS analysis was performed on an instrument equipped with a ZB-5 capillary column (0.25 mm i.d. × 15 m with 0.25 μm film). One microliter of the sample was injected in splitless mode at injection port temperature of 220 °C. The oven temperature was kept at 50 °C for 2 min followed by a ramp of 10 °C min−1 to 240 °C followed by an additional ramp of 30 °C min−1 to 280 °C and finally kept for 2 min. Helium at a flow rate of 1.5 mL min−1 served as carrier gas. Ionization potential was set to 70 eV, and scanning was performed from 40 to 250 amu. Compounds were identified by comparing their mass spectra and Kováts indices (retention indices) with published reference spectra (Garms et al.11) For quantification of enzyme products, the compounds were first separated on a gas chromatograph (H2 carrier gas 1.5 mL min−1, injection volume 2 μL) under the conditions described above and subsequently analyzed on a flame ionization detector (FID) (250 °C). Correction of the different response factors of sesquiterpene hydrocarbons and alcohols was achieved using calibration curves obtained from samples with different concentrations of (E)-β-caryophyllene and torreyol. The average and standard deviations of relative ratios were determined by at least four independent samples setting the sum of identified compounds to 100%.Kinetic characterization of 3-bromo analogues as inhibitors of MtTPS5
For kinetic studies, terpene synthase activity was determined by monitoring the decrease in absorbance at 340 nm as a consequence of the consumption of NADPH coupled to the release of pyrophosphate. Pyrophosphate was detected using a coupled-enzyme system supplied as a pyrophosphate reagent by Sigma-Aldrich. This reagent was reconstituted in buffer (16.7 mg in 1 ml 10 mM Tris-HCl, 10 mM MgCl2, 1 mM β-mercaptoethanol, pH 8). Assay reaction mixtures were prepared in 96-well microplates with 50 μl of pyrophosphate reagent, 90 μl of buffer (0–16 μM inhibitor, 25 mM HEPES, pH 7.5, 10% glycerol, 10 mM MgCl2, 1 mM DTT) and 10 μl of various concentrations of FPP. Similarly, a blank reaction mixture without FPP (instead, 10 μl of buffer was used) was prepared, and both reaction mixtures were preheated at 30 °C for 5 min. Next, 5 μl of enzyme (0.2 mg ml−1) was added to both mixtures to start the reactions. The activity was determined as the difference between the decrease in absorbance per minute of the sample and of the blank.Apart from spectrophotometry based measurements, Assays (1 ml final volume) were initiated by addition of purified MTTPS5 solution (600 nM). Assays contained 0.1–5 μM farnesyl diphosphate, 0–3 μM inhibitor, 25 mM HEPES, pH 7.5, 10% glycerol, 10 mM MgCl2, 1 mM DTT and were warmed to 30 °C prior to addition of enzyme solution. After incubation for 90 min. each assay was stopped by addition of 100 mM EDTA and 100 μL of pentane containing 1 ng μL−1 of dodecane as an internal standard to trap the reaction products. After vortexing for 10 s, the pooled hexane extracts were vortexed with silica (50 mg) the sample was centrifuged at 13000 rpm for 5 min and then the hexane was decanted into a vial and the activity quantified. KM and KM(app) values were determined by a nonlinear fit of the data to the equation V = Vmax[S]/(KM + [S]) using Origin 8G. Mode of action of the inhibitors was determined by examination of double reciprocal plots of 1/v versus 1/[S]. Ki values were determined using plots of [I] versus KM(app) once each inhibitor was observed to be competitive.
Modelling studies
Synthetic procedure
:1 hexanes–diethyl ether) to give 1.264 g of 2a (63%) as a colorless oil data: (lit.34) 1H NMR (400 MHz, CDCl3) δ: 5.12–5.14 (1H, m), 4.79 (1H, t, J = 3.3 Hz), 4.22 (2H, app q), 3.79–3.85 (1H, m), 3.49–3.53 (1H, m), 2.17–2.21 (4H, m), 1.68 (3H, s), 1.60 (3H, s), 1.50–1.91 (6H, m); 13C NMR (400 MHz, CDCl3) δ: 132.9, 122.8, 96.5, 86.4, 75.7, 61.9, 54.5, 30.2, 27.3, 24.6, 25.3, 19.3, 19.1, 17.7. IR (neat) cm−1: ν 2942, 2870, 1441, 1117, 1024.
:1 hexanes–diethyl ether) to give 1.051 g of 2a (51%) as a colorless oil data: (lit.36) 1H NMR (400 MHz, CDCl3) δ: 5.18 (s, 1H), 5.07 (s, lH), 4.76 (s, lH), 4.2 (t, J = 2 Hz, 2H), 3.6 (m, 2H), 2.9 (d, J = 7 Hz, 2H), 1.98 (m, 4H), 1.62 (m, 1H); 13C NMR (400 MHz, CDCl3) δ: 135.9, 132.6, 122.8, 123.5, 100.2, 87.8, 75.7, 63.3, 54.5, 39.7, 30.2, 27.3, 25.9, 25.4, 24.6, 20.8, 19.3, 18.9, 17.3. IR (neat) cm−1: ν 3060, 2960, 2240, 1140.
:1 hexanes–diethyl ether) to give 0.828 g of the alcohol 4a (80%) as a colorless oil. (bp: 93–5 °C) as a colorless oil. Rf 0.43 (1:l hexanes–ether); data: 1H NMR (400 MHz, CDCl3) δ: 5.18–5.13 (m, 1H), 4.25 (s, 2H), 2.26–2.17 (m, 4H), 1.70 (s, 3H), 1.62 (s, 3H), 1.52 (s, 1H); 13C NMR (400 MHz, CDCl3) δ: 133.1, 122.6, 86.4, 78.3, 51.4, 27.3, 25.6, 19.2, 17.7; HRMS calcd for C9H140 138.1045, found 138.1041. IR (neat) cm−1: ν 3550–3160, 2970, 2920, 2860, 2280, 2220, 1470, 1370, 1130, 1100, 1010 cm−1.
:1 hexanes–diethyl ether) to give 1.282 g of the alcohol 4a (83%) as a colorless oil. (bp 120–124 °C (0.5 mm)) as a colorless oil. Rf 0.40 (1:l hexanes–ether); data: (lit.37) 1H NMR (400 MHz, CDCl3) δ: 5.16 (m, 1H), 5.09 (m, 1H), 4.25 (d, 2H, J = 10.8), 2.22 (narrow m, 4H), 2.07 (m, 2H), 1.99 (m, 2H), 1.68 (s, 3H), 1.61 (s, 3H), 1.60 (s, 3H), 1.50 (t, 1H, J = 10.8), 13C NMR (400 MHz, CDCl3) δ: 133.1, 122.6, 86.4, 78.3, 51.4, 27.3, 25.6, 19.2, 17.7; HRMS calcd for C14H1220: 206.1671, found 206.1667 IR (neat) cm−1 3334, 2287, 2224, 1670, 1020 cm−1.
:1 (v/v) hexane–ether to yield 200 mg (42%) of a colorless oil (GC: 96% pure). Rf = 0.43 (1:l hexanes–ether); data: 1H NMR (400 MHz, CDCl3) δ: 5.93 (tt, 1H), 5.08 (t, 1H), 4.26 (d, 2H), 2.50 (t, 2H), 2.25 (dt, 2H), 1.78 (s, 1H), 1.70 (s, 3H), 1.64 (s, 3H); 13C NMR (400 MHz, CDCl3): δ: 133.44, 130.2, 128.0, 122.5, 62.8, 42.0, 27.1, 26.0, 18.1. HRMS calcd for C14H22BrO (M − H2O) 200.0195, found 200.0196. IR (neat) cm−1: v 3540–3100, 2970, 2930, 2850, 1705, 1635, 1445, 1375, 1080, 1020, 825.
:1 (v/v) hexane–ether to yield 257 mg (39%) of a colorless oil (GC: 96% pure). Rf = 0.40 (1:l hexanes–ether); Data: 1H NMR (400 MHz, CDCl3) δ: 5.83 (t, J = 5.8 Hz, 1H) 5.08 (t, J = 5.7 Hz, 2H), 4.18 (d, J = 5.8 Hz, 2H), 2.52 (m, 2H), 2.21 (m, 2H), 2.06–1.95 (m, 4H), 1.73 (s, 1H), 1.68 (s, 3H), 1.62 (s, 3H), 1.59 (s, 3H) 13C NMR (400 MHz, CDCl3) δ: 136.9, 133.8, 131.6, 124.4, 122.1, 110.3, 67.5, 45.6, 39.8, 28.1, 26.9, 25.9, 17.9, 16.4: HRMS calcd for C14H22BrO (M − H2O) 268.0821, found 268.0820. IR (neat) cm−1: v 3540–3100, 2970, 2930, 2850, 1705, 1635, 1445, 1375, 1080, 1020, 825.
General procedure for the preparation of trisammonium diphosphates
Trisammonium diphosphates were prepared according to the modified method of Woodside et al.38 To a solution of N-chlorosuccinimide (11.39 mmol) in CH2Cl2 (45 mL) at −30 °C under argon was added dropwise freshly distilled dimethyl sulfide (1.1 eq. mol). The mixture was warmed to 0 °C, stirred at this temperature for 10 min and cooled to −40 °C. A solution of alcohol 4a or 4b (1 eq. mol) in CH2Cl2 (5 mL) was slowly added before the reaction mixture was warmed to 0 °C. Stirring was continued for 2 h at 0 °C and 15 min at rt. The clear solution was then washed with cold saturated NaCl (25 mL). The aqueous phase was extracted with pentane (2 × 20 mL). The combined organic layers were washed with cold saturated NaCl (20 mL), dried (MgSO4), concentrated under reduced pressure (no water bath) and completely removed under high vacuum for 2 h. Corresponding alkyl chlorides were used without further purification. Freshly prepared tris(tetrabutylammonium) hydrogen pyrophosphate (1.2 eq. mol) was dissolved in ACN (5 mL) at rt under argon before dropwise addition of alkyl chloride in ACN (2 mL). Stirring was continued at rt overnight. The mixture was concentrated under reduced pressure. The residue was dissolved in (NH4)2CO3 (3 mL) (0.25 mM, 2% isopropyl alcohol), loaded onto a 2 × 30 cm column of Dowex 50WX8-200 (NH4+ form) before elution of two volumes column of (NH4)2CO3 (0.25 mM, 2% isopropyl alcohol). The eluent was lyophilized and the resulting white powder was purified by chromatography on cellulose (1:9 (v/v) water in ACN). Fractions were monitored by TLC (silica gel, iPr-OH–water–AcOEt 6:3:1) and those containing trisammonium diphosphate were combined. Solvents were removed under reduced pressure and the resulting solution was lyophilized to afford 5a or 5b.
Acknowledgements
We would like acknowledge Max Planck Society for funding. We would also like to acknowledge Stefan Garms and Stephan von Reuss for helpful discussions. We also like to thank Peter Rahfeld and Rita Buchler for their help with protein purification.Notes and references
- H. V. Thulasiram, H. K. Erickson and C. D. Poulter, Science, 2007, 316, 73–76 CrossRef CAS PubMed.
- J. H. Langenheim, J. Chem. Ecol., 1994, 20, 1223–1280 CrossRef CAS PubMed.
- E. Pichersky and J. Gershenzon, Curr. Opin. Plant Biol., 2002, 5, 237–243 CrossRef CAS.
- E. Nambara and A. Marion-Poll, Annu. Rev. Plant Biol., 2005, 56, 165–185 CrossRef CAS PubMed.
- M. Hilker, C. Kobs, M. Varama and K. Schrank, J. Exp. Biol., 2002, 205, 455–461 Search PubMed.
- J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman & Hall, New York, 1992 Search PubMed.
- C. A. Lesburg, J. M. Caruthers, C. M. Paschall and D. W. Christianson, Curr. Opin. Struct. Biol., 1998, 8, 695 CrossRef CAS.
- E. M. Davis and R. Croteau, in Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Springer-Verlag, Berlin, 2000, vol. 209, pp. 53–95 Search PubMed.
- J. P. Jones, K. R. Korzekwa, A. E. Rettie and W. F. Trager, J. Am. Chem. Soc., 1986, 108, 7074 CrossRef CAS.
- C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, J. Biol. Chem., 1998, 273, 2078 CrossRef CAS PubMed.
- S. Garms, T. G. Köllner and W. Boland, J. Org. Chem., 2010, 75, 5590–5600 CrossRef CAS PubMed.
- G. I. Arimura, S. Garms, M. Maffei, S. Bossi, B. Schulze, M. Leitner, A. Mithöfer and W. Boland, Planta, 2008, 227, 453–464 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2006, 106, 3412–3442 CrossRef CAS PubMed.
- N. A. Heaps and C. D. Poulter, J. Org. Chem., 2011, 76, 1838–1843 CrossRef CAS PubMed.
- F. Y. David, J. Miller, D. W. Knight and R. K. Allemann, Org. Biomol. Chem., 2009, 7, 962–975 Search PubMed.
- D. J. Hosfield, Y. Zhang, D. R. Dougan, A. Broun, L. W. Tari, R. V. Swanson and J. Finn, J. Biol. Chem., 2004, 279, 8526–8529 CrossRef CAS PubMed.
- F.-Y. Lin, C.-I. Liu, Y.-L. Liu, Y. Zhang, K. Wang, W.-Y. Jeng, T.-P. Ko, R. Cao, A. H.-J. Wang and E. Oldfield, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21337–21342 CrossRef CAS PubMed.
- J. A. Faraldos and R. K. Allemann, Org. Lett., 2011, 13, 1202–1205 CrossRef CAS PubMed.
- D. J. Miller, F. Yu, D. W. Knight and R. K. Allemann, Org. Biomol. Chem., 2009, 7, 962–975 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Chen, N. Al-lami, M. Janvier, E. L. D'Antonio, J. A. Faraldos, D. E. Cane, R. K. Allemann and D. W. Christianson, Biochemistry, 2013, 52, 5441–5453 CrossRef CAS PubMed.
- C. RA, Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists, John Wiley & Sons, Hoboken, NJ, 2005 Search PubMed.
- C. D. Poulter, J. C. Argyle and E. A. Mash, J. Biol. Chem., 1978, 253, 7227–7233 CAS.
- C. Steffen, K. Thomas, U. Huniar, A. Hellweg, O. Rubner and A. Schroer, J. Comput. Chem., 2010, 31, 2967–2970 CAS.
- K. Kim and K. D. Jordan, J. Phys. Chem., 1994, 98, 10089–10094 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Frick, R. Nagel, A. Schmidt, R. R. Bodemann, P. Rahfeld, G. Pauls, W. Brandt, J. Gershenzon, W. Boland and A. Burse, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4194–4199 CrossRef CAS PubMed.
- C. M. Starks, K. W. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815 CrossRef CAS.
- E. Krieger, G. Koraimann and G. Vriend, Proteins: Struct., Funct., Bioinf., 2002, 47, 393–402 CrossRef CAS PubMed.
- S. Sirimulla, J. B. Bailey, R. Vegesna and M. Narayan, J. Chem. Inf. Model., 2013, 53, 2781–2791 CrossRef CAS PubMed.
- Z. Dauter, M. Dauter and K. R. Rajashankar, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 232–237 CrossRef CAS.
- J. M. Bijvoet, A. F. Peerdeman and A. J. van Bommel, Nature, 1951, 168, 271–272 CrossRef CAS.
- M. E. Jung and M. H. Parker, J. Org. Chem., 1997, 62, 7094–7095 CrossRef CAS.
- A. C. Oehlschlager, S. M. Singh and S. Sharma, J. Org. Chem., 1991, 56, 3856–3861 CrossRef CAS.
- S. Ghosal, M. Nirmal, J. C. Medina and K. S. Kyler, Synth. Commun., 1987, 17, 1683–1694 CrossRef CAS.
- J. Hooz, J. Cabezas, S. Musmanni and J. Calzada, Org. Synth., 1990, 69, 120–128 CrossRef CAS.
- A. B. Woodside, H. Zheng and C. D. Poulter, Org. Synth., 1988, 66, 211–219 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and spectra are included. See DOI: 10.1039/c5ob00506j |
| This journal is © The Royal Society of Chemistry 2015 |