
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis of (R)-harmonine – the toxic principle of the multicolored Asian lady beetle (Harmonia axyridis)†
Nadja C.
Nagel
a,
Anita
Masic
b,
Uta
Schurigt
b and
Wilhelm
Boland
*a
aMax Planck Institute for Chemical Ecology, Beutenberg Campus, Hans-Knoell-Straße 8, D-07745 Jena, Germany. E-mail: boland@ice.mpg.de
bInstitute for Molecular Infection Biology, Josef-Schneider-Str. 2/D15, D-97080 Wuerzburg, Germany
First published on 24th March 2015
Abstract
A flexible synthetic route to (R)-harmonine ((R)-1), the toxic principle of the Asian lady beetle Harmonia axyridis (H. axyridis), via reductive olefination of the macrocyclic lactone (S)-5, is reported. High enantiomeric purity is achieved by enantioselective saponification of the lactone rac-5 with horse liver esterase. Minor modifications in the synthetic route give access to racemic and chiral harmonine (1), analogs and putative biosynthetic precursors. In addition, the antimicrobial activity of harmonine against Leishmania major (L. major) is demonstrated and provides the rationale for harmonine-based drug development against parasitic diseases.
Introduction
The multicolored Asian lady beetle H. axyridis, also known as the harlequin ladybird, is natively distributed from southern Siberia to southern China and from the Altai mountains to the Pacific coast.1 This tree dwelling beetle, of the family Coccinellidae, is an important predator of aphids and scale insects and has been introduced for biological control in many countries. Over time populations began to establish and two decades ago H. axyridis became an invasive species in North America, Europe and South America threatening the native lady beetles.1–3 The invasive success arises from intra-guild predation2 and from H. axyridis’ resistance to pathogens.3 When a lady beetle is disturbed or attacked it releases droplets of hemolymph from the tibio-femoral joints of its legs. This behavior is referred to as reflex-bleeding.4,5 The repellent and sometimes toxic properties of the hemolymph are due to some alkaloids, which are considered to be synthesized de novo by the beetles.6H. axyridis produces (R)-harmonine ((R)-1) ((17R,9Z)-octadec-9-ene-1,17-diamine) as the major defense compound (Fig. 1).7 | ||
| Fig. 1 (R)-Harmonine ((R)-1) and (9Z)-18-aminooctadec-9-en-2-ol (rac-2). | ||
(R)-Harmonine ((R)-1) displays antibacterial activity against fast-growing mycobacteria, Mycobacterium tuberculosis, and Plasmodium falciparum (P. falciparum), demonstrates multi-stage antimalarial activity,7 and exhibits cytotoxicity against human tumor cell lines.8 Recently microsporidia from the hemolymph of H. axyridis were shown to infect intra-guild predators.9 In this context, (R)-harmonine ((R)-1) was postulated to protect the harlequin beetle against self-infection.10 Currently, harmonine ((R)-1) is considered a promising lead for clinical and agricultural use (yellow biotechnology).11 Although harmonine ((R)-1) has been isolated12 and synthesized previously,12–14 there is a need for rapid and efficient syntheses of harmonine ((R)-1) and related molecules, since the mode of action is still unknown. In particular, for bioassays larger quantities are needed.
L. major is the causative agent of cutaneous leishmaniasis with an estimated annual incidence of 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–1.3 million new infections worldwide.15,16 Current chemotherapy against leishmaniasis is limited due to the continuous development of drug resistance accompanied by severe side effects.17 Therefore, naturally derived compounds like harmonine are investigated to identify and develop new therapies against leishmaniasis. The antiparasitic activity of harmonine against P. falciparum has already been reported and prompted the assessment of its activity against L. major.7
000–1.3 million new infections worldwide.15,16 Current chemotherapy against leishmaniasis is limited due to the continuous development of drug resistance accompanied by severe side effects.17 Therefore, naturally derived compounds like harmonine are investigated to identify and develop new therapies against leishmaniasis. The antiparasitic activity of harmonine against P. falciparum has already been reported and prompted the assessment of its activity against L. major.7
Here, we report a short and flexible synthetic route to harmonine ((R)-1) in only a few steps starting from the readily available macrocyclic lactone (S)-5. One-pot olefination of (S)-5 directly provides the basic skeleton of harmonine ((R)-1) and allows the synthesis of biosynthetic precursors and structural analogs via functional group modifications. Furthermore, we report the antiparasitic activity of harmonine against the causative agent of cutaneous leishmaniasis, L. major.18 Cultivation in the presence of (R)-1 leads to the inhibition of parasitic proliferation with a consequent early necrotic cell death phenotype.
Results and discussion
According to Scheme 1 the harmonine backbone can be obtained from the lactone (S)-5 and the phosphonium salt 4 by a Wittig-type reaction. The chiral lactone (S)-5 is available with high ee and excellent yield by enantioselective hydrolysis with horse liver esterase. Furthermore, synthetic intermediates like ((S)-3) are promising candidates for biosynthetic and pharmaceutical studies.19 By modifying the chain length of the phosphonium salt or the ring size of the lactone, analogs with different positions of the double bond and molecular size become available, thus opening a new synthetic route to harmonine-like compounds using a unified procedure.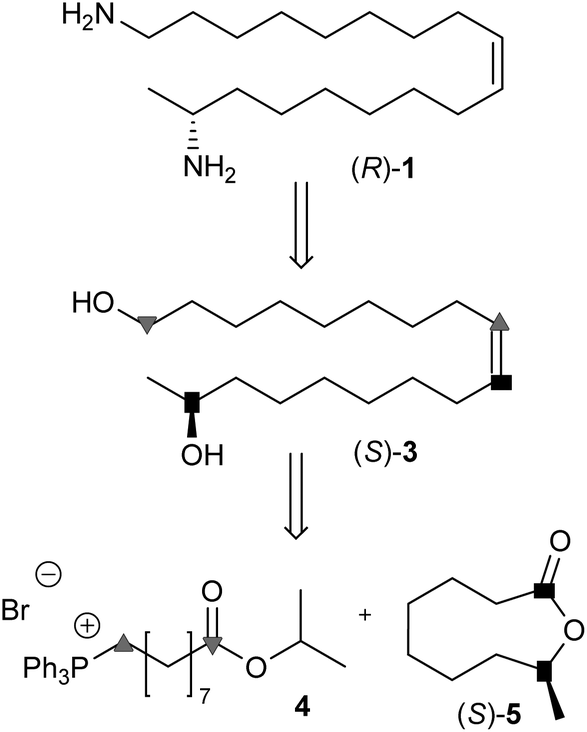 | ||
| Scheme 1 Retrosynthesis of harmonine ((R)-1). | ||
Following the retrosynthetic strategy the backbone of harmonine (rac-1) was assembled by reductive olefination17 from rac-5 and the ylide of (9-isopropoxy-9-oxononyl)triphenylphosphonium bromide (4) in a single operation. The resulting isopropylester rac-6 was converted into the amide rac-8 or directly reduced to the diol rac-3 (Scheme 2). The lactone rac-5 was obtained from commercial cyclooctanone20 and the phosphonium salt 4 from 9-bromononanoic acid after esterification and reaction with triphenylphosphine.21,22
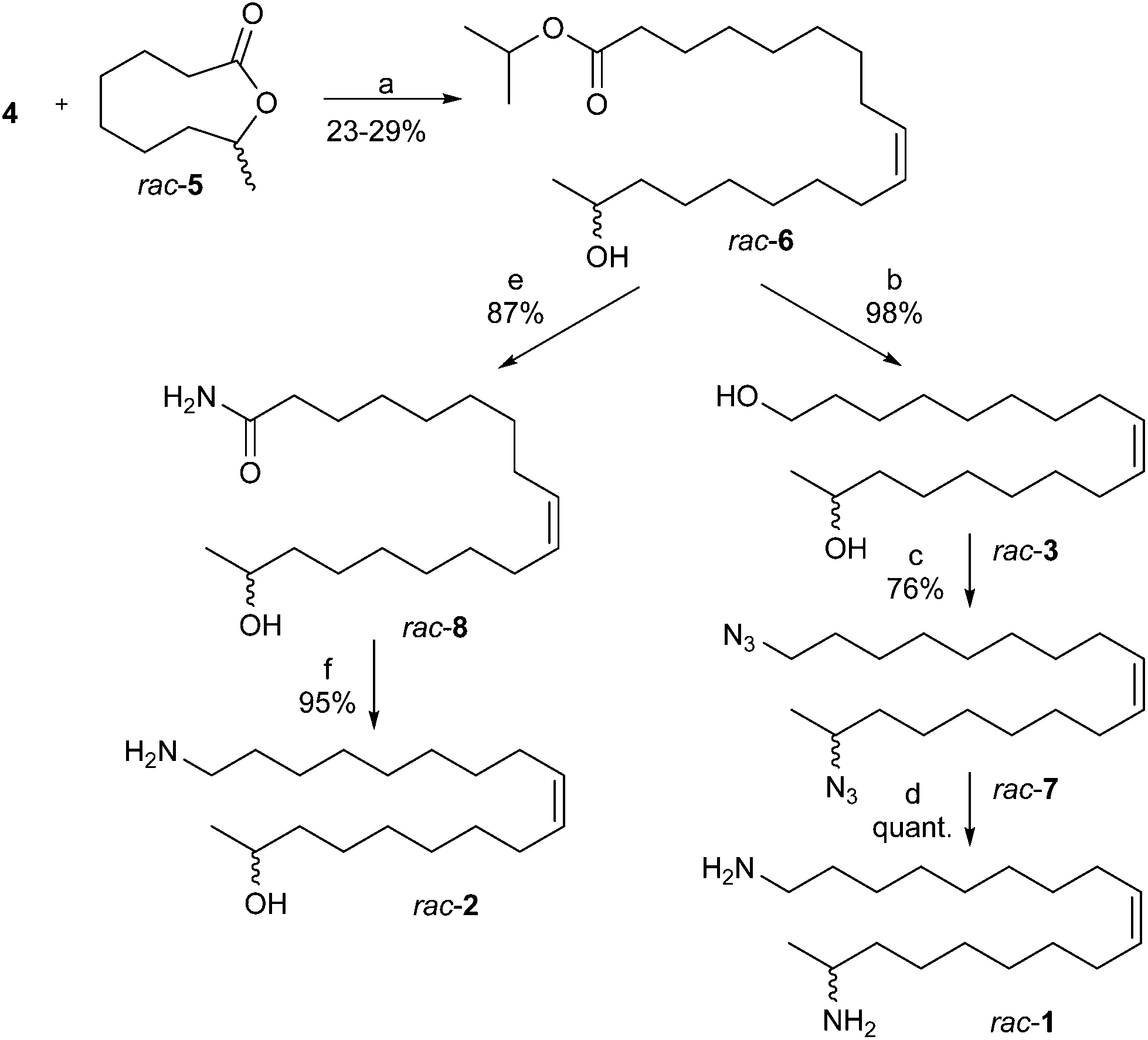 | ||
| Scheme 2 Synthetic sequence to harmonine (rac-1) and a putative biosynthetic precursor rac-2. Reagents and conditions: (a) (i) DiBAlH, MeOH, toluene, −78 °C; (ii) KHMDS, THF, −78 °C; (iii) −78 °C to rt in 1 h, 1 h rt; (b) LiAlH4, THF, rt; (c) PPh3, DPPA, DIAD, THF, rt; (d) LiAlH4, THF, rt; (e) Mg3N2, MeOH, 80 °C; (f) LiAlH4, THF, 45 °C. | ||
The one-pot “reductive olefination” generates from the lactone rac-5 and DiBAlH at low temperatures an organo aluminium acetal that decomposes above −40 °C to a hydroxy aldehyde which reacts in situ with the phosphorane to the hydroxyester rac-6 ((Z)/(E) > 98/2). The isopropylester was used to minimize side reactions during the Wittig olefination. Reduction of rac-6 with lithium aluminum hydride afforded the diol rac-3 in quantitative yield.23 Following the Mitsunobu protocol the diol rac-3 was converted into the diazide rac-7 using triphenylphosphine (PPh3), diphenyl phosphorazidate (DPPA) and diisopropyl azodicarboxylate (DIAD).24 The final reduction of rac-7 with an excess of LiAlH4 afforded racemic harmonine (rac-1) in quantitative yield.24 Overall, racemic harmonine (rac-1) could be prepared in only four steps and 22% overall yield starting from rac-5 and 4. Spectral data of rac-1 were identical to literature values.8,12
For biosynthetic studies19 and pharmaceutical assays7 the (Z)-18-aminooctadec-9-en-2-ol (rac-2) was of interest. Conversion of the isopropylester rac-6 into the amide rac-8 was readily achieved using magnesium nitride in refluxing methanol (Scheme 2).23 Subsequent reduction with LiAlH425 provided the aminoalcohol rac-2 in nearly quantitative yields over two steps.
The synthetic protocol was easily extended to chiral (17R,9Z)-1,17-diaminooctadec-9-ene ((R)-1). The key step was a kinetically controlled enantioselective saponification of the lactone rac-5 which left behind unreacted (S)-lactone ((S)-5) (ee > 99.5%) in 43% yield (Scheme 3). For hydrolysis the lactone (rac-5) was suspended in a NaH2PO4-buffer (0.1 M, pH = 7.2) and horse liver esterase (HLE) was added as lyophilized powder.26 The hydrolysis started immediately and the pH was kept constant by addition of dil. NaOH (0.5 M) to avoid spontaneous hydrolysis.
 | ||
| Scheme 3 Enantioselective, enzymatic saponification of rac-5. Reagents and conditions: (a) HLE, NaH2PO4, NaOH, rt. | ||
The progress of the reaction was monitored using a GC-MS equipped with a chiral column (β-6TBDM) for resolution of the enantiomers (Fig. 2).
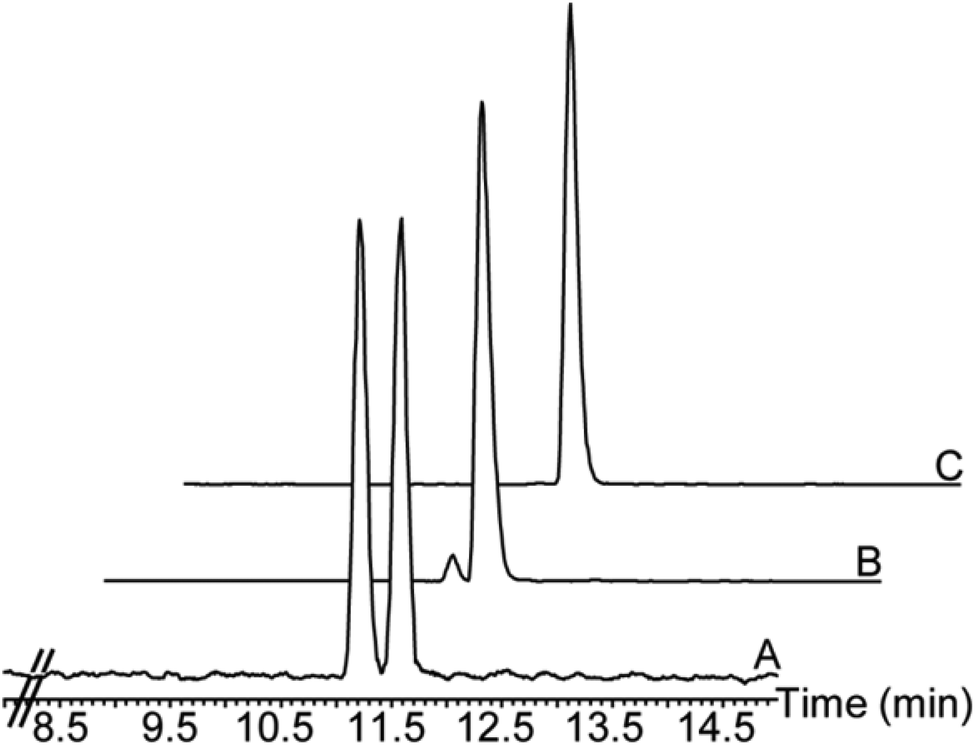 | ||
| Fig. 2 Enantiomeric analysis on a β-6TBDM column. (A) Racemic lactone rac-5. (B) 98% hydrolysis of (R)-lactone. (C) Isolated lactone (S)-5 (ee ≥ 99.5%). | ||
After hydrolysis of the (R)-lactone, the reaction stopped and both the remaining lactone (S)-5 and the hydroxyacid (R)-9 were obtained with good yield and excellent ee (S)-5 (>99.5%), (R)-9 (>99%).
The chiral lactone (S)-5 was converted into (R)-harmonine ((R)-1) using the same sequence of reactions as shown in Scheme 2. The spectral data of (R)-1 were identical to literature values.8,12,13
The ee of (R)-harmonine ((R)-1) was determined by 19F NMR using (S)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA chloride) for derivatization.27,28 Due to the large chemical shifts of the α-CF3 groups of the MTPA-diastereomers the high ee of (R)-1 was reliably confirmed (ee > 97%).
For the evaluation of the antileishmanial activity of harmonine, the AlamarBlue® assay with L. major promastigotes and the Britelite™ assay with Luciferase-transgenic L. major amastigotes were employed (Table 1). The cytotoxic effect of harmonine against host cells was investigated in bone marrow-derived macrophages (BMDM). The half maximal inhibitory concentration (IC50) of harmonine was detected at 14.2 μM against L. major promastigotes and at 2.4 μM against the intracellular and clinically relevant L. major amastigote form. When compared to Miltefosine (1-hexadecylphosphocholine, positive control) with an IC50 value of 36.2 μM against L. major promastigotes and 33.0 μM against L. major amastigotes,29 harmonine showed leishmanicidal activities at significantly lower concentrations. Miltefosine showed an IC50 value of 65.5 μM and harmonine showed a value of 36.5 μM against BMDM. The antileishmanial efficacy of a tested drug compared to its cytotoxicity against host cells is defined as the selectivity index (SI). SIs > 20 are considered excellent, as high antileishmanial activity and low cytotoxicity are desirable prerequisites for drug development.30 Here, harmonine shows a very good SI of 15.2 towards L. major which is considerably higher than the SI of 2.0 of Miltefosine.
| IC50 (μM)a | ||||
|---|---|---|---|---|
| L. major promastigotes | L. major amastigotes | BMDMb | SIc | |
| a IC50: half maximal inhibitory concentration. b BMDM: bone marrow-derived macrophages. c SI (Selectivity Index): IC50 for BMDM/IC50 for L. major. d IC50 synthetic rac-harmonine: 13.2. | ||||
| (R)-Harmonine | 14.2d | 2.4 | 36.5 | 15.2 |
| Miltefosine | 36.2 | 33.0 | 65.5 | 2.0 |
The antileishmanial effect of harmonine as shown in Table 1 was further investigated. The cell morphology of L. major promastigotes was studied by means of transmitted light microscopy upon treatment with harmonine for 6 h, 10 h, and 24 h (Fig. 3). The visual examination of promastigotes incubated with harmonine showed rapid changes of the cell morphology. After 24 h only dead cells were observed upon harmonine-treatment whereas in the presence of dimethyl sulfoxide (DMSO) no significant changes in the cell morphology of the parasites were observed. Rounding of cells upon harmonine treatment was induced after 6 h of culture and dead cells were visible after 10 h of cultivation. DMSO-treated control L. major promastigotes show upon 6 h and 10 h of culture the characteristic flagellated and slender shape of the viable and unaffected parasite (Fig. 3).
 | ||
| Fig. 3 Harmonine induced cell death in L. major promastigotes. L. major promastigotes were treated with 30 μM harmonine or 1% DMSO for 6 h, 10 h, and 24 h. Annexin V and PI double staining was performed for the determination of the cell death phenotype (dot-blots) and Diff-Quick staining was performed to investigate cell morphological changes via transmission light microscopy (pictures). Scale bars = 10 μm. Numbers in the quadrants represent cells (%) showing early necrotic, late necrotic, or apoptotic cell death. | ||
The type of cell death induced by harmonine was investigated using flow cytometric approaches. The loss of membrane integrity in necrotic cells and the translocation of phosphatidylserine (PS) to the outside of the cellular membrane of apoptotic cells can be determined by Annexin V (AV, binds PS) and PI (DNA-binding) staining.31 Double staining with AV-fluorescein isothiocyanate (FITC) and PI allows the discrimination between four Leishmania cell death phenotypes as described elsewhere: live (AV−/PI−), late necrotic/late apoptotic (AV+/PI+), early necrotic (AV−/PI+) and early apoptotic cells (AV+/PI−).31,32
Harmonine-treatment for 24 h induced early necrotic cell death in 31.6% and late necrotic/late apoptotic cell death in 51.2% of all the treated cells (Fig. 3). This means that after 24 h of harmonine-treatment a total of 82.8% of cells were dead. At the same time under normal growth conditions only 26.6% of cells were not viable (Fig. 3). Cell death during harmonine-treatment was observed to increase over time, as after 6 h a total of 26.2% and after 10 h a total of 40.3% of cells were dead, as indicated by PI staining. Early necrosis is characterized by PI-binding to the DNA of cells which have lost their cell membrane integrity. Harmonine was found to induce early necrotic cell death in L. major, as a clear population of 24.5% early necrotic cells was detected after 10 h of cultivation (Fig. 3). The increase of late necrotic/late apoptotic cells to 51.15% after 24 h is a consequence of an early necrotic cell death phenotype, as the rupture of the cell membrane allows AV to bind to PS in the dead parasite.
Conclusion
In conclusion a highly efficient and flexible synthetic route to chiral (R)-harmonine ((R)-1) is reported. A highly enantioselective hydrolysis of rac-5 affords both the remaining lactone (S)-5 and the hydroxy acid (R)-9 in high yield and excellent enantiomeric purities. Reductive olefination of the lactone (S)-5 with readily available phosphonium ylides gives direct access to the backbone of harmonine ((R)-1). Subsequent functional group modification provides derivatives and analogs of the natural product for structure–activity studies and mechanistic analyses to unravel the mode of action of the ladybeetle alkaloid harmonine ((R)-1). The pronounced activity of harmonine ((R)-1) against mycobacteria or the malaria parasite P. falciparum as is reported elsewhere,7 and the activity against L. major as is shown for the first time in the present study are already encouraging observations for further investigations. Readily available synthetic harmonine and analogs may provide a base for the development of novel anti-parasitic drugs with both parasitocidal and transmission-blocking activities.7Experimental
Synthesis
:1) for elution. Removal of the solvents yielded rac-6 as a colorless oil (49 mg, 0.13 mmol, 26%, (Z)/(E) > 98/2; ylide preparation with n-BuLi: 29%, (Z)/(E) 80/20). Rf = 0.18 (hexane–ethyl acetate 5:1).
HRMS m/z calcd for C21H40O3 340.2978 [M]+, found 340.2985; IR (thin film, cm−1) ν 3437 (br, m), 2977 (m), 2964 (m), 2928 (s), 2855 (s), 1733 (s), 1655 (w), 1464 (m), 1374 (m), 1249 (m), 1181 (m), 1145 (m), 1110 (s); 1H NMR (400 MHz, CDCl3) δ 5.40–5.31 (m, 2 H, H-10/H-9), 5.01 (sp, 3J1′,2′ = 3J1′,2′′ = 6.2 Hz, 1 H, H-1′), 3.80 (tq, 3J17,18 = 3J17,16 = 6.1 Hz, 1 H, H-17), 2.26 (t, 3J2,3 = 7.7 Hz, 2 H, H-2), 2.04–1.96 (m, 4 H, H-11/H-8), 1.61 (mc, 2 H, H-3), 1.48–1.38 (m, 2 H, H-16), 1.36–1.27 (m, 16 H, H-15–H-12/H-7–H-4), 1.23 (d, 3J2′,1′ = 3J2′′,1′ = 6.3 Hz, 6 H, H-2′/H-2′′), 1.19 (d, 3J18,17 = 6.2 Hz, 3 H, H-18); 13C NMR (100 MHz, CDCl3) δ 173.4 (C-1), 129.8 (C-10/C-9), 68.1 (C-17), 67.3 (C-1′), 39.4 (C-16), 34.7 (C-2), 29.7, 29.5, 29.2, 29.1, 29.1, 25.7 (C-15–C-12/C-7–C-4), 27.1 (C-11/C-8), 25.0 (C-3), 23.5 (C-18), 21.8 (C-2′/C-2′′).
:1) for elution. After removal of the solvents, rac-3 was obtained as a slightly yellow oil (243 mg, 0.85 mmol, 98%). Rf = 0.35 (hexane–ethyl acetate 1:1).
HRMS m/z calcd for C18H36O2 284.2715 [M]+, found 284.2705; IR (thin film, cm−1) ν 3332 (br, m), 3004 (w), 2926 (s), 2854 (s), 1654 (w), 1462 (m), 1373 (m); 1H NMR (400 MHz, CDCl3) δ 5.40–5.30 (m, 2 H, H-10/H-9), 3.79 (tq, 3J17,18 = 3J17,16 = 5.9 Hz, 1 H, H-17), 3.63 (t, 3J1,2 = 6.6 Hz, 2 H, H-1), 2.06–1.95 (m, 4 H, H-11/H-8), 1.95–1.80 (br, 2 H, O–H), 1.56 (tt, 3J2,3 = 3J2,1 = 6.7 Hz, 2 H, H-2), 1.49–1.38 (m, 2 H, H-16), 1.38–1.25 (m, 18 H, H-14–H-12/H-7–H-4), 1.18 (d, 3J18,17 = 6.0 Hz, 3 H, H-18). 13C NMR (100 MHz, CDCl3) δ 129.9, 129.8 (C-10/C-9), 68.2 (C-17), 63.0 (C-1), 39.3 (C-16), 32.7 (C-2), 29.7, 29.7, 29.5, 29.4, 29.4, 29.2, 29.1 (C-14–C-12/C-7–C-4), 27.1 (C-11/C-8), 25.7 (C-15/C-3), 23.4 (C-18).
:1) for elution. Removal of solvents afforded rac-7 as a colorless oil (195 mg, 0.58 mmol, 76%). Rf = 0.30 (hexane–DCM 3:1).
HRMS m/z calcd for C18H34N6Na 357.2737 [M + Na]+, found 357.2737; IR (thin film, cm−1) ν 3004 (w), 2928 (s), 2855 (s), 2094 (s), 1653 (w), 1459 (m), 1378 (w), 1249 (s); 1H NMR (400 MHz, CDCl3) δ 5.42–5.30 (m, 2 H, H-10/H-9), 3.42 (tq, 3J17,18 = 3J17,16 = 6.5 Hz, 1 H, H-17), 3.26 (t, 3J1,2 = 7.0 Hz, 2 H, H-1), 2.07–1.94 (m, 4 H, H-11/H-8), 1.61 (tt, 3J2,3 = 3J2,1 = 7.0 Hz, 2 H, H-2), 1.57–1.28 (m, 20 H, H-16–H-12/H-7–H-3), 1.25 (d, 3J18,17 = 6.6 Hz, 3 H, H-18). 13C NMR (100 MHz, CDCl3) δ 129.9, 129.8 (C-10/C-9), 58.0 (C-17), 51.5 (C-1), 36.2 (C-16), 29.7, 29.6, 29.4, 29.3, 29.2, 29.1, 29.1 (C-14–C-12/C-7–C-4), 28.8 (C-2), 27.2, 27.1 (C-11/C-8), 26.7 (C-3), 26.1 (C-15), 19.5 (C-18).
HRMS m/z calcd for C18H39N2 283.3108 [M + H]+, found 283.3106; IR (thin film, cm−1) ν 3320 (m), 3004 (w), 2921 (s), 2851 (s), 1639 (w), 1564 (m), 1468 (m), 1430 (w), 1390 (m), 1331 (m); 1H NMR (400 MHz, CDCl3) δ 5.38–5.27 (m, 2 H, H-10/H-9), 2.85 (tq, 3J17,18 = 3J17,16 = 6.0 Hz, 1 H, H-17), 2.66 (t, 3J1,2 = 7.0 Hz, 2 H, H-1), 2.04–1.91 (m, 4 H, H-11/H-8), 1.50–1.38 (m, 6 H, H-2, N–H2), 1.36–1.21 (m, 20 H, H-16–H-12/H-7–H-3), 1.03 (d, 3J18,17 = 6.2 Hz, 3 H, H-18). 13C NMR (100 MHz, CDCl3) δ 129.8, 129.8 (C10/C-9), 46.9 (C-17), 42.1 (C-1), 40.1 (C-16), 33.7 (C-2), 29.7, 29.6, 29.6, 29.4, 29.4, 29.2, 29.2 (C-14–C-12/C-7–C-4), 27.1 (C11/C-8), 26.8 (C-3), 26.3 (C-15), 23.8 (C-18).
:9) for elution. CHCl3 was added and the solvents were removed under reduced pressure. This procedure was repeated three times with CHCl3 and three times with benzene to remove the last traces of water. The residue was concentrated under high vacuum to yield rac-8 as a colorless, sticky oil. (182 mg, 0.61 mmol, 87%). Rf = 0.43 (H2O–MeOH 1:9, RP-18).
HRMS m/z calcd for C18H36NO2 298.2741 [M + H]+, found 298.2734; IR (thin film, cm−1) ν 3358 (s), 3191 (br, m), 3003 (w), 2967 (w), 2924 (s), 2852 (s), 1703 (w), 1659 (s), 1633 (s), 1468 (m), 1423 (m), 1412 (m); 1H NMR (400 MHz, CDCl3) δ 5.68 (d, 2JNH,NH′ = 81.9 Hz, 2 H, N–H2), 5.40–5.27 (m, 2 H, H-10/H-9), 3.78 (tq, 3J17,18 = 3J17,16 = 5.7 Hz, 1 H, H-17), 2.20 (t, 3J2,3 = 7.5 Hz, 2 H, H-2), 2.05–1.92 (m, 4 H, H-11/H-8), 1.84 (brs, 1 H, O–H), 1.62 (tt, 3J3,4 = 3J3,2 = 7.1 Hz, 2 H, H-3), 1.51–1.37 (m, 2 H, H-16), 1.37–1.23 (m, 16 H, H-15–H-12/H-7–H-4), 1.17 (d, 3J18,17 = 6.1 Hz, 3 H, H-18). 13C NMR (100 MHz, CDCl3) δ 175.8 (C-1), 130.3, 130.3 (C-10/C-9 (E)), 129.9, 129.8 (C-10/C-9), 68.0 (C-17), 39.3 (C-16), 35.9 (C-2), 32.5, 32.4 (C-11/C-8 (E)), 29.6, 29.6, 29.5, 29.5, 29.2, 29.2, 29.0 (C-14–C-12/C-7–C-4), 27.1, 27.1 (C-11/C-8), 25.7 (C-15), 25.5 (C-3), 23.4 (C-18).
HRMS m/z calcd for C18H38NO 284.2948 [M + H]+, found 284.2935; IR (thin film, cm−1) ν 3332 (br, m), 3004 (m), 2925 (s), 2853 (s), 1632 (m), 1572 (m), 1464 (m), 1372 (m), 1315 (m); 1H NMR (400 MHz, CDCl3) δ 5.38–5.28 (m, 2 H, H-10/H-9), 3.75 (tq, 3J17,18 = 3J17,16 = 6.1 Hz, 1 H, H-17), 2.66 (t, 3J1,2 = 7.0 Hz, 2 H, H-1), 2.04–1.91 (m, 4 H, H-11/H-8), 1.82–1.76 (m, 3 H, N–H2, O–H), 1.48–1.35 (m, 5 H, H-16/H-15a/H-2), 1.35–1.23 (m, 17 H, H-15b/H-14–H-12/H-7–H-3), 1.15 (d, 3J18,17 = 6.1 Hz, 3 H, H-18). 13C-NMR (100 MHz, CDCl3) δ 130.3, 130.2 (C-10/C-9 (E)), 129.8, 129.8 (C-10/C-9), 67.7 (C-17 (E)), 67.7 (C-17), 42.1 (C-1), 39.4 (C-16), 33.6 (C-2 (E)), 33.6 (C-2), 32.5 (C-11/C-8 (E)), 29.6, 29.6, 29.5, 29.4, 29.4, 29.2, 29.1, (C-14–C-12/C-7–C-4), 27.1 (C-11/C-8), 26.8 (C-3), 26.8 (C-3 (E)), 25.7 (C-15), 23.4 (C-18).
Enzymatic hydrolysis of lactone (rac-5)
Racemic 9-methyloxonan-2-one (rac-5) (3.50 g, 22.4 mmol) was suspended in NaH2PO4 buffer (0.1 M, pH = 7.2, 100 ml) and stirred for 15 minutes. Then horse liver esterase (350 mg, lyophilized powder) was added and the pH was kept constant by dropwise addition of 0.5 M NaOH during the whole reaction time. After 12 hours, another portion of horse liver esterase (70 mg) was added and stirring was continued for 4 hours. Then Celite (3.50 g) and ice (7.00 g) were added, the suspension was stirred for 5 minutes and the solids were filtered off. The filter cake was washed with Et2O (2 × 50 ml). The layers of the filtrate were separated and the aqueous phase was extracted with Et2O (3 × 50 ml). The combined organic extracts were washed with a saturated NaHCO3 solution (100 ml) and dried over Na2SO4. Evaporation of the solvent yielded (9S)-9-methyloxonan-2-one ((S)-5) as a colorless liquid.HRMS m/z calcd for C9H16O2 156.1150 [M]+, found 156.1152; IR (thin film, cm−1) ν 2929 (s), 2857 (m), 1726 (s), 1575 (w), 1464 (w), 1427 (w), 1377 (w), 1286 (m), 1254 (s); 1H NMR (400 MHz, CDCl3) δ 5.07 (mc, 1 H, H-8), 2.25 (mc, 2 H, H-2), 1.93 (mc, 1 H, H-4a), 1.82–1.74 (m, 1 H, H-7a), 1.72–1.64 (m, 1 H, H-3a), 1.67–1.55 (m, 2 H, H-6a/H-3b), 1.55–1.43 (m, 3 H, H-7b/H-5a/H-4b), 1.37–1.27 (m, 2 H, H-6b/H-5b), 1.25 (d, 3J9,8 = 6.5 Hz, 3 H, H-9); 13C NMR (100 MHz, CDCl3) δ 175.5 (C-1), 71.5 (C-8), 35.7 (C-2), 35.1 (C-7), 29.4 (C-5), 25.0 (C-4), 23.9 (C-3), 21.8 (C-6), 20.7 (C-9).
HRMS m/z calcd for C9H18O3Na 197.1148 [M + Na]+, found 197.1146; IR (thin film, cm−1) ν 3391 (br, s), 2933 (s), 2858 (s), 1711 (s), 1462 (m), 1410 (m), 1376 (m), 1261 (m), 1127 (m), 1103 (m); 1H NMR (400 MHz, CDCl3) δ 5.25 (br, 2 H, O–H, COO–H), 3.81 (tq, 3J8,9 = 3J8,7 = 6.1 Hz, 1 H, H-8), 2.36 (t, 3J2,3 = 7.5 Hz, 2 H, H-2), 1.65 (tt, 3J3,4 = 3J3,2 = 7.5 Hz, 2 H, H-3), 1.50–1.40 (m, 3 H, H-7/H-6a), 1.40–1.30 (m, 5 H, H-6b/H-5/H-4), 1.20 (d, 3J9,8 = 6.1 Hz, 3 H, H-9); 13C-NMR (100 MHz, CDCl3) δ 179.0 (C-1), 68.2 (C-8), 39.2 (C-7), 33.9 (C-2), 29.2, 29.0 (C-5/C-4), 25.5 (C-6), 24.6 (C-3), 23.5 (C-9).
HRMS m/z calcd for C12H23O2BrNa, C12H23O281BrNa 301.0774, 303.0753 [M + Na]+, found 301.0776, 303.0753; IR (thin film, cm−1) ν 2979 (m), 2932 (s), 2857 (m), 1731 (s), 1466 (m), 1374 (m), 1256 (m), 1181 (m), 1145 (w), 1110 (s), 964 (w); 1H NMR (400 MHz, CDCl3) δ 5.00 (sp, 3J1′,2′ = 3J1′,2′′ = 6.2 Hz, 1 H, H-1′), 3.40 (t, 3J9,8 = 6.9 Hz, 2 H, H-9), 2.26 (t, 3J2,3 = 7.6 Hz, 2 H, H-2), 1.85 (mc, 2 H, H-8), 1.61 (mc, 2 H, H-3), 1.42 (mc, 2 H, H-7), 1.34–1.28 (m, 6 H, H-6–H-4), 1.22 (d, 3J2′,1′ = 3J2′′,1′ = 6.2 Hz, 6 H, H-2′/H-2′′); 13C NMR (100 MHz, CDCl3) δ 173.3 (C-1), 67.3 (C-1′), 34.6 (C-2), 33.9 (C-9), 32.7 (C-8), 29.0, 29.0 (C-5/C-4), 28.5 (C-6), 28.1 (C-7), 24.9 (C-3), 21.8 (C-2′, C-2′′).
:1). The solvents were removed under reduced pressure and 4 was obtained as a sticky syrup (4.33 g, 8.00 mmol, 77%). Rf = 0.51 (DCM–MeOH 14:1).
HRMS m/z calcd for C30H38O2P 461.2604 [M]+, found 461.2601; IR (thin film, cm−1) ν 3057 (w), 2977 (w), 2855 (s), 2802 (w), 1720 (s), 1587 (w), 1522 (w), 1485 (m), 1104 (m), 856 (s), 842 (s); 1H NMR (400 MHz, CDCl3) δ 7.88–7.76 (m, 9 H, Har-6/Har-2/Har-4), 7.74–7.67 (m, Har-5/Har-3), 4.96 (sp, 3J1′,2′ = 3J1′,2′′ = 6.2 Hz, 1 H, H-1′), 3.82–3.71 (m, 2 H, H-9), 2.20 (t, 3J2,3 = 7.5 Hz, 2 H, H-2), 1.68–1.56 (m, 4 H, H-8/H-7), 1.53 (mc, 2 H, H-3), 1.30–1.21 (m, 6 H, H-6–H-4), 1.20 (d, 3J2′,1′ = 3J2′′,1′ = 6.2 Hz, 6 H, H-2′′/H-2′); 13C NMR (100 MHz, CDCl3) δ 173.3 (C-1), 135.0, 135.0 (Car-4), 133.7, 133.6 (Car-6/Car-2), 130.5, 130.4 (Car-5/Car-3), 118.8, 118.0 (Car-1), 67.3 (C-1′), 34.6 (C-2), 30.3, 30.2 (C-7), 28.9, 28.9, 28.7 (C-6–C-4), 24.8 (C-3), 23.0, 22.6 (C-9), 22.6, 22.5 (C-8), 21.8 (C-2′/C-2′′).
Antileishmanial activity
The virulent L. major isolate (MHOM/IL/81/FE/BNI) and Luciferase-transgenic (Luc-tg.) L. major were maintained by continuous passage in female BALB/c mice. L. major amastigotes were isolated from lesions as described previously33,34 and promastigotes were grown in vitro in blood-agar cultures at 27 °C, 5% CO2, and 95% humidity. AlamarBlue® assays for the determination of antileishmanial activities against L. major promastigotes and BMDM cytotoxicity, and Britelite™ plus (PerkinElmer, Waltham, MA, USA) assays against intracellular Luc-tg. L. major amastigotes were performed as previously reported.34Diff-Quick staining for transmitted light microscopy
After incubation for 6 h, 10 h, and 24 h in the presence of 30 μM harmonine or 1% DMSO as the solvent control, L. major promastigotes were harvested and centrifuged using a Cytospin 3 centrifuge (Shandon, Frankfurt, Germany) on microscopic slides. Cytospin preparations were stained using the Differential Quick stain (Diff-Quick) dye (Medion Diagnostics AG, Duedingen, Switzerland), according to the manufacturer's protocol. Diff-Quik stains the leishmanial nuclei, the kinetoplasts dark purple and the cytoplasm light purple allowing the observation of phenotypic changes within the parasite. The stained cells were analyzed by transmitted light microscopy under a 50× objective on a Nikon ECLIPSE 50i microscope equipped with a digital camera (Nikon, Tokyo, Japan). The images were processed using NIS Elements D software (Nikon).Determination of the cell death phenotype by flow cytometric analysis
L. major promastigotes were either treated for 6 h, 10 h, and 24 h with 30 μM harmonine or 1% DMSO as the solvent control. Cell staining was performed using an Annexin V-FITC Apoptosis detection kit (Sigma-Aldrich, Saint Louis, MO, USA) according to the manufacturer's protocol. The stained samples were immediately analyzed by flow cytometry using a MACS Quant Analyzer (Miltenyi Biotech, Bergisch Gladbach, Germany).Acknowledgements
We thank Dr Christian Paetz (MPI CE, Jena) for recording the 19F-NMR spectra and Sybille Lorenz (MPI CE, Jena) for HR-MS measurements. Furthermore, the authors thank Martina Schultheis (Institute for Molecular Infection Biology, Wuerzburg) for the determination of IC50 and cytotoxicity values, and excellent technical support. This work was supported and financed by the Max Planck Society and by a grant of the Deutsche Forschungsgemeinschaft (DFG), Collaborative Research Center 630 (SFB 630/TP B3), “Recognition, Preparation and Functional Analysis of Agents against Infectious Diseases”.References
- R. L. Koch, J. Insect Sci., 2003, 3, 32 CrossRef CAS PubMed.
- H. Roy and E. Wajnberg, BioControl, 2008, 53, 1 CrossRef.
- H. Roy, P. J. Brown, P. Rothery, R. Ware and M. N. Majerus, BioControl, 2008, 53, 265 CrossRef.
- S. Al Abassi, M. A. Birkett, J. Pettersson, J. A. Pickett and C. M. Woodcock, CMLS, Cell. Mol. Life Sci., 1998, 54, 876 CrossRef CAS.
- T. L. Galvan, S. Kells and W. D. Hutchison, J. Agric. Food Chem., 2008, 56, 1065 CrossRef CAS PubMed.
- D. Daloze, J.-C. Braekman and J. Pasteels, Chemoecology, 1994, 5–6, 173 CrossRef.
- C. R. Röhrich, C. J. Ngwa, J. Wiesner, H. Schmidtberg, T. Degenkolb, C. Kollewe, R. Fischer, G. Pradel and A. Vilcinskas, Biol. Lett., 2012, 8, 308 CrossRef PubMed.
- N. C. Alam, I. S. Choi, K.-S. Song, J.-K. Hong, C. O. Lee and J. H. Jung, Bull. Korean Chem. Soc., 2002, 23, 497 CrossRef CAS.
- A. Vilcinskas, K. Stoecker, H. Schmidtberg, C. R. Röhrich and H. Vogel, Science, 2013, 340, 862 CrossRef CAS PubMed.
- A. Vilcinskas, H. Schmidtberg, A. Estoup, A. Tayeh, B. Facon and H. Vogel, Insect Sci., 2014 DOI:10.1111/1744-7917.12159.
- A. Vilcinskas and H. Schmidtberg, Biol. Unserer Zeit, 2014, 44, 386 CrossRef CAS.
- M. F. Braconnier, J. C. Braekman and D. Daloze, Bull. Soc. Chim. Belg., 1985, 94, 605 CrossRef CAS.
- D. Enders and D. Bartzen, Liebigs Ann. Chem., 1991, 1991, 569 CrossRef.
- S. Chandra Philkhana, P. Dhasaiyan, B. L. V. Prasad and D. S. Reddy, RSC Adv., 2014, 4, 30923 RSC.
- J. Alvar, I. D. Velez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CAS.
- World Health Organization, 2014.
- S. L. Croft, S. Sundar and A. H. Fairlamb, Clin. Microbiol. Rev., 2006, 19, 111 CrossRef CAS PubMed.
- H. J. de Vries, S. H. Reedijk and H. D. Schallig, Am. J. Clin. Dermatol., 2015, 16, 99–109 CrossRef PubMed.
- E. Haulotte, P. Laurent and J.-C. Braekman, Eur. J. Org. Chem., 2012, 2012, 1907 CrossRef CAS.
- J. van Buijtenen, B. A. C. van As, M. Verbruggen, L. Roumen, J. A. J. M. Vekemans, K. Pieterse, P. A. J. Hilbers, L. A. Hulshof, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 7393 CrossRef CAS PubMed.
- Y. Yang, M. R. Mannion, L. N. Dawe, C. M. Kraml, R. A. Pascal and G. J. Bodwell, J. Org. Chem., 2011, 77, 57 CrossRef PubMed.
- S. H. Woo, S. Frechette, E. A. Khalil, G. Bouchain, A. Vaisburg, N. Bernstein, O. Moradei, S. Leit, M. Allan, M. Fournel, M.-C. Trachy-Bourget, Z. Li, J. M. Besterman and D. Delorme, J. Med. Chem., 2002, 45, 2877 CrossRef CAS PubMed.
- T. Ooi, Y. Uematsu and K. Maruoka, J. Org. Chem., 2003, 68, 4576 CrossRef CAS PubMed.
- L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563 CrossRef CAS PubMed.
- Z. Csíki and P. Fügedi, Tetrahedron, 2010, 66, 7821 CrossRef.
- E. Fouque and G. Rousseau, Synthesis, 1989, 661 CrossRef CAS.
- J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512 CrossRef CAS.
- G. R. Sullivan, J. A. Dale and H. S. Mosher, J. Org. Chem., 1973, 38, 2143 CrossRef CAS.
- J. Glaser, M. Schultheis, S. Hazra, B. Hazra, H. Moll, U. Schurigt and U. Holzgrabe, Molecules, 2014, 19, 1394 CrossRef PubMed.
- S. Nwaka and A. Hudson, Nat. Rev. Drug Discovery, 2006, 5, 941 CrossRef CAS PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1995, 184, 39 CrossRef CAS PubMed.
- G. Zhang, V. Gurtu, S. R. Kain and G. Yan, BioTechniques, 1997, 23, 525 CAS.
- C. Bogdan, H. Moll, W. Solbach and M. Rollinghoff, Eur. J. Immunol., 1990, 20, 1131 CrossRef CAS PubMed.
- G. Bringmann, K. Thomale, S. Bischof, C. Schneider, M. Schultheis, T. Schwarz, H. Moll and U. Schurigt, Antimicrob. Agents Chemother., 2013, 57, 3003 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00461f |
| This journal is © The Royal Society of Chemistry 2015 |