
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triazolophostins: a library of novel and potent agonists of IP3 receptors†
Amol M.
Vibhute
a,
Vera
Konieczny
b,
Colin W.
Taylor
b and
Kana M.
Sureshan
*a
aSchool of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram, Kerala-695016, India. E-mail: kms@iisertvm.ac.in; Web: http://www.iisertvm.ac.in/scientists/kana-m-sureshan/personal-information.html Fax: (+) 914712597427
bDepartment of Pharmacology, Tennis Court Road, University of Cambridge, Cambridge CB2 1PD, UK
First published on 26th March 2015
Abstract
IP3 receptors are channels that mediate the release of Ca2+ from the intracellular stores of cells stimulated by hormones or neurotransmitters. Adenophostin A (AdA) is the most potent agonist of IP3 receptors, with the β-anomeric adenine contributing to the increased potency. The potency of AdA and its stability towards the enzymes that degrade IP3 have aroused interest in AdA analogs for biological studies. The complex structure of AdA poses problems that have necessitated optimization of synthetic conditions for each analog. Such lengthy one-at-a-time syntheses limit access to AdA analogs. We have addressed this problem by synthesizing a library of triazole-based AdA analogs, triazolophostins, by employing click chemistry. An advanced intermediate having all the necessary phosphates and a β-azide at the anomeric position was reacted with various alkynes under Cu(I) catalysis to yield triazoles, which upon deprotection gave triazolophostins. All eleven triazolophostins synthesized are more potent than IP3 and some are equipotent with AdA in functional analyses of IP3 receptors. We show that a triazole ring can replace adenine without compromising the potency of AdA and provide facile routes to novel AdA analogs.
I. Introduction
Many biological processes are regulated by changes in the intracellular concentration of Ca2+.1 A major pathway for these Ca2+ signals is the release of Ca2+ from intracellular stores within the endoplasmic reticulum (ER) via IP3 receptors (IP3R).2 IP3R are a family of intracellular Ca2+ channels that are expressed largely in ER membranes.3 IP3 (Fig. 1A, 1) is produced when cell-surface receptors stimulate phospholipase C activity. IP3 then binds to IP3R causing its channel to open and release Ca2+ into the cytosol.4 The ability of IP3 to interact with IP3R is terminated by its dephosphorylation or phosphorylation by specific 5-phosphatase and 3-kinase enzymes.5 While terminating the Ca2+-mobilizing ability of IP3, these steps also initiate recycling of inositol to the lipid from which IP3 is generated and the synthesis of more complex bioactive phosphoinositols including the pyrophosphates,6 many of which have been recent synthetic targets.7 Adenophostin A (AdA), a fungal metabolite isolated from the culture broth of Penicillium brevicompactum, is a more potent agonist than IP3 of IP3R.8 Moreover, AdA is resistant to the enzymes that metabolize IP3. Many analogs of AdA have been synthesized to address structure–activity relationships and the determinants of the increased potency of AdA.9 These studies revealed that the three phosphates and 2′′-OH (color coded in Fig. 1A, 2), which structurally mimic the essential pharmacophore of IP3,10 are essential for AdA activity. The nucleobase/surrogate with β-stereochemistry is indispensible for the enhanced potency.9b,11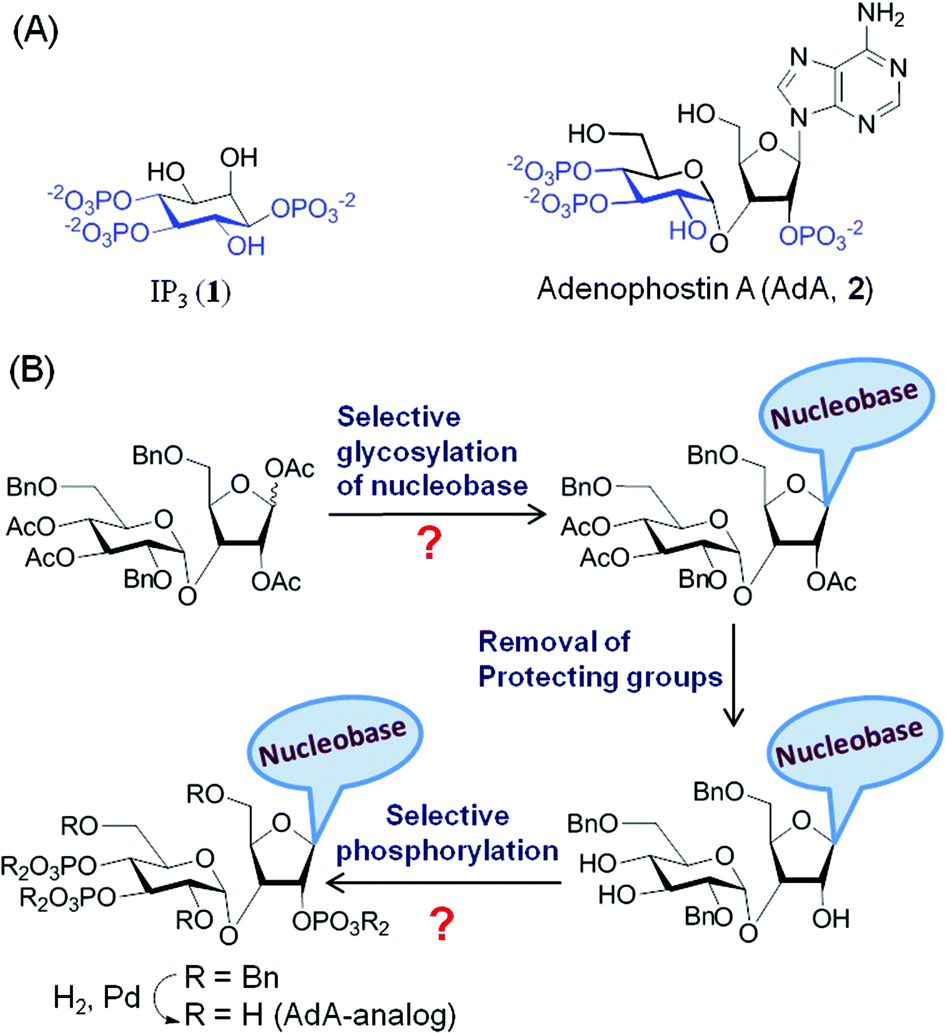 | ||
| Fig. 1 (A) Molecular structures of IP3 (1) and AdA (2); (B) conventional synthesis of AdA-analogs. The question marks represent the complexity of conventional syntheses. | ||
Recent studies have suggested that a cation–π interaction between adenine and a cationic residue (Arg504) of the IP3R might be responsible for the increased potency of AdA.9b,12 Additional base-modified analogs of AdA would be useful in both testing this proposal and as potent and metabolically stable agonists of IP3R to probe Ca2+ signaling mechanisms.
The major hurdle in developing such AdA analogs lies in the synthesis of these structurally complex base-modified analogs (disaccharide nucleotides).9e,13 The traditional synthesis involves N-glycosylation of a nucleobase with an orthogonally protected disaccharide derivative followed by chemoselective demasking and phosphorylation of specific hydroxyl groups in the nucleoside (Fig. 1B). This early introduction of the nucleobase necessitates an almost individually tailored synthesis (branching at an early stage of synthesis) for each base-modified analog. Further complications arise from the lack of a generally applicable method for glycosylation and phosphorylation of such complex molecules. For instance glycosylation of nucleobases is challenging with respect to yield14 and regioselectivity,13b,15 and each glycosylation condition has to be optimized individually. Chemoselective O-phosphorylation in the presence of nucleobases having nucleophilic groups/sites is also challenging.16 Thus synthesis of analogs by a one-at-a-time method is cumbersome, time-consuming and impractical. We herein report a general strategy for making a combinatorial library of structurally diverse AdA analogs that are potent agonists of IP3R.
II. Results and discussion
Imidophostin (Fig. 2A), though weaker than AdA, is more potent than IP3, suggesting that even a 5-membered aromatic heterocyclic ring can be a partial functional mimic of adenine. As triazole is isosteric with imidazole, we envisioned that triazole-based AdA analogs would also be potent agonists of IP3R. This and the earlier reports on the use of click reaction in biologically important molecules17 prompted us to adopt azide–alkyne click chemistry to make a library of triazolophostins, AdA analogs wherein the nucleobase is replaced by substituted triazoles. These molecules can easily be prepared by reacting appropriately protected tris-phosphate derivative II having an azide at the anomeric position with aryl or heteroaryl alkynes under Cu(I) catalysis (Fig. 2B). Such an approach would solve three major issues associated with conventional AdA analog synthesis: (i) ensure the necessary β-anomeric stereochemistry of the base-surrogate, (ii) as the triazole-based base-surrogate is introduced only at the penultimate step (after phosphorylation), the interference of nucleobases during phosphorylation can be avoided and (iii) diversification at the penultimate step provides access to many structurally diverse analogs from a common advanced intermediate.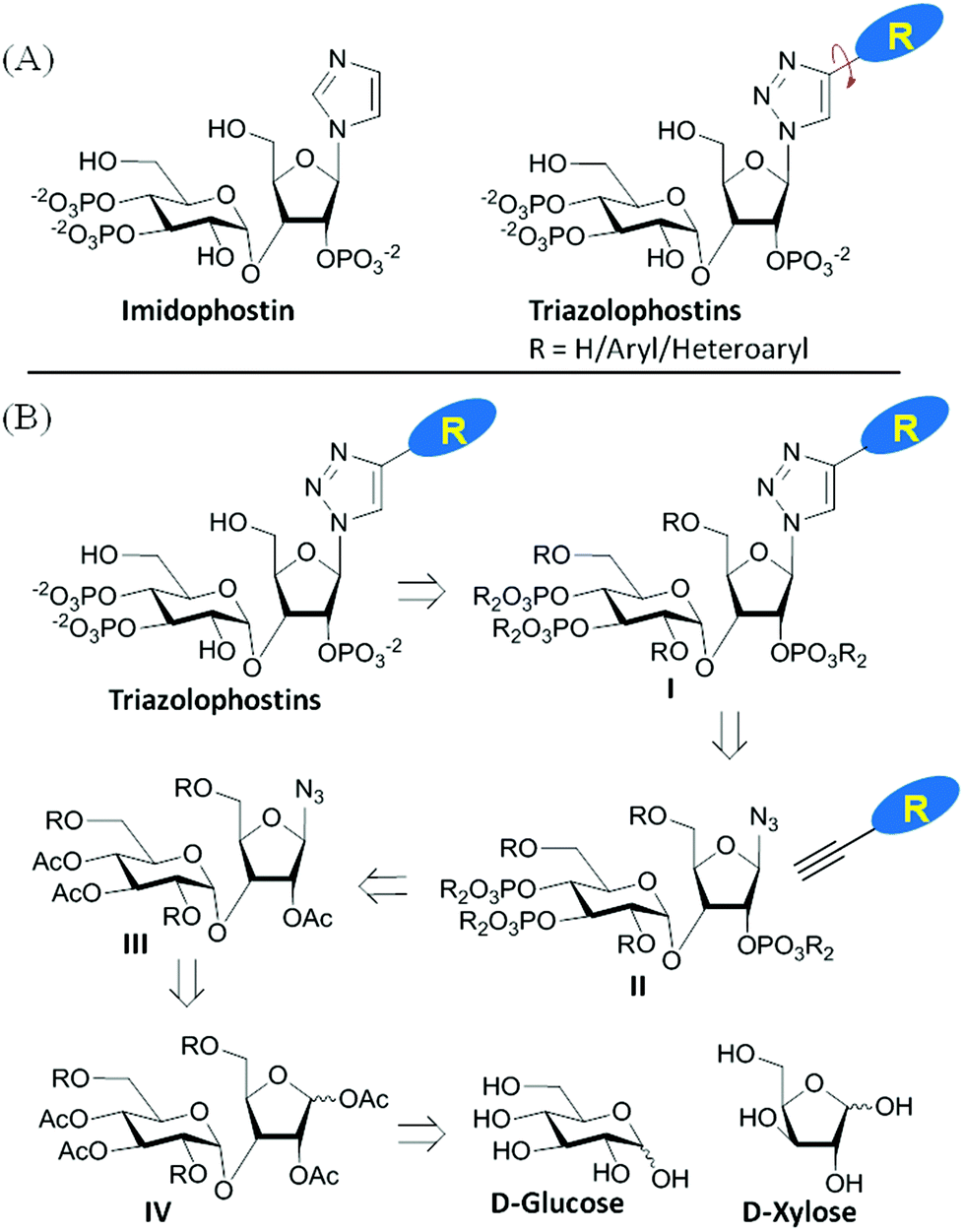 | ||
| Fig. 2 (A) Imidophostin and triazolophostins. (B) Retrosynthetic analysis of triazolophostins. | ||
We have synthesized orthogonally protected disaccharide 4 in thirteen steps from D-glucose and D-xylose in a convergent manner as reported previously.18 Acetolysis19 of the ketal 4 gave tetraacetate 5 as anomeric mixtures in very good yield. Lewis acid catalysed azidation of the acetate 5 gave anomerically pure β-azide 6.20 Methanolysis of the acetate groups in 6 provided the triol 7 in excellent yield. Phosphitylation of the triol 7 using phosphoramidite followed by in situ oxidation using m-CPBA provided the fully protected trisphosphate 8. Compound 8 with three phosphates in their appropriate relative positions and β-oriented azide is the common advanced intermediate for coupling with different alkynes. Copper(I) catalysed click reaction of azide 8 with various alkynes provided fully protected triazolophostins 9a–k. As the reaction is catalyzed by copper(I), 1,4-substituted triazoles21 were obtained exclusively in all the cases. Global removal of the benzyl protecting groups by transfer hydrogenolysis provided triazolophostins 10a–k in very good yields (Scheme 1). Though in a recent elegant communication, Potter et al. reported a functionally active triazole based cADPR analog,22 ours is the first report on the use of click chemistry to generate a library of IP3R agonists.
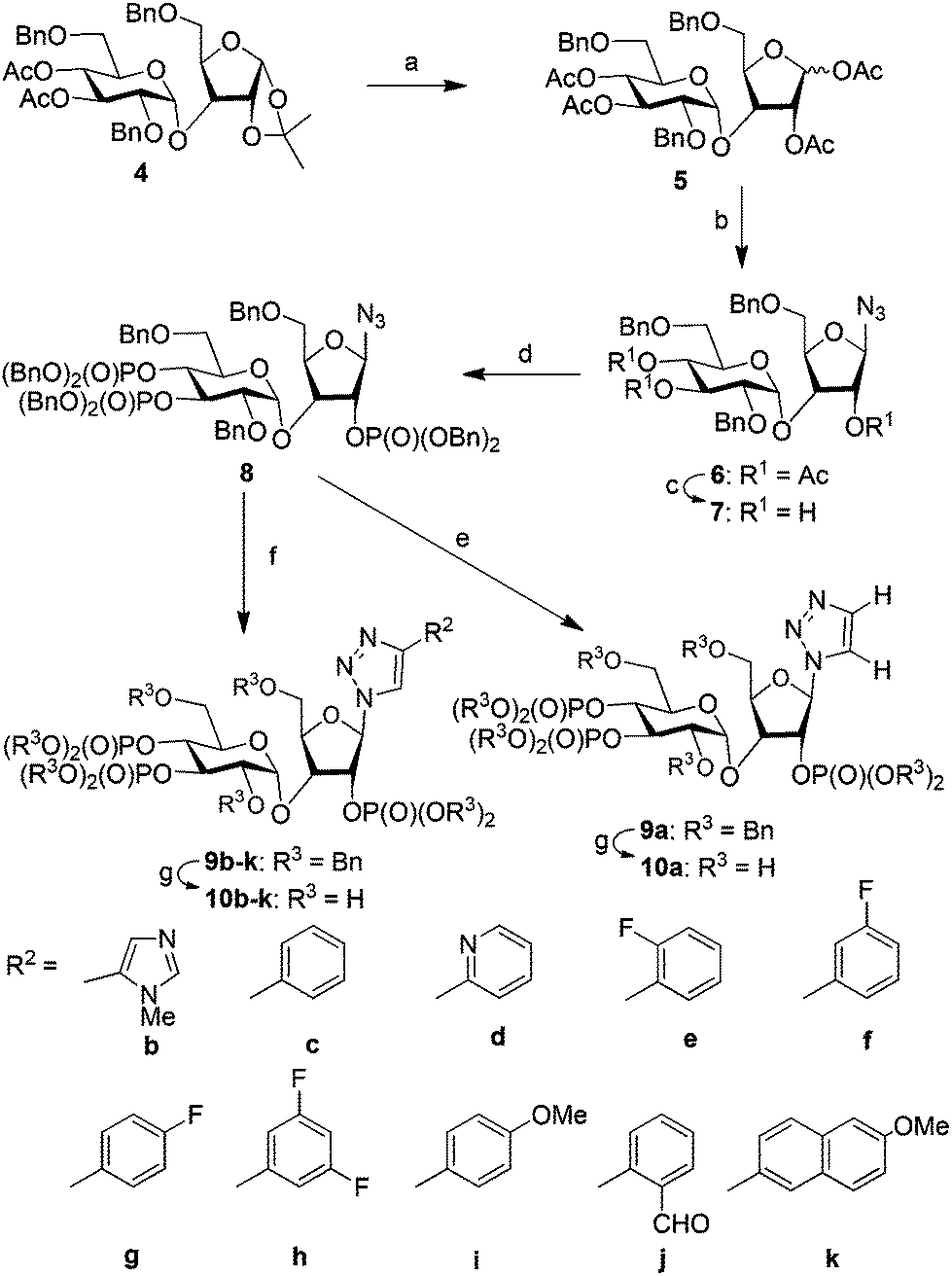 | ||
Scheme 1 Synthesis of triazolophostins. (a) H2SO4-silica, Ac2O, DCM, rt, 12 h, 90%; (b) TMSN3, AlCl3, DCM, 0 °C, 10 min, 99%; (c) NaOMe, MeOH, rt, 30 min, 96%; (d) (i) (iPr)2NP(OBn)2, ImOTf, DCM, rt, 1 h, (ii) m-CPBA, −78 °C, 45 min, 90%; (e) (i) trimethylsilylacetylene, Cu, CuSO4, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tBuOH, rt, 12 h; (ii) TBAF, THF, rt, 2 h, (95% for two steps); (f) alkyne, Cu, CuSO4, H2O:tBuOH, rt, 12 h, 48–97%; (g) cyclohexene, Pd(OH)2, MeOH:H2O, 80 °C, 4 h, Bn = benzyl, Ac = acetyl, Me = methyl. :tBuOH, rt, 12 h; (ii) TBAF, THF, rt, 2 h, (95% for two steps); (f) alkyne, Cu, CuSO4, H2O:tBuOH, rt, 12 h, 48–97%; (g) cyclohexene, Pd(OH)2, MeOH:H2O, 80 °C, 4 h, Bn = benzyl, Ac = acetyl, Me = methyl. | ||
The abilities of the newly synthesized ligands to evoke Ca2+ release via IP3R were assessed using permeabilized cells that express only type 1 IP3R.23 A low-affinity Ca2+ indicator trapped within the ER allowed the effects of IP3 to be directly measured using a fluorescence plate-reader equipped to allow automated additions. All the triazolophostins fully release the IP3-sensitive Ca2+ stores and all are more potent than IP3 (Table 1, Fig. 3). It is interesting to note that the parent analog triazolophostin 10a having only the five-membered ring is 13-fold more potent than IP3. This remarkable AdA-like potency of 10a suggests that a single triazole ring can effectively replace the adenine base in AdA without compromising potency. In contrast, imidophostin was previously reported to be only slightly (1.3-fold) more potent than IP3, and it was therefore suggested that a fused bicyclic nucleobase/surrogate is essential for AdA-like potency.18a Among all triazolophostins, the 3-fluoro derivative 10f is the most potent, and equipotent to AdA: it is the most potent AdA analog without a purine nucleobase. Of the several AdA analogs reported in the literature, very few match the potency of AdA.9c,12b,13b,14,18a,24,25 In this context, our late-stage diversification method is important because it allows many potent analogs to be made in an easy and combinatorial way from the same advanced intermediate.
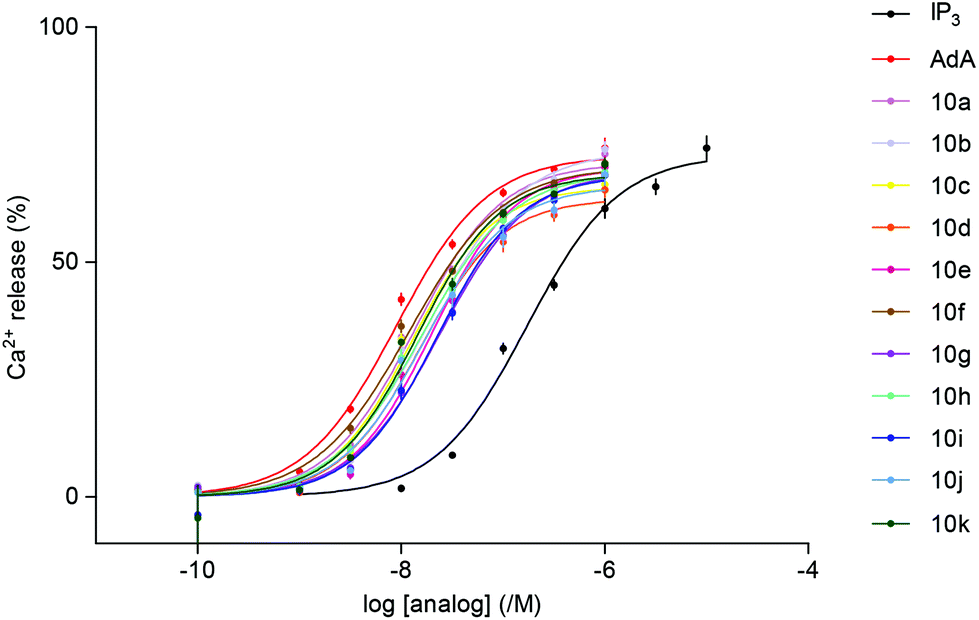 | ||
| Fig. 3 Concentration-dependent release of Ca2+ (%) by IP3, AdA and triazolophostins from the intracellular stores of cells expressing IP3R1. Results show means ± SEM from at least 3 experiments. | ||
|
|
|||||
|---|---|---|---|---|---|
| Ligand | R = | pEC50 | EC50 (nM) | EC50 w.r.t. 1 | Max. response (%) |
| a Results show the maximal Ca2+ release (% of Ca2+ content of intracellular stores), EC50 and pEC50 values (means ± SEM from at least 3 experiments), and EC50 relative to 1 (ECIP350/ECanalog50). Hill coefficients were not significantly different from 1. | |||||
| IP3 (1) | 6.75 ± 0.08 | 178 | 1 | 73 ± 3 | |
| AdA (2) | 8.05 ± 0.04 | 9 | 20 | 73 ± 2 | |
| 10a | H | 7.87 ± 0.00 | 13 | 13 | 71 ± 1 |
| 10b |
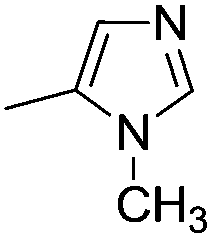
|
7.75 ± 0.03 | 18 | 10 | 72 ± 2 |
| 10c |
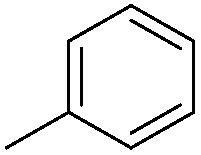
|
7.81 ± 0.05 | 15 | 11 | 65 ± 3 |
| 10d |
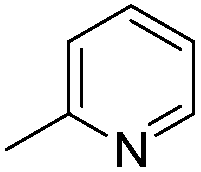
|
7.77 ± 0.03 | 17 | 10 | 64 ± 1 |
| 10e |
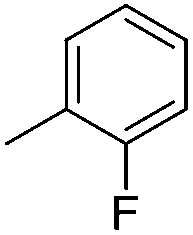
|
7.70 ± 0.07 | 20 | 9 | 75 ± 4 |
| 10f |
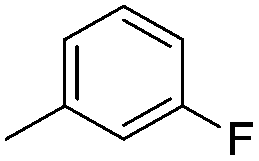
|
8.00 ± 0.03 | 10 | 18 | 69 ± 1 |
| 10g |
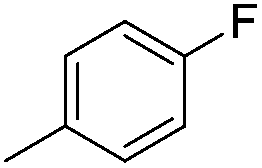
|
7.86 ± 0.04 | 14 | 13 | 71 ± 2 |
| 10h |
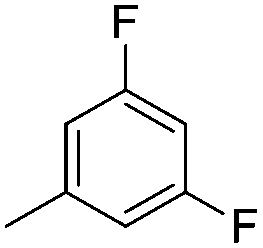
|
7.64 ± 0.01 | 23 | 8 | 67 ± 2 |
| 10i |
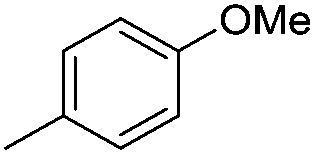
|
7.45 ± 0.03 | 35 | 5 | 65 ± 1 |
| 10j |
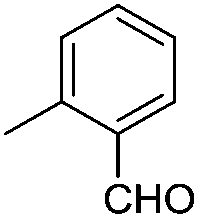
|
7.78 ± 0.03 | 17 | 11 | 68 ± 2 |
| 10k |
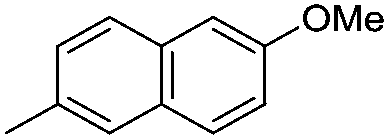
|
7.89 ± 0.02 | 13 | 14 | 70 ± 1 |
The docking study suggests that triazolophostin 10a and AdA exhibit similar interactions with the IP3-binding core of IP3R1 (IBC, residues 224–604).24 The triazolyl ring of 10a forms a cation–π interaction with Arg504 similar to the proposed interaction between the adenine of AdA and Arg504. This supports previous reports that a cation–π interaction between the base and IP3R contributes to the increased potency of AdA and analogs.9c,24 All other triazolophostin analogs showed a similar interaction. The small differences in activity among these analogs seem to arise from steric, rather than electronic factors. For instance, attaching aromatic rings with different electrostatic surface potentials to the triazole ring did not cause any systematic change in biological activity. The fact that all the analogs are both more potent than IP3, and show a conserved cation–π interaction between the triazole ring and Arg504 suggests that this critical interaction is unaffected by aromatic substitution, irrespective of the electronic nature of the aromatic substituent. The single bond connection between the triazole ring and the substituent (aromatic ring) allow torsional freedom (freely rotatable) and hence the sterically favorable non-coplanar conformation of the two aromatic rings. As a result of such non-coplanarity (lack of conjugation), the electron density on the triazole ring cannot be influenced by the aromatic unit. This could be the reason why there is no clear trend in activity when the aromatic ring is modified with electron-deficient or electron-rich substituents.
One reason for the increased potency of AdA is believed to be its conformational flexibility allowing it to adopt an optimal conformation for binding to IP3R.26 The additional conformational freedom due to the single bond linkage of this hetero-biaryl unit (rather than fused rings in AdA) allows the second ring to orient to an electronically favorable position, such that it can have additional electrostatic interactions. Docking results suggest that the most potent analog, 10f, has additional interactions between F and IP3R side-chains (D566, Q565 and Q507) that may enhance its activity (Fig. 4).
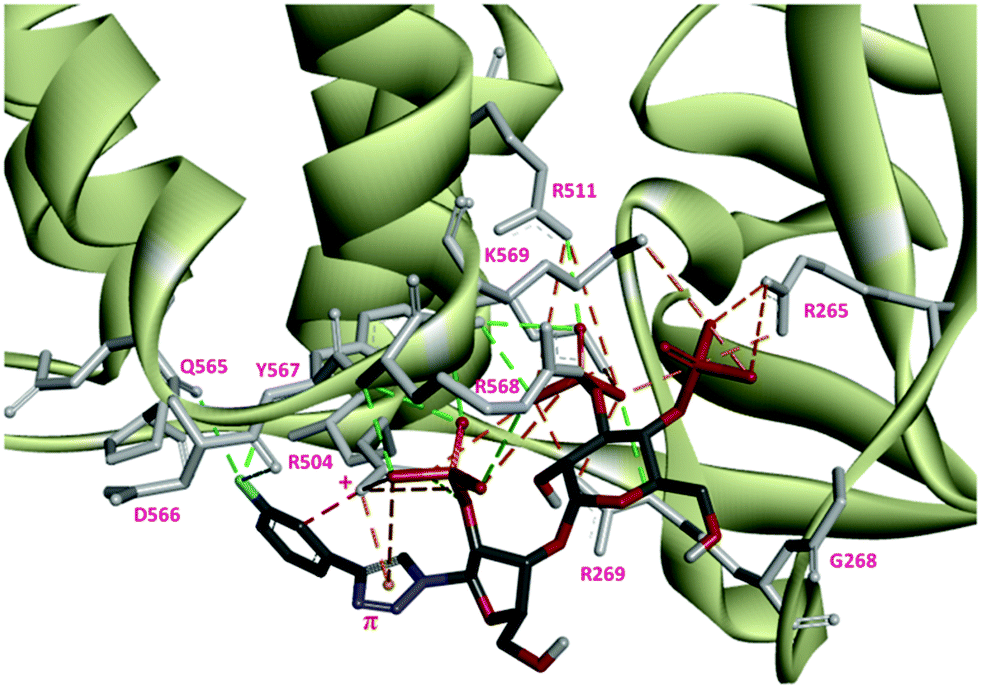 | ||
| Fig. 4 Docking of 3-fluorophenyl triazolophostin with IBC showing preferred binding mode and the important cation–π and fluorine–oxygen interactions with residues Arg504 and Asp566, respectively. The cation–π distance r = 2.97 Å. The fluorophenyl moiety orients perpendicular to the triazole ring. Color code for ligands: carbon, gray; oxygen, red; nitrogen, blue; phosphorus, yellow; fluorine, cyan. | ||
III. Conclusions
In conclusion, addressing the problems associated with conventional one-at-a-time synthesis of AdA analogs, we have synthesized a library of new analogs using azide–alkyne click chemistry. The simple reaction conditions, high yield and atom-economy of the click reaction to install base-surrogates are advantageous over the conventional Vorbruggen method of installation of a nucleobase. By this easy and rapid method, we have generated a variety of ligands with diverse structures from a common advanced intermediate. All the analogs synthesized by this method are more potent than IP3 and some (10a, 10f, 10g, 10k) are as potent as AdA. Though replacement of purines with pyrimidines or other aromatic groups is reported to give agonists that are more potent than IP3, none of the previously reported non-purinated AdA analogs was as potent as AdA. We have shown that AdA-like potency can be achieved without a nucleobase. Our study reveals that a five-membered aromatic ring, triazole, is sufficient as a base surrogate to exhibit AdA-like potency. Easy access to potent, non-metabolizable ligands of IP3R and the ease of conjugation with reporter groups such as fluorescent probes, photoaffinity probes, etc. offers avenues for advanced biological exploration. This report that triazoles can be functional mimics of nucleobases has broader implication and might trigger such attempts in other nucleobase-derived signaling molecules.IV. Experimental section
1. General methods
Chromatograms were visualized under UV light and by dipping plates into either phosphomolybdic acid in MeOH or anisaldehyde in ethanol, followed by heating. 1H NMR, COSY, NOESY and HMQC spectra were recorded on a 500 MHz NMR spectrometer. Proton chemical shifts are reported in ppm (δ) relative to the internal standard tetramethylsilane (TMS, δ 0.0 ppm) or with the solvent reference relative to TMS employed as the internal standard (CDCl3, δ 7.26 ppm; D2O, δ 4.79 ppm). Data are reported as follows: chemical shift (multiplicity [singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), and multiplet (m)], coupling constants [Hz], integration and peak identification). All NMR signals were assigned on the basis of 1H NMR, 13C NMR, COSY and HMQC experiments. 13C spectra were recorded with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to TMS with the respective solvent resonance as the internal standard. All NMR data were collected at 25 °C. The concentration of the compounds for 1H NMR was 5 mg per 0.5 mL and for 13C NMR it was 20 mg per 0.5 mL for protected compounds and 5–7 mg per 0.5 mL for final compounds in the case of 1H and 13C NMR. Each triazolophostin (10a–k) was quantified using the Briggs's/Ames's phosphate assay. Melting point was determined using melting point apparatus and is uncorrected. Flash column chromatography was performed using a silica gel 230–400 mesh. Wherever needed, the reactions were carried out under an argon or nitrogen atmosphere employing oven dried glassware.X-ray intensity data measurements of freshly grown crystals of 4 were carried out at 293–296 K on a Bruker-KAPPA APEX II CCD diffractometer with graphite-monochromatized (MoKα = 0.71073 Å) radiation. The X-ray generator was operated at 50 kV and 30 mA. Data were collected with a scan width of 0.3° at different settings of φ (0°, 90° and 180°) keeping the sample to detector distance fixed at 40 mm and the detector position (2θ) fixed at 24°. The X-ray data collection was monitored by SMART program. All the data were corrected for Lorentzian, polarization and absorption effects using SAINT and SADABS programs. SHELX-97 was used for structure solution and full matrix least-squares refinement on F2. Molecular and packing diagrams were generated using Mercury-3.1. Geometrical calculations were performed using SHELXTL and PLATON.
2. Synthesis of 1,2,3′,4′-tetra-O-acetyl-2′,5,6′-tri-O-benzyl-3-O-α-D-glucopyranosyl-D-ribofuranose (5)
To a solution of disaccharide 4 (1.0 g, 1.41 mmol) and Ac2O (0.4 mL, 4.24 mmol) in dry DCM (20 mL), 0.05 g of freshly prepared H2SO4-silica19 was added. The reaction mixture was stirred at room temperature for 12 h under a nitrogen atmosphere and the reaction was monitored using TLC. When the starting material disappeared, the reaction mixture was quenched by the addition of solid NaHCO3. The solid material was filtered off and the organic layer was washed with saturated aqueous NaHCO3 solution. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The crude product thus obtained was purified by flash column chromatography using 40% ethyl acetate in petroleum ether as the eluent to obtain the known18a tetraacetate 5 (0.96 g, 90%) as a sticky mass.3. Synthesis of 2,3′,4′-tri-O-acetyl-2′,5,6′-tri-O-benzyl-3-O-α-D-glucopyranosyl-β-D-ribofuranosyl azide (6)
To a solution of tetraacetate 5 (0.51 g, 0.68 mmol) and TMSN3 (0.44 mL, 3.4 mmol) in anhydrous dichloromethane (10 mL) was added AlCl3 (0.09 g, 0.68 mmol) at 0 °C. The reaction mixture was stirred at 0 °C under a nitrogen atmosphere for 10 min. When the starting material disappeared (TLC), the reaction mixture was quenched by the addition of aqueous NaHCO3. The reaction mixture was partitioned between dichloromethane and aqueous layers in a separating funnel. The dichloromethane layer was washed with brine, dried over Na2SO4 and evaporated under reduced pressure. The crude product thus obtained was purified by flash column chromatography using 25% ethyl acetate in petroleum ether as the eluent to obtain triacetate azide 6 (0.495 g, 99%) as a colourless gum. 1H NMR (500 MHz, CDCl3) δ: 1.79 (s, 6H, 2 × COCH3), 1.86 (s, 3H, COCH3), 3.22 (dd, J = 10.0, 4.0 Hz, 1H, H-6A′), 3.26 (dd, J = 10.0, 2.0 Hz, 1H, H-6B′), 3.46 (dd, J = 10.0, 3.4 Hz, 1H, H-2′), 3.52 (dd, J = 11.0, 4.0 Hz, 1H, H-5A), 3.61 (dd, J = 11.0, 3.0 Hz, 1H, H-5B), 3.77 (d, J = 9.8, Hz, 1H, H-5′), 4.22 (d, J = 12.0, Hz, 1H, 0.5 × PhCH2), 4.29–4.30 (m, 1H, H-4), 4.38–4.43 (m, 3H, 1.5 × PhCH2), 4.46–4.54 (m, 3H, H-3 and 1 × PhCH2), 4.88 (d, J = 3.3 Hz, 1H, H-1′), 4.95 (t, J = 9.8 Hz, 1H, H-4′), 5.02 (d, J = 2.6 Hz, 1H, H-2), 5.27–5.29 (m, 2H, H-1 and H-3′), 7.16–7.22 (m, 15H, 3 × Ph); 13C NMR (125 MHz, CDCl3) δ: 20.5, 20.7, 20.9 (COCH3), 67.6 (C-6′), 69.0 (C-4′ and C-5′), 69.5 (C-5), 71.9 (C-3′), 73.3, 73.5, 73.5 (3 × CH2Ph), 73.9 (C-2), 74.5 (C-3), 76.6 (C-2′), 81.4 (C-4), 92.77 (C-1), 96.7 (C-1′), 127.5, 127.7, 127.7, 127.8, 128.0, 128.4, 128.4, 128.5 (Ar-C), 137.5, 137.7, 137.9 (ipso Ar–C), 169.8, 170.5, 170.5 (3 × COCH3); Anal. calcd for C38H43N3O12: C, 62.20; H, 5.91; N, 5.73. found: C, 62.12; H, 6.09; N, 5.71.4. Synthesis of 2′,5,6′-tri-O-benzyl-3-O-α-D-glucopyranosyl-β-D-ribofuranosyl azide (7)
To the suspension of triacetate 6 (0.42 g, 0.57 mmol) in MeOH (10 mL) was added sodium methoxide (0.015 g, 0.28 mmol) and stirred at room temperature. The reaction mixture became clear after 30 min. When TLC showed complete disappearance of the starting material, the reaction was quenched by adding Dowex 50-ion exchange resin. The reaction mixture was then filtered through a filter paper and washed successively with methanol (2 times). The filtrate was concentrated under reduced pressure to obtain a thick mass. The thick mass was then purified by flash column chromatography using 50% ethyl acetate in petroleum ether as the eluent to obtain triol azide 7 (0.34 g, 96%) as a white solid. mp: 69–71 °C; 1H NMR (500 MHz, CDCl3) δ: 3.33 (dd, J = 9.5, 3.5 Hz, 1H, H-2′), 3.43–3.54 (m, 5H, H-4′, H-5A, H-5B, H-6A′, H-6B′), 3.66 (ddd, J = 9.8, 8.7, 3.9, 1H, H-5′), 3.82–3.85 (m, 2H, H-3′ and H-2), 4.16–4.18 (m, 2H, 3-H and H-4), 4.38–4.48 (m, 4H, 2 × CH2Ph), 4.61–4.70 (m, 3H, H-1′ and 1 × CH2Ph), 5.20 (d, J = 1.4 Hz, 1H, H-1), 7.18–7.29 (m, 15H, 3 × Ar-H); 13C NMR (125 MHz, CDCl3) δ: 69.2 (C-5 or C-6′), 70.2 (C-5 or C-6′), 70.6 (C-5′), 71.1 (C-4′), 73.3 (C-3′), 73.5 and 73.6 (2 × CH2Ph), 74.0 (C-2), 74.3 (1 × CH2Ph), 78.3 (C-3 or C-4), 78.4 (C-2′), 81.4 (C-3 or C-4), 95.1 (C-1), 98.0 (C-1′), 127.6, 127.7, 127.8, 127.9, 128.4, 128.5, 128.6, 128.8 (Ar-C), 137.1, 137.6, 137.9 (ipso Ar-C); Anal. calcd for C32H37N3O9: C, 63.25; H, 6.14; N, 6.92; found: C, 63.23; H, 6.16; N, 6.90.5. Synthesis of 2′,5,6′-tri-O-benzyl-2,3′,4′-tris(dibenzyloxyphosphoryl)-3-O-α-D-glucopyranosyl-β-D-ribofuranosyl azide (8)
A solution of the triol 7 (0.1 g, 0.16 mmol), bis(benzyloxy) N,N-diisopropylaminophosphine (0.27 mL, 0.82 mmol), and imidazolium triflate (0.20 g, 0.90 mmol) in anhydrous DCM (10 mL) was stirred at room temperature under a N2 atmosphere for 30 min. The reaction mixture was then cooled to −78 °C, and then m-CPBA (0.093 g, 0.54 mmol) was added. The reaction mixture was then stirred for 45 min at −78 °C and then at room temperature for an additional 30 min. The reaction was quenched by the addition of aqueous NaHCO3. The reaction mixture was partitioned between DCM and the aqueous layer in a separating funnel. The DCM layer was washed with brine, dried over Na2SO4 and evaporated under reduced pressure. The crude product thus obtained was purified by flash column chromatography using 25% acetone in petroleum ether as the eluent to obtain pure 8 (0.206 g, 90%) as a colourless gum: 1H NMR (500 MHz, CDCl3) δ: 3.43–3.58 (m, 5H, H-6A′, H-6B′, H-5B, H-5A, H-2′), 3.70–3.72 (m, 1H, H-5′), 4.17–4.22 (m, 2H, H-4, and 0.5 × PhCH2), 4.27–4.34 (m, 3H, H-3 and 1 × PhCH2), 4.37–4.42 (m, 2H, 1 × PhCH2), 4.47 (dd, 19.05, 9.75 Hz, 1H, H-4′), 4.55–4.59 (m, 2H, H-2, 0.5 × PhCH2), 4.65 (dd, 11.95, 8.5 Hz, 1H, 0.5 × PhCH2), 4.78–4.94 (m, 12H, H-3′ and 5.5 × PhCH2), 5.01 (d, 3.55 Hz, 1H, H-1′), 5.30 (d, 2.6 Hz, 1H, H-1), 6.96–7.25 (m, 45H, 9 × Ph), 13C NMR (125 MHz, CDCl3) δ: 68.1 (C-6′), 69.1 (C-5), 69.3, 69.3, 69.4, 69.5, 69.8, 69.83, 69.9, (31P coupled, PhCH2OP), 70.0 (C-5′), 72.0 (1 × PhCH2OC), 73.2 (C-3), 73.3, 73.5 (2 × PhCH2OC), 74.4 (31P coupled, C-4′), 76.9 (C-2′), 77.6 (C-3′, 31P coupled), 78.1 (C-2, 31P coupled), 81.3 (C-4), 92.4 (C-1), 95.2 (C-1′), 127.6, 127.65, 127.70, 127.8, 128.06, 128.08, 128.26, 128.28, 128.31, 128.34, 128.39, 128.44, 128.49, 128.52, 128.62, 128.64, 128.68, 128.69 (aromatic carbons), 135.40, 135.45, 135.70, 135.77, 135.85, 135.90, 136.14, 136.16, 136.20, 136.23, 137.6, 137.9, 138.1, (31P coupled, ipso carbons of POCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.20, −1.90, −2.15; Anal. calcd for C74H76N3O18P3: C, 64.02; H, 5.52; N, 3.03. Found: C, 64.33; H, 5.45; N, 3.19.6. Synthesis of fully protected triazolophostins
:7 v/v) as the eluent to obtain pure 9a (0.098 g, 97%) as a colourless gum. 1H NMR (500 MHz, CDCl3) δ: 3.43–3.45 (m, 1H, H-5A′), 3.47–3.57 (m, 3H, H-2′′, H-5B′, H-6A′′), 3.59–3.61 (m, 1H, H-6B′′), 3.75 (ddd, J = 9.2, 4.65, 4.2 Hz, 1H, H-5′′), 4.21–4.23 (m, 1H, 0.5 × PhCH2), 4.29–4.32 (m, 4H, H-4′, 1.5 × PhCH2), 4.39–4.46 (m, 3H, H-3′, H-4′′ and 0.5 × PhCH2), 4.58–4.66 (m, 3H, 1.5 × PhCH2), 4.68–4.72 (m, 3H, 1.5 × PhCH2), 4.80–4.96 (m, 8H, H-3′′, 3.5 × PhCH2), 5.13 (d, J = 3.0 Hz, 1H, H-1′′), 5.27 (ddd, J = 8.75, 8.2, 4.6 Hz, 1H, H-2′), 6.31 (d, 4.9 Hz, 1H, H-1′), 6.99–7.27 (m, 45H, 9 × C6H5), 7.48 (s, 1H, H-5), 7.61 (s, 1H, H-4); 13C NMR (125 MHz, CDCl3) δ: 68.5 (C-6′′), 69.1 (C-5′), 69.2, 69.36, 69.40, 69.5, 69.6, 69.8, 69.83, 69.9, 70.0, 70.02 (31P coupled, PhCH2OP), 70.3 (C-5′′), 71.9, 73.4, 73.6 (3 × PhCH2OC), 73.9 (C-3′), 74.5 (31P coupled, C-4′′), 76.9 (C-2′), 78.0 (C-3′′, 31P coupled), 78.6 (C-2′, 31P coupled), 82.81 (C-4′), 90.0 (C-1′), 95.6 (C-1′′), 122.4 (C-5), 127.6, 127.7, 127.74, 127.77, 127.8, 127.9, 128.0, 128.07, 128.1, 128.2, 128.3, 128.34, 128.4, 128.5, 128.52, 128.55, 128.59, 128.7, (aromatic carbons), 135.2, 135.28, 135.7, 135.74, 135.8, 135.87, 136.1, 136.2, 137.4, 137.6, 138.0 (31P coupled, ipso carbons of POCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.427, −1.908, −2.128; Anal. calcd for C76H78N3O18P3: C, 64.54; H, 5.56; N, 2.97. Found: C, 64.31; H, 5.42; N, 3.01.
:1:15 v/v/v) as the eluent to obtain pure 9b (0.205 g, 95%) as a colourless gum. 1H NMR (500 MHz, CDCl3) δ: 3.55–3.76 (m, 8H, 1 × N-CH3, 6A′′, H-6B′′, H-5B′, H-2′′ and H-5A′), 3.88 (bs, 1H, H-5′′), 4.31–4.45 (m, 5H, H-4′ and 2 × PhCH2), 4.51–4.54 (m, 3H, H-4′′ and 1 × PhCH2), 4.70–4.82 (m, 6H, H-3′, and 2.5 × PhCH2), 4.94–5.03 (m, 8H, H-3′′ and 3.5 × PhCH2), 5.26 (bs, 1H, H-1′′), 5.41 (bs, 1H, H-2′), 6.42 (s, 1H, H-1′), 6.97–7.34 (m, 47H, Ar-H), 7.75 (s, 1H, H-5); 13C NMR (125 MHz, CDCl3) δ: 33.4 (N-CH3), 68.6 (C-6′′), 69.2 (C-5′), 69.4, 69.42, 69.5, 69.59, 69.8, 69.9, 70.03 (31P coupled PhCH2OP), 70.3 (C-5′′), 71.92, 73.4, 73.7 (3 × PhCH2OC), 74.3 (C-3′), 74.5 (C-4′′, 31P coupled), 77.0 (C-2′, 31P coupled), 78.0 (C-2′′), 78.8 (C-3′′, 31P coupled), 83.1 (C-4′), 90.4 (C-1′, 31P coupled), 95.77 (C-1′′), 119.41 (C-5), 127.64, 127.67, 127.71, 127.80, 127.8, 127.96, 128.0, 128.1, 128.14, 128.3, 128.5, 128.52, 128.6, 128.67 (aromatic carbons), 135.1, 135.2, 135.6, 135.76, 135.80, 135.9, 136.1, 136.18, 137.2, 137.6 (31P coupled, ipso carbons of POCH2Ph), 138.0, 138.8 (ipso carbons of COCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.453, −1.894, −2.080; Anal. calcd for C80H82N5O18P3: C, 64.29; H, 5.53; N, 4.69. Found: C, 63.99; H, 5.23; N, 4.81.
:7 v/v) as the eluent to obtain pure 9c (0.101 g, 94%) as a colourless gum. 1H NMR (500 MHz, CDCl3) δ: 3.46–3.52 (m, 2H, H-2′′ and H-5A′), 3.55–3.58 (m, 2H, H-6B′′ and H-5B′), 3.64–3.66 (m, 1H, H-6A′′), 3.77–3.80 (m, 1H, H-5′′), 4.21–4.35 (m, 5H, H-4′ and 2 × PhCH2), 4.41–4.46 (m, 3H, H-3′, H-4′′ and 0.5 × PhCH2), 4.58–4.64 (m, 3H, 1.5 × PhCH2), 4.68–4.72 (m, 3H, 1.5 × PhCH2), 4.81–4.94 (m, 8H, H-3′′, and 3.5 × PhCH2), 5.16 (d, J = 3.5 Hz, 1H, H-1′′), 5.33 (ddd, J = 9.4, 8.55, 5.0, Hz, 1H, H-2′), 6.33 (d, 5.4 Hz, 1H, H-1′), 6.99–7.47 (m, 50H, 10 × C6H5), 7.83 (s, 1H, H-5); 13C NMR (125 MHz, CDCl3) δ: 68.7 (C-6′′), 69.2 (C-5′), 69.2, 69.4, 69.45, 69.5, 69.55, 69.6, 69.85, 69.88, 70.0, 70.02, 70.1 (31P coupled, PhCH2OP), 70.4 (C-5′′), 72.0, 73.5, 73.7 (3 × PhCH2OC), 74.5 (C-3′), 74.6 (31P coupled, C-4′′), 77.0 (31P coupled, C-2′′), 78.1 (C-3′′, 31P coupled), 78.8 (C-2′), 83.0 (C-4′), 90.1 (C-1′), 95.9 (C-1′′), 118.2 (C-5), 125.8 (Ar-C), 127.6, 127.7, 127.8, 127.86, 128.0, 128.1, 128.2, 128.3, 128.4, 128.45, 128.50, 128.53, 128.6, 128.7 (aromatic carbons), 130.3, 135.2, 135.23, 135.3, 135.7, 135.8, 135.87, 135.93, 136.2, 136.25, 137.4, 137.6, 138.1, 148.1; (31P coupled, ipso carbons of POCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.551, −1.932, −2.105; Anal. calcd for C82H82N3O18P3: C, 66.08; H, 5.55; N, 2.82. Found: C, 65.95; H, 5.72; N, 2.97.
:7 v/v) as the eluent to obtain pure 9d (0.104 g, 48%) as a colourless gum. 1H NMR (500 MHz, CDCl3) δ: 3.43–3.45 (m, 1H, H-5A′), 3.48–3.59 (m, 4H, H-2′′, H-5B′, H-6B′′and H-6A′′), 3.64–3.71–3.76 (m, 1H, H-5′′), 4.20–4.23 (m, 1H, 0.5 × PhCH2), 4.27–4.37 (m, 4H, H-4′ and 1.5 × PhCH2), 4.41–4.45 (m, 3H, H-3′, H-4′′ and 0.5 × PhCH2), 4.60–4.62 (m, 1H, 0.5 × PhCH2), 4.66–4.72 (m, 5H, 2.5 × PhCH2), 4.80–4.93 (m, 8H, H-3′′, and 3.5 × PhCH2), 5.15 (d, J = 2.9 Hz, 1H, H-1′′), 5.34–5.35 (m, 1H, H-2′), 6.30 (d, J = 5.0 Hz, 1H, H-1′), 6.99–7.19 (m, 45H, 9 × C6H5), 7.28 (d, J = 7.0 Hz, 1H, pyr-H), 7.72 (t, J = 7.0 Hz, 1H, pyr-H), 8.03 (d, J = 7.0 Hz, 1H, pyr-H), 8.44 (s, 1H, H-5), 8.52 (d, J = 4.0 Hz, 1H, pyr-H); 13C NMR (125 MHz, CDCl3) δ: 68.4 (C-6′′), 69.1 (C-5′), 69.13, 69.18, 69.4, 69.42, 69.5, 69.55, 69.8, 69.85, 69.87, 69.9, 70.0, 70.04 (31P coupled, PhCH2OP), 70.3 (C-5′′), 72.0, 73.38, 73.6 (3 × PhCH2OC), 74.2 (C-3′), 74.5 (31P coupled, C-4′′), 77.1 (31P coupled, C-2′′), 78.1 (C-3′′, 31P coupled), 78.7 (C-2′), 83.1 (C-4′), 90.4 (C-1′), 95.7 (C-1′′), 120.6 (C-5), 123.0 (Ar-C), 127.6, 127.7, 127.8, 127.9, 128.0, 128.1, 128.2, 128.3, 128.4, 128.43, 128.49, 128.5, 128.55 (aromatic carbons), 135.2, 135.25, 135.28, 135.30, 136.19, 136.25, 137.37, 137.60, 138.11; (31P coupled, ipso carbons of POCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.60, −1.95, −2.13; Anal. calcd for C81H81N4O18P3: C, 65.23; H, 5.47; N, 3.76. Found: C, 65.01; H, 5.21; N, 3.92.
:3 v/v) as the eluent to obtain 9e (0.203 g, 93%) as a colourless gum. 1H NMR (500 MHz, CDCl3) δ: 3.53–3.55 (m, 1H, H-5A′), 3.61–3.73 (m, 4H, H-2′′, H-5B′, H-6A′′ and H-6B′′), 3.88 (bs, 1H, H-5′′), 4.33–4.35 (m, 1H, 0.5 × PhCH2), 4.42–4.48 (m, 4H, H-4′ and 1.5 × PhCH2), 4.53–4.58 (m, 3H, H-3′, H-4′′ and 0.5 × PhCH2), 4.72–4.81 (m, 6H, 3 × PhCH2), 4.96–5.06 (m, 8H, H-3′′, 3.5 × PhCH2), 5.27 (bs, 1H, H-1′′), 5.46 (bs, 1H, H-2′), 6.42 (bs, 1H, H-1′), 7.13–7.40 (m, 48H, Ar-H), 8.21 (s, 1H, H-5), 8.30 (bs, 1H, Ar-H); 13C NMR (125 MHz, CDCl3) δ: 68.4 (C-6′′), 69.0 (C-5′), 69.1, 69.2, 69.4, 69.42, 69.5, 69.6, 69.8, 69.85, 69.89, 70.0, 70.05 (31P coupled, PhCH2OP), 70.2 (C-5′′), 71.9, 73.4, 73.6 (3 × PhCH2OC), 74.2 (C-3′), 74.4 (31P coupled, C-4′′), 77.1 (31P coupled, C-2′′), 78.0 (C-3′′, 31P coupled), 78.6 (C-2′), 83.0 (C-4′), 90.2 (C-1′), 95.7 (C-1′′), 115.7 (C-5), 121.6, 121.7, 124.5, 124.6, 127.6, 127.9, 128.0, 128.1, 128.5, 128.6 (aromatic carbons), 129.4, 129.5, 135.2, 135.22, 135.3, 135.7, 135.8, 135.84, 135.89, 136.2, 136.22, 137.3, 137.6, 138.1, 141.48, 141.5, 158.3, 160.3 (31P coupled, ipso carbons of POCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.632, −1.930, −2.134; 19F NMR (470.68 MHz, CDCl3) δ: −114.22; Anal. calcd for C82H81FN3O18P3: C, 65.29; H, 5.41; N, 2.79. Found: C, 65.43; H, 5.59; N, 2.67.
:3 v/v) as the eluent to obtain 9f (0.188 g, 86%) as a colourless gum; 1H NMR (500 MHz, CDCl3) δ: 3.58 (dd, J = 10.85, 2.45 Hz, 1H, H-5A′), 3.62 (dd, J = 9.65, 3.65 Hz, 1H, H-2′′), 3.66–3.69 (m, 2H, H-6A′′, H-5B′), 3.78 (dd, J = 10.8, 1.5 Hz, 1H, H-6B′′) 3.90 (ddd, J = 10.8, 9.2, 4.2 Hz, 1H, H-5′′), 4.33–4.38 (m, 2H, 1 × PhCH2), 4.41–4.44 (m, 2H, 1 × PhCH2), 4.46–4.47 (m, 1H, H-4′), 4.52–4.56 (m, 3H, H-3′, H-4′′ and 0.5 × PhCH2), 4.70–4.75 (m, 3H, 1.5 × PhCH2), 4.80–4.84 (m, 3H, 1.5 × PhCH2), 4.92–5.06 (m, 8H, H-3′′, 3.5 × PhCH2), 5.28 (d, J = 3.6 Hz, 1H, H-1′′), 5.43 (ddd, J = 8.85, 5.2, 4.65 Hz, 1H, H-2′), 6.44 (d, 5.6 Hz, 1H, H-1′), 7.09–7.39 (m, 49H, Ar-H), 7.90 (s, 1H, H-5); 13C NMR (125 MHz, CDCl3) δ: 68.7 (C-6′′), 69.1 (C-5′), 69.4, 69.5, 69.8, 69.6, 69.7, 69.87, 69.9, 69.94, 70.07, 70.1 (31P coupled, PhCH2OP), 72.0 (C-5′′), 72.1, 73.5, 73.7 (3 × PhCH2OC), 74.5 (C-3′), 74.53 (31P coupled, C-4′′), 77.1 (31P coupled, C-2′′), 78.1 (C-3′′, 31P coupled), 78.9 (C-2′), 83.1 (C-4′), 90.3 (C-1′), 95.9 (C-1′′), 112.7, 114.8 (Ar-C), 118.5 (C-5), 121.4 (Ar-C), 127.6, 127.7, 127.8, 127.83, 127.9, 128.0, 128.07, 128.14, 128.31, 128.34, 128.43, 128.48, 128.5, 128.51, 128.69 (aromatic carbons), 130.2, 130.27, 136.2, 136.23, 137.31, 137.61, 138.1, 148.1, 162.3, 164.2 (31P coupled, ipso carbons of POCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.515, −1.920, −2.064; 19F NMR (470.68 MHz, CDCl3) δ: −112.766; Anal. calcd for C82H81FN3O18P3: C, 65.29; H, 5.41; N, 2.79. Found: C, 65.50; H, 5.21; N, 2.66.
:3 v/v) as the eluent to obtain 9i (0.193 g, 88%) as a colourless gum; 1H NMR (500 MHz, CDCl3) δ: 3.49–3.56 (m, 5H, 6A′′, H-6B′′, H-5B′, H-2′′ and H-5A′), 3.77 (bs, 4H, 1 × OCH3 and H-5′′), 4.21–4.35 (m, 5H, H-4′ and 2 × PhCH2), 4.40–4.45 (m, 3H, H-3′, H-4′′ and 0.5 × PhCH2), 4.58–4.72 (m, 6H, and 3 × PhCH2), 4.84–4.93 (m, 8H, H-3′′ and 3.5 × PhCH2), 5.16 (bs, 1H, H-1′′), 5.32 (bs, 1H, H-2′), 6.33 (d, J = 3.2 Hz, 1H, H-1′), 6.80 (d, J = 7.5 Hz, 2H, Ar-H), 7.00–7.28 (m, 45H, 9 × C6H5), 6.38 (d, J = 7.5 Hz, 2H, Ar-H), 7.75 (s, 1H, H-5); 13C NMR (125 MHz, CDCl3) δ: 55.4 (OCH3), 68.6 (C-6′′), 69.16 (C-5′), 69.2, 69.4, 69.44, 69.5, 69.58, 69.85, 69.9, 70.07, (31P coupled PhCH2OP), 70.3 (C-5′′), 72.0, 73.4, 73.6 (3 × PhCH2OC), 74.4 (C-3′ and C-4′′, 31P coupled), 77.0 (C-2′, 31P coupled), 78.0 (C-2′′), 78.6 (C-3′′, 31P coupled), 83.0 (C-4′), 90.3 (C-1′, 31P coupled), 95.9 (C-1′′), 114.2, 117.5 (C-5), 127.1, 127.2, 127.6, 127.7, 127.8, 127.82, 127.85, 128.0, 128.03, 128.07, 128.1, 128.2, 128.3, 128.4, 128.45, 128.5, 128.52, 128.6, 128.64 (aromatic carbons), 135.1, 135.15, 135.2, 135.65, 135.70, 135.8, 135.84, 136.1, 136.16, 137.4, 137.6, (31P coupled, ipso carbons of POCH2Ph), 138.0 (ipso carbons of COCH2Ph); 31P NMR (202.4 MHz, CDCl3) δ: −1.576, −1.941, −2.118; Anal. calcd for C83H84N3O19P3: C, 65.56; H, 5.57; N, 2.76. Found: C, 65.66; H, 5.77; N, 2.95.
:3 v/v) as the eluent to obtain pure 9j (0.19 g, 87%) as a colourless gum;. 1H NMR (500 MHz, CDCl3) δ: 3.57–3.64 (m, 2H, H-2′′ and H-5A′), 3.68–3.70 (m, 2H, H-5B′ and H-6A′′), 3.76–3.78 (m, 1H, H-6B′′), 3.90 (bs, 1H, H-5′′), 4.33–4.44 (m, 4H, 2 × PhCH2), 4.49–4.58 (m, 4H, H-4′, H-4′′ and 1 × PhCH2), 4.72–4.86 (m, 6H, H-3′ and 2.5 × PhCH2), 4.93–5.07 (m, 8H, H-3′′, 3.5 × PhCH2), 5.29 (bs, 1H, H-1′′), 5.47 (bs, 1H, H-2′), 6.48 (d, 5.0 Hz, 1H, H-1′), 7.13–7.41 (m, 46H, Ar-H), 7.51–7.59 (m, 2H, Ar-H), 8.00 (s, 1H, H-5), 8.05 (d, J = 7.4 Hz, 1H, Ar-H), 10.35 (s, 1H, CHO); 13C NMR (125 MHz, CDCl3) δ: 68.6 (C-6′′), 68.66 (C-5′), 69.2, 69.22, 69.3, 69.4, 69.45, 69.6, 69.61, 69.87, 69.9, 69.94, 70.07, 70.1 (31P coupled, PhCH2OP), 70.4 (C-5′′), 72.0, 73.5, 73.7 (3 × PhCH2OC), 74.3 (C-3′), 74.5 (31P coupled, C-4′′), 77.3 (31P coupled, C-2′′), 78.0 (C-3′′, 31P coupled), 78.9 (C-2′), 83.2 (C-4′), 90.5 (C-1′), 95.8 (C-1′′), 121.7 (C-5), 127.7, 127.71, 127.73, 127.8, 127.83, 127.86, 128.0, 128.1, 128.14, 128.19, 128.35, 128.38, 128.5, 128.54, 128.59, 128.69 (aromatic carbons), 129.9, 132.8, 134.1, 136.2, 137.2, 137.6, 138.1, 145.6 (31P coupled, ipso carbons of POCH2Ph), 192.21 (CHO); 31P NMR (202.4 MHz, CDCl3) δ: −1.32, −1.89, −2.07; Anal. calcd for C83H82N3O19P3: C, 65.65; H, 5.44; N, 2.77. Found: C, 65.41; H, 5.73; N, 2.90.
7. General procedure for the synthesis of triazolophostins
To a solution of protected triazolophostins 9a–k (80–200 mg, 0.06–0.13 mmol) and cyclohexene (2 mL) in a mixture of methanol and water (9:1 v/v, 5 mL), Pd(OH)2 (20% on carbon, 50 mg) was added and the mixture was stirred at 80 °C for 4 h. The reaction mixture was then cooled, filtered through a membrane filter by washing successively with methanol and water. The combined filtrate was evaporated under reduced pressure. The crude product thus obtained was purified by ion-exchange column chromatography on a Q-Sepharose matrix using 0→1.0 M triethylammonium bicarbonate (TEAB) as the eluent to obtain pure triazolophostins 10a–k.
8. Biological assay
Ca2+ release from the intracellular stores of saponin-permeabilized DT40 cells expressing only type 1 IP3R was measured using a low-affinity Ca2+ indicator (Mag-fluo-4) as described previously.23 Briefly, the ER was loaded with Ca2+ to the steady state for ∼120 s by addition of 1.5 mM MgATP in medium containing p-trifluoromethoxyphenylhydrazone (FCCP) to inhibit mitochondria. IP3, AdA or triazolophostins were added with cyclopiazonic acid (10 μM) to inhibit further Ca2+ uptake. Ca2+ release was assessed 10–20 s after addition of the analog, and expressed as a fraction of the ATP-dependent Ca2+ uptake.Acknowledgements
A. M. V. thanks the University Grants Commission (UGC) India for a Senior Research Fellowship (SRF) during this work. K. M. S. thanks the Department of Science and Technology (DST) India for Swarnajayanti Fellowship, Ramanujan Fellowship and for financial support. C. W. T. and V. K. were supported by the Wellcome Trust, Biotechnology and Biological Sciences Research Council, and the German Academic Exchange Service (V. K.).Notes and references
- (a) M. J. Berridge, Cell Signalling Biol., 2012 DOI:10.1042/csb0001002; (b) M. J. Berridge, P. Lipp and M. D. Bootman, Nat. Rev. Mol. Cell Biol., 2000, 1, 11 CrossRef CAS PubMed; (c) A. H. Guse, Curr. Mol. Med., 2004, 4, 239 CrossRef CAS.
- (a) M. Nebel, A. P. Schwoerer, D. Warszta, C. C. Siebrands, A.-C. Limbrock, J. M. Swarbrick, R. Fliegert, K. Weber, S. Bruhn, M. Hohenegger, A. Geisler, L. Herich, S. Schlegel, L. Carrier, T. Eschenhagen, B. V. L. Potter, H. Ehmke and A. H. Guse, J. Biol. Chem., 2013, 288, 16017 CrossRef CAS PubMed; (b) A. H. Guse, Sci. Signaling, 2012, 5, 18 CrossRef PubMed; (c) A. Galione and A. White, Trends Cell Biol., 1994, 4, 431 CrossRef CAS; (d) S.-Y. Hua, T. Tokimasa, S. Takasawa, S. Furuya, M. Nohmi, H. Okamoto and K. Kuba, Neuron, 1994, 12, 1073 CrossRef CAS; (e) H. C. Lee, Physiol. Rev., 1997, 77, 1133 CAS; (f) V. Lukyanenko, I. Györke, T. F. Wiesner and S. Györke, Circ. Res., 2001, 89, 614 CrossRef CAS; (g) H. C. Lee, Curr. Biol., 2003, 13, R186 CrossRef CAS; (h) S. Patel, G. C. Churchill and A. Galione, Trends Biochem. Sci., 2001, 26, 482 CrossRef CAS; (i) M.-D. Seo, S. Velamakanni, N. Ishiyama, P. B. Stathopulos, A. M. Rossi, S. A. Khan, P. Dale, C. Li, J. B. Ames, M. Ikura and C. W. Taylor, Nature, 2012, 483, 108 CrossRef CAS PubMed.
- (a) C. W. Taylor and S. C. Tovey, Cold Spring Harbor Perspect. Biol., 2010, 2, a004010 CAS; (b) C. W. Taylor, A. A. Genazzani and S. A. Morris, Cell Calcium, 1999, 26, 237 CrossRef CAS PubMed.
- (a) K. Mikoshiba, Curr. Opin. Neurobiol., 1997, 7, 339 CrossRef CAS; (b) C. W. Taylor and S. C. Tovey, Cold Spring Harbor Perspect. Biol., 2010, 2, a004010 CAS; (c) H. Saleem, S. C. Tovey, T. Rahman, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2013, 8, e54877 CAS; (d) J. K. Foskett, C. White, K. H. Cheung and D. O. Mak, Physiol. Rev., 2007, 87, 593 CrossRef CAS PubMed; (e) A. M. Rossi, A. M. Reley, S. C. Tovey, T. Rahman, O. Dellis, E. J. A. Taylor, V. G. Veresov, B. V. L. Potter and C. W. Taylor, Nat. Chem. Biol., 2009, 5, 631 CrossRef CAS PubMed.
- T. Wundenberg and G. W. Mayr, Biol. Chem., 2012, 393, 979 CrossRef CAS PubMed.
- (a) B. V. L. Potter and D. Lampe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1933 CrossRef CAS PubMed; (b) R. F. Irvine and M. J. Schell, Nat. Rev. Mol. Cell Biol., 2001, 2, 327 CrossRef CAS PubMed; (c) M. D. Best, H. Zhang and G. D. Prestwich, Nat. Prod. Rep., 2010, 27, 1403 RSC.
- (a) S. Capolicchio, H. Wang, D. T. Thakor, S. B. Shears and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 9508 CrossRef CAS PubMed; (b) M. Wu, L. S. Chong, S. Capolicchio, H. J. Jessen, A. C. Resnick and D. Fiedler, Angew. Chem., Int. Ed., 2014, 53, 7192 CrossRef CAS PubMed; (c) S. Capolicchio, D. T. Thakor, A. Linden and H. J. Jessen, Angew. Chem., Int. Ed., 2013, 52, 6912 CrossRef CAS PubMed; (d) A. M. Marmelstein, L. M. Yates, J. H. Conway and D. Fiedler, J. Am. Chem. Soc., 2014, 136, 108 CrossRef CAS PubMed; (e) M. Wu, B. E. Dul, A. J. Trevisan and D. Fiedler, Chem. Sci., 2013, 4, 405 RSC.
- M. Takahashi, T. Kagasaki, T. Hosoya and S. Takahashi, J. Antibiot., 1993, 46, 1643 CrossRef CAS.
- (a) A. M. Rossi, A. M. Riley, B. V. L. Potter and C. W. Taylor, Curr. Top. Membr., 2010, 66, 209 CAS; (b) H. Saleem, S. C. Tovey, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2013, 8, e58027 CAS; (c) C. N. Borissow, S. J. Black, M. Paul, S. C. Tovey, S. G. Dedos, C. W. Taylor and B. V. L. Potter, Org. Biomol. Chem., 2005, 3, 245 RSC; (d) J. S. Marchant, M. D. Beecroft, A. M. Riley, D. J. Jenkins, R. D. Marwood, C. W. Taylor and B. V. L. Potter, Biochemistry, 1997, 36, 12780 CrossRef CAS PubMed; (e) S. Shuto, K. Tatani, Y. Ueno and A. Matsuda, J. Org. Chem., 1998, 63, 8815 CrossRef CAS; (f) R. D. Marwood, A. M. Riley, V. Correa, C. W. Taylor and B. V. L. Potter, Bioorg. Med. Chem. Lett., 1999, 9, 453 CrossRef CAS; (g) H. Hotoda, K. Murayama, S. Miyamoto, Y. Iwata, M. Takahashi, Y. Kawase, K. Tanzawa and M. Kaneko, Biochemistry, 1999, 38, 9234 CrossRef CAS PubMed; (h) M. Kashiwayanagi, K. Tatani, S. Shuto and A. Matsuda, Eur. J. Neurosci., 2000, 12, 606 CrossRef CAS PubMed; (i) S. Shuto, M. Terauchi, Y. Yahiro, H. Abe, S. Ichikawa and A. Matsuda, Tetrahedron Lett., 2000, 41, 4151 CrossRef CAS; (j) F. Chretien, N. Moitessier, F. Roussel, J.-P. Mauger and Y. Chapleur, Curr. Org. Chem., 2000, 4, 513 CrossRef CAS; (k) H. Abe, S. Shuto and A. Matsuda, J. Org. Chem., 2000, 65, 4315 CrossRef CAS PubMed; (l) T. Mochizuki, Y. Kondo, H. Abe, S. C. Tovey, S. G. Dedos, C. W. Taylor, M. Paul, B. V. L. Potter, A. Matsuda and S. Shuto, J. Med. Chem., 2006, 49, 5750 CrossRef CAS PubMed.
- I. Bosanac, J.-R. Alattia, T. K. Mal, J. Chan, S. Talarico, F. K. Tong, K. I. Tong, F. Yoshikawa, T. Furuichi, M. Iwai, T. Michikawa, K. Mikoshiba and M. Ikura, Nature, 2002, 420, 696 CrossRef CAS PubMed.
- V. Correa, A. M. Riley, S. Shuto, G. Horne, E. P. Nerou, R. D. Marwood, B. V. L. Potter and C. W. Taylor, Mol. Pharmacol., 2001, 59, 1206 CAS.
- (a) K. M. Sureshan, A. M. Riley, M. P. Thomas, S. C. Tovey, C. W. Taylor and B. V. L. Potter, J. Med. Chem., 2012, 55, 1706 CrossRef CAS PubMed; (b) K. M. Sureshan, M. Trusselle, S. C. Tovey, C. W. Taylor and B. V. L. Potter, J. Org. Chem., 2008, 73, 1682 CrossRef CAS PubMed; (c) A. M. Rossi, K. M. Sureshan, A. M. Riley, B. V. L. Potter and C. W. Taylor, Br. J. Pharmacol., 2010, 161, 1070 CrossRef CAS PubMed.
- (a) R. D. Marwood, S. Shuto, D. J. Jenkins and B. V. L. Potter, Chem. Commun., 2000, 219 RSC; (b) T. Mochizuki, Y. Kondo, H. Abe, C. W. Taylor, B. V. L. Potter, A. Matsuda and S. Shuto, Org. Lett., 2006, 8, 1455 CrossRef CAS PubMed.
- S. Shuto, G. Horne, R. D. Marwood and B. V. L. Potter, Chem. – Eur. J., 2001, 7, 4937 CrossRef CAS.
- (a) A. A. Koshkin, J. Fensholdt, H. M. Pfundheller and C. Lomholt, J. Org. Chem., 2001, 66, 8504 CrossRef CAS PubMed; (b) M. J. Robins, R. Zou, Z. Guo and S. F. Wnuk, J. Org. Chem., 1996, 61, 9207 CrossRef CAS; (c) M. Zhong and M. J. Robins, Tetrahedron Lett., 2003, 44, 9327 CrossRef CAS PubMed.
- Y. Hayakawa and M. Kataoka, J. Am. Chem. Soc., 1998, 120, 12395 CrossRef CAS.
- (a) P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442 CrossRef CAS PubMed; (b) G. A. Burley, J. Gierlich, M. R. Mofid, H. Nir, S. Tal, Y. Eichen and T. Carell, J. Am. Chem. Soc., 2006, 128, 1398 CrossRef CAS PubMed; (c) J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639 CrossRef CAS PubMed; (d) F. Seela and V. R. Sirivolu, Chem. Biodiversity, 2006, 3, 509 CrossRef CAS PubMed; (e) B. M. Swarts and Z. Guo, Chem. Sci., 2011, 2, 2342 RSC.
- (a) R. D. Marwood, D. J. Jenkins, V. Correa, C. W. Taylor and B. V. L. Potter, J. Med. Chem., 2000, 43, 4278 CrossRef CAS PubMed; (b) M. de Kort, V. Correa, A. R. P. M. Valentijn, G. A. van der Marel, B. V. L. Potter, C. W. Taylor and J. H. van Boom, J. Med. Chem., 2000, 43, 3295 CrossRef CAS PubMed.
- A. Vidyasagar, A. Pathigoolla and K. M. Sureshan, Org. Biomol. Chem., 2013, 11, 5443 CAS.
- A. Stimac and J. Kobe, Carbohydr. Res., 1992, 232, 359 CrossRef CAS.
- (a) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- J. M. Swarbrick, R. Graeff, C. Garnham, M. P. Thomas, A. Galione and B. V. L. Potter, Chem. Commun., 2014, 50, 2458 RSC.
- H. Saleem, S. C. Tovey, T. F. Molinski and C. W. Taylor, Br. J. Pharmacol., 2014, 171, 3298 CrossRef CAS PubMed.
- H. J. Rosenberg, A. M. Riley, A. J. Laude, C. W. Taylor and B. V. L. Potter, J. Med. Chem., 2003, 46, 4860 CrossRef CAS PubMed.
- K. M. Sureshan, M. Trusselle, S. C. Tovey, C. W. Taylor and B. V. L. Potter, Chem. Commun., 2006, 2015 RSC.
- M. de Kort, A. D. Regenbogen, H. S. Overkleeft, R. A. John Challiss, Y. Iwata, S. Miyamoto, G. A. van der Marela and J. H. van Boom, Tetrahedron, 2000, 56, 5915 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and spectral data for all new compounds, crystal data for disaccharide 4 and details of the docking study. CCDC 1022279. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00440c |
| This journal is © The Royal Society of Chemistry 2015 |