
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalyzed asymmetric addition of arylboronic acids to cyclic N-sulfonyl ketimines towards the synthesis of α,α-diaryl-α-amino acid derivatives†
Ryosuke
Takechi
and
Takahiro
Nishimura
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8502, Japan. E-mail: tnishi@kuchem.kyoto-u.ac.jp
First published on 18th March 2015
Abstract
Rhodium/chiral diene complex-catalyzed asymmetric addition of arylboronic acids to cyclic ketimines having an ester group proceeded to give the corresponding α-amino acid derivatives in high yields with high enantioselectivity. The cyclic amino acid derivative was transformed into a linear α,α-diaryl-substituted α-N-methylamino acid ester.
Introduction
Transition metal-catalyzed addition reaction of organometallic reagents to imines is one of the most powerful methods for the selective formation of a carbon–carbon bond, which provides an easy access to α-chiral amines.1 Although many successful reports on the asymmetric alkylation or arylation to aldimines have been reported,2 the addition to ketimines giving chiral α-tert-amines is still a challenging objective in organic chemistry.1d,3 Pioneering studies of the transition metal-catalyzed asymmetric addition of organometallic reagents to ketimines have been attained by Kanai and Shibasaki (allylboronates, Cu),3a Charette (dialkylzinc, Cu),3b and Snapper and Hoveyda (dialkylzinc, Zr).3c The rhodium-catalyzed asymmetric addition of arylboron reagents to N-sulfonyl ketimines reported by Hayashi and co-workers is an another approach leading to chiral α-tert-amines.4,5 On the other hand, cyclic N-sulfonyl and N-acyl ketimines are good substrates for the asymmetric addition to give chiral α-tert-amines with high enantioselectivity, because the geometry (E or Z) of the imine is fixed, and thus the enantioface of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond is selectively differentiated without an influence of the isomerization.6,7 For example, the rhodium-catalyzed asymmetric arylation of diaryl-substituted cyclic ketimines has been reported by us in 20126a and Xu in 2013.7a Asymmetric alkenylation and allylation were also reported by Lam and co-workers.8 Recently, Pd-catalyzed asymmetric addition of arylboronic acids to the same types of cyclic N-sulfonyl ketimines was reported.9,10
N bond is selectively differentiated without an influence of the isomerization.6,7 For example, the rhodium-catalyzed asymmetric arylation of diaryl-substituted cyclic ketimines has been reported by us in 20126a and Xu in 2013.7a Asymmetric alkenylation and allylation were also reported by Lam and co-workers.8 Recently, Pd-catalyzed asymmetric addition of arylboronic acids to the same types of cyclic N-sulfonyl ketimines was reported.9,10
α,α-Disubstituted α-amino acids have been focused on as non-proteinogenic amino acids in medicinal chemistry, and the stereoselective synthesis giving α,α-dialkyl and α,α-alkyl-aryl substituted α-amino acid derivatives has been developed.11 We were interested in the development of general synthetic methods for chiral α,α-diaryl-substituted amino acid derivatives in an enantioselective manner. Although it was reported that rhodium can catalyze the asymmetric addition of arylboronic acids to five-membered cyclic N-sulfonyl ketimines substituted with an ester group to give α,α-diaryl-substituted amino acid derivatives,7a the transformation of the cyclic structure to the linear α-amino acid derivatives has not been achieved probably due to the difficulty of cleavage of the cyclic structure. Our approach leading to chiral α,α-diaryl-α-amino acid derivatives is the asymmetric arylation of cyclic ketimines giving benzosulfamidates and the subsequent breaking the cyclic structure (Scheme 1). Here we report that the rhodium/chiral diene complex can catalyze the asymmetric addition of arylboronic acids to cyclic aromatic ketimines having an ester group to give the corresponding α-amino acid derivatives in high yields with high enantioselectivity.12 The cyclic amino acid derivative was successfully transformed into a linear α,α-diaryl-α-N-methylamino acid ester by reductive cleavage of a carbon–oxygen bond constituting the cyclic structure.
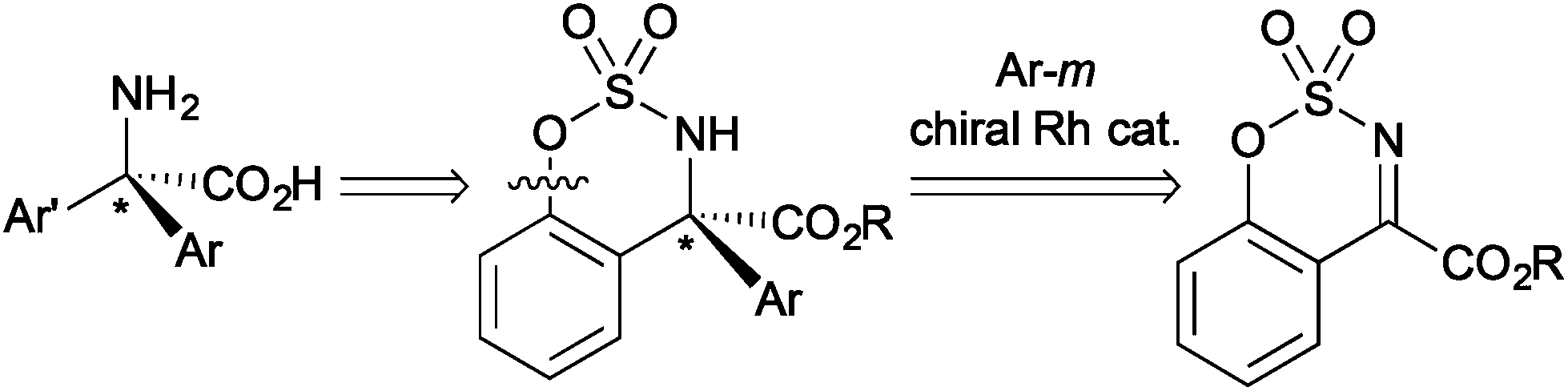 | ||
| Scheme 1 Synthetic strategy for diaryl-substituted α-amino acids. | ||
Results and discussion
Our initial studies were focused on the addition of phenylboronic acid to cyclic ketimine 1a in the presence of rhodium complexes for the catalytic synthesis of sulfamidate 3aa substituted with an ester group (Table 1). Treatment of 1a with phenylboronic acid (2a, 3 equiv.) in the presence of [Rh(OH)(cod)]2 (5 mol% of Rh) in 1,4-dioxane at 60 °C for 12 h gave 3aa in 99% yield (entry 1). The use of [RhCl(cod)]2 as a catalyst in the presence of K3PO4 was found to decrease the yield of 3aa (67% yield, entry 2). We have recently developed chiral diene ligands base on a tetrafluorobenzobarrelene (tfb) framework,13 which display high coordination ability to Rh(I) giving stable hydroxorhodium complexes. A ferrocenyl-substituted tfb-ligand (Fc-tfb*) displayed high enantioselectivity (93% ee), but the yield of 3aa was low (34% yield, entry 3). Higher catalytic activity was observed with phenyl-substituted tfb (Ph-tfb*) and benzyl-substituted tfb (Bn-tfb*) giving 3aa in 73% and 99% yield, respectively (entries 4 and 5), with high enantioselectivity (96% ee). The use of ligand L1,14 which is effective in catalyzing the asymmetric addition of arylboronic acids to aldimines, gave 3aa in 20% yield with 96% ee (entry 6). A hydroxorhodium complex coordinated with binap [Rh(OH)((S)-binap)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 efficiently catalyzed the present reaction to give a 92% yield of 3aa, but the enantioselectivity was modest (61% ee; entry 7). The reaction in the presence of [Rh(OH)((S,S)-Bn-tfb*)]2 using 2 equiv. of phenylboronic acid (2a) proceeded as well to give 3aa in 99% yield with 96% ee within 3 h (entry 8).
15 efficiently catalyzed the present reaction to give a 92% yield of 3aa, but the enantioselectivity was modest (61% ee; entry 7). The reaction in the presence of [Rh(OH)((S,S)-Bn-tfb*)]2 using 2 equiv. of phenylboronic acid (2a) proceeded as well to give 3aa in 99% yield with 96% ee within 3 h (entry 8). | (1) |
|
|
|||
|---|---|---|---|
| Entry | Rh catalyst | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.10 mmol), PhB(OH)2 (0.30 mmol), and Rh catalyst (3 mol% of Rh) in 1,4-dioxane (0.4 mL) at 60 °C for 12 h. b Isolated yields. c Determined by chiral HPLC analysis. d 5 mol% of Rh was used. e K3PO4 (1 equiv.). f Performed with 2 equiv. of PhB(OH)2 (2a, 0.20 mmol) for 3 h. | |||
| 1 | [Rh(OH)(cod)]2d |
99 | — |
| 2 | [RhCl(cod)]2/K3PO4d,e |
67 | — |
| 3 | [Rh(OH)((S,S)-Fc-tfb*)]2 | 34 | 93 |
| 4 | [Rh(OH)((R,R)-Ph-tfb*)]2 | 73 | 96 |
| 5 | [Rh(OH)((S,S)-Bn-tfb*)]2 | 99 | 96 |
| 6 | [RhCl((R)-L1)]2/K3PO4d |
20 | 96 |
| 7 | [Rh(OH)((S)-binap)]2 | 92 | 61 |
| 8f | [Rh(OH)((S,S)-Bn-tfb*)]2 | 99 | 96 |

The addition of p-tolylboronic acid (2b) to ketimine 1 having ethyl (1a), benzyl (1b), and tert-butyl ester (1c) proceeded to give the corresponding sulfamidates 3ab–3cb in high yields with high enantioselectivity (92–94% ee, eqn (1)). In the reaction of tert-butyl ester 1c, the use of K3PO4 improved the yield of 3cb to avoid the formation of a small amount of an arylated carboxylic acid observed in the absence of the base. We selected the tert-butyl esters as suitable substrates, which are tolerant to a basic condition and are readily transformed into carboxylic acids.
The results obtained for the reaction of ketimine 1 having a tert-butyl ester group with a variety of arylboronic acids 2 are summarized in Table 2. Aryl groups having electron-donating and -withdrawing substituents (2a–2j) were successfully introduced into ketimine 1c with 86–96% ee (entries 1–10). The addition of p-tolylboronic acid (2b) to ketimines 1d–1h possessing methyl, methoxy, and methylenedioxy proceeded to give the corresponding sulfamidates 3db–3hb with high enantioselectivity (93–97% ee, entries 11–15). A slight decrease of the enantioselectivity was observed in the addition to ketimines 1i and 1j substituted with electron-withdrawing groups (F, Cl) at the 6-position (entries 16 and 17).16
|
|
||||
|---|---|---|---|---|
| Entry | X | Ar | Yieldb (%) | eec (%) |
| a Reaction conditions: 1 (0.20 mmol), ArB(OH)2 (0.40 mmol), [Rh(OH)((S,S)-Bn-tfb*)]2 (3 mol% of Rh) and K3PO4 (1 equiv.) in 1,4-dioxane (0.8 mL) at 60 °C. Reaction time: 3 h (entries 1 and 2), 6 h (entries 6, 11–17), 12 h (entries 3–5, 7–10). b Isolated yields. c Determined by chiral HPLC analysis. d Performed with 4 equiv. of 2b and 2 equiv. of K3PO4. | ||||
| 1 | H (1c) | Ph (2a) | 98 (3ca) | 96 |
| 2 | H (1c) | 4-MeC6H4 (2b) | 99 (3cb) | 94 |
| 3 | H (1c) | 4-MeOC6H4 (2c) | 96 (3cc) | 86 |
| 4 | H (1c) | 3,4-(OCH2O)C6H3 (2d) | 93 (3cd) | 94 |
| 5 | H (1c) | 3,4,5-(MeO)3C6H2 (2e) | 94 (3ce) | 86 |
| 6 | H (1c) | 4-FC6H4 (2f) | 95 (3cf) | 94 |
| 7 | H (1c) | 4-ClC6H4 (2g) | 95 (3cg) | 95 |
| 8 | H (1c) | 4-BrC6H4 (2h) | 97 (3ch) | 94 |
| 9 | H (1c) | 4-CF3C6H4 (2i) | 96 (3ci) | 94 |
| 10 | H (1c) | 2-Naphthyl (2j) | 96 (3cj) | 95 |
| 11 | 8-Me (1d) | 4-MeC6H4 (2b) | 94 (3db) | 94 |
| 12 | 7-Me (1e) | 4-MeC6H4 (2b) | 93 (3eb) | 96 |
| 13 | 6-Me (1f) | 4-MeC6H4 (2b) | 95 (3fb) | 93 |
| 14 | 6-MeO (1g) | 4-MeC6H4 (2b) | 99 (3gb) | 93 |
| 15d | 5,6-(OCH2O) (1h) | 4-MeC6H4 (2b) | 84 (3hb) | 97 |
| 16 | 6-F (1i) | 4-MeC6H4 (2b) | 95 (3ib) | 83 |
| 17 | 6-Cl (1j) | 4-MeC6H4 (2b) | 99 (3jb) | 84 |

The reductive cleavage of the O–S bond of benzosulfamidates by LiAlH4 has been reported, where 2-(aminomethyl)phenols are formed.6a,9b On the other hand, nickel-catalyzed cross-coupling of benzosulfamidates with phenylboronic acid was reported to give ortho-phenylated product involving the cleavage of the C–O bond.8a,17 A nickel catalysis using alkylmagnesium reagents was also reported by Du Bois and co-workers,18 but the catalytic systems are not applicable to the benzosulfamidates having tetra-substituted α-carbon of the amino group. After screening and modifying the reported catalytic conditions18 to cleave a C–O bond of the benzosulfamidate formed in the present arylation, it was found that the reductive cleavage of the C–O bond proceeds using diethylzinc as a reducing reagent to give a linear α,α-diaryl-α-amino acid derivative (eqn (2)). Thus, introduction of a methyl group on the nitrogen atom of 3cb (94% ee) followed by reductive cleavage of the C–O bond by use of diethylzinc in the presence of NiCl2(dippe) in N,N-dimethylacetamide (DMA) at 100 °C for 24 h gave α,α-diaryl-α-N-methylamino acid ester 4 in 70% yield in two steps without loss of the enantiomeric purity.
 | (2) |
Conclusions
In summary, we have developed the asymmetric synthesis of α,α-diaryl-α-amino acid derivatives in the enantioselective addition of arylboronic acids to cyclic aromatic ketimines having an ester group catalyzed by a hydroxorhodium/chiral diene complex. The cyclic amino acid derivative was successfully transformed into a linear α,α-diaryl-α-N-methylamino acid ester by reductive cleavage of a carbon–oxygen bond constituting the cyclic structure.Experimental
General remarks
All anaerobic and moisture-sensitive manipulations were carried out with standard Schlenk techniques under predried nitrogen. NMR spectra were recorded on JEOL JNM ECA-600 spectrometer (600 MHz for 1H, 150 MHz for 13C). Chemical shifts are reported in δ (ppm) referenced to the residual peak of CDCl3 (δ 7.26) for 1H NMR, and CDCl3 (δ 77.00) for 13C NMR. The following abbreviations are used; s: singlet, d: doublet, t: triplet, m: multiplet. Optical rotations were measured on a JASCO P-2200 polarimeter. High-resolution mass spectra were obtained with a Bruker micrOTOF spectrometer. Flash column chromatography was performed with Silica Gel 60 N (spherical, neutral) (Cica-Reagent). Preparative thin-layer chromatography was performed with Silica Gel 60 PF254 (Merck). Alumina (activated 200) for column chromatography was purchased from Nacalai Tesque.1,4-Dioxane was purified by passing through a neutral alumina column under N2. Rhodium complexes, [RhCl(cod)]219 (CAS: 12092-47-6), [Rh(OH)(cod)]220 (CAS: 73468-85-6), [Rh(OH)((S)-binap)]215 (CAS: 805323-12-0 for (R)-complex), [Rh(OH)((S,S)-Bn-tfb*)]213b (CAS: 1204591-09-2), [Rh(OH)((R,R)-Ph-tfb*)]221 (CAS: 1235989-05-5), and [Rh(OH)((S,S)-Fc-tfb*)]222 (CAS: 1204591-10-5) were prepared according to the reported procedures.
A diene ligand L16a (CAS: 1365284-85-0) were prepared according to the reported procedures. Ketimines were prepared as shown in the ESI.† All other chemicals were purchased from commercial suppliers and used as received.
General procedure for Table 1
A rhodium catalyst (0.0030 mmol of Rh), 1a (25.5 mg, 0.10 mmol), and PhB(OH)2 (36.6 mg, 0.30 mmol) were placed in a Schlenk tube under nitrogen. 1,4-Dioxane (0.4 mL) was added and the mixture was stirred at 60 °C for 12 h. The mixture was passed through a short column of silica gel with ethyl acetate as eluent. The solvent was removed on a rotary evaporator and the residue was subjected to preparative TLC on silica gel with hexane–ethyl acetate (3:1) to give 3aa. The ee of 3aa was determined by chiral HPLC (Daicel Chiralpak AD-H).
General procedure for eqn (1)
[Rh(OH)((S,S)-Bn-tfb*)]2 (1.6 mg, 0.0030 mmol of Rh), 1a,b or c (0.10 mmol), and p-tolylboronic acid (24.4 mg, 0.20 mmol) were placed in a Schlenk tube under nitrogen. 1,4-Dioxane (0.4 mL) was added and the mixture was stirred at 60 °C for 3 h. The mixture was passed through a short column of silica gel with ethyl acetate as eluent. The solvent was removed on a rotary evaporator and the residue was subjected to preparative TLC on silica gel with hexane–ethyl acetate (3:1) to give 3. The ee of 3 was determined by chiral HPLC.
General procedure for Table 2, entries 1–10
[Rh(OH)((S,S)-Bn-tfb*)]2 (3.2 mg, 0.0060 mmol of Rh), 1c (56.6 mg, 0.20 mmol), and arylboronic acids 2 (0.40 mmol) were placed in a Schlenk tube under nitrogen. 1,4-Dioxane (0.8 mL) was added and the mixture was stirred at 60 °C. The mixture was passed through a short column of silica gel with ethyl acetate as eluent. The solvent was removed on a rotary evaporator and the residue was subjected to preparative TLC on silica gel with hexane–ethyl acetate (3:1) to give 3.
General procedure for Table 2, entries 11–17
[Rh(OH)((S,S)-Bn-tfb*)]2 (3.2 mg, 0.0060 mmol of Rh), 1 (0.20 mmol), and p-tolylboronic acid (2b, 48.8 mg, 0.40 mmol) were placed in a Schlenk tube under nitrogen. 1,4-Dioxane (0.8 mL) was added and the mixture was stirred at 60 °C for 3 h. The mixture was passed through a short column of silica gel with ethyl acetate as eluent. The solvent was removed on a rotary evaporator and the residue was subjected to preparative TLC on silica gel with hexane–ethyl acetate (3:1) to give 3.
:20, flow 0.5 mL min−1, 254 nm, t1 = 14.9 min (R), t2 = 18.9 min (S)); [α]20D −45 (c 0.82, CHCl3) for 96% ee (S). 1H NMR (CDCl3) δ 1.28 (t, J = 7.1 Hz, 3H), 4.32–4.41 (m, 2H), 6.45 (s, 1H), 7.14 (d, J = 7.5 Hz, 1H), 7.21–7.26 (m, 3H), 7.34–7.38 (m, 3H), 7.43 (td, J = 8.1, 1.4 Hz, 1H), 7.51 (dd, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 13.8, 64.1, 76.8, 118.9, 119.7, 124.9, 127.5, 128.5, 128.9, 130.5, 130.8, 139.3, 151.1, 170.0. HRMS (ESI) calcd for C16H15NNaO5S (M + Na)+ 356.0563, found 356.0564.
:20, flow 0.5 mL min−1, 254 nm, t1 = 16.5 min (R), t2 = 21.7 min (S)); [α]20D −40 (c 0.69, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.28 (t, J = 6.8 Hz, 3H), 2.35 (s, 3H), 4.32–4.41 (m, 2H), 6.41 (s, 1H), 7.11 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 8.1 Hz, 1H), 7.16 (d, J = 8.1 Hz, 2H), 7.22 (td, J = 8.1, 1.4 Hz, 1H), 7.42 (td, J = 8.1, 2.0 Hz, 1H), 7.50 (dd, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 13.8, 21.1, 64.0, 71.0, 119.2, 119.6, 124.8, 127.5, 129.2, 130.5, 130.7, 134.0, 136.5, 138.9, 151.0, 170.1. HRMS (ESI) calcd for C17H17NNaO5S (M + Na)+ 370.0720, found 370.0714.
:20, flow 0.5 mL min−1, 254 nm, t1 = 21.0 min (R), t2 = 27.7 min (S)); [α]20D −41 (c 0.88, CHCl3) for 92% ee (S). 1H NMR (CDCl3) δ 2.34 (s, 3H), 5.29 (d, J = 12.2 Hz, 1H), 5.32 (d, J = 12.2 Hz, 1H), 6.35 (s, 1H), 7.06 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 8.2 Hz, 2H), 7.13 (t, J = 7.3 Hz, 2H), 7.23–7.27 (m, 2H), 7.31–7.36 (m, 3H), 7.38–7.43 (m, 2H); 13C NMR (CDCl3) δ 21.1, 69.4, 71.1, 119.2, 119.5, 124.8, 127.4, 128.57, 128.63, 128.9, 129.2, 130.67, 130.69, 134.0, 136.3, 139.0, 150.9, 169.9. HRMS (ESI) calcd for C22H19NNaO5S (M + Na)+ 432.0876, found 432.0867.
:20, flow 0.5 mL min−1, 254 nm, t1 = 14.0 min (S), t2 = 22.4 min (R)); [α]20D −30 (c 0.61, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.47 (s, 9H), 2.35 (s, 3H), 6.41 (s, 1H), 7.10–7.16 (m, 5H), 7.21 (td, J = 7.5, 1.4 Hz, 1H), 7.41 (td, J = 7.8, 2.1 Hz, 1H), 7.49 (dd, J = 7.5, 2.1 Hz, 1H); 13C NMR (CDCl3) δ 21.1, 27.6, 71.1, 85.7, 119.5, 124.6, 127.4, 129.1, 130.5, 130.6, 137.0, 138.5, 151.0, 168.8. HRMS (ESI) calcd for C19H21NNaO5S (M + Na)+ 398.1033, found 398.1024.
:20, flow 0.5 mL min−1, 254 nm, t1 = 13.9 min (S), t2 = 21.7 min (R)); [α]20D −29 (c 1.13, CHCl3) for 96% ee (S). 1H NMR (CDCl3) δ 1.46 (s, 9H), 6.45 (s, 1H), 7.13 (dd, J = 8.9 Hz, 1H), 7.02–7.29 (m, 3H), 7.32–7.39 (m, 3H), 7.42 (t, J = 7.5 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 71.2, 85.8, 119.2, 119.6, 124.6, 127.5, 128.4, 128.6, 130.5, 130.6, 139.9, 151.1, 168.7. HRMS (ESI) calcd for C18H19NNaO5S (M + Na)+ 384.0876, found 384.0870.
:20, flow 0.5 mL min−1, 254 nm, t1 = 16.0 min (S), t2 = 24.1 min (S)); [α]20D −26 (c 1.06, CHCl3) for 86% ee (S). 1H NMR (CDCl3) δ 1.47 (s, 9H), 3.80 (s, 3H), 6.41 (s, 1H), 6.85 (d, J = 8.9 Hz, 2H), 7.12 (dd, J = 8.1, 1.4 Hz, 1H), 7.15 (d, J = 8.9 Hz, 2H), 7.21 (td, J = 8.1, 1.4 Hz, 1H), 7.41 (td, J = 8.1, 1.4 Hz, 1H), 7.50 (dd, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 55.2, 71.0, 85.7, 113.7, 119.57, 119.63, 124.6, 128.8, 130.5, 130.6, 132.0, 151.0, 159.6, 168.8. HRMS (ESI) calcd for C19H21NNaO6S (M + Na)+ 414.0982, found 414.0979.
:20, flow 0.5 mL min−1, 254 nm, t1 = 20.1 min (S), t2 = 28.4 min (R)); [α]20D −33 (c 0.94, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.47 (s, 9H), 5.97 (d, J = 1.4 Hz, 1H), 5.98 (d, J = 1.4 Hz, 1H), 6.42 (s, 1H), 6.68–6.75 (m, 3H), 7.11 (dd, J = 8.1, 1.4 Hz, 1H), 7.22 (td, J = 7.5, 1.4 Hz, 1H), 7.42 (td, J = 7.8, 1.4 Hz, 1H), 7.53 (dd, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 71.0, 85.9, 101.5, 107.8, 108.1, 119.2, 119.6, 121.4, 124.7, 130.3, 130.7, 133.7, 147.80, 147.83, 151.0, 168.6. HRMS (ESI) calcd for C19H19NNaO7S (M + Na)+ 428.0774, found 428.0768.
:20, flow 0.5 mL min−1, 254 nm, t1 = 13.7 min (S), t2 = 28.6 min (R)); [α]20D −9 (c 0.94, CHCl3) for 85% ee (S). 1H NMR (CDCl3) δ 1.48 (s, 9H), 3.75 (s, 6H), 3.85 (s, 3H), 6.40 (s, 1H), 6.48 (s, 2H), 7.12 (dd, J = 8.1, 1.4 Hz, 1H), 7.21 (td, J = 8.1, 1.4 Hz, 1H), 7.41 (td, J = 7.8, 1.4 Hz, 1H), 7.50 (dd, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 56.1, 60.8, 71.3, 85.7, 119.5, 119.6, 124.6, 130.7, 130.8, 135.2, 138.2, 150.8, 153.0, 168.5. HRMS (ESI) calcd for C21H25NNaO8S (M + Na)+ 474.1193, found 474.1187.
:20, flow 0.5 mL min−1, 254 nm, t1 = 11.9 min (S), t2 = 17.1 min (R)); [α]20D −53 (c 1.09, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.46 (s, 9H), 6.50 (s, 1H), 7.02 (t, J = 8.5 Hz, 2H), 7.14 (dd, J = 8.1, 1.4 Hz, 1H), 7.19–7.26 (m, 3H), 7.44 (dt, J = 7.8, 1.4 Hz, 1H), 7.53 (dd, J = 8.1, 2.1 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 70.6, 86.2, 105.0, 115.3 (d, JF–C = 21 Hz), 118.7, 119.9, 124.8, 129.5 (d, JF–C = 9 Hz), 129.9, 130.9, 135.7, 151.2, 162.5 (d, JF–C = 247 Hz), 168.5. HRMS (ESI) calcd for C18H18FNNaO5S (M + Na)+ 402.0782, found 402.0776.
:20, flow 0.5 mL min−1, 254 nm, t1 = 11.3 min (S), t2 = 14.6 min (R)); [α]20D −48 (c 1.18, CHCl3) for 95% ee (S). 1H NMR (CDCl3) δ 1.45 (s, 9H), 6.53 (s, 1H), 7.13 (d, J = 7.8 Hz, 1H), 7.17 (d, J = 8.6 Hz, 2H), 7.25 (td, J = 7.8, 1.4 Hz, 1H), 7.31 (d, J = 8.6 Hz, 2H), 7.45 (dd, J = 7.8, 1.4 Hz, 1H), 7.54 (dd, J = 7.8, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 70.6, 86.3, 118.3, 119.9, 124.8, 128.5, 128.9, 129.9, 131.0, 134.6, 138.4, 151.2, 168.3. HRMS (ESI) calcd for C18H18ClNNaO5S (M + Na)+ 418.0486, found 418.0481.
:20, flow 0.5 mL min−1, 254 nm, t1 = 13.9 min (S), t2 = 18.1 min (R)); [α]20D −38 (c 0.70, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.45 (s, 9H), 6.51 (s, 1H), 7.10 (d, J = 8.9 Hz, 2H), 7.17 (dd, J = 8.1, 1.4 Hz, 1H), 7.25 (td, J = 8.1, 1.4 Hz, 1H), 7.45 (td, J = 8.1, 1.4 Hz, 1H), 7.47 (d, J = 8.9 Hz, 2H), 7.53 (dd, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 27.6, 70.7, 86.4, 118.3, 119.9, 122.9, 124.8, 129.2, 129.8, 131.0, 131.5, 138.9, 151.2, 168.2. HRMS (ESI) calcd for C18H18BrNNaO5S (M + Na)+ 461.9981, found 461.9980.
:20, flow 0.5 mL min−1, 254 nm, t1 = 9.8 min (S), t2 = 11.4 min (R)); [α]20D −60 (c 0.65, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.45 (s, 9H), 6.58 (s, 1H), 7.16 (dd, J = 8.2, 1.4 Hz, 1H), 7.28 (td, J = 8.2, 1.4 Hz, 1H), 7.36 (d, J = 8.1 Hz, 2H), 7.48 (td, J = 8.1, 1.4 Hz, 1H), 7.56 (dd, J = 8.1, 1.4 Hz, 1H), 7.60 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3) δ 27.6, 70.7, 86.7, 117.9, 120.1, 123.8 (q, JF–C = 273 Hz), 124.9, 125.3 (q, JF–C = 3 Hz), 128.0, 129.7, 130.7 (q, JF–C = 31 Hz), 131.2, 143.6, 151.4, 168.0. HRMS (ESI) calcd for C19H18F3NNaO5S (M + Na)+ 452.0750, found 452.0744.
:20, flow 0.5 mL min−1, 254 nm, t1 = 16.4 min (S), t2 = 22.6 min (R)); [α]20D −26 (c 1.05, CHCl3) for 95% ee (S). 1H NMR (CDCl3) δ 1.47 (s, 9H), 6.54 (s, 1H), 7.18 (dd, J = 8.2, 1.4 Hz, 1H), 7.26 (td, J = 7.8, 1.4 Hz, 1H), 7.38 (dd, J = 8.9, 2.0 Hz, 1H), 7.44–7.54 (m, 3H), 7.59 (dd, J = 8.1, 1.4 Hz, 1H), 7.63 (d, J = 1.4 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.84 (t, J = 8.9 Hz, 2H); 13C NMR (CDCl3) δ 27.6, 71.4, 86.0, 118.9, 119.8, 124.7, 124.8, 126.5, 126.9, 127.1, 127.6, 128.5, 128.6, 130.5, 130.8, 132.6, 133.0, 137.0, 151.2, 168.7. HRMS (ESI) calcd for C22H21NNaO5S (M + Na)+ 434.1033, found 434.1025.
:20, flow 0.5 mL min−1, 254 nm, t1 = 12.5 min (S), t2 = 20.2 min (R)); [α]20D −12 (c 0.57, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.46 (s, 9H), 2.31 (s, 3H), 2.34 (s, 3H), 6.39 (s, 1H), 7.09 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 8.9 Hz, 2H), 7.14 (d, J = 8.9 Hz, 2H), 7.26 (d, J = 8.1 Hz, 1H), 7.30 (d, J = 8.1 Hz, 1H); 13C NMR (CDCl3) δ 15.8, 21.1, 27.6, 71.2, 85.5, 119.5, 123.9, 127.5, 127.9, 128.8, 129.0, 131.9, 137.1, 138.4, 149.5, 168.9. HRMS (ESI) calcd for C20H23NNaO5S (M + Na)+ 412.1189, found 412.1186.
:20, flow 0.5 mL min−1, 254 nm, t1 = 13.7 min (S), t2 = 20.4 min (R)); [α]20D −15 (c 0.70, CHCl3) for 96% ee (S). 1H NMR (CDCl3) δ 1.46 (s, 9H), 2.34 (s, 3H), 2.38 (s, 3H), 6.39 (s, 1H), 6.92 (s, 1H), 7.01 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 8.9 Hz, 2H), 7.13 (d, J = 8.9 Hz, 2H), 7.36 (d, J = 8.2 Hz, 1H); 13C NMR (CDCl3) δ 21.0, 21.1, 27.6, 70.9, 85.5, 116.3, 119.7, 125.6, 127.4, 129.1, 130.2, 137.2, 138.4, 141.3, 150.9, 168.9. HRMS (ESI) calcd for C20H23NNaO5S (M + Na)+ 412.1189, found 412.1182.
:20, flow 0.5 mL min−1, 254 nm, t1 = 13.7 min (S), t2 = 20.5 min (R)); [α]20D −20 (c 1.03, CHCl3) for 93% ee (S). 1H NMR (CDCl3) δ 1.47 (s, 9H), 2.32 (s, 3H), 2.35 (s, 3H), 6.39 (s, 1H), 7.00 (d, J = 8.9 Hz, 1H), 7.13 (d, J = 8.5 Hz, 2H), 7.16 (d, J = 8.5 Hz, 2H), 7.20 (dd, J = 8.9, 1.4 Hz, 1H), 7.28 (d, J = 1.4 Hz, 1H); 13C NMR (CDCl3) δ 20.9, 21.1, 27.6, 71.1, 85.5, 119.0, 119.1, 127.4, 129.1, 130.7, 131.2, 134.3, 137.0, 138.5, 148.9, 168.8. HRMS (ESI) calcd for C20H23NNaO5S (M + Na)+ 412.1189, found 412.1185.
:20, flow 0.5 mL min−1, 254 nm, t1 = 18.5 min (S), t2 = 34.7 min (R)); [α]20D −47 (c 1.10, CHCl3) for 93% ee (S). 1H NMR (CDCl3) δ 1.48 (s, 9H), 2.34 (s, 3H), 3.75 (s, 3H), 6.35 (s, 1H), 6.95 (dd, J = 8.8, 2.7 Hz, 1H), 7.02 (d, J = 2.7 Hz, 1H), 7.05 (d, J = 2.7 Hz, 1H), 7.12 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3) δ 21.1, 27.7, 55.7, 71.1, 85.6, 115.0, 116.3, 120.1, 120.3, 127.4, 129.1, 136.9, 138.5, 144.9, 155.9, 168.7. HRMS (ESI) calcd for C20H23NNaO6S (M + Na)+ 428.1138, found 428.1135.
:20, flow 0.5 mL min−1, 254 nm, t1 = 17.4 min (S), t2 = 49.1 min (R)); [α]20D +34 (c 1.04, CHCl3) for 97% ee (S). 1H NMR (CDCl3) δ 1.51 (s, 9H), 2.35 (s, 3H), 5.74 (d, J = 1.4 Hz, 1H), 5.85 (d, J = 1.4 Hz, 1H), 5.86 (s, 1H), 6.62 (d, J = 8.6 Hz, 1H), 6.82 (d, J = 8.6 Hz, 1H), 7.14 (d, J = 8.6 Hz, 2H), 7.21 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3) δ 21.1, 27.7, 70.1, 84.9, 101.8, 106.8, 109.0, 111.2, 127.3, 129.1, 134.7, 138.9, 145.0, 145.2, 146.1, 167.8. HRMS (ESI) calcd for C20H21NNaO7S (M + Na)+ 442.0931, found 442.0940.
:20, flow 0.5 mL min−1, 254 nm, t1 = 15.4 min (S), t2 = 26.1 min (R)); [α]20D −8 (c 0.80, CHCl3) for 88% ee (S). 1H NMR (CDCl3) δ 1.48 (s, 9H), 2.36 (s, 3H), 6.37 (s, 1H), 7.05–7.15 (m, 4H), 7.16 (d, J = 8.2 Hz, 2H), 7.21 (dd, J = 8.2, 2.7 Hz, 1H); 13C NMR (CDCl3) δ 21.1, 27.6, 71.0, 86.1, 117.2 (d, JF–C = 27 Hz), 117.8 (d, JF–C = 23 Hz), 120.9 (d, JF–C = 9 Hz), 121.2 (d, JF–C = 7 Hz), 127.2, 129.3, 136.5, 138.9, 146.9, 158.6 (d, JF–C = 245 Hz), 168.3. HRMS (ESI) calcd for C19H20FNNaO5S (M + Na)+ 416.0938, found 416.0936.
:20, flow 0.5 mL min−1, 254 nm, t1 = 14.1 min (S), t2 = 26.9 min (R)); [α]20D −19 (c 1.03, CHCl3) for 84% ee (S). 1H NMR (CDCl3) δ 1.48 (s, 9H), 2.36 (s, 3H), 6.39 (s, 1H), 7.06 (d, J = 8.9 Hz, 1H), 7.11 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.38 (dd, J = 8.9, 2.7 Hz, 1H), 7.49 (d, J = 2.7 Hz, 1H); 13C NMR (CDCl3) δ 21.1, 27.6, 70.9, 86.2, 120.8, 121.1, 127.2, 129.4, 129.9, 130.56, 130.60, 136.4, 138.9, 149.5, 168.2. HRMS (ESI) calcd for C19H20ClNNaO5S (M + Na)+ 432.0643, found 432.0641.
Transformations of 3cb
:1) to give 3cb′ (467 mg, 1.19 mmol, 99% yield). The ee was measured by HPLC (Chiralpak AD-H, hexane–2-propanol = 80:20, flow 0.5 mL min−1, 254 nm, t1 = 10.7 min (R), t2 = 11.5 min (S)); [α]20D +117 (c 1.01, CHCl3) for 94% ee (S). 1H NMR (CDCl3) δ 1.56 (s, 9H), 2.36 (s, 3H), 2.91 (s, 3H), 6.93 (dd, J = 8.1, 1.4 Hz, 1H), 7.02 (dd, J = 8.1, 1.4 Hz, 1H), 7.03 (td, J = 8.1, 1.4 Hz, 1H), 7.15 (d, J = 8.5 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 7.27 (td, J = 8.1, 1.4 Hz, 1H); 13C NMR (CDCl3) δ 21.1, 27.9, 34.0, 75.7, 84.3, 117.9, 124.1, 124.4, 128.5, 129.5, 129.6, 133.3, 136.0, 139.1, 148.9, 168.8. HRMS (ESI) calcd for C20H23NNaO5S (M + Na)+ 412.1189, found 412.1180.
:1) to give 4 (21.8 mg, 0.070 mmol, 70% yield). The ee was measured by HPLC (Chiralpak AD-H × 2, hexane–2-propanol = 90:10, flow 0.5 mL min−1, 254 nm, t1 = 17.3 min (S), t2 = 18.1 min (R)); [α]20D −1 (c 1.02, CHCl3) for 94% ee (R). 1H NMR (CDCl3) δ 1.39 (s, 9H), 2.13 (s, 3H), 2.33 (s, 3H), 7.11 (d, J = 8.1 Hz, 2H), 7.22–7.26 (m, 1H), 7.28–7.32 (m, 4H), 8.14 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3) δ 21.0, 27.8, 30.6, 73.2, 81.8, 127.0, 127.6, 128.4, 128.5, 128.6, 136.6, 138.5, 141.6, 172.7. HRMS (ESI) calcd for C20H25NNaO2 (M + Na)+ 334.1778, found 334.1774.
Acknowledgements
This work has been supported by a Grant-in-Aid for Scientific Research from the MEXT, Japan. We thank Prof. A. Osuka for X-ray crystallographic analysis of compound 3ch.Notes and references
- For recent reviews, see: (a) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed; (b) C. S. Marques and A. J. Burke, ChemCatChem, 2011, 3, 635 CrossRef CAS; (c) K. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874 CrossRef CAS PubMed; (d) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853 CrossRef CAS PubMed; (e) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541 CrossRef CAS.
- For selected examples, see: Cu: (a) I. Bonnaventure and A. B. Charette, Tetrahedron, 2009, 65, 4968 CrossRef CAS; (b) Q. Perron and A. Alexakis, Tetrahedron: Asymmetry, 2008, 19, 1871 CrossRef CAS; (c) M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2008, 73, 940 CrossRef CAS PubMed; (d) M. Shi, Z.-Y. Lei and Q. Xu, Adv. Synth. Catal., 2006, 348, 2237 CrossRef CAS; Zr: (e) J. R. Porter, J. F. Traverse, A. H. Hoveyda and M. L. Snapper, J. Am. Chem. Soc., 2001, 123, 984 CrossRef CAS PubMed; Rh: (f) T. Hayashi and M. Ishigedani, J. Am. Chem. Soc., 2000, 122, 976 CrossRef CAS; (g) M. Kuriyama, T. Soeta, X. Hao, X. Chen and K. Tomioka, J. Am. Chem. Soc., 2004, 126, 8128 CrossRef CAS PubMed; (h) H.-F. Duan, Y.-X. Jia, L.-X. Wang and Q.-L. Zhou, Org. Lett., 2006, 8, 2567 CrossRef CAS PubMed; (i) R. B. C. Jagt, P. Y. Toullec, D. Geerdink, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Angew. Chem., Int. Ed., 2006, 45, 2789 CrossRef CAS PubMed; (j) C. Marelli, C. Monti, C. Gennari and U. Piarulli, Synlett, 2007, 2213 CAS; (k) Z.-Q. Wang, C.-G. Feng, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2007, 129, 5336 CrossRef CAS PubMed; (l) K. Kurihara, Y. Yamamoto and N. Miyaura, Adv. Synth. Catal., 2009, 351, 260 CrossRef CAS; (m) D. J. Weix, Y. Shi and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 1092 CrossRef CAS PubMed; (n) H. Wang and M.-H. Xu, Synthesis, 2013, 2125 CAS; Pd: (o) G.-N. Ma, T. Zhang and M. Shi, Org. Lett., 2009, 11, 875 CrossRef CAS PubMed; (p) C. S. Marques and A. J. Burke, Eur. J. Org. Chem., 2010, 1639 CrossRef CAS.
- (a) R. Wada, T. Shibuguchi, S. Makino, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7687 CrossRef CAS PubMed; (b) C. Lauzon and A. B. Charette, Org. Lett., 2006, 8, 2743 CrossRef CAS PubMed; (c) P. Fu, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 5530 CrossRef CAS PubMed.
- R. Shintani, M. Takeda, T. Tsuji and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 13168 CrossRef CAS PubMed.
- For examples of rhodium-catalyzed asymmetric synthesis of α-tertiary amines, see: (a) H. H. Jung, A. W. Buesking and J. A. Ellman, Org. Lett., 2011, 13, 3912 CrossRef CAS PubMed; (b) J. S. Arnold and H. M. Nguyen, J. Am. Chem. Soc., 2012, 134, 8380 CrossRef CAS PubMed; (c) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098 CrossRef CAS PubMed.
- (a) T. Nishimura, A. Noishiki, G. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056 CrossRef CAS PubMed; (b) T. Nishimura, A. Noishiki, Y. Ebe and T. Hayashi, Angew. Chem., Int. Ed., 2013, 52, 1777 CrossRef CAS PubMed; (c) T. Nishimura, Y. Ebe, H. Fujimoto and T. Hayashi, Chem. Commun., 2013, 49, 5504 RSC.
- (a) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (b) Y.-J. Chen, Y.-H. Chen, C.-G. Feng and G.-Q. Lin, Org. Lett., 2014, 16, 3400 CrossRef CAS PubMed; (c) H. Wang, Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 3962 CrossRef CAS PubMed; (d) T. Jiang, Z. Wang and M.-H. Xu, Org. Lett., 2015, 17, 528 CrossRef CAS PubMed.
- (a) Y. Luo, A. J. Carnell and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 6762 CrossRef CAS PubMed; (b) Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309 CrossRef CAS PubMed. For examples of asymmetric allylation, see: (c) H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605 CrossRef CAS PubMed; (d) H. B. Hepburn, N. Chotsaeng, Y. Luo and H. W. Lam, Synthesis, 2013, 2649 CAS.
- (a) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540 CrossRef CAS PubMed; (b) C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 53, 9936 CrossRef CAS PubMed.
- For a recent example of organocatalytic asymmetric Mannich reaction of cyclic ketimines, see: S. Zhang, L. Li, Y. Hu, Z. Zha, Z. Wang and T.-P. Loh, Org. Lett., 2015, 17, 1050 CrossRef CAS PubMed.
- For reviews, see: (a) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 1998, 9, 3517 CrossRef CAS; (b) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2000, 11, 645 CrossRef CAS; (c) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127 CrossRef CAS; (d) H. Vogt and S. Bräse, Org. Biomol. Chem., 2007, 5, 406 RSC For a selected example of α-arylation of aldimine Schiff bases, see: S. Shirakawa, K. Yamamoto, T. Tokuda and K. Maruoka, Asian J. Org. Chem., 2014, 3, 433 CrossRef CAS and references therein.
- For examples of asymmetric synthesis of α-mono-substituted α-amino acid derivatives by metal-catalyzed addition of arylboronic acids to α-imino esters, see: (a) J. Chen, X. Lu, W. Lou, Y. Ye, H. Jiang and W. Zeng, J. Org. Chem., 2012, 77, 8541 CrossRef CAS PubMed; (b) H. Dai, M. Yang and X. Lu, Adv. Synth. Catal., 2008, 350, 249 CrossRef CAS; (c) M. A. Beenen, D. J. Weix and J. A. Ellman, J. Am. Chem. Soc., 2006, 128, 6304 CrossRef CAS PubMed.
- (a) T. Nishimura, H. Kumamoto, M. Nagaosa and T. Hayashi, Chem. Commun., 2009, 5713 RSC; (b) T. Nishimura, J. Wang, M. Nagaosa, K. Okamoto, R. Shintani, F. Kwong, W. Yu, A. S. C. Chan and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 464 CrossRef CAS PubMed.
- K. Okamoto, T. Hayashi and V. H. Rawal, Chem. Commun., 2009, 4815 RSC.
- T. Hayashi, M. Takahashi, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 2002, 124, 5052 CrossRef CAS PubMed.
- The absolute configuration of 3ch produced by use of (S,S)-Bn-tfb* was determined to be S by X-ray crystallographic analysis.
- K. W. Quasdorf, M. Riener, K. V. Petrova and N. K. Garg, J. Am. Chem. Soc., 2009, 131, 17748 CrossRef CAS PubMed.
- P. M. Wehn and J. Du Bois, Org. Lett., 2005, 7, 4685 CrossRef CAS PubMed.
- R. Uson, L. A. Oro and J. A. Cabeza, Inorg. Synth., 1985, 23, 129 CrossRef.
- G. Giordano and R. H. Crabtree, Inorg. Synth., 1979, 19, 218 CrossRef CAS.
- R. Shintani, M. Takeda, T. Nishimura and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 3969 CrossRef CAS PubMed.
- T. Nishimura, A. Kasai, M. Nagaosa and T. Hayashi, Chem. Commun., 2011, 47, 10488 RSC.
- F. Scott, C. Kriiger and P. Betz, J. Organomet. Chem., 1990, 387, 113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and X-ray crystallographic data of compound 3ch. CCDC 1044727. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00431d |
| This journal is © The Royal Society of Chemistry 2015 |