
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroxyethylene isosteres introduced in type II collagen fragments substantially alter the structure and dynamics of class II MHC Aq/glycopeptide complexes†
Cecilia
Lindgren
a,
Ida E.
Andersson
ab,
Lotta
Berg
a,
Doreen
Dobritzsch
c,
Changrong
Ge
b,
Sabrina
Haag
b,
Urszula
Uciechowska
a,
Rikard
Holmdahl
b,
Jan
Kihlberg
*c and
Anna
Linusson
*a
aDepartment of Chemistry, Umeå University, SE-901 87 Umeå, Sweden. E-mail: anna.linusson@umu.se; Tel: +46-90-7866890
bMedical Inflammation Research, Department of Medical Biochemistry and Biophysics, Karolinska Institute, SE-171 77 Stockholm, Sweden
cDepartment of Chemistry – BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden. E-mail: jan.kihlberg@kemi.uu.se; Tel: +46-18-4713801
First published on 21st April 2015
Abstract
Class II major histocompatibility complex (MHC) proteins are involved in initiation of immune responses to foreign antigens via presentation of peptides to receptors of CD4+ T-cells. An analogous presentation of self-peptides may lead to autoimmune diseases, such as rheumatoid arthritis (RA). The glycopeptide fragment CII259–273, derived from type II collagen, is presented by Aq MHCII molecules in the mouse and has a key role in development of collagen induced arthritis (CIA), a validated model for RA. We have introduced hydroxyethylene amide bond isosteres at the Ala261–Gly262 position of CII259–273. Biological evaluation showed that Aq binding and T cell recognition were dramatically reduced for the modified glycopeptides, although static models predicted similar binding modes as the native type II collagen fragment. Molecular dynamics (MD) simulations demonstrated that introduction of the hydroxyethylene isosteres disturbed the entire hydrogen bond network between the glycopeptides and Aq. As a consequence the hydroxyethylene isosteric glycopeptides were prone to dissociation from Aq and unfolding of the β1-helix. Thus, the isostere induced adjustment of the hydrogen bond network altered the structure and dynamics of Aq/glycopeptide complexes leading to the loss of Aq affinity and subsequent T cell response.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease that mainly affects peripheral joints by causing inflammation that destroys cartilage and bone. The cause of the disease is not yet known, but a genetic link has been found to the expression of the class II major histocompatibility complex (MHC) proteins HLA-DR1 and -DR4.1 In collagen-induced arthritis (CIA), a validated mouse model for studies of RA,2 development of disease is linked to expression of Aq MHC II molecules.3 Class II MHC proteins are found in antigen presenting cells (APC) where they are associated with peptides derived from endocytosed protein antigens. The complexes are then transported to the surface of the APC and presented to T-cell receptors (TCR) on circulating CD4+ T-cells, leading to initiation of an immune response towards the original antigen.CII259–273 1 (Fig. 1) – a glycopeptide fragment derived from type II collagen – is presented to CD4+ T-cells by Aq MHC molecules in CIA.4 Glycopeptide 1 has also been proven to work as a vaccine that effectively prevents development of disease in CIA when administered as a complex with Aq.5 The complex also reduces disease progression and severity in a mouse model of chronic relapsing arthritis.5 In humans, glycopeptide 1 binds to DR4 and the complex is recognized by T-cells isolated from patients suffering from severe RA, suggesting 1 to have a central role in RA just as in CIA.6 The minimal epitope in 1 required for binding to Aq, and stimulation of CII restricted T cell hybridomas obtained in CIA, consists of amino acid residues Ile260–Gln267.7 Antigenic peptides bind between the α1- and β1-helices of class II MHC proteins through a hydrogen bond network and by anchoring of sidechains in pockets in the binding groove.8 Synthesis and biological evaluation of a comprehensive panel of synthetic analogues of 1 has revealed Ile260 and Phe263 to be crucial anchor residues that bind in hydrophobic pockets of Aq (Fig. 2).9,10 Moreover, the galactosylated hydroxylysine at position 264 protrudes out of the binding site and is recognized by the TCR with exquisite specificity.11
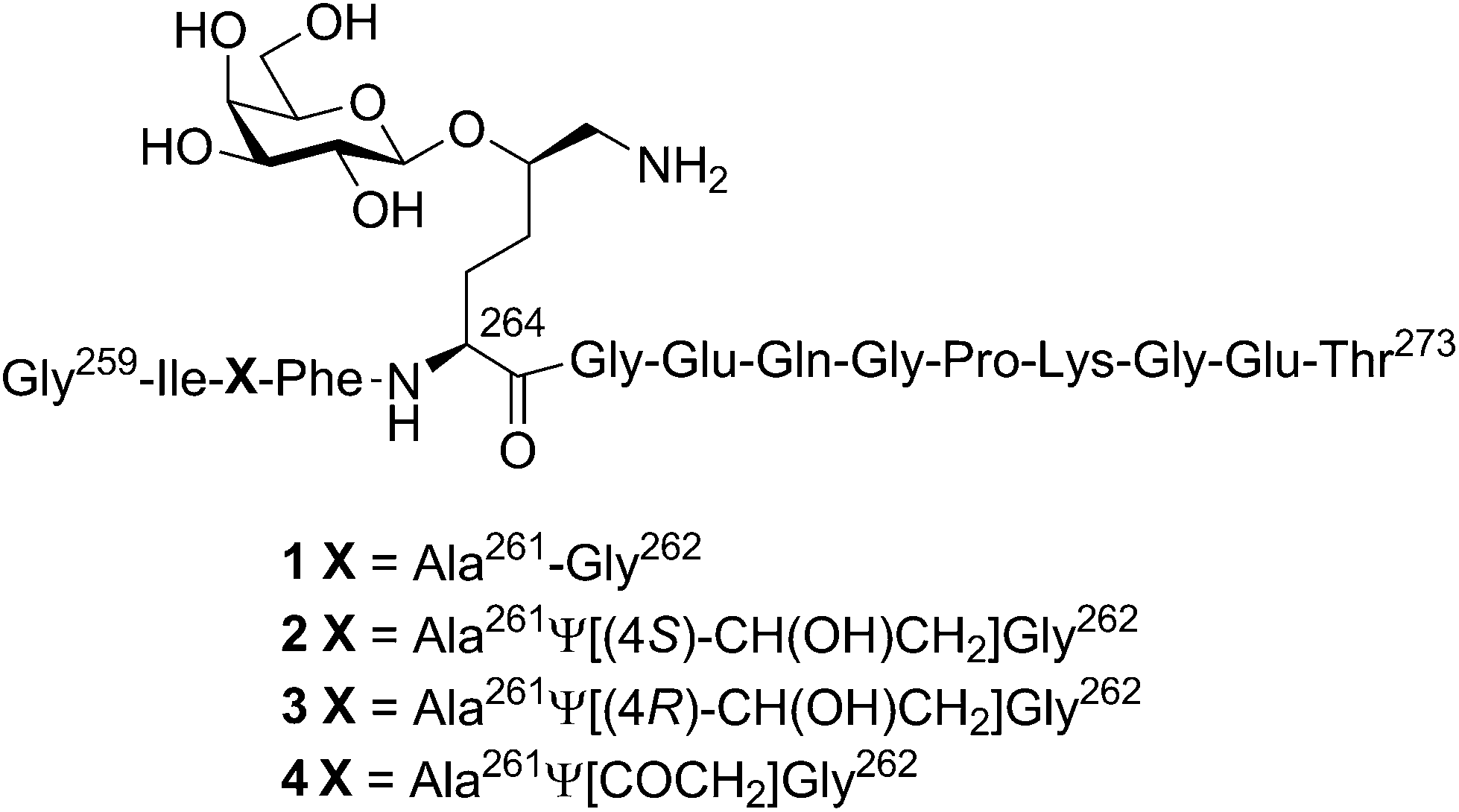 | ||
| Fig. 1 Structures of the four investigated glycopeptides 1–4. Chemical structure of the T-cell epitope CII259–273, 1, and the modified glycopeptides 2–4. | ||
 | ||
| Fig. 2 CII259–273 1 in complex with Aq. A 3D stick model displaying the general binding mode of the glycopeptide 1 between the α1- and β1-helix of Aq. The important anchor residues Ile260 and Phe263 are positioned in their respective binding pockets. The carbohydrate moiety is displayed in ball and stick with oxygen in red and carbon in green. The picture is based on a homology model of Aq.14 | ||
Modification of glycopeptides, through the incorporation of amide bond isosteres, has been used to probe molecular interactions in the MHC/glycopeptide/TCR system.12–17 For instance, aza-amino acids,15 (E)-alkene and ethylene isosteres13 and oxazole modifications12 have been employed. The Ile260–GalHyl264 fragment of glycopeptide 1 has been the target for the majority of these modifications as it contains essential features, such as the two MHC anchor residues9 and the galactose moiety crucial for TCR recognition.4,11 Incorporation of isosteres into 1 revealed the importance of having an intact network of hydrogen bonds for the stability of the complex with Aq.13 Furthermore, the dynamics of the complex were found to govern the response from the TCR; complexes that were more rigid than those of Aq and 1 resulted in weaker T cell responses.10,13
In this study, we further explore the molecular interactions in the MHC/glycopeptide/TCR system that are crucial for the development of CIA, and most likely also for RA, by synthesis and evaluation of two additional amide bond isosteres incorporated into glycopeptide 1.
Results
Amide bond isosteres
Hydroxyethylene isosteres were selected for incorporation at the Ala261–Gly262 position into glycopeptide 1 (Fig. 1, compounds 2 and 3). The choice of isosteres and their location were based on previous investigations, where we explored replacement of this amide bond with ketomethylene, methyleneamine and (E)-alkene isosteres.14 Evaluation of these glycopeptides showed that only ketomethylene-isostere modified glycopeptide 4 displayed comparable affinity for Aq and T-cell responses to 1, whereas both binding and cellular responses were lost for the others. We thus concluded that a hydrogen bond acceptor was required to maintain binding to Aq and T-cell responses, whereas the hydrogen bond donor of the amide bond could be omitted. The hydroxyethylene isosteres utilized in this study introduce a chiral centre and a hydrogen bond donor capability at the Ala261–Gly262 position, while retaining the hydrogen bond acceptor capability.Synthesis
The diastereomeric Ala261–Gly262 hydroxyethylene isosteres were synthesized as dipeptide building blocks suitably protected for use in Fmoc-based solid-phase peptide synthesis (SPPS).18 In short, ketomethylene derivative 514 was first diastereoselectively reduced to the respective alcohol, followed by protective group manipulations and separation of diastereomeric mixtures. Diastereoselective reduction of ketomethylene derivative 514 was performed using conditions guided by the work of Hoffman et al.19 and Våbenø et al.20 (Scheme 1). Treatment of 5 with LiAlH(O-tBu)3 in EtOH19,20 gave alcohol 6 in 75% yield and with an excellent diastereomeric ratio (dr) 4S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4R of 1:99 according to 1H NMR analysis. When 5 was instead treated with (S)-alpine-hydride in THF,20 alcohol 7 was obtained in 84% yield although with a disappointing 4S:4R dr of 2:1. The diastereomeric mixture 7 was difficult to separate by column chromatography and therefore additional transformations were performed. Both 6 and 7 were deprotected using TFA to remove the Boc and tBu protecting groups followed by Fmoc protection, which produced the lactones 8 and 9 (4S:4R of 2:1), respectively. The latter mixture of lactones was at this stage separated by chiral HPLC chromatography to afford 9 with a 4S:4R dr of 99:1. In the final step, reaction with tBuSH-AlMe3 converted the lactones into the corresponding γ-hydroxy thioesters that were immediately silylated with TBDMSCl to give 10 and 12 in order to prevent relactonization.21 Finally, base-catalysed hydrolysis of the thioesters provided the target hydroxyethylene isostere building blocks 11 and 13.
:4R of 1:99 according to 1H NMR analysis. When 5 was instead treated with (S)-alpine-hydride in THF,20 alcohol 7 was obtained in 84% yield although with a disappointing 4S:4R dr of 2:1. The diastereomeric mixture 7 was difficult to separate by column chromatography and therefore additional transformations were performed. Both 6 and 7 were deprotected using TFA to remove the Boc and tBu protecting groups followed by Fmoc protection, which produced the lactones 8 and 9 (4S:4R of 2:1), respectively. The latter mixture of lactones was at this stage separated by chiral HPLC chromatography to afford 9 with a 4S:4R dr of 99:1. In the final step, reaction with tBuSH-AlMe3 converted the lactones into the corresponding γ-hydroxy thioesters that were immediately silylated with TBDMSCl to give 10 and 12 in order to prevent relactonization.21 Finally, base-catalysed hydrolysis of the thioesters provided the target hydroxyethylene isostere building blocks 11 and 13.
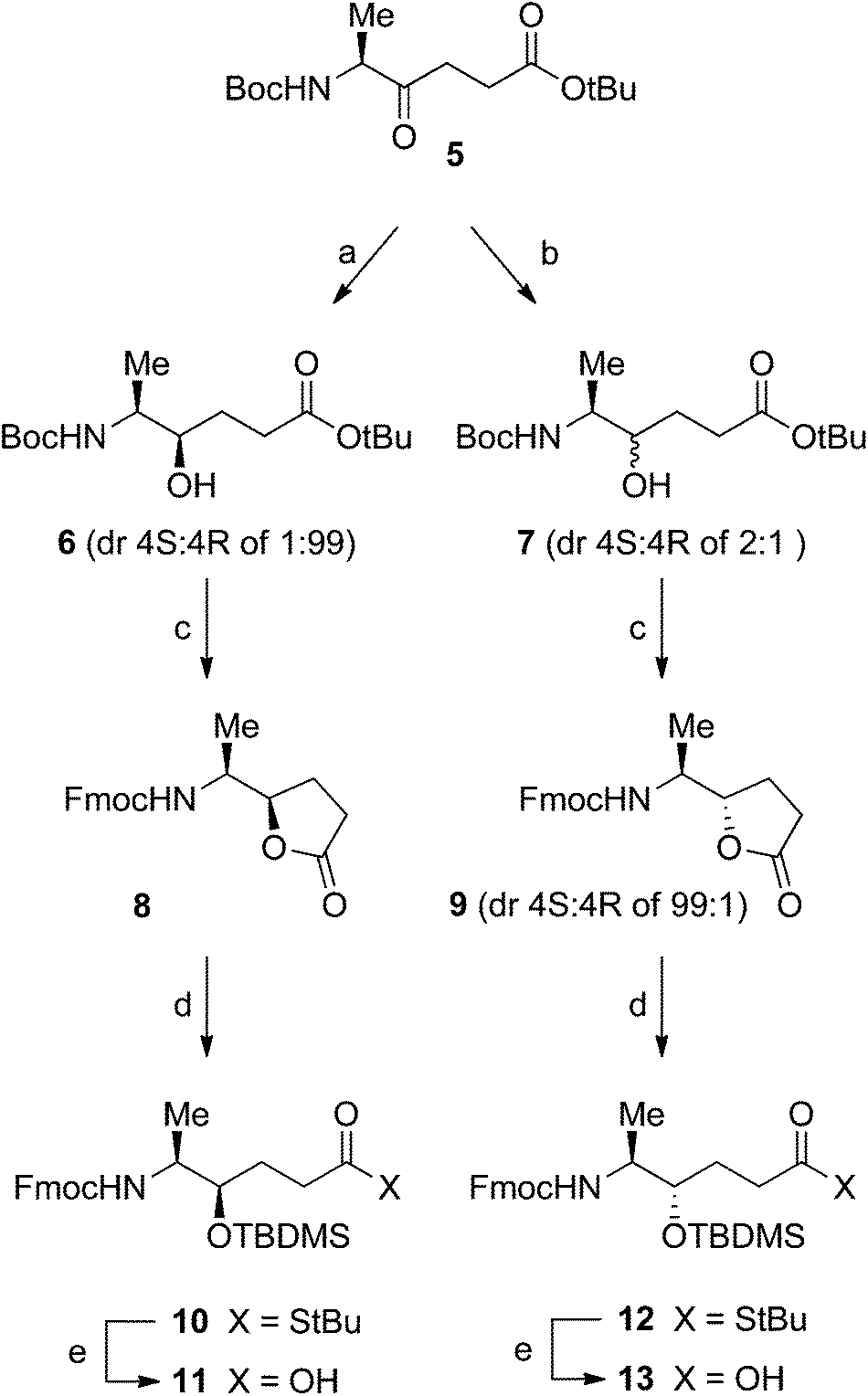 | ||
| Scheme 1 Reagents and conditions: (a) LiAlH(O-tBu)3, EtOH, −78 °C, 75%; (b) (S)-alpine-hydride, THF, −78 °C, 84%; (c) (i) TFA, CH2Cl2, rt; (ii) FmocOSu, Na2CO3, dioxane, H2O, 0 °C → rt, 86% for 8 from 6, 48% for 9 from 7 (after separation of the diastereomers by chiral chromatography); (d) (i) tBuSH, AlMe3, CH2Cl2, 0 °C; (ii) TBDMSCl, imidazole, DMF, rt, 55% for 10 from 8 and 85% for 12 from 9; (e) LiOH, H2O2, THF, 0 °C → rt, 75% for 11 from 10 and 82% for 13 from 12. | ||
The isostere building blocks 11 and 13 were incorporated into the CII259–273 glycopeptide sequence using Fmoc-based SPPS18 under standard conditions. The isosteric glycopeptides were cleaved from the solid support, deacetylated, and purified by reversed-phase HPLC to give the trifluoroacetate salts of 2 and 3 (Fig. 1) with overall yields of 18 and 28%, respectively, and in >98% purity. All glycopeptides were homogeneous, according to analytical reversed-phase HPLC and their structures were confirmed by 1H NMR spectroscopy and MALDI-TOF mass spectrometry.
Binding to the Aq protein and recognition by T-cell hybridomas
The ability of the hydroxyethylene-modified glycopeptides 2 and 3 to bind to the Aq protein compared to 1 was assessed in a competitive ELISA binding assay. Significant optimisation was undertaken to obtain a satisfactory signal to noise ratio in the ELISA, which led to the use of rather high concentrations of the biotinylated class II-associated invariant chain peptide (CLIP) tracer. As the CLIP binds with high affinity to Aq higher concentrations of weaker inhibitors, such as native 1, had to be employed in the ELISA. No binding could be detected for 2 and 3 even at the highest concentration of 1 mM where 1 completely competed out of the biotinylated CLIP tracer (Fig. 3). However, it is likely that a weak binding of 2 and 3 to Aq would not be detected under the conditions that had to be employed in the ELISA. Replacement of the Ala261–Gly262 amide bond with the two different hydroxyethylene isosteres thus had severe effects on the glycopeptide abilities to bind to Aq. These results underscore our previous results that this particular amide bond is highly sensitive to modifications. At this position the corresponding ketomethylene-isostere 4 is the only modified glycopeptide so far that has been shown to bind equally well to Aq as 1.12–14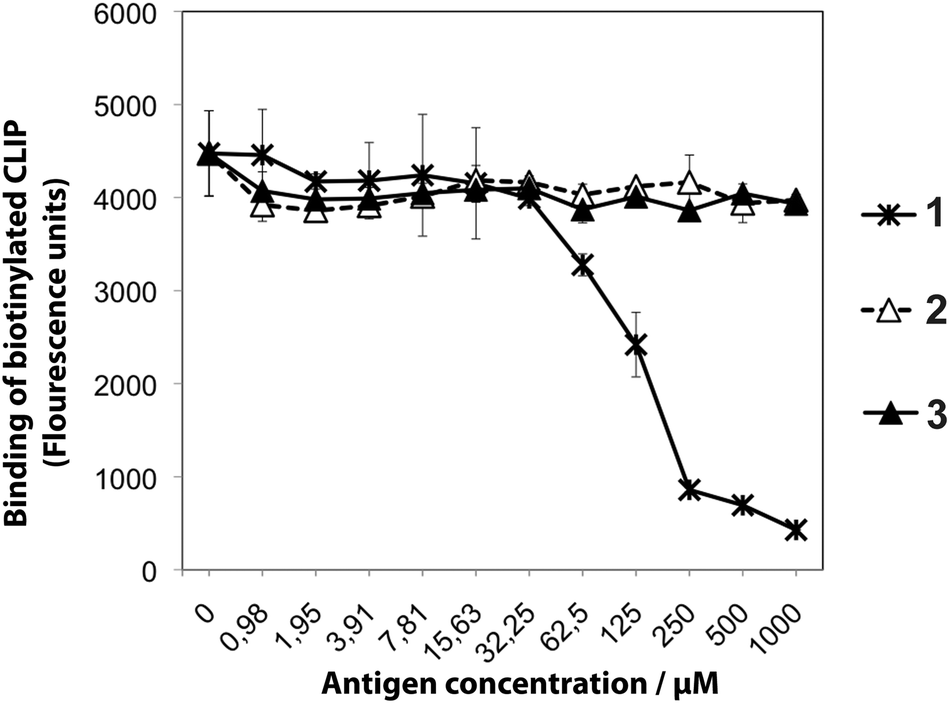 | ||
| Fig. 3 Binding of native glycopeptide 1 and hydroxyethylene isosteres 2 and 3 to Aq. In the competitive binding assay, increasing concentrations of 1, 2, or 3 were incubated with Aq protein and a fixed concentration of a biotinylated CLIP peptide. After the incubation, the Aq-bound CLIP peptide was detected in a time-resolved fluoroimmunoassay using europium-labeled streptavidin. The points represent the average of duplicates and error bars are set to ±one standard deviation. | ||
The T-cell response was investigated in a cell-based assay, where IL-2 secretion of Aq restricted T-cell hybridoma lines was measured after incubation with antigen-presenting spleen cells expressing Aq and increasing concentrations of the glycopeptides 1, 2 or 3 (Fig. 4). Four Aq-restricted T-cell hybridomas that differed in their TCR specificity for the hydroxyl groups of the galactose moiety were investigated for their ability to recognize the glycopeptides. Despite the fact that no binding of 2 and 3 to Aq was detected by the competition assay, both were recognized and stimulated IL-2 secretion by HCQ.3 and HM1R.2, while the other two hybridomas HCQ.10 and 22a1-7e did not give any response (Fig. 4). The HCQ.3 and HM1R.2 responses of 2 and 3 were significantly weaker compared to the responses elicited by both the non-modified 1 and ketomethylene-isosteric glycopeptide 4; the latter of which elicited intermediate to strong responses from three of the four hybridomas.14 This suggests that 2 and 3 do in fact bind to Aq and were presented to the T-cell receptor in the functional assay, although too weakly to be detected in the binding assay.
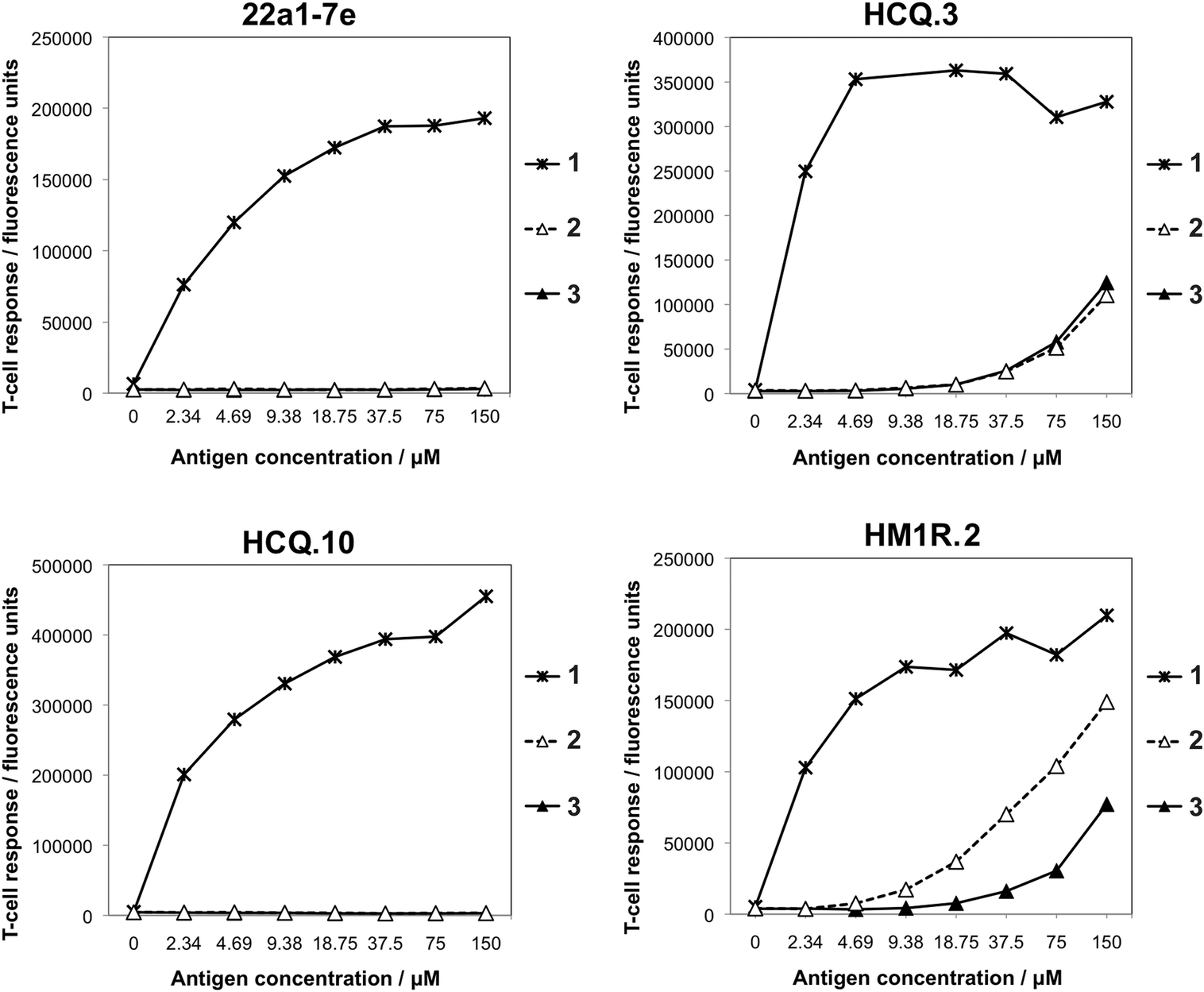 | ||
| Fig. 4 Recognition of glycopeptides 1–3 by the Aq-restricted T-cell hybridomas HCQ.3, HCQ.10, HM1R.2, and 22a1-7e after incubation with antigen-presenting spleen cells expressing Aq and increasing concentrations of 1–3. Recognition of the peptide/MHC complex by the T-cell hybridomas results in secretion of IL-2, which was quantified by a sandwich ELISA using the DELFIA system. | ||
That HCQ.10 and 22a1-7e were more sensitive towards the introduced modifications in the backbone compared to HCQ.3 and HMIR.2 has also been seen in previous studies.13,14
Analysis of glycopeptide/Aq-complexes by MD simulations
Glycopeptides 1–4 were modelled into Aq to understand if the structures of the complexes correlated with the binding affinity for Aq and with the subsequent T cell responses. We found that the modified glycopeptides 2–4 displayed a binding mode almost identical to that of the native glycopeptide 1 in their respective complexes. Similar interactions were retained for all four glycopeptides although the position of the hydrogen bond acceptor had been altered for the hydroxylethylene-isosteric glycopeptides 2 and 3 compared to the carbonyl oxygen in 1 and 4. As the static structures were unable to explain the observed differences in Aq binding and T cell responses the dynamics of the isostere modified glycopeptide/Aq-complexes and the native system were studied over time using MD simulations. Repeated 60 ns MD simulations at different initial velocities were conducted for 1 (two times) and the modified glycopeptides 2–4 (three times) in a complex with Aq, using the static models as starting structures. The repeated runs will hereafter be referred to as (I), (II) and (III) and can be seen as samples from the complete conformational landscape of the systems. In general, the simulations reached equilibrium after 40 ns according to the root-mean-square deviation (RMSD) values (see the ESI† for RMSD plots). The analyses of the MD simulations were then focused on the binding mode and flexibility of the glycopeptides, and the hydrogen bonding network between the modified glycopeptides and Aq over the last 20 ns of the repeated runs. The system of 1/Aq served as a reference to which the modified glycopeptides 2–4 have been compared.Flexibility of glycopeptide binding to Aq
During the simulations, glycopeptide 1 was bound in the groove-shaped binding site of Aq (Fig. 5). The N-terminus remained firmly anchored in the binding groove, mediated by a network of intermolecular hydrogen bonds and by positioning of the anchor residues Ile260 and Phe263 in the P1 and P4 pockets, respectively. The C-terminus on the other hand proved to be much more flexible and did not adopt a stable binding mode during the course of the simulations. This is consistent with previous MD simulations of the system,10,13 as well as the fact that the minimal epitope required for binding to Aq consists of the N-terminal octapeptide Ile260–Gln267.7 | ||
| Fig. 5 Snapshots from the MD-simulations comparing the mobility of the Aq/glycopeptide complexes of 1–3. (A) Superposition of frames extracted over the last 20 ns of the MD simulations, comparing native 1 (I) (in green, left) to 2 (I) (in orange, right). Glycopeptides 2 and 3 (data not shown) both display a higher mobility than the native glycopeptide 1. In order to extract representative snapshots, frames have been randomly selected from each of the top 10 clusters for the minimal epitope (Ile260–Gln267) over the last 20 ns of the MD simulations. (B) Comparison of structures of glycopeptides 1–3 bound to Aq. The representative snapshots have been randomly selected from the most highly populated cluster for the minimal epitope (Ile260–Gln267) over the last 20 ns of the MD simulations. In contrast to native 1, isosteric glycopeptides 2 and 3 both dissociate from the binding groove of Aq, here illustrated by comparison of the structure of 1 (I) (green) to 2 (III) (in orange, left) and 3 (II) (in purple, right). A complete list with figures displaying superposed frames of the complexes from the last 20 ns of each MD simulations can be found in the ESI†. See the Experimental section for details on the clustering. The roman numbers I, II and III refer to the repeated MD simulations conducted for the same glycopeptide. | ||
The S- and R-hydroxyethylene-isosteric glycopeptides 2 and 3 displayed a different dynamic pattern compared to 1, with large variations between the repeated MD simulations (Table 1 and ESI†). The S-hydroxyethylene-isostere 2 showed larger movements; the N-terminus of the glycopeptide was not as firmly anchored in the binding groove as for 1 and displayed a high degree of flexibility (Fig. 5 and the ESI†). In fact, the anchor residue Phe263 and occasionally also Ile260 were leaving their respective binding pockets. In addition, a clear unfolding was observed in the β1-helix at the point of the modification of the peptide. The R-hydroxyethylene-isostere 3 also displayed larger flexibility; again both anchor residues were leaving their respective binding pockets and the β1-helix was unfolded adjacent to the isostere in 3 (Fig. 5). The simulations of the ketomethylene isostere 4 in complex with Aq displayed a similar dynamic behaviour as the native glycopeptide 1, including firmly anchored side chains of Phe263 and Ile260 and a stable β1-helix.

The results of the repeated MD simulations are different scenarios or time periods. While some of the MD simulations for 2 and 3 displayed a behaviour indicative of a poor binder about to completely dissociate from Aq other simulations indicated a plausible binder instead. It should be noted that in previous MD simulations10,13 of the native glycopeptide 1, we have not observed a behaviour similar to 2 and 3, with the peptide dissociating from the binding groove, anchor residues leaving the P1 and P4 pockets, or unfolding of the β1-helix.
Hydrogen bond network
An extensive hydrogen bond network anchored glycopeptide 1 into Aq during the molecular dynamics simulations. In total, 13 different hydrogen bonds were observed between the backbone of 1 and Aq, and 7 of these had over 40% occupancy during the simulation (Tables 1 and 2). The majority of hydrogen bonds were formed between the N-terminal part of 1 (Gly259–Gly265) and Aq, which was consistent with previous findings.13,14 In this sequence, 8 out of 13 intermolecular hydrogen bonds were present, of which 5 were considered strong bonds. In particular two hydrogen bonds showed a high presence during the MD simulations, Ala261(a)–Asn82(s) and Gln267(a)–Trp61(s) (Table 2).| Glycopeptide residue (backbone)a | Aq residueb | 1 | 2 | 3 | 4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ic | II | I | II | III | I | II | III | I | II | III | ||
| a a = hydrogen bond acceptor, d = hydrogen bond donor. b s = sidechain in Aq, m = main chain in Aq. c I, II and III represent the different MD simulations conducted for the same glycopeptide. d The hydrogen bond occupancy has been calculated with the aid of the hydrogen bond extension tool in VMD23 with a donor–acceptor length of less than 3.3 Å and a donor–H–acceptor angle of minimum 160°. Only hydrogen bonds with an occupancy >10% are reported. e — = hydrogen bond occupancy <10%, the occupancy is therefore not reported. f The two hydrogen bonds that showed a high presence during the MD simulations; Ala261(a)–Asn82(s) and Gln267(a)–Trp61(s), are marked in bold. g * = not applicable. h [OH] represents the hydroxyl group in the hydroxyethylene-isosteric glycopeptides 2 and 3. | ||||||||||||
| Gly259 (a) | βHis81 (s) | 22d | 46 | —e | — | 11 | — | — | — | 34 | — | 19 |
| Ile260 (a) | αAsp56 (m) | — | — | — | — | — | — | 22 | — | — | — | — |
| Ile260 (d) | αThr53 (m) | — | — | — | — | 13 | — | — | — | — | — | — |
| Ile260 (d) | αLeu52 (m) | — | — | — | — | — | — | — | — | — | — | 14 |
| Ile260 (d) | αSer54 (m) | 16 | 16 | — | — | — | — | 17 | 23 | 29 | 23 | — |
| Ala 261 (a) | βAsn 82 (s) | 67 | 70 | *g | * | * | * | * | * | 66 | 66 | 74 |
| Ala261 [OH]h (a) | βAsn82 (s) | * | * | — | 33 | — | — | — | — | * | * | * |
| Ala261 [OH] (d) | βThr77 (m) | * | * | 24 | — | — | — | 10 | — | * | * | * |
| Ala261 [OH] (d) | βVal78 (m) | * | * | — | 57 | — | — | — | — | * | * | * |
| Ala261 (d) | βHis81 (s) | — | — | — | — | — | — | — | — | — | 11 | — |
| Ala261 (d) | βAsn82 (s) | 26 | 18 | — | 36 | — | 26 | — | — | 40 | — | 61 |
| Ala261 [OH] (d) | βAsn82 (s) | * | * | — | — | — | 42 | — | — | * | * | * |
| Phe263 (a) | αTyr23 (s) | 70 | 19 | — | — | — | 67 | — | 39 | — | — | — |
| Phe263 (a) | αAsn63 (s) | — | — | 22 | — | 49 | — | 16 | — | — | — | — |
| Phe263 (d) | αTyr9 (m) | 42 | 39 | 22 | 40 | — | 18 | — | 51 | 21 | — | 57 |
| Lys264 (d) | βGlu74 (s) | — | — | — | — | — | — | — | — | 36 | — | — |
| Gly265 (a) | αThr66 (s) | — | — | 18 | — | — | — | — | — | — | 20 | — |
| Gly265 (a) | αAsn63 (s) | 25 | — | — | — | — | — | — | — | — | — | — |
| Gly265 (a) | βArg70 (s) | — | — | — | — | — | — | — | — | — | — | 11 |
| Gly265 (d) | αAsn63 (s) | 26 | 46 | 18 | 33 | — | 44 | — | 31 | — | 48 | — |
| Glu266 (a) | αThr66 (s) | — | — | — | — | 14 | — | — | — | — | — | 57 |
| Glu266 (a) | αAsn70 (s) | 34 | — | — | 51 | 39 | 70 | 36 | 63 | 65 | 52 | — |
| Glu266 (d) | βTyr30 (s) | 11 | — | — | 18 | — | 24 | 16 | — | — | 41 | — |
| Gln 267 (a) | βTrp 61 (s) | 61 | 60 | 24 | 38 | 52 | 67 | 65 | 57 | 61 | 64 | — |
| Gly268 (a) | αHis69 (s) | — | — | — | — | — | 18 | — | 15 | 21 | 10 | — |
| Gly268 (d) | αGln62 (m) | — | — | — | — | — | — | — | — | — | — | 29 |
| Gly268 (d) | αHis69 (s) | |||||||||||
| Gly268 (d) | αAsn70 (s) | 49 | — | — | — | 36 | 47 | 21 | 36 | 38 | 38 | — |
| Pro269 (a) | αHis69 (s) | 23 | — | 13 | — | — | — | 12 | — | — | 11 | — |
In the case of the modified glycopeptides 2 and 3, insertion of the hydroxyethylene isostere at the Ala261–Gly262 position disturbed the overall hydrogen bond pattern during the MD simulations (Tables 1 and 2). Although hydrogen bonds could still be formed with the hydroxyl group of the isostere, these were weak, and the remaining hydrogen bond network between the two glycopeptides and Aq was disrupted causing the number of strong hydrogen bonds to show a large variation over time between MD simulations. A weak hydrogen bond was formed with Asn82(s) only in one MD-run, and thus no strong hydrogen bond corresponding to the strong Ala261(a)–Asn82(s) hydrogen bond in the 1/Aq complex was observed. The ketomethylene isostere introduced in 4, on the other hand, maintained the Ala261(a)–Asn82(s) hydrogen bond (Table 2). The pattern of strong hydrogen bonds observed for 4 was more consistent with the pattern seen in the complex of 1 and Aq, despite the loss of hydrogen bond donation ability of Gly262(d); a finding that agrees well with the observation that no hydrogen bond was formed with this residue in the MD simulations of native 1.14
The disrupted hydrogen bond networks seen in the dynamics analyses of 2 and 3, as compared with 1 and 4, resulted in the larger flexibility as seen for those glycopeptides and their dissociation from the binding groove of Aq discussed in the previous section. It is also likely that the altered hydrogen bonding was connected to the unfolding of the β1-helix observed in the complexes of 2 and 3. Thus, the dynamics calculations point to the fact that loss of one important hydrogen bond has a tremendous effect on both the structure and the stability of the entire complex. This loss most likely explains the dramatic difference in binding to Aq between isosteres 2 and 3 and native 1 as seen in the competitive binding assay. Retention of the hydrogen bond between Gln267(a)–Trp61(s), which has a high occupancy for all four glycopeptides (Table 2), may explain the weak binding of 2 and 3 to Aq as revealed by the stimulation of two of the four T cell hybridomas.
In conclusion, the MD-simulations displayed a far more detailed picture than the static models, and emphasized the importance of studying the dynamics to understand the behaviour and evolution over time of the Aq–glycopeptide complexes. While the binding mode for the native glycopeptide 1 and the ketomethylene-isosteric glycopeptide 4 was similar to the static model, the hydroxyethylene-isosteres 2 and 3 displayed a behaviour deviating from what could be seen in static models. Studies of the dynamics of Aq–glycopeptide complexes, but not static models, were therefore crucial to explain the results from the in vitro studies of class II MHC binding and T cell recognition.
Alteration of the epitope presented to the T cell
As described above, the T-cell response patterns elicited by the modified glycopeptides 2 and 3 differed from that of 1, being reduced for two hybridomas and not detectable for the other two. A reduced or eliminated T-cell response may be due to either a weaker binding to Aq, and/or that the epitope presented to the TCR has been changed. We conclude that the finding that two of the hybridomas respond weakly to 2 and 3, while the other two do not respond at all, suggests that both effects are in operation. This conclusion is further supported by the MD simulations that showed increased flexibility of 2 and 3, relative to 1, in their complexes with Aq and dissociation from the MHC binding groove indicating a reduced affinity. The increased flexibility, and the predicted unfolding of the β1-helix of Aq, also suggests that the epitope presented to the T-cell has been altered, including the position of the GalHyl264, an important TCR contact point.4,11 Thus, even a small modification at the Ala261–Gly262 amide bond, not itself directly in contact with the TCR, had a major impact on the T-cell response pattern.Conclusions
Different amide bond isosteres display different properties and are valuable tools to probe interactions in complexes between peptides and proteins. With this in mind, we have further explored the effect of replacing the Ala261–Gly262 amide bond in the glycopeptide CII259–273 (1) with isosteres upon binding to the Aq class II MHC molecule and subsequent T cell recognition of the complex. The results revealed an unexpected fine specificity as hydroxyethylene-isosteric glycopeptides 2 and 3 displayed a substantial loss of Aq binding and T-cell response compared with 1. In contrast, the previous introduction of a ketomethylene-isostere at the same position, to give glycopeptide 4, resulted in maintained affinity for Aq and T-cell responses was maintained.14 Static models of the complexes of 1 and isosteric glycopeptides 2–4 with Aq displayed comparable binding modes and thus did not provide any insight into the observed effects on isostere introduction. MD simulations, however, demonstrated that hydroxyethylene isosteric glycopeptides 2 and 3, in contrast to 1 and 4, were more flexible in their complexes with Aq and also prone to dissociation from the binding groove. In addition, glycopeptides 2 and 3 led to unfolding of the β1-helix of Aq. Analysis of the Aq/glycopeptide complexes revealed that replacement of the Ala261–Gly262 amide bond with hydroxyethylene-isosteres disrupted the hydrogen bond network in the complexes. This, in turn, led to the altered structure and dynamics and explains the dramatic effects on the Aq binding and the T cell recognition.Experimental section
Chemistry
:1) with H2O (δH 4.76) as an internal standard. HRMS data were recorded with fast atom bombardment (FAB+) ionization. Analytical reversed-phase HPLC was performed on a Beckman System Gold HPLC equipped with a Supelco Discovery® Bio Wide Pore C18 column (250 × 4.6 mm, 5 μm) using a flow-rate of 1.5 mL min−1 and detection at 214 nm. Preparative reversed-phase HPLC was performed by using a Supelco Discovery® Bio Wide Pore C18 column (250 × 21.2 mm, 5 μm) using the same eluent as for the analytical HPLC, a flow rate of 11 mL min−1, and detection at 214 nm. Compound 514,24 and glycopeptide 14 were synthesized as described in the cited references.
(4R,5S)-5-tert-Butoxycarbonylamino-4-hydroxy-hexanoic acid tert-butyl ester (6). Ketone 514,24 (160 mg, 0.531 mmol) dissolved in EtOH (8.0 mL) was added dropwise to a solution of LiAlH(O-tBu)3 (278 mg, 1.06 mmol) in EtOH (5.5 mL) at −78 °C. After stirring for 150 min at −78 °C, a second portion of LiAlH(O-tBu)3 (28 mg, 0.107 mmol) was added. After stirring for additional 60 min, citric acid (10%, aq.) was added and the mixture was warmed to rt followed by addition of EtOAc and H2O. The aqueous phase was extracted with EtOAc and the combined organic layers were washed with H2O and brine, dried and concentrated under reduced pressure. The crude was purified by flash chromatography (n-heptane/EtOAc 3
:1) to afford 6 (4S:4R 1:99, 122 mg, 75%) as a white solid. [α]20D = −19.5 (c = 1.0, CHCl3); 1H NMR (CDCl3) δ 4.77 (br s, 1H, NH), 3.76–3.63 (m, 1H, NCH), 3.63–3.56 (m, 1H, HOCH), 2.48–2.32 (m, 2H, CH2CO2), 1.73–1.57 (m, 2H, CH2CH2CO2), 1.43 (s, 9H, C(CH3)3), 1.42 (s, 9H, C(CH3)3), 1.10 (d, J = 6.8 Hz, 3H, CH3); 13C NMR (CDCl3) δ 173.8 (CO2), 156.0 (NCO), 80.6 (C(CH3)3), 79.5 (C(CH3)3), 74.3 (HOCH), 51.0 (NCH), 32.4 (CH2CO2), 28.4 (C(CH3)3), 28.1 (CH2CH2CO2), 28.1 (C(CH3)3), 14.9 (CH3).
(4S/R,5S)-5-tert-Butoxycarbonylamino-4-hydroxy-hexanoic acid tert-butyl ester (7). Ketone 5 (269 mg, 0.892 mmol) was dissolved in dry THF (22 mL) and cooled to −78 °C followed by dropwise addition of (S)-alpine-hydride (0.5 M in THF, 1.96 mL, 0.980 mmol). After stirring for 80 min at −78 °C, citric acid (10%, aq.) was added and the mixture was allowed to reach rt followed by the addition of Et2O and brine. The aqueous phase was extracted with Et2O and the combined organic layers were dried and concentrated under reduced pressure. The crude was purified by flash chromatography (n-heptane/EtOAc 5
:1 → 2:1) to afford 7 (4S:4R 2:1, 227 mg, 84%) as a colorless oil. (4S)-7 (major isomer): 1H NMR (CDCl3) δ 4.74 (br s, 1H, NH), 3.66–3.57 (m, 1H, NCH), 3.53–3.47 (m, 1H, HOCH), 2.47–2.33 (m, 2H, CH2CO2), 1.81–1.60 (m, 2H, CH2CH2CO2), 1.43 (s, 18H, C(CH3)3), 1.17 (d, J = 6.8 Hz, 3H, CH3); 13C NMR (CDCl3) δ 173.9 (CO2), 156.2 (NCO), 80.7 (C(CH3)3), 79.4 (C(CH3)3), 74.6 (HOCH), 50.7 (NCH), 32.3 (CH2CO2), 29.3 (CH2CH2CO2), 28.4 (C(CH3)3), 28.1 (C(CH3)3), 18.3 (CH3). (4R)-7 (minor isomer): 1H NMR (CDCl3) δ 4.74 (br s, 1H, NH), 3.75–3.66 (m, 1H, NCH), 3.66–3.57 (m, 1H, HOCH), 2.47–2.20 (m, 2H, CH2CO2), 1.81–1.60 (m, 2H, CH2CH2CO2), 1.43 (s, 18H, C(CH3)3), 1.10 (d, J = 6.8 Hz, 3H, CH3); 13C NMR (CDCl3) δ 173.8 (CO2), 156.2 (NCO), 80.7 (C(CH3)3), 79.5 (C(CH3)3), 74.4 (HOCH), 51.0 (NCH), 32.5 (CH2CO2), 29.3 (CH2CH2CO2), 28.4 (C(CH3)3), 28.1 (C(CH3)3), 14.9 (CH3).
(5S)-5-[(1R)-1-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethyl]-dihydro-furan-2-one (12). Alcohol 6 (137 mg, 0.452 mmol) dissolved in CH2Cl2 (2.2 mL) was treated with TFA (0.9 mL) at rt for 24 h. The solution was concentrated under reduced pressure, the resulting residue was concentrated from CHCl3 three times and was thereafter dissolved in dioxane (2.2 mL) and H2O (0.8 mL). FmocOSu (187 mg, 0.542 mmol) was added and the mixture was cooled to 0 °C followed by dropwise addition of Na2CO3 (58 mg, 0.542 mmol) dissolved in H2O (0.35 mL). The mixture was allowed to reach rt and was stirred for 24 h before dioxane was removed under reduced pressure. CHCl3 and H2O were added and the aqueous phase was acidified by the addition of HCl (10%, aq.) at 0 °C to pH 1. The phases were separated and the aqueous phase was extracted with CHCl3. The combined organic layers were washed with brine, dried and concentrated under reduced pressure. The resulting crude was purified by flash chromatography (n-heptane/EtOAc/AcOH 2
:1:1% → 1:1:1%) to afford 8 (136 mg, 86%). [α]20D = −32.8 (c = 1.0, CHCl3); 1H NMR (CD3OD) δ 7.78 (d, J = 7.4 Hz, 2H, Fmoc-arom), 7.62 (d, J = 7.4 Hz, 2H, Fmoc-arom), 7.40–7.34 (m, 2H, Fmoc-arom), 7.33–7.26 (m, 2H, Fmoc-arom), 4.47–4.31 (m, 3H, Fmoc-CH2, OCH), 4.18 (t, J = 6.4 Hz, 1H, Fmoc-CH), 3.85–3.75 (m, 1H, NCH), 2.56–2.46 (m, 2H, CH2CO2), 2.28–2.14 (m, 1H, OCHCH2), 2.05–1.92 (m, 1H, OCHCH2), 1.17 (d, J = 7.0 Hz, 3H, CH3); 13C NMR (CD3OD) δ 179.5 (CO2), 158.2 (NCO), 145.1 (splitted), 142.5, 128.6, 128.0, 126.0 (splitted), 120.8, 84.2 (OCH), 67.4 (Fmoc-CH2), 50.7 (NCH), 48.4 (Fmoc-CH), 29.0 (CH2CO2), 24.8 (OCHCH2), 16.5 (CH3).
(5S)-5-[(1S/R)-1-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethyl]-dihydro-furan-2-one (9). Alcohol 7 (188 mg, 0.621 mmol) dissolved in CH2Cl2 (3.0 mL) was treated with TFA (1.3 mL) at rt for 19 h. The solution was concentrated under reduced pressure, the resulting residue was concentrated from CHCl3 three times and was thereafter dissolved in dioxane (3.0 mL) and H2O (1.2 mL). FmocOSu (257 mg, 0.746 mmol) was added and the mixture was cooled to 0 °C followed by dropwise addition of Na2CO3 (79 mg, 0.746 mmol) dissolved in H2O (0.3 mL). The mixture was allowed to reach rt and was stirred for 22 h before dioxane was removed under reduced pressure. H2O and CHCl3 were added, the phases were separated and the aqueous phase was extracted with CHCl3. The combined organic layers were washed with brine, dried and concentrated under reduced pressure. The resulting crude was purified by flash chromatography (n-heptane/EtOAc 2
:1 → 1:1) followed by purification by chiral preparative HPLC (Chiralpak AD column, 250 × 20 mm, 5 μm, with n-heptane/EtOH 70:30 as the eluent, 18 mL min−1 flow rate and UV detection at 300 nm) to afford 9 (4S:4R 99:1, 108 mg, 48%). H1 NMR (CD3OD) δ 7.79 (d, J = 7.6 Hz, 2H, Fmoc-arom), 7.65 (d, J = 7.5 Hz, 2H, Fmoc-arom), 7.41–7.36 (m, 2H, Fmoc-arom), 7.34–7.27 (m, 2H, Fmoc-arom), 4.52–4.45 (m, 2H, Fmoc-CH2, OCH), 4.39–4.32 (m, 1H, Fmoc-CH2), 4.25–4.18 (m, 1H, Fmoc-CH), 3.87–3.79 (m, 1H, NCH), 2.61–2.47 (m, 1H, CH2CO2), 2.46–2.35 (m, 1H, CH2CO2), 2.26–2.15 (m, 1H, CH2CH2CO2), 1.99–1.85 (m, 1H, CH2CH2CO2), 1.19 (d, J = 6.9 Hz, 3H, CH3). A 13C NMR spectrum could not be obtained due to degradation during NMR analysis.
(4R,5S)-4-(tert-Butyl-dimethyl-silanyloxy)-5-(9H-fluoren-9-ylmethoxy-carbonylamino)-hexanethioic acid tert-butyl ester (10). AlMe3 (1 M in heptane, 2.50 mL, 2.50 mmol) was added dropwise to a solution of t-BuSH (0.28 mL, 2.53 mmol) in CH2Cl2 (3.6 mL) at 0 °C. The mixture was stirred for 10 min at 0 °C, 10 min at rt followed by 10 min at 0 °C. Lactone 8 (106 mg, 0.301 mmol) dissolved in CH2Cl2 (1.9 m L) was added dropwise followed by stirring for 3 h and 30 min at 0 °C. The reaction was cooled to −78 °C and Et2O (11 mL) was added followed by dropwise addition of HCl (1 M, 3 mL). The resulting mixture was then stirred for 10 min before it was allowed to reach 0 °C. The workup was performed under cold conditions to prevent relactonization using ice-cold solvent and aqueous solutions. After addition of Et2O and HCl (1 M), the layers were separated, and the aqueous layer was extracted with Et2O. The combined organic layers were washed with HCl (1 M, aq.), NaHCO3 (satd, aq.) and brine, dried and concentrated under reduced pressure under cold conditions. The resulting crude product was mixed with TBDMSCl (197 mg 1.27 mmol) and imidazole (173 mg, 2.54 mmol), and dissolved in DMF (0.27 mL). After stirring for 20 h and 30 min at rt, NaHCO3 was added followed by the addition of H2O and CH2Cl2. The phases were separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with H2O and brine, dried, and concentrated under reduced pressure. Purification by flash chromatography twice (first using n-heptane/EtOAc 8
:1, then using n-heptane/EtOAc 1:0 → 8:1) gave 10 (93 mg, 55%) as a colorless oil. [α]20D = −7.2 (c = 1.0, CHCl3); 1H NMR (CDCl3) δ 7.76 (d, J = 7.4 Hz, 2H, Fmoc-arom), 7.62–7.56 (m, 2H, Fmoc-arom), 7.43–7.37 (m, 2H, Fmoc-arom), 7.35–7.28 (m, 2H, Fmoc-arom), 4.85–4.76 (m, 1H, NH), 4.44–4.32 (m, 2H, Fmoc-CH2), 4.21 (t, J = 6.7 Hz, 1H, Fmoc-CH), 3.78–3.65 (m, 2H, NCH, OCH), 2.60–2.39 (m, 2H, CH2COS), 1.80–1.70 (m, 2H, CH2CH2COS), 1.46 (s, 9H, C(CH3)3), 1.10 (d, J = 5.5 Hz, 3H, CH3), 0.91 (s, 9H, C(CH3)3), 0.08 (s, 3H, SiCH3), 0.04 (s, 3H, SiCH3); 13C NMR (CDCl3) δ 199.7 (COS), 155.6 (NCO), 144.0 (splitted), 141.3, 127.6, 127.0, 125.0, 120.0, 73.5 (SiOCH), 66.5 (Fmoc-CH2), 50.1 (NCH), 48.0 (C(CH3)3), 47.3 (Fmoc-CH), 40.5 (CH2COS), 29.8 (C(CH3)3), 29.4 (CH2CH2COS), 25.9 (C(CH3)3), 18.1 (C(CH3)3), 14.2 (CH3), −4.2 (SiCH3), −4.6 (SiCH3).
(4S,5S)-4-(tert-Butyl-dimethyl-silanyloxy)-5-(9H-fluoren-9-ylmethoxy-carbonylamino)-hexanethioic acid tert-butyl ester (12). AlMe3 (2 M in heptane, 1.15 mL, 2.30 mmol) was added dropwise to a solution of t-BuSH (0.26 mL, 2.31 mmol) in CH2Cl2 (2.5 mL) at 0 °C. The mixture was stirred for 10 min at 0 °C, 10 min at rt followed by 10 min at 0 °C. Lactone 9 (96 mg, 0.273 mmol) dissolved in CH2Cl2 (3.5 mL) was added dropwise followed by stirring for 3 h at 0 °C. The solution was cooled to −78 °C and Et2O (10 mL) was added followed by dropwise addition of HCl (1 M, 2.7 mL). The resulting mixture was then stirred for 10 min before it was allowed to reach 0 °C. The workup was performed using ice-cold solvent and aqueous solutions to prevent relactonization. After addition of Et2O and HCl (1 M), the phases were separated, and the aqueous layer was extracted with Et2O. The combined organic layers were washed with HCl (1 M, aq.), NaHCO3 (satd, aq.) and brine, dried and concentrated under reduced pressure and cold conditions. The resulting crude product was immediately mixed with TBDMSCl (182 mg 1.20 mmol) and imidazole (157 mg, 2.30 mmol) and dissolved in DMF (1.0 mL). After stirring for 16 h at rt, NaHCO3 (aq., satd.) was added followed by the addition of H2O and CH2Cl2. The phases were separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with H2O and brine, dried, and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc 1
:0 → 8:1) gave 12 (102 mg, 85%) as a colorless oil. [α]20D = +8.9 (c = 1.0, CHCl3); 1H NMR (CDCl3) δ 7.77 (d, J = 7.5 Hz, 2H, Fmoc-arom), 7.60 (d, J = 7.5 Hz, 2H, Fmoc-arom), 7.43–7.37 (m, 2H, Fmoc-arom), 7.34–7.29 (m, 2H, Fmoc-arom), 4.87 (d, J = 8.9 Hz, 1H, NH), 4.46–4.39 (m, 1H, Fmoc-CH2), 4.39–4.31 (m, 1H, Fmoc-CH2), 4.25 (t, J = 7.0 Hz, 1H, Fmoc-CH), 3.83–3.73 (m, 1H, NCH), 3.68–3.61 (m, 1H, OCH), 2.50 (t, J = 7.5 Hz, 2H, CH2CO), 1.82–1.67 (m, 2H, CH2CH2O), 1.45 (s, 9H, SC(CH3)3), 1.15 (d, J = 6.7 Hz, 3H, CH3), 0.93 (s, 9H, SiC(CH3)3), 0.11 (s, 3H, SiCH3), 0.10 (s, 3H, SiCH3); 13C NMR (CDCl3) δ 199.9 (COS), 155.9 (NCO), 144.0, 141.3, 127.6, 127.0, 125.1 (splitted), 119.9, 73.4 (CHO), 66.6 (Fmoc-CH2), 49.1 (NCH), 47.9 (SC(CH3)3), 47.3 (Fmoc-CH), 40.5 (CH2CO), 29.8 (SC(CH3)3), 29.2 (CH2CH2CO), 25.9 (SiC(CH3)3), 18.1 (SiC(CH3)3), 17.9 (CH3), −4.3 (SiCH3), −4.6 (SiCH3).
(4R,5S)-4-(tert-Butyl-dimethyl-silanyloxy)-5-(9H-fluoren-9-ylmethoxy-carbonylamino)-hexanoic acid (11). A premixed solution of H2O2 (30% aq. solution, 2.70 mL) and LiOH (0.2 M, 1.38 mL, 0.276 mmol) was added dropwise to 10 (81 mg, 0.145 mmol) dissolved in THF (10 mL) at 0 °C. The mixture was allowed to reach rt followed by stirring for 3.5 h and then it was cooled to 0 °C. Na2SO3 (2 M, 5 mL) was added dropwise followed by stirring for 40 min at 0 °C. The mixture was acidified by the addition of HCl (10%, aq.), Et2O was added, the phases were separated and the aqueous layer was extracted with Et2O. The combined organic layers were washed with NaHCO3 (satd, aq.), dried and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc/AcOH 6
:1:1% → 3:1:1%) gave 11 (53 mg, 75%) as a white solid. HRMS (FAB+) calcd for C27H36NNa2O5Si+ (M − H + 2Na)+ 528.2153, found 528.2178. [α]20D = −7.7 (c = 0.5, CHCl3); H1 NMR (CD3OD) δ 7.78 (d, J = 7.4 Hz, 2H, Fmoc-arom), 7.65 (d, J = 7.4 Hz, 2H, Fmoc-arom), 7.41–7.35 (m, 2H, Fmoc-arom), 7.33–7.27 (m, 2H, Fmoc-arom), 6.73 (d, J = 8.0 Hz, 1H, NH), 4.43–4.28 (m, 2H, Fmoc-CH2), 4.19 (t, J = 6.6 Hz, 1H, Fmoc-CH), 3.79–3.70 (m, 1H, OCH), 3.66–3.55 (m, 1H, NCH), 2.43–2.30 (m, 2H, CH2CO2), 1.80–1.72 (m, 2H, CH2CH2CO2), 1.10 (d, J = 6.6 Hz, 3H, CH3), 0.90 (s, 9H, C(CH3)3), 0.07 (s, 3H, SiCH3), 0.05 (s, 3H, SiCH3); 13C NMR (MeOD) δ 177.3 (CO2H), 158.2 (splitted, NCO), 145.4, 142.6, 128.7, 128.1, 126.2, 120.9, 74.8 (OCH), 67.5 (Fmoc-CH2), 51.5 (splitted, NCH), 48.5 (Fmoc-CH), 30.3 (CH2CO2), 29.7 (CH2CH2CO2), 26.4 (C(CH3)3), 18.9 (C(CH3)3), 16.1 (CH3), −4.2 (SiCH3), −4.3 (SiCH3).
(4S,5S)-4-(tert-Butyl-dimethyl-silanyloxy)-5-(9H-fluoren-9-ylmethoxy-carbonylamino)-hexanoic acid (13). A premixed solution of H2O2 (30% aq., 3.0 mL) and LiOH (0.2 M, 1.5 mL, 0.300 mmol) was added dropwise to 12 (91 mg, 0.165 mmol) in THF (11 mL) at 0 °C. The mixture was allowed to reach rt followed by stirring for 3.5 h and then it was cooled to 0 °C. Na2SO3 (2 M, 5.6 mL) was added dropwise followed by stirring for 40 min at 0 °C. The mixture was acidified by addition of HCl (10% aq.), Et2O was added, the phases were separated and the aqueous phase was extracted with Et2O. The combined organic layers were washed with NaHCO3 (satd, aq.), dried and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc/AcOH 6
:1:1%) gave 13 (65 mg, 82%) as a white solid. HRMS (FAB+) calcd for C27H36NNa2O5Si+ (M − H + 2Na)+ 528.2153, found 528.2175. [α]20D = +5.5 (c = 1.0, CHCl3); H1 NMR (CD3OD) δ 7.79 (d, J = 7.5 Hz, 2H, Fmoc-arom), 7.68–7.63 (m, 2H, Fmoc-arom), 7.41–7.35 (m, 2H, Fmoc-arom), 7.33–7.27 (m, 2H, Fmoc-arom), 6.73 (d, J = 8.1 Hz, 1H, NH), 4.42 (dd, J = 10.5, 6.8 Hz, 1H, Fmoc-CH2), 4.32 (dd, J = 10.4, 6.8 Hz, 1H, Fmoc-CH2), 4.23–4.18 (m, 1H, Fmoc-CH), 3.76–3.67 (m, 2H, NCH and NCHCH), 2.42–2.26 (m, 2H, CH2CO2), 1.80–1.69 (m, 1H, CH2CH2CO2), 1.69–1.58 (m, 1H, CH2CH2CO2), 1.09 (d, J = 6.5 Hz, 3H, CH3), 0.92 (s, 9H, C(CH3)3), 0.11 (s, 3H, SiCH3), 0.10 (s, 3H, SiCH3); 13C NMR (MeOD) δ 177.4 (CO2H), 158.2 (NCO, splitted), 145.3 (splitted), 142.6, 128.7, 128.1 (splitted), 126.2 (splitted), 120.9, 74.5 (NCHCH), 67.5 (Fmoc-CH2), 51.4 (NCH, splitted), 48.6 (Fmoc-CH), 31.3 (CH2CO2), 28.4 (CH2CH2CO2), 26.4 (C(CH3)3), 18.9 (C(CH3)3), 15.4 (CH3), −4.3 (SiCH3), −4.4 (SiCH3).
:2:2:1, 40 mL) for 3 h at 40 °C with workup performed essentially as described elsewhere.25 Purification by reversed-phase HPLC was followed by deacetylation with NaOMe in MeOH (20 mM, 1 mL per mg peptide) for 2–3 h at rt (monitored by analytical reversed-phase HPLC). The mixture was neutralized by addition of AcOH and concentrated under reduced pressure, and the residue was finally purified using reversed-phase HPLC followed by lyophilization.
Glycyl-L-isoleucyl-L-alanylψ[(4S)-CHOHCH2]glycyl-L-phenylalanyl-(5R)-5-O-(β-D-galactopyranosyl)-5-hydroxy-L-lysylglycyl-Lglutam-1-yl-L-glutaminylglycyl-L-prolyl-L-lysylglycyl-L-glutam-1-yl-L-threonine (2). Synthesis was performed with building block 13 according to the general procedure described above. This afforded the trifluoroacetate salt of 2 (12.4 mg, 18% yield based on the amount of resin used) as a white amorphous solid after lyophilization. MS (MALDI-TOF) calcd 1654.82 for [M + H]+, found 1654.82. 1H NMR data are given in Table 3.
| Residue | NH | Hα | Hβ | Hγ | Others |
|---|---|---|---|---|---|
| a Measured at 500 MHz and 298 K in water containing 10% D2O with H2O (δH 4.76 ppm) as an internal standard. b Degeneracy has been assumed. c Chemical shifts for the galactose moiety: δ 4.43 (H1), 3.91 (H4), 3.76 (H6), 3.68 (H5), 3.64 (H3), and 3.52 (H2). | |||||
| Gly259 | 3.86b | ||||
| Ile260 | 8.39 | 4.17 | 1.81 | 1.41, 1.14, 0.88 (CH3) | 0.84 (Hδ) |
| Ala261ψ[CHOHCH2] | 7.92 | 3.86 | 1.10 | 3.41 ψ[CHOHCH2], 1.55b ψ[CHOHCH2] | |
| Gly262 | 2.31b | ||||
| Phe263 | 8.20 | 4.62 | 3.10, 2.98 | 7.25 (Hδ), 7.35 (Hε), 7.31 (Hζ) | |
| Hyl264 | 8.40 | 4.29 | 2.00, 1.75 | 1.61b | 4.02 (Hδ), 3.18 and 2.98 (Hε), 7.62 (εNH2)c |
| Gly265 | 8.00 | 3.92b | |||
| Glu266 | 8.23 | 4.37 | 2.10, 1.94 | 2.43b | |
| Gln267 | 8.49 | 4.37 | 2.10, 1.96 | 2.35b | 7.50 and 6.83 (δNH2) |
| Gly268 | 8.27 | 4.11, 3.98 | |||
| Pro269 | 4.40 | 2.26, 1.90 | 2.00b | 3.58b (Hδ) | |
| Lys270 | 8.45 | 4.29 | 1.84, 1.75 | 1.43b | 1.65b (Hδ), 3.00b (Hε), 7.50 (εNH2) |
| Gly271 | 8.35 | 3.94b | |||
| Glu272 | 8.21 | 4.47 | 2.14, 1.96 | 2.45b | |
| Thr273 | 8.09 | 4.33 | 4.33 | 1.16 | |
Glycyl-L-isoleucyl-L-alanylψ[(4R)-CHOHCH2]glycyl-L-phenylalanyl-(5R)-5-O-(β-D-galactopyranosyl)-5-hydroxy-L-lysylglycyl-L-glutam-1-yl-L-glutaminylglycyl-L-prolyl-L-lysylglycyl-L-glutam-1-yl-L-threonine (3). Synthesis was performed with building block 11 according to the general procedure described above. This afforded the trifluoroacetate salt of 3 (22.5 mg, 28% yield based on the amount of resin used) as a white amorphous solid after lyophilization. MS (MALDI-TOF) calcd 1654.82 for [M + H]+, found 1654.81. 1H NMR data are given in Table 4.
| Residue | NH | Hα | Hβ | Hγ | Others |
|---|---|---|---|---|---|
| a Measured at 500 MHz and 298 K in water containing 10% D2O with H2O (δH 4.76 ppm) as an internal standard. b Degeneracy has been assumed. c Chemical shifts for the galactose moiety: δ 4.43 (H1), 3.92 (H4), 3.76 (H6), 3.68 (H5), 3.64 (H3), and 3.52 (H2). | |||||
| Gly259 | 3.84b | ||||
| Ile260 | 8.41 | 4.13 | 1.81 | 1.43, 1.16, 0.88 (CH3) | 0.84 (Hδ) |
| Ala261ψ[CHOHCH2] | 8.11 | 3.82 | 1.08 | 3.49 ψ[CHOHCH2], 1.67 and 1.47 ψ[CHOHCH2] | |
| Gly262 | 2.33, 2.24 | ||||
| Phe263 | 8.19 | 4.60 | 3.06, 2.98 | 7.25 (Hδ), 7.34 (Hε), 7.30 (Hζ) | |
| Hyl264 | 8.39 | 4.29 | 2.00, 1.75 | 1.59b | 4.02 (Hδ), 3.16 and 2.98 (Hε), 7.62 (εNH2)c |
| Gly265 | 7.94 | 3.88b | |||
| Glu266 | 8.22 | 4.37 | 2.10, 1.94 | 2.43b | |
| Gln267 | 8.49 | 4.37 | 2.10, 1.96 | 2.35b | 7.50 and 6.83 (δNH2) |
| Gly268 | 8.27 | 4.13, 4.00 | |||
| Pro269 | 4.40 | 2.26, 1.90 | 1.98b | 3.58b (Hδ) | |
| Lys270 | 8.45 | 4.29 | 1.84, 1.75 | 1.43b | 1.65b (Hδ), 2.98b (Hε), 7.50 (εNH2) |
| Gly271 | 8.35 | 3.94b | |||
| Glu272 | 8.20 | 4.47 | 2.14, 1.96 | 2.45b | |
| Thr273 | 8.10 | 4.33 | 4.33 | 1.16 | |
Aq binding assay
The abilities of the backbone-modified glycopeptides 2 and 3 and non-modified CII259–273 14 to bind to Aq were determined relative to a biotinylated CLIP peptide with a competitive inhibition assay performed using 96-well microtiter assay plates essentially as described elsewhere.9,27 Briefly, purified soluble recombinant Aq proteins (1 μM) expressed in Drosophila melanogaster SL2 cells were incubated with a fixed concentration of the CLIP-bio (3 μM, sequence KPVSKMRMATPLLMQALPM) and increasing concentrations of test peptides 1, 2 and 3 (0, 0.98, 1.95, 3.91, 7.81, 15.63, 32.25, 62.5, 125, 250, 500 and 1000 μM) in PBS for 48 h at room temperature. This mixture also contained a cocktail of protease inhibitors (Complete™, Boehringer, Mannheim). During this incubation, new 96-well microtiter assay plates were precoated with Y3P monoclonal antibodies (mAb) (10 μg mL−1) by incubation overnight at 4 °C followed by blocking with PBS containing 2% low fat milk and washing with PBS containing 0.1% Tween 20. The incubated mixtures containing Aq proteins, CLIP-bio, and test peptides (90 μL) were transferred to the plates precoated with mAb followed by incubation overnight at 4 °C. After washing with PBS (0.1% Tween 20) the amount of CLIP-bio bound to the Aq proteins captured by the mAb in the wells was quantified using the dissociation-enhanced lanthanide fluoroimmunoassay (DELFIA®) system based on the time-resolved fluoroimmunoassay technique with europium labeled streptavidin (Wallac, Turku), according to the manufacturer's instructions. The experiments were performed in duplicates.T-cell activation assays
Responses of the Aq restricted T-cell hybridoma lines (i.e., the amount of IL-2 secreted following incubation of the hybridoma with antigen-presenting spleen cells expressing Aq and increasing concentrations of the glycopeptides) were determined essentially as described elsewhere,28 with slight modifications. Glycopeptides 1–3 were evaluated with the Aq-restricted T-cell hybridomas16,29 HCQ.3, HCQ.10, HM1R.2 and 22a1-7E. In brief, T-cell hybridomas (5 × 104) were co-cultured with syngeneic spleen cells (5 × 105) and increasing concentrations of the test glycopeptides (0, 0.01, 0.048, 0.24, 1.2, 6.0, 30, and 150 μM) in a volume of 200 μL in 96-well U-bottom microtiter plates. After 24 h, 140 μL supernatant was removed, transferred to a V-bottom microtiter plate, and spun down to avoid transfer of T-cell hybridomas to the ELISA plate. The contents of IL-2 in the culture supernatant (100 μL) were measured by sandwich ELISA (capturing mAb: purified rat anti-mouse IL-2, JES6-IA 12; detecting mAb, biotinylated anti-Mouse Interleukin-2 mAb 5H4, Mabtech AB) using the DELFIA® system (Wallac, Turku, Finland), according to the manufacturer's instructions. The culture supernatant from ConA activated splenocytes served as a positive control.Molecular modelling
The GLYCAM force field atom types were assigned for the carbohydrate part and AMBER force field atom types for the amino acid part.
Analysis was performed with the aid of the Visual Molecular Dynamics (VMD)23 software. Frames were extracted and visualized with a time step of 2 ps. RMSD values were calculated with the RMSD trajectory tool to confirm stability of the complexes. The hydrogen bond occupancy was calculated with the hydrogen bond extension tool, with a donor–acceptor length of less than 3.3 Å and a donor–H–acceptor angle of minimum 160°. Only hydrogen bonds with an occupancy >10% are reported, and hydrogen bonds with an occupancy >40% are considered strong. Cluster analysis was performed with the clustering plugin40 over the last 20 ns. Clustering was based on the minimal epitope (Ile260–Gln267) after superposing the complex against the binding groove, defined as αAla3–Asn85, βGlu4–Gln97. An RMSD cut-off of 0.8 Å was used and a maximum number of 20 clusters were extracted. Representative snapshots were thereafter randomly selected from each cluster. Figures have been prepared with MOE.41
Acknowledgements
This study was supported by the Swedish Research Council, Swedish Foundation for Strategic Research, Knut and Alice Wallenberg Foundation, and the EU IMI Project BeTheCure. The access to synchrotron radiation and data collection support by the beamline staff at the European Synchrotron Radiation Facility (Grenoble, France) are gratefully acknowledged. We also thank HPC2N (High Performance Computing Center North) for HPC resources and technical support.Notes and references
- P. K. Gregersen, J. Silver and R. J. Winchester, Arthritis Rheum., 1987, 30, 1205–1213 CrossRef CAS PubMed.
- M. Hegen, J. C. Keith, M. Collins and C. L. Nickerson-Nutter, Ann. Rheum. Dis., 2008, 67, 1505–1515 CrossRef CAS PubMed.
- U. Brunsberg, K. Gustafsson, L. Jansson, E. Michaelsson, L. Ahrlund-Richter, S. Pettersson, R. Mattsson and R. Holmdahl, Eur. J. Immunol., 1994, 24, 1698–1702 CrossRef CAS PubMed.
- J. Broddefalk, J. Bäcklund, F. Almqvist, M. Johansson, R. Holmdahl and J. Kihlberg, J. Am. Chem. Soc., 1998, 120, 7676–7683 CrossRef CAS.
- B. Dzhambazov, K. S. Nandakumar, J. Kihlberg, L. Fugger, R. Holmdahl and M. Vestberg, J. Immunol., 2006, 176, 1525–1533 CrossRef CAS.
- J. Backlund, S. Carlsen, T. Hoger, B. Holm, L. Fugger, J. Kihlberg, H. Burkhardt and R. Holmdahl, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9960–9965 CrossRef CAS PubMed.
- L. Holm, P. Kjellen, R. Holmdahl and J. Kihlberg, Bioorg. Med. Chem., 2005, 13, 473–482 CrossRef CAS PubMed.
- E. F. Rosloniec, K. B. Whittington, D. D. Brand, L. K. Myers and J. M. Stuart, Cell. Immunol., 1996, 172, 21–28 CrossRef CAS PubMed.
- P. Kjellen, U. Brunsberg, J. Broddefalk, B. Hansen, M. Vestberg, I. Ivarsson, A. Engstrom, A. Svejgaard, J. Kihlberg, L. Fugger and R. Holmdahl, Eur. J. Immunol., 1998, 28, 755–767 CrossRef CAS.
- I. E. Andersson, C. D. Andersson, T. Batsalova, B. Dzhambazov, R. Holmdahl, J. Kihlberg and A. Linusson, PLoS One, 2011, 6 Search PubMed.
- B. Holm, J. Backlund, M. A. F. Recio, R. Holmdahl and J. Kihlberg, ChemBioChem, 2002, 3, 1209–1222 CrossRef CAS.
- I. E. Andersson, T. Batsalova, B. Dzhambazov, L. Edvinsson, R. Holmdahl, J. Kihlberg and A. Linusson, Org. Biomol. Chem., 2010, 8, 2931–2940 CAS.
- I. E. Andersson, T. Batsalova, S. Haag, B. Dzhambazov, R. Holmdahl, J. Kihlberg and A. Linusson, J. Am. Chem. Soc., 2011, 133, 14368–14378 CrossRef CAS PubMed.
- I. E. Andersson, B. Dzhambazov, R. Holmdahl, A. Linusson and J. Kihlberg, J. Med. Chem., 2007, 50, 5627–5643 CrossRef CAS PubMed.
- M. Hart and C. Beeson, J. Med. Chem., 2001, 44, 3700–3709 CrossRef CAS PubMed.
- L. Holm, R. Bockermann, E. Wellner, J. Bäcklund, R. Holmdahl and J. Kihlberg, Bioorg. Med. Chem., 2006, 14, 5921–5932 CrossRef CAS PubMed.
- A. M. H. Boots, H. Hubers, M. Kouwijzer, L. D. Zandbrink, B. M. Westrek-Esselink, C. van Doorn, R. Stenger, E. S. Bos, M. J. C. van Lierop, G. F. Verheijden, C. M. Timmers and C. J. van Staveren, Arthritis Res. Ther., 2007, 9, R71 Search PubMed.
- E. Atherton and R. C. Sheppard, Solid phase peptide synthesis, IRL Press at Oxford University Press, Oxford, England, 1989 Search PubMed.
- R. V. Hoffman, N. Maslouh and F. Cervantes-Lee, J. Org. Chem., 2002, 67, 1045–1056 CrossRef CAS PubMed.
- J. Våbenø, M. Brisander, T. Lejon and K. Luthman, J. Org. Chem., 2002, 67, 9186–9191 CrossRef PubMed.
- T. M. Gierasch, Z. J. Shi and G. L. Verdine, Org. Lett., 2003, 5, 621–624 CrossRef CAS PubMed.
- D. Dobritzsch, H. Uysal, C. Ge and R. Holmdahl, unpublished results.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- R. Déziel, R. Plante, V. Caron, L. Grenier, M. Llinas-Brunet, J. S. Duceppe, E. Malenfant and N. Moss, J. Org. Chem., 1996, 61, 2901–2903 CrossRef.
- J. Broddefalk, M. Forsgren, I. Sethson and J. Kihlberg, J. Org. Chem., 1999, 64, 8948–8953 CrossRef CAS.
- B. M. Syed, T. Gustafsson and J. Kihlberg, Tetrahedron, 2004, 60, 5571–5575 CrossRef CAS PubMed.
- C. M. Hill, A. Liu, K. W. Marshall, J. Mayer, B. Jorgensen, B. Yuan, R. M. Cubbon, E. A. Nichols, L. S. Wicker and J. B. Rothbard, J. Immunol., 1994, 152, 2890–2898 CAS.
- E. Michaëlsson, M. Andersson, A. Engström and R. Holmdahl, Eur. J. Immunol., 1992, 22, 1819–1825 CrossRef PubMed.
- A. Corthay, J. Bäcklund, J. Broddefalk, E. Michaëlsson, T. J. Goldschmidt, J. Kihlberg and R. Holmdahl, Eur. J. Immunol., 1998, 28, 2580–2590 CrossRef CAS.
- Schrödinger Suite 2009 Protein Preparation Wizard; Epik version 2.0, Schrödinger, LLC, New York, NY, 2009; Impact version 5.5, Schrödinger, LLC, New York, NY, 2009; Prime version 2.1, Schrödinger, LLC, New York, NY, 2009.
- Maestro, version 9.1, Schrödinger, LLC, New York, NY, 2010 Search PubMed.
- J. L. Banks, H. S. Beard, Y. X. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- MacroModel, version 9.8, Schrödinger, LLC, New York, NY, 2010 Search PubMed.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F.-Y. Dupradeau, Nucleic Acids Res., 2011, 39, W511–W517 CrossRef CAS PubMed.
- F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821–7839 RSC.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Fransisco, 2012 Search PubMed.
- High Performance Computer Center North (HPC2N); HPC2N: Umeå, Sweden; http://www.hpc2n.umu.se/.
- L. Gracia, Weill Cornell Medical College, 1300 York Ave, New York, 10065 NY.
- Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc., 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2012.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of all new isolated compounds, RMSD for Aqvs. simulation time, RMSD for the peptide binding groove vs. simulation time, RMSD for glycopeptides 1–4vs. simulation time, RMSD for Ile260–Glu266 sequences vs. simulation time, RMSD for the α1 helix vs. simulation time, RMSD for the β1 helix vs. simulation time, RMSD for the β-sheet of the α1β1 domain vs. simulation time, the number of frames occupying each cluster for the MD simulations, superposition of frames from the cluster analysis displaying the glycopeptide/Aq complex, superposition of frames from the cluster analysis displaying the glycopeptides 1–4, visualization of hydrogen bonds formed between Aq and glycopeptides 1–4. See DOI: 10.1039/c5ob00395d |
| This journal is © The Royal Society of Chemistry 2015 |