
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
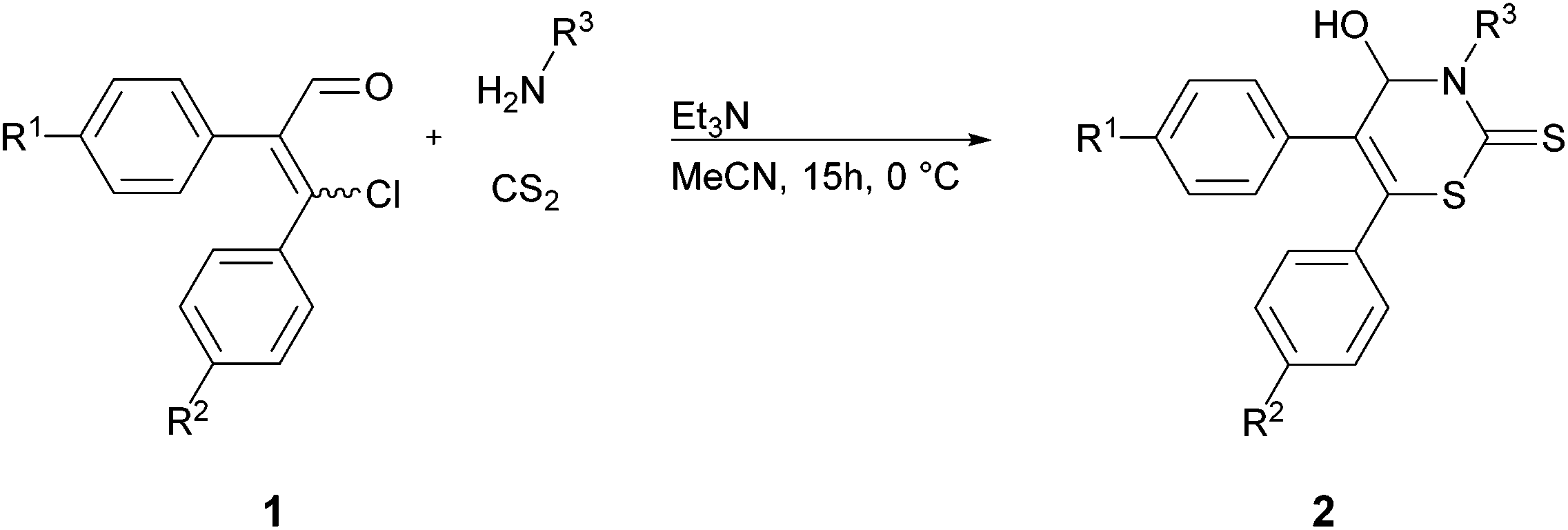
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA three-component reaction for rapid access to underexplored 1,3-thiazine-2-thiones†
Denis
Kröger
,
Fabian
Brockmeyer
* and
Christoph
Kahrs
Institut für Chemie, Carl von Ossietzky Universität Oldenburg, P. O. Box 2503, Carl-von-Ossietzky-Str. 9–11, 26111 Oldenburg, Germany. E-mail: fabian.brockmeyer@uni-oldenburg.de
First published on 20th May 2015
Abstract
Driven by the shortage of known effective possibilities for the synthesis of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones on the one hand and the promising potential of these structures as novel drug candidates on the other hand, synthetic access to 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones was developed. The desired products could be synthesized effectively and facilely starting from β-chlorovinyl aldehydes with the aid of a new MCR (multicomponent reaction). Furthermore, the 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones are shown to be appropriate substrates in the preparation of diverse annulated polycyclic systems.
Introduction
Thiazines are six-membered heterocycles containing sulfur and nitrogen. Apart from being used as dyes,1 several thiazine derivatives are known for their biological activity and are applied in various medical areas.2Recently, we described the synthesis of theretofore not accessible 2,2-dialkyl- and 2-alkyl-2-aralkyl-5,6-diaryl-2H-1,3-thiazines starting from β-chlorovinyl aldehydes.3 Following the synthesis protocol of the newly established MCR (multicomponent reaction), the targeted products were prepared by the conversion of a β-chlorovinyl aldehyde with ammonia, sodium hydrosulfide and a second carbonyl compound. This reaction is an example for the general opportunity to synthesize various heterocyclic compounds by simultaneous formation of at least two or more bonds with the aid of MCRs.4
Herein, we report the use of β-chlorovinyl aldehydes as starting materials in a further new MCR for the preparation of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones. Up to now this class of heterocycles has not been sufficiently investigated.
A few inefficient synthesis routes to different 3,4-dihydro-2H-1,3-thiazine-2-thiones have been reported.5 However, apart from some 3′-hydroxy-pyrazolo[1,5-c][1,3]thiazine-7-thiones6 only two examples of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones have been prepared yet (Fig. 1).7
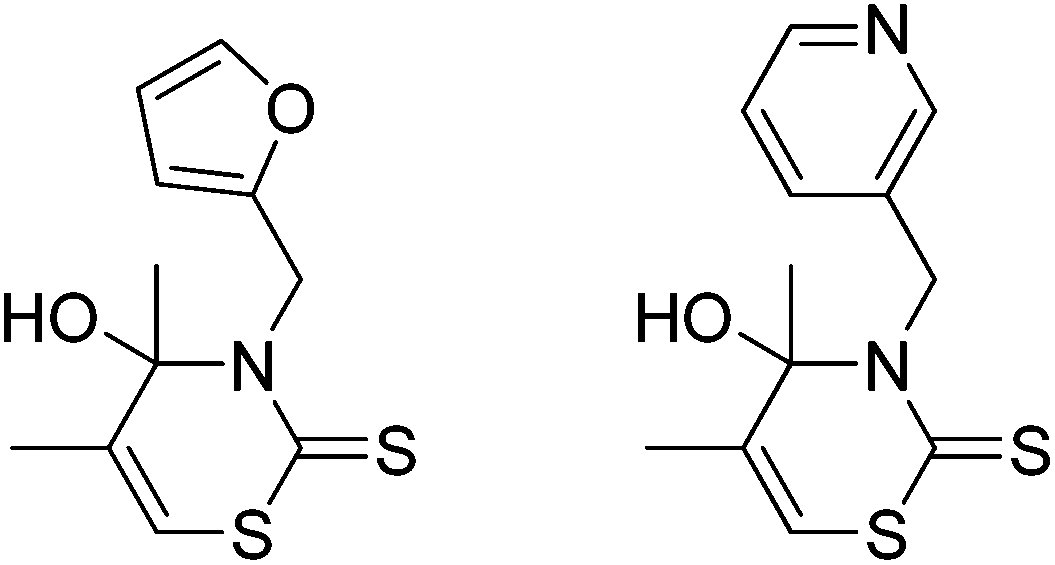 | ||
| Fig. 1 Known 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones.7 | ||
They were synthesized by the reaction of acetone with dimethylamine hydrochloride and paraformaldehyde in isopropanol. A subsequent treatment of the intermediate with carbon disulfide and an amine led to the formation of the desired 1,3-thiazine-2-thiones. It was shown that both examples are characterized by an inhibitory effect on the proliferation of different tumor cells and therefore are potential antitumor agents.7
In view of the pharmacological potential as well as the lack of efficient synthetic routes and our ongoing interest in developing practical methods for synthetic chemistry, we designed a facile synthesis of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones 2 under mild reaction conditions.
In consideration of our experience in the use of β-chlorovinyl aldehydes 1 and the known preparation of 4-hydroxythiazolidine-2-thiones, we envisioned that the formation of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones 2 could be realized by a new MCR, reacting a β-chlorovinyl aldehyde 1 with carbon disulfide and a primary amine (Fig. 2).
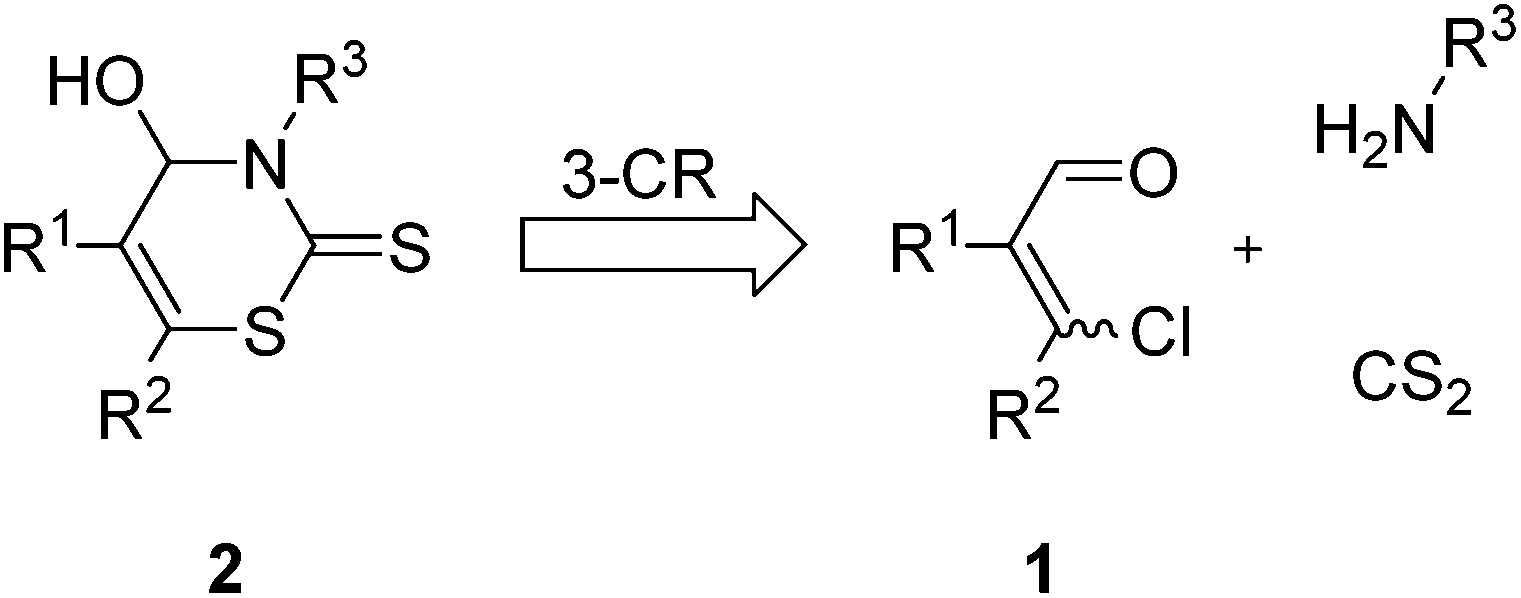 | ||
| Fig. 2 Retrosynthetic consideration of the targeted structures. | ||
Combining a MCR employed to synthesize a relatively complex heterocyclic scaffold with a subsequent post-transformation has been abundantly highlighted as an advantageous method for the synthesis of heterocyclic compounds.8 Thus, we also investigated the conversion of the synthesized 1,3-thiazine-2-thiones to annulated bicycles by exploiting the variable substituent at the nitrogen atom and the hydroxy group, which is formed during the synthesis of 1,3-thiazine-2-thiones.
Results and discussion
Synthesis of the substrates
Combining our previous published concept of synthesis of 2,2-dialkyl- and 2-alkyl-2-aralkyl-5,6-diaryl-2H-1,3-thiazines3 and thiazolidinethiones9 the targeted 3,4-dihydro-2H-1,3-thiazine-2-thiones should be obtained in a MCR using β-chlorovinyl aldehydes, primary amines and carbon disulfide. The β-chlorovinyl aldehydes are received by the conversion of α-methylene ketones with DMF and POCl3.10 We were able to synthesize (EZ)-3-chloro-2,3-diphenylacrylaldehyde11 (1a), (EZ)-3-chloro-3-(4-hydroxyphenyl)-2-phenylacrylaldehyde3 (1b), (EZ)-3-chloro-3-(4-chlorophenyl)-2-phenylacrylaldehyde12 (1c), (EZ)-3-chloro-2,3-bis(4-nitrophenyl)acrylaldehyde3 (1e), 2-chloro-1-cyclohexene-1-carboxaldehyde13 (1f), and (EZ)-3-chloro-3-methyl-2-phenylacrylaldehyde14 (1g).Screening of reaction conditions
We started our investigation with (EZ)-3-chloro-2,3-diphenylacrylaldehyde (1a), allylamine, and carbon disulfide to investigate the optimal conditions for the preparation of 2H-1,3-thiazine-2-thione 2a (Table 1). Our previous research revealed the feasibility of using both isomers of the β-chlorovinyl aldehydes in a (EZ)-mixture as the substrate.3 Thus, we used the (EZ)-mixture of substrate 1 in all synthesis, with the exception of (E)-1d. The stoichiometry of the substrates was chosen as reported recently.9 As related to our previous studies, H2O (in the case of thiazolidinethiones) and MeOH (in the case of 2,2-dialkyl- and 2-alkyl-2-aralkyl-5,6-diaryl-2H-1,3-thiazines) are the solvents of choice. Thus, we tested H2O as well as MeOH in combination with different bases—i.e. K2CO3 and Et3N—which are necessary to trap the liberated hydrogen chloride.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Temperature | Reaction time [h] | Yieldb of 2a [%] |
| a All reactions were performed using 1.00 mmol β-chlorovinyl aldehyde 1a, 1.50 mmol allylamine, 3.00 mmol carbon disulfide, and 0.50 mmol base in 5 mL solvent followed by column chromatography. b All yields are isolated yields. c The reaction was carried out two times using both 0.50 mmol and 1.00 mmol Et3N, which led to identical yields in both cases. d After completion of the reaction the solvent was removed by high-vacuum low-temperature distillation. | |||||
| 1 | H2O | K2CO3 | r.t. | 24 | 0 |
| 2 | MeOH | K2CO3 | r.t. | 24 | 25 |
| 3 | MeOH | Et3N | r.t. | 24 | 22 |
| 4 | MeOH/THF | K2CO3 | r.t. | 24 | 62 |
| 5 | MeOH/THF | Et3N | r.t. | 24 | 46 |
| 6 | THF | K2CO3 | r.t. | 24 | 37 |
| 7 | THF | Et3N | r.t. | 24 | 45 |
| 8 | DCM | K2CO3 | r.t. | 24 | 10 |
| 9 | DCM | Pyridine | r.t. | 24 | 0 |
| 10 | DCM | Et3N | r.t. | 24 | 64 |
| 11 | DMF | Et3N | r.t. | 24 | 58 |
| 12 | MeCN | K2CO3 | r.t. | 24 | 22 |
| 13 | MeCN | Pyridine | r.t. | 24 | 6 |
| 14 | MeCN | Et3N | r.t. | 24 | 67c |
| 15 | MeCN | Et3N | r.t. | 5 | 46 |
| 16 | MeCN | Et3N | r.t. | 15 | 55 |
| 17 | MeCN | Et3N | r.t. | 48 | 47 |
| 18 | MeCN | Et 3 N | 0 °C | 15 | 90 |
| 19 | MeCN | Et3N | 0 °C | 24 | 90 |
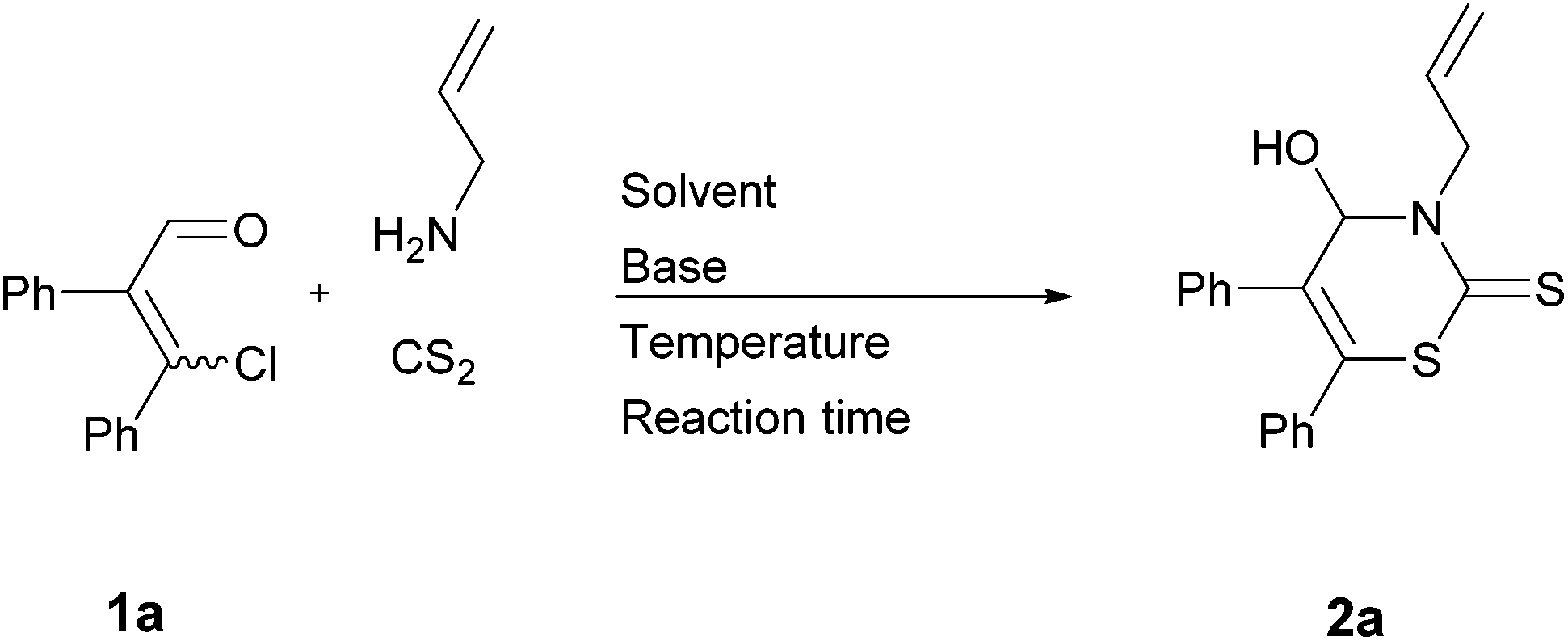
H2O turned out to be an inappropriate solvent, whereas the use of MeOH led to the formation of the targeted 2H-1,3-thiazine-2-thione 2a in low yields (Table 1, entries 2 and 3). This may be reasoned by a (extremely) low solubility of 1a in H2O and MeOH. For this reason, we screened further different solvents and bases. The highest yield so far was obtained using Et3N in MeCN at r.t. (Table 1, entry 14).
Next, we investigated the influence of the reaction time. A reaction time of 15 hours led to the highest yield of 2a (55%; Table 1, entry 16). Since an extended reaction time resulted in a lower yield, we had reason to suppose that a part of the already formed product decomposes under the tested conditions. Some NMR spectra suggested the assumption that enaminothiones are the products of decomposition.15 Thus, we performed the reaction in MeCN with Et3N again, but this time for 15 hours at 0 °C. In this way, 2H-1,3-thiazine-2-thione 2a was isolated in 90% yield. For this reason the optimal reaction was performed at 0 °C for 15 hours using Et3N in MeCN (Table 1, entry 18). It is worth mentioning that MeCN is distilled off via high-vacuum low-temperature distillation after the reaction is complete. Removal of the solvent at a rotary evaporator at 40 °C also causes partial decomposition.
The proposed mechanism for the formation of 2H-1,3-thiazine-2-thione 2 is shown in Scheme 1. First, the addition of the primary amine to the carbon disulfide leads to the N-monosubstituted carbamic acid A. The dithiocarbamate B is formed by the substitution of the chloride in the β-chlorovinyl aldehyde 1 with carbamic acid. The hydrogen chloride, which is formed as a co-product, is trapped by triethylamine. A subsequent ring-closure, occurring via a nucleophilic attack of the nitrogen atom of the dithiocarbamate group on the carbonyl group and a migration of a proton results in the formation of the product 2.
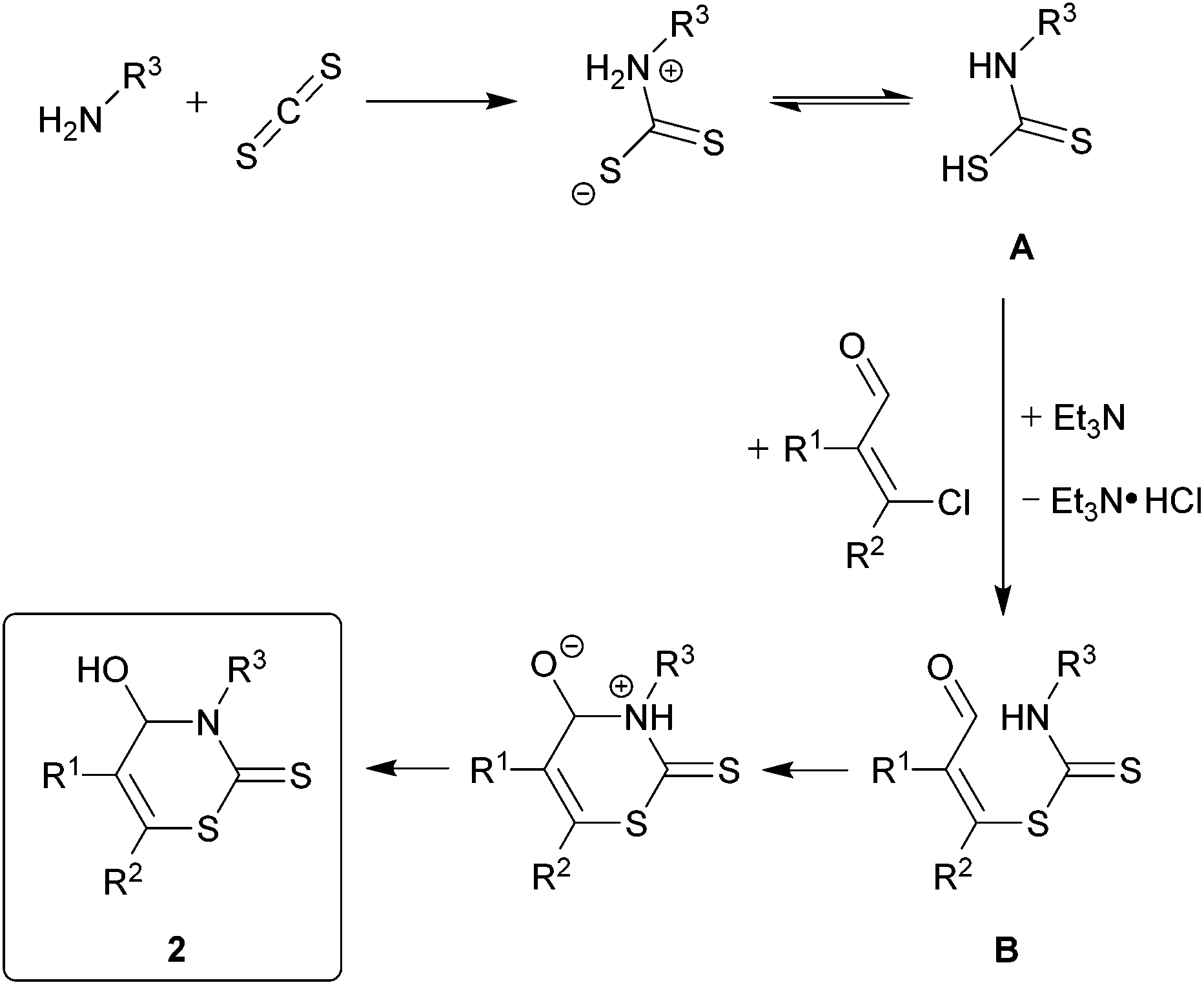 | ||
| Scheme 1 Proposed mechanism for the synthesis of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones 2. | ||
Reaction scope
In order to explore the scope of the reaction under the optimized conditions, a number of β-chlorovinyl aldehydes 1a–g and primary amines were investigated. The results are summarized in Table 2. In almost all cases the performed reactions lead to the desired 2H-1,3-thiazine-2-thiones 2 in moderate to good yields. Referring to our model reaction with allylamine (Table 2, entry 1), we first investigated the influence of different unsaturated amines in the reaction with aldehyde 1a and carbon disulfide.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aldehyde | Product | R1 | R2 | R3 | Yieldb [%] |
| a All reactions were performed using 1.00 eq. β-chlorovinyl aldehyde 1, 1.50 eq. primary amine, 3.00 eq. carbon disulfide, and 0.50 eq. Et3N in MeCN (5 ml per mmol β-chlorovinyl aldehyde) followed by high-vacuum low-temperature distillation to remove the solvent and column chromatography. b All yields are isolated yields. c The 2H-1,3-thiazine-2-thione 2v was observed in the NMR of the crude product, but could not be isolated. | ||||||
| 1 | 1a | 2a | H | H | CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
90 |
| 2 | 1a | 2b | H | H | CH2C(CH3)CH2 |
79 |
| 3 | 1a | 2c | H | H | (CH2)2CHCH2 |
64 |
| 4 | 1a | 2d | H | H | (CH2)2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
33 |
| 5 | 1a | 2e | H | H | (CH2)3CCH |
53 |
| 6 | 1a | 2f | H | H | (CH2)3CH3 | 48 |
| 7 | 1a | 2g | H | H | Cy | 70 |
| 8 | 1a | 2h | H | H | CH2Ph | 90 |
| 9 | 1a | 2i | H | H | CH2(4-OMe–C6H4) | 53 |
| 10 | 1a | 2j | H | H | (CH2)2(2-Br–C6H4) | 89 |
| 11 | 1b | 2k | H | OH | CH2CHCH2 |
43 |
| 12 | 1c | 2l | H | Cl | CH2CHCH2 |
38 |
| 13 | 1d | 2m | OCH3 | OCH3 | CH2CHCH2 |
78 |
| 14 | 1e | 2n | NO2 | NO2 | CH2CHCH2 |
40 |
| 15 | 1d | 2o | OCH3 | OCH3 | CH2C(CH3)CH2 |
67 |
| 16 | 1d | 2p | OCH3 | OCH3 | (CH2)2CHCH2 |
56 |
| 17 | 1d | 2q | OCH3 | OCH3 | CH(CH3)2 | 60 |
| 18 | 1d | 2r | OCH3 | OCH3 | C(CH3)3 | — |
| 19 | 1d | 2s | OCH3 | OCH3 | CH2CH2OCH3 | 81 |
| 20 | 1d | 2t | OCH3 | OCH3 | CH2CH2OPh | 65 |
| 21 | 1d | 2u | OCH3 | OCH3 | CH2Ph | 65 |
| 22 | 1d | 2v | OCH3 | OCH3 | 4-CH3–C6H4 | —c |
| 23 | 1d | 2w | OCH3 | OCH3 | 4-NO2–C6H4 | — |
The products 2d and 2e designed from alkyne derivatives (Table 2, entries 4 and 5) are isolated in lower yields than those products synthesized from alkene derivatives (Table 2, entries 1–3).
Moreover, alkyl amines bearing a methylene group next to the nitrogen atom such as n-butylamine (Table 2, entry 6) and aliphatic amines with a methine group at this position—i.e. cyclohexyl amine and isopropyl amine (Table 2, entries 7 and 17)—were feasible substrates in the reaction, whereby n-butylamine seemed to react reluctantly. Only amines with a quaternary carbon atom next to the nitrogen atom—e.g. tert-butylamine (Table 2, entry 18)—seem to be not feasible substrates in this new multicomponent reaction. However, using p-toluidine as the substrate in the MCR the corresponding 2H-1,3-thiazine-2-thione 2v was generated. This could be confirmed by the NMR spectra of the crude product (see Fig. S3 in the ESI†). The isolation of 2v was attempted in different ways without success. This could probably be the result of a decomposition based on the instability of the resulting product 2v. In contrast to the conversion of p-toluidine, electron-poor aromatics did not even form the 2H-1,3-thiazine-2-thiones (Table 2, entry 23), with the result that only the educts of the reaction could be isolated.
But if aralkyl amines with a methylene group next to the nitrogen atom are used instead of aromatic amines the developed MCR successfully leads to 2H-1,3-thiazine-2-thiones 2 (Table 2, entries 8–10 and 21).
Noteworthily, amines with aromatics bearing functional groups such as bromine substituents (Table 2, entry 10) or with different kinds of aliphatic and aromatic ethers (Table 2, entries 9, 19 and 20) are also tolerated. The conversions of benzylamine with 1a and carbon disulfide to 2h (Table 2, entry 8) and the product 2a (Table 2, entry 1) of our model reaction, using allylamine, revealed the highest yield (90%).
The structure of 2a was confirmed by X-ray diffraction analysis (see Fig. S1 in the ESI†).16 Besides model substrate 1a, other β-chlorovinyl aldehydes were investigated likewise (Table 2, entries 11–23). Both substrates with electron-donating (Table 2, entries 11–13) and electron-withdrawing (Table 2, entry 14) groups at the para-position provided the desired thiones 2. Thus, the MCR tolerates the usage of aldehydes with different kinds of functional groups such as chlorine substituents, hydroxy, methoxy and nitro groups (Table 2, entries 11–14).
Analogous to our research results on synthesizing 2,2-dialkyl- and 2-alkyl-2-aralkyl-5,6-diaryl-2H-1,3-thiazines3 the formation of 2H-1,3-thiazine-2-thiones 2 is also limited to 2,3-diaryl-β-chlorovinyl aldehydes 1a–e as substrates. The conversions of 2-chloro-1-cyclohexene-1-carboxaldehyde (1f) and 3-chloro-3-methyl-2-phenylacrylaldehyde (1g) each with allylamine and carbon disulfide did not lead to the corresponding products 2.
However, the presented results (Table 2) documented that the novel MCR allows the preparation of manifold 2H-1,3-thiazine-2-thiones 2 in moderate to excellent yields starting from readily accessible substrates.
In order to screen the reaction scope the applicability of various tryptamine derivatives, containing the pharmaceutically interesting indole, in the developed MCR was also examined (Fig. 3). Surprisingly, a product resulting from a twofold cyclization could be isolated after column chromatography.
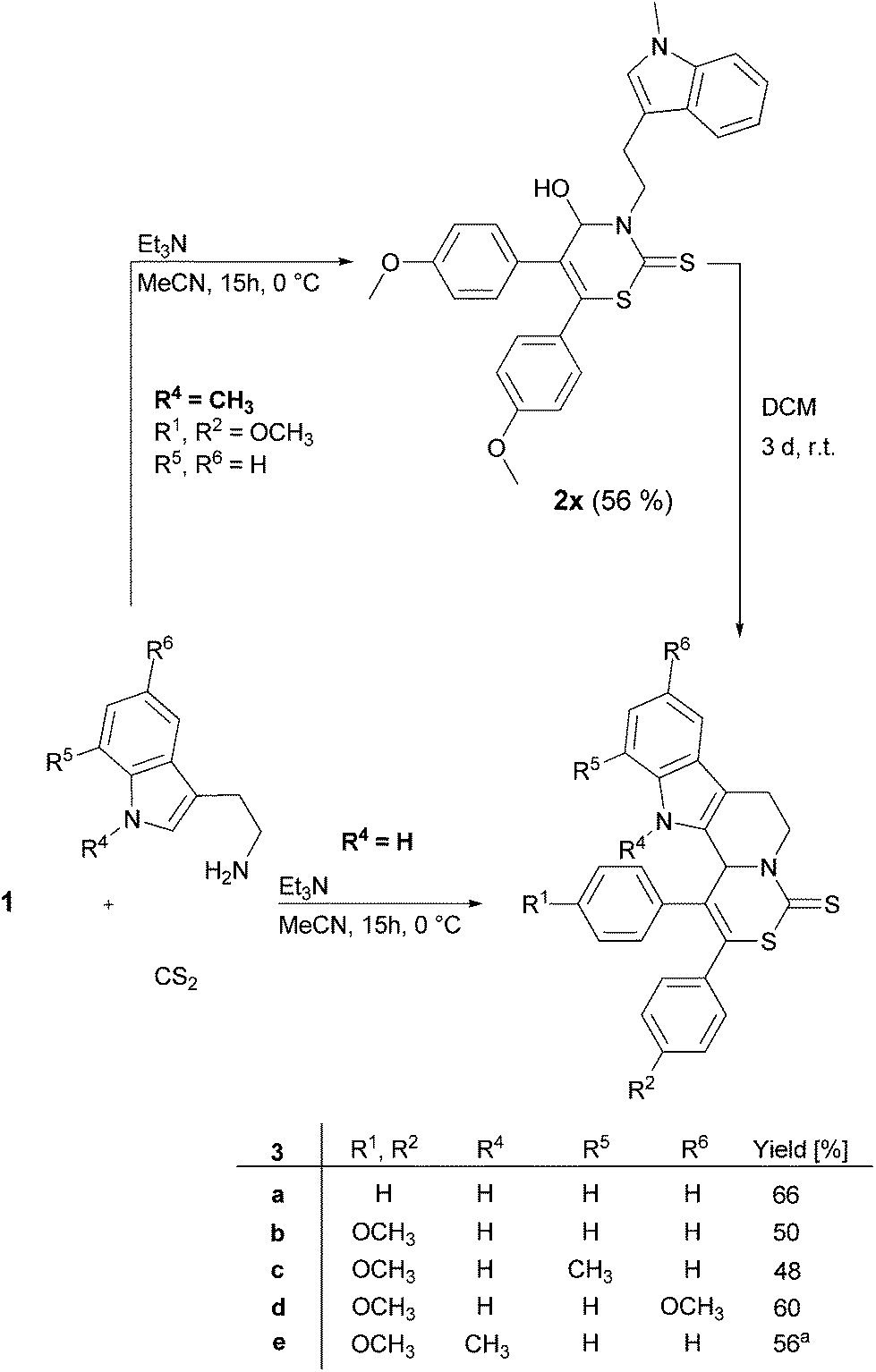 | ||
| Fig. 3 Cascade reaction to indole derivatives 3. aOverall yield after both reactions (via2x). | ||
At first, the formation of 2H-1,3-thiazine-2-thione occurred followed by an intramolecular electrophilic aromatic substitution (Pictet–Spengler-type reaction) at the heteroaromatic indole cycle and simultaneous substitution of the hydroxy group. The indole annulated 2H-1,3-thiazine-2-thiones 3 were obtained in moderate yields of up to 66% (Fig. 3). When 1-methyltryptamine was used as the substrate the second cyclization via the Pictet–Spengler-type reaction did not take place directly and 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thione 2x with an N-methylated indole could be isolated (Fig. 3). However, the cyclization of 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thione 2x to 2H-1,3-thiazine-2-thiones 3e can be realized by placing the substrate in DCM for 3 days at r.t. The transformation occurs quantitatively.
Derivatization
Based on the described results concerning 2H-1,3-thiazine-2-thiones 2 and our ongoing interest in using them as precursors in subsequent reactions (Scheme 2), we designed derivatives 2c and 2p to examine the conversion in a Lewis-acid mediated ring-closing reaction17 to pyridothiazinethiones 4. To the best of our knowledge this kind of ring-closing reaction was performed using InCl3 for the first time. 2H-1,3-thiazine-2-thiones 2c and 2p were therefore stirred for 15 hours at r.t. in a DCM solution containing equimolar amounts of InCl3.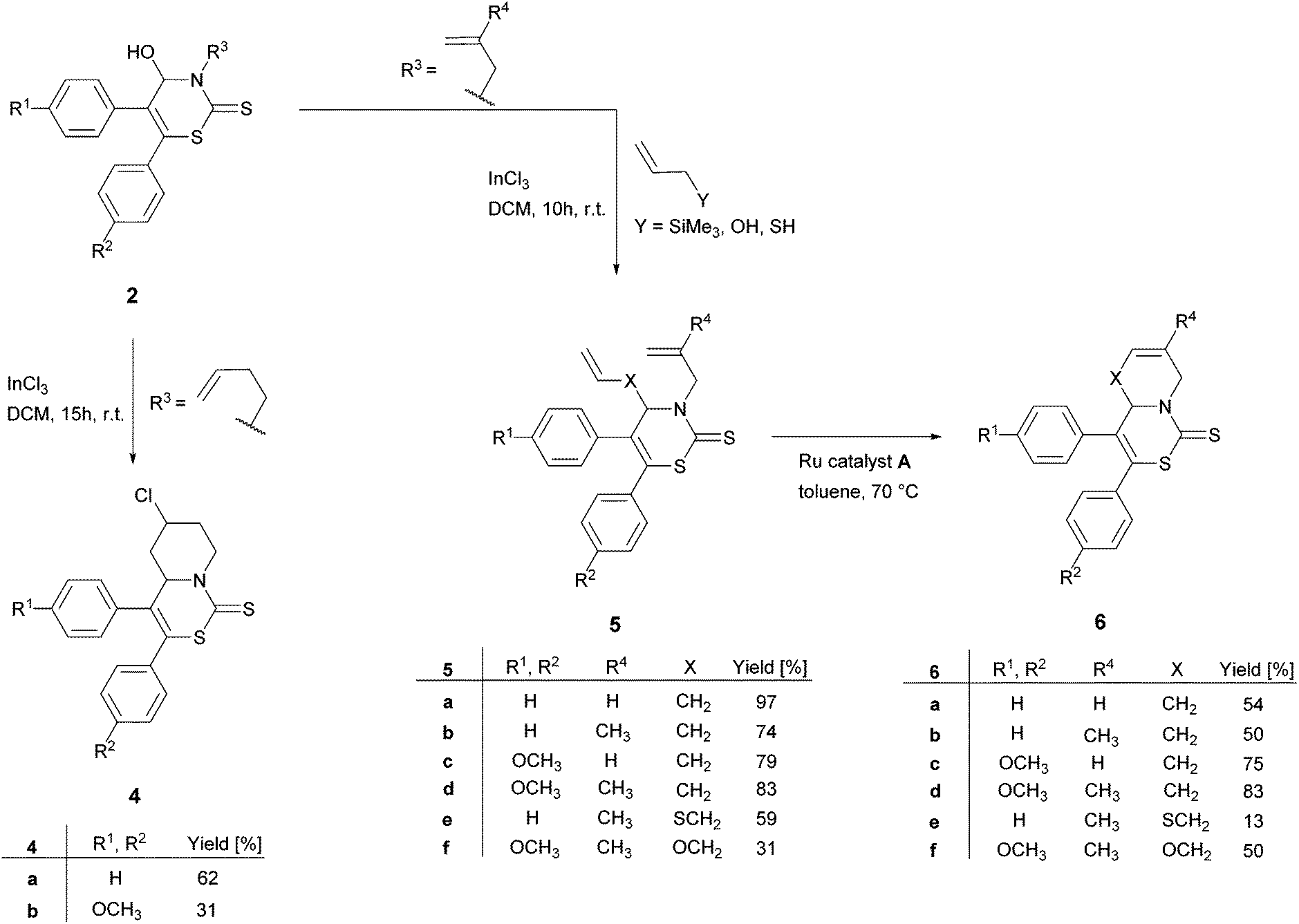 | ||
| Scheme 2 Subsequent reactions to various 2H-1,3-thiazine-2-thiones derivatives: pyridothiazinethiones 4, dialkenes 5, unsaturated pyridothiazinethiones and azepinethiazinethiones 6. | ||
The mixtures of the racemic diastereomers of the targeted pyridothiazinethiones 4 were isolated in moderate yields (Scheme 2, products 4a and 4b).
Treatmeant of alcohols with catalytic amounts of InCl3 and allyltrimethylsilane, allyl alcohol or allyl mercaptan, respectively, led to C–C, C–O or C–S bond formation by direct substitution of the hydroxy group.18 Using allylamine, which is the analogous nitrogen nucleophile, no C–N bond formation was observed. However, the products starting from allyltrimethylsilane could be obtained in good to excellent yields (Scheme 2, products 5a–d). Also the corresponding sulfur and oxygen containing dialkenes were synthesized in this way (Scheme 2, products 5e and 5f). Noteworthily, all generated products without a hydroxyl group (this extends to compound 3–6) were more stable than the 2H-1,3-thiazine-2-thiones 2. This may be reasoned with the low steadiness of N,O-hemiacetal in 2. Thus, a reaction temperature beyond 0 °C in subsequent reactions is allowed.
Due to their terminal alkene functional groups, compounds 5a–f are ideal substrates for ring-closing metathesis (RCM). Because of the positive outcome of previously reported similar procedures by our group we chose to employ the Ru catalyst A (Fig. 4).9
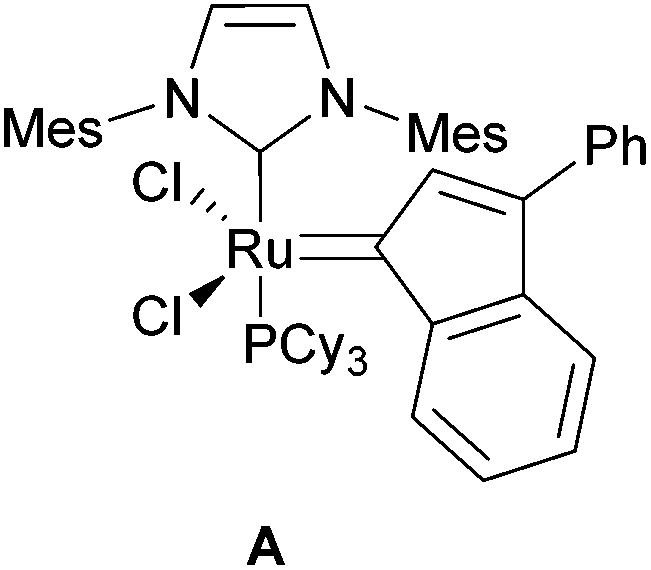 | ||
| Fig. 4 Ru catalyst A used for RCM. | ||
The RCMs were performed in toluene using 5 mol% of Ru catalyst A to generate the six- and seven-membered annulated bicycles 6 in yields up to 83% (Scheme 2, products 6a–f).
Remarkably, the average yields of unsaturated pyridothiazinethiones 6a–d were higher than those of unsaturated azepinethiazinethiones 6e and 6f. For example, a comparison of the yield of the annulated bicycle 6d (83% yield) with the comparable oxygen containing seven membered bicycle 6f (50% yield) supports this observation. The structure of 6b was established by X-ray diffraction analysis (see Fig. S2 in the ESI†).19 Thus, several compounds containing the 2H-1,3-thiazine-2-thione substructures in addition to another biologically active structure could be formed based on the synthesized 2H-1,3-thiazine-2-thiones 2.
The subsequent conversions to annulated bicycles shown in Scheme 2 exemplify the feasibility to create a plethora of 2H-1,3-thiazine-2-thione-containing compounds starting from 2H-1,3-thiazine-2-thiones 2.
Conclusions
In conclusion, we developed a novel multicomponent reaction, which allows efficient and facile access to 4-hydroxy-3,4-dihydro-2H-1,3-thiazine-2-thiones. Starting from readily accessible β-chlorovinyl aldehydes, the targeted products can be prepared with the aid of primary amines and carbon disulfide under mild conditions. Moreover, the use of tryptamine derivatives as amine components leads to a cascade consisting of the 2H-1,3-thiazine-2-thione formation and a Pictet–Spengler-type cyclization. The shown potential of the 2H-1,3-thiazine-2-thiones in subsequent reactions provides the preparation of various annulated polycycles.Having developed the novel MCR, it is now possible to synthesize a broad compound library of 2H-1,3-thiazine-2-thiones, which can be used for screening to identify new biologically active structures.
Experimental
Synthesis of (RS)-3-allyl-4-hydroxy-5,6-diphenyl-3,4-dihydro-2H-1,3-thiazine-2-thione (2a) as a representative example for the synthesis of all 2H-1,3-thiazine-2-thiones
The aldehyde (EZ)-1a (243 mg, 1.00 mmol) was dissolved in 2 mL anhydrous MeCN. Allylamine (86 mg, 1.50 mmol), dissolved in 3 mL anhydrous MeCN, CS2 (228 mg, 3.00 mmol) and anhydrous Et3N (51 mg, 0.50 mmol) were added at 0 °C. After stirring overnight at 0 °C the solvent was removed via high-vacuum low-temperature distillation. Column chromatography (DCM; Rf = 0.23) afforded the desired thiazinethione 2a (304 mg, 90%) as a yellow solid, mp 110–112 °C (from DCM/n-hexane); IR (ATR):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 3272, 2054, 3019, 2923, 1643, 1156, 1127, 760, 731 cm−1; 1H NMR (500.1 MHz, CDCl3): δ 3.89–3.91 (1 H, m, OH), 4.68–4.72 (1 H, m, NCH2), 5.18–5.22 (1 H, m, NCH2), 5.26–5.28 (1 H, m, CHCH2), 5.30–5.33 (1 H, m, CHCH2), 5.75 (1 H, d, 3J = 9.2 Hz, NCH), 5.94–6.02 (1 H, m, CHCH2), 7.20–7.28 (10 H, m, 10 CHAr) ppm; 13C NMR (125.8 MHz, CDCl3): δ 55.41 (NCH2), 84.72 (NCH), 119.51 (CHCH2), 127.41 (CHCC), 127.91 (p-CHAr), 128.54, 128.65, 128.89 (6 CHAr), 129.22 (p-CHAr), 130.05 (2 CHAr), 131.49 (NCH2CH), 134.08 (CArCS), 135.79 (CArCCH), 137.09 (CHCC), 189.48 (CS) ppm; MS (ESI): m/z 362.1 (M + Na+, 10%), 322.0 (M − OH+, 100%), 279.0 (M − COS+, 10%); HRMS (ESI): found 362.0660; calc. for C19H17NNaOS2 [M + Na]+ 362.0649.
3272, 2054, 3019, 2923, 1643, 1156, 1127, 760, 731 cm−1; 1H NMR (500.1 MHz, CDCl3): δ 3.89–3.91 (1 H, m, OH), 4.68–4.72 (1 H, m, NCH2), 5.18–5.22 (1 H, m, NCH2), 5.26–5.28 (1 H, m, CHCH2), 5.30–5.33 (1 H, m, CHCH2), 5.75 (1 H, d, 3J = 9.2 Hz, NCH), 5.94–6.02 (1 H, m, CHCH2), 7.20–7.28 (10 H, m, 10 CHAr) ppm; 13C NMR (125.8 MHz, CDCl3): δ 55.41 (NCH2), 84.72 (NCH), 119.51 (CHCH2), 127.41 (CHCC), 127.91 (p-CHAr), 128.54, 128.65, 128.89 (6 CHAr), 129.22 (p-CHAr), 130.05 (2 CHAr), 131.49 (NCH2CH), 134.08 (CArCS), 135.79 (CArCCH), 137.09 (CHCC), 189.48 (CS) ppm; MS (ESI): m/z 362.1 (M + Na+, 10%), 322.0 (M − OH+, 100%), 279.0 (M − COS+, 10%); HRMS (ESI): found 362.0660; calc. for C19H17NNaOS2 [M + Na]+ 362.0649.
Single crystals of 2a obtained by crystallization from DCM and n-hexane were mounted in inert oil and transferred to the cold gas stream of the diffractometer.
Crystal structure determination of 2a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 499 reflections measured, 7596 unique (Rint = 0.0308), which were used in all calculations. The final wR(F2) was 0.1039 (all data).
499 reflections measured, 7596 unique (Rint = 0.0308), which were used in all calculations. The final wR(F2) was 0.1039 (all data).
Acknowledgements
The authors gratefully acknowledge Marc Schmidtmann for X-ray crystallography. The ruthenium catalyst A was generously supplied by Evonik Degussa GmbH.Notes and references
- (a) M. Havelcová, P. Kubát and I. Nĕmcová, Dyes Pigm., 1999, 44, 49 CrossRef; (b) S.-K. Lee and A. Mills, J. Fluoresc., 2003, 13, 375 CrossRef CAS.
- (a) S. A. Greene and J. C. Thurmon, J. Vet. Pharmacol. Ther., 1988, 11, 295 CrossRef CAS PubMed; (b) S. Shweta, S. N. Pandeya, V. Anupam and Y. Deepika, Int. J. Res. Ayurveda Pharm., 2011, 2, 1130 Search PubMed; (c) J. Srikanth, K. Padmini, P. J. Preethi, P. V. P. D. Bharadwaj and P. V. Rao, Asian J. Res. Pharm. Sci., 2013, 3, 170 Search PubMed; (d) G. Vincent, B. Mathew, J. Joseph, M. Chandran, A. R. Bhat and K. K. Kumar, Int. J. Pharm. Chem. Sci., 2014, 3, 341 Search PubMed.
- F. Brockmeyer, R. Shoemaker, M. Schmidtmann and J. Martens, Org. Biomol. Chem., 2014, 12, 5168 CAS.
- (a) B. B. Rouré and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef PubMed; (b) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., 2011, 123, 6358; ( Angew. Chem., Int. Ed. , 2011 , 50 , 6234 ) CrossRef; (c) O. Pando, S. Stark, A. Denkert, A. Porzel, R. Preusentanz and L. A. Wessjohann, J. Am. Chem. Soc., 2011, 133, 7692 CrossRef CAS PubMed.
- For examples, see: (a) J. Imrich and P. Kristian, Collect. Czech. Chem. Commun., 1982, 47, 3268 CrossRef CAS; (b) W. Hanefeld, Arch. Pharm., 1984, 317, 297 CrossRef CAS; (c) A. S. Fisyuk, A. S. Moskovkin, I. V. Miroshnichenko, M. Y. Botnikov and B. V. Unkovskii, Chem. Heterocycl. Compd., 1992, 28, 823 CrossRef.
- M. G. Marei and M. El-ghanam, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 107, 1 CrossRef CAS.
- R. Li, Z. Ge, J. Cui, X. Sun, Z. Wang and X. Yan, Patent, EP2562157A1, 2013 Search PubMed.
- For great and detailed reviews, see: (a) S. Marcaccini and T. Torroba, in Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, Weinheim, 2005, ch. 2 Search PubMed; (b) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef PubMed; (c) G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544 CrossRef PubMed.
- T. Stalling, W. Saak and J. Martens, Eur. J. Org. Chem., 2013, 8022 CrossRef CAS.
- F. Barth, J.-P. Ducoux, M. Rinaldi-Carmona and C. Congy, US Patent, US264470, 2006 Search PubMed.
- D. Prim, A. Fuss, G. Kirsch and A. M. S. Silva, J. Chem. Soc., Perkin Trans. 2, 1999, 1175 RSC.
- M. Weißenfels and M. Pulst, Tetrahedron Lett., 1968, 9, 3045 CrossRef.
- B. J. Stokes, C. V. Vogel, L. K. Urnezis, M. Pan and T. G. Driver, Org. Lett., 2010, 12, 2884 CrossRef CAS PubMed.
- A. Chakraborty and J. K. Ray, Synth. Commun., 1995, 25, 1869 CrossRef CAS.
- The enaminothiones can be directly synthesized from β-chlorovinyl aldehydes: M. Weißenfels and M. Pulst, J. Prakt. Chem., 1973, 315, 873 CrossRef.
- CCDC-1049355 contains the supplementary crystallographic data for compound 2a.
- (a) K. D. Moeller, S. L. Rothfus and P. L. Wong, Tetrahedron, 1991, 47, 583 CrossRef CAS; (b) F. Brockmeyer, T. Stalling and J. Martens, Synthesis, 2012, 44, 2947 CrossRef CAS.
- M. Yasuda, T. Saito, M. Ueba and A. Baba, Angew. Chem., 2004, 116, 1438 ( Angew. Chem., Int. Ed. , 2004 , 43 , 1414 ) CrossRef.
- CCDC-1049334 contains the supplementary crystallographic data for compound 6b.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for all isolated new compounds as well as crystal structures of compounds 2a and 6b. CCDC 1049355 and 1049334. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00377f |
| This journal is © The Royal Society of Chemistry 2015 |