
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A Maitland–Japp inspired synthesis of dihydropyran-4-ones and their stereoselective conversion to functionalised tetrahydropyran-4-ones†
Paul A.
Clarke
 *,
Philip B.
Sellars
and
Nadiah Mad
Nasir
*,
Philip B.
Sellars
and
Nadiah Mad
Nasir
Department of Chemistry, University of York, Heslington, York, North Yorkshire YO10 5DD, UK. E-mail: paul.clarke@york.ac.uk; Fax: +44 (0)1904 322516; Tel: +44 (0)1904 322614
First published on 12th March 2015
Abstract
The Maitland–Japp reaction has been extended to the synthesis of highly functionalised dihydropyran-4-ones. These dihydropyran-4-ones can in turn be converted stereoselectively into tetrahydropyran-4-ones with tertiary and quaternary stereocentres via the one-pot addition of hydride or carbon nucleophiles and trapping with carbon electrophiles. The utility of this method is demonstrated by providing access to the functionalised tetrahydropyran units present in a component of the Civet fragrance and the anticancer polyketide lasonolide A.
Introduction
Tetrahydropyran (THP) containing natural products, such as (−)-centrolobine, (+)-phorboxazole A and B, (−)-lasonolide A, and Civet cat secretion (Fig. 1), are an important class of synthetic targets because of their challenging architectural features, their biological activities and their limited availability from natural sources. In the cases of (−)-centrolobine, (+)-phorboxazole A and B, and (−)-lasonolide A, each has potent activity against a human disease, with (−)-centrolobine showing activity against the parasite responsible for leishmaniasis1 and the phorboxazoles and lasonolide A showing potent anticancer activity.2,3 As such, these molecules have the potential to become the next generation of therapeutic agents if enough material can be provided to complete the required biological studies and satisfy the supply problem. The challenging molecular architectures of these compounds, coupled with their biological activities, have prompted many groups around the world to embark upon research programs aimed at the development of new methods for the construction of the tetrahydropyran rings found within them.4 There have been significant developments in the formation of tetrahydropyrans by the Prins reaction5 and the hetero-Diels–Alder reaction,6 and these strategies have been applied with varying degrees of success to the synthesis of tetrahydropyran-containing natural products including (−)-centrolobine, (+)-phorboxazole A and B, and (−)-lasonolide A.4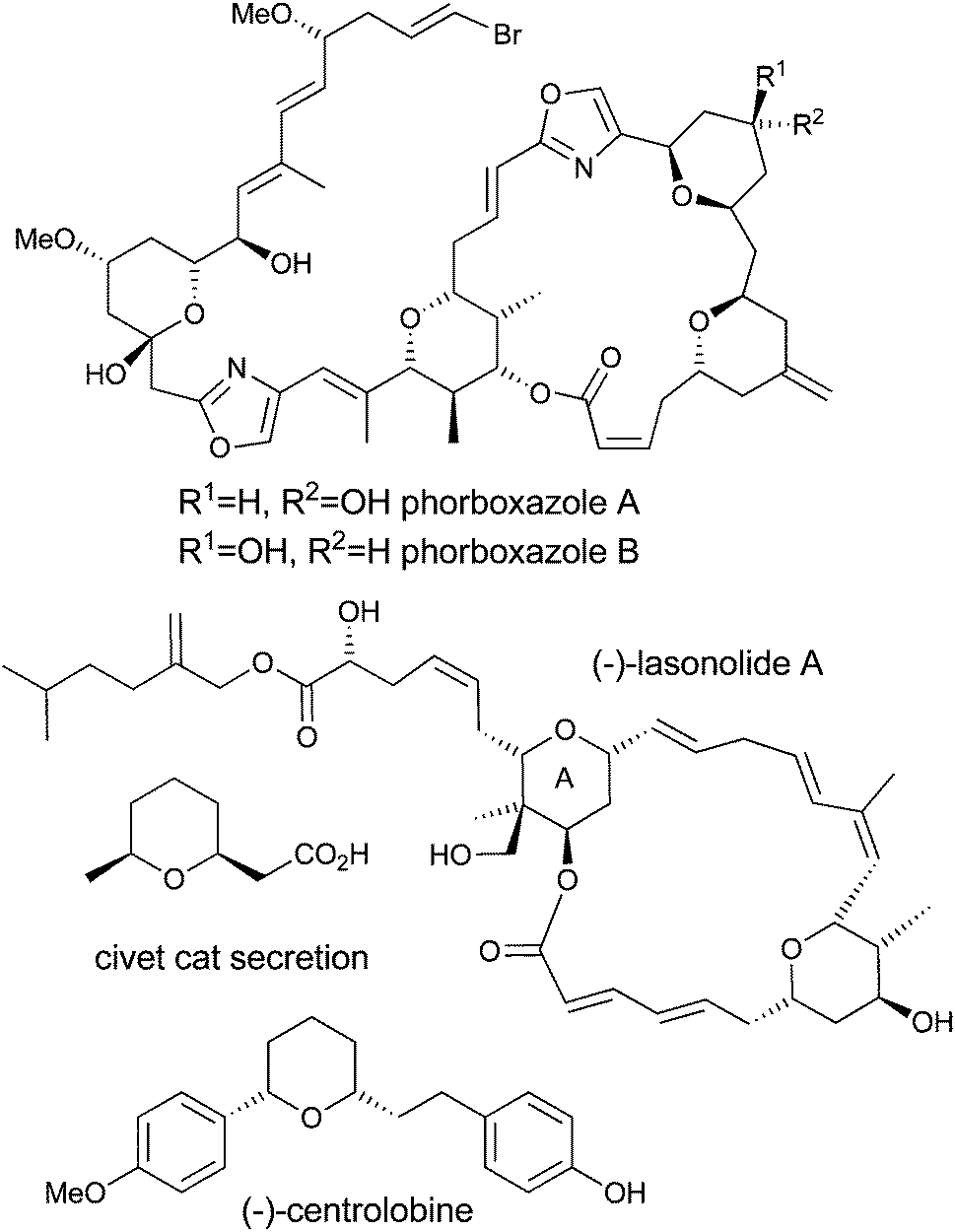 | ||
| Fig. 1 Tetrahydropyran containing natural products. | ||
Over the last few years we have been interested in developing new methods for the synthesis of functionalised tetrahydropyran-4-ones7 and the application of these methods to the total synthesis of tetrahydropyran containing natural products such as (−)-centrolobine8 and (+)-phorboxazole B.9,10 Our work in this area focused on updating the venerable Maitland–Japp reaction,11 initially as a two-pot process involving the addition of the Weiler dianion to an aldehyde in the first step, to be followed by the Lewis acid catalysed Knoevenagel reaction and oxy-Michael cyclisation in the second step.12 This in turn led to the development of a one-pot procedure. When Chan's diene was used as the nucleophile, we found that we could effect a Lewis acid catalysed Mukaiyama aldol reaction and follow it with the Knoevenagel reaction and oxy-Michael cyclisation, without the need for isolation of the intermediate δ-hydroxy-β-ketoester adduct. This generated mixtures of 2,6-cis and 2,6-trans-tetrahydropyran-4-ones in good yields.13 Later we replaced Chan's diene with diketene and made the reaction pot, atom and step economic (PASE),14 as well as asymmetric.14,15
However, despite the utility of the Chan's diene and diketene versions of the Maitland–Japp reaction, it became apparent that there were a number of difficulties associated with them. Of primary concern was the formation of mixtures of the 2,6-cis and 2,6-trans diastereomers 1 and 2, which interconverted under the reaction conditions (Scheme 1).13b While these diastereomers could be separated via flash column chromatography and re-equilibrated to give the desired diastereomer, such a procedure was not ideal. Of secondary concern was the inherent difficulty in functionalising either the 3- or 5-positions of the tetrahydropyran-4-one ring. Treating the tetrahydropyran-4-one products with a base resulted in a retro-Michael reaction affording 3;8 furthermore, after decarboxylation, it proved impossible to control the regioselectivity of enolate formation in the resulting decarboxylated tetrahydropyran-4-one 4 and hence formation of products 5 and 6 (Scheme 2). As such, the tetrahydropyran-4-one products from the Maitland–Japp reaction cannot be readily converted into the tetrahydropyrans found in the C20–C32 fragment of the phorboxazoles16 or the A-ring of lasonolide A.
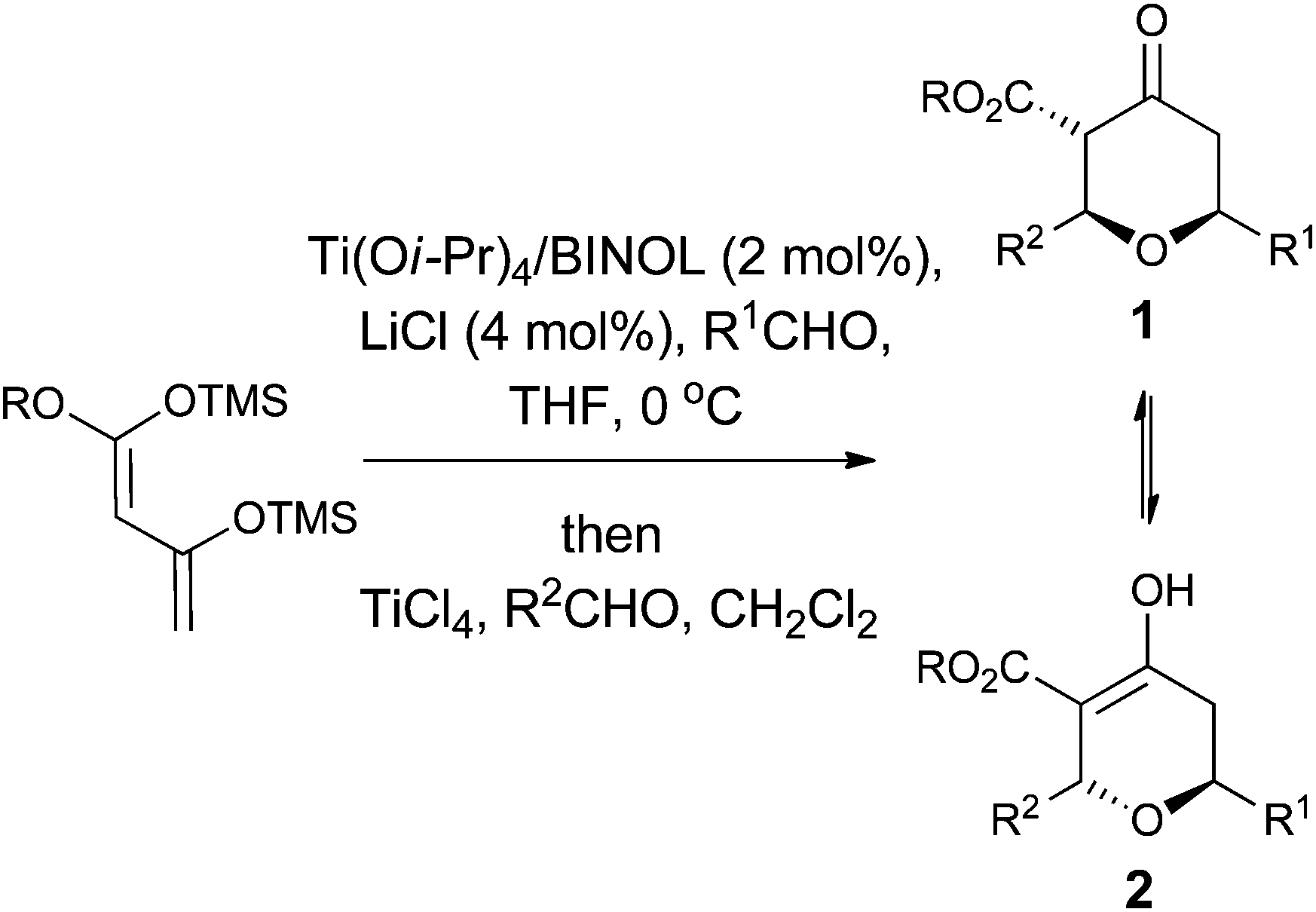 | ||
| Scheme 1 The Maitland–Japp reaction. | ||
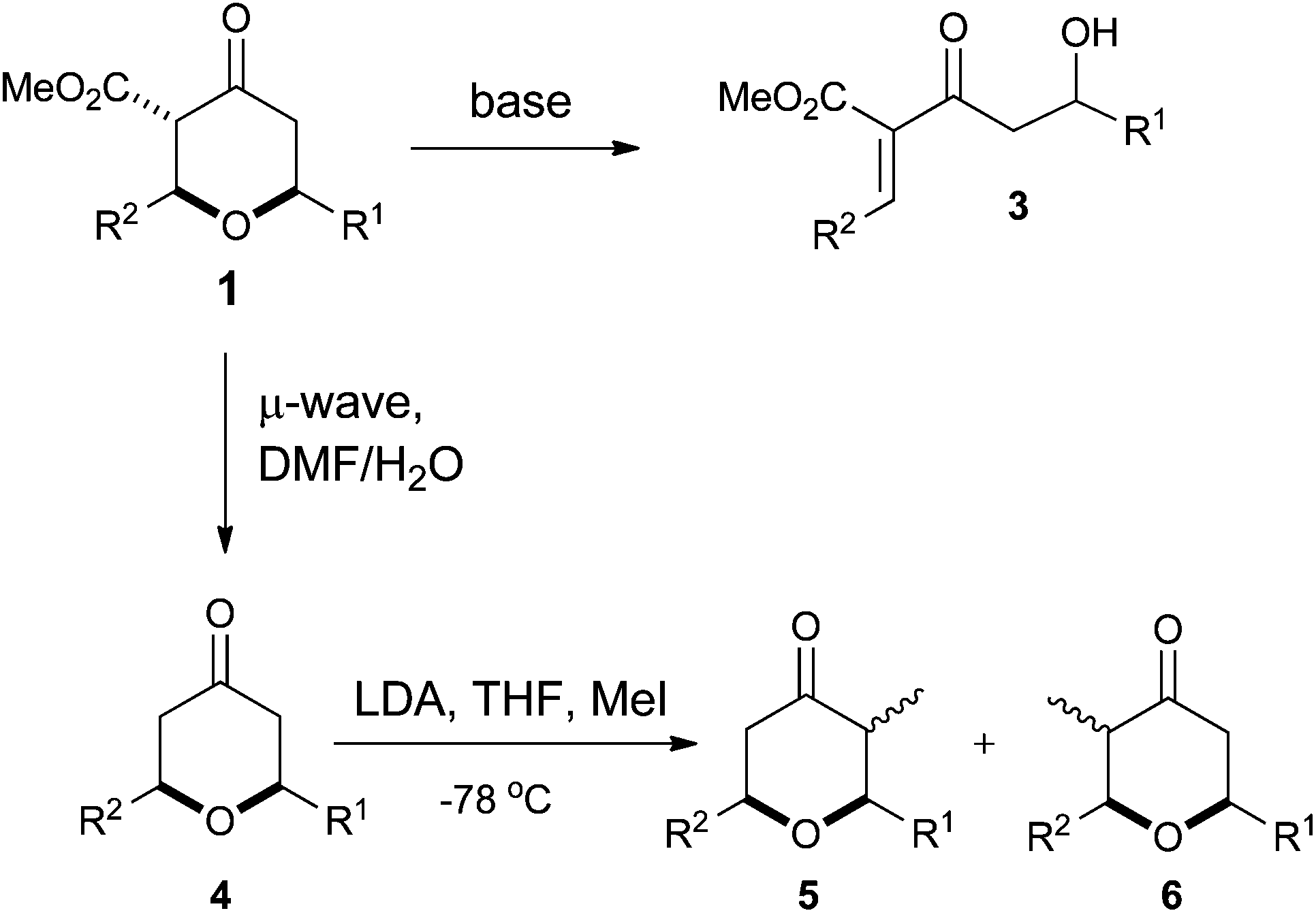 | ||
| Scheme 2 Problems of functionalising Maitland–Japp products. | ||
In order to overcome these problems we considered the possibility of developing a procedure to generate dihydropyran-4-ones 8, which would be more amenable to further functionalisation. Conjugate addition of a nucleophile to the double bond of the dihydropyran-4-one would generate an enolate which we hoped we could trap with an appropriate electrophile, thus generating a quaternary stereocentre. If the nucleophile was a hydride, then the resulting tetrahydropyran-4-one 9 would have the 2,6-cis relationship, and if the nucleophile was an organometallic reagent the resulting tetrahydropyran-4-one would have a tertiary stereocentre at C2 (Scheme 3). This paper builds on our earlier communication and fully details our studies in this area.17
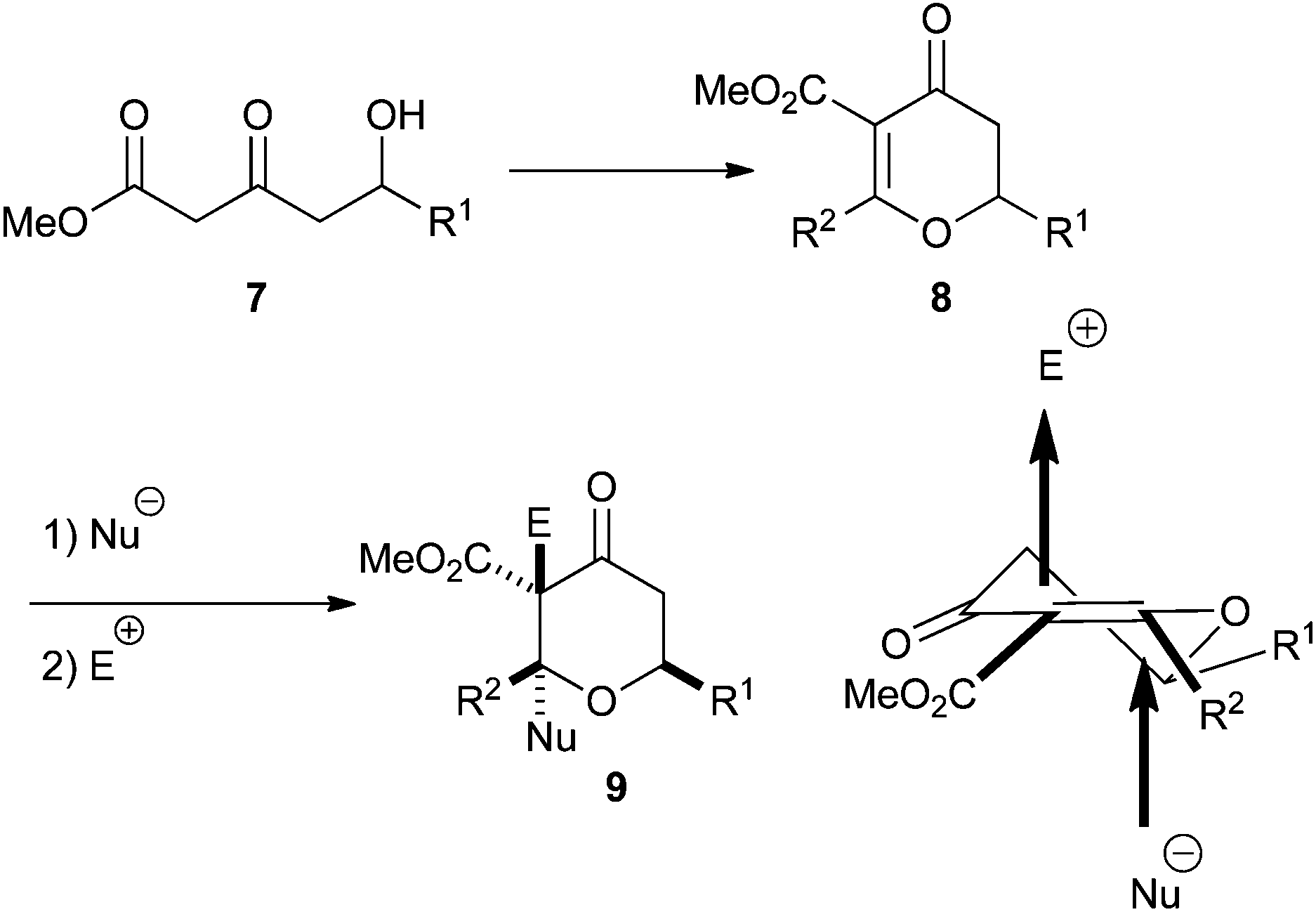 | ||
| Scheme 3 Proposed dihydropyran route. | ||
Results and discussion
Formation of dihydropyran-4-ones
Our initial investigations focused on modifying the Maitland–Japp cyclisation to produce dihydropyran-4-ones 10. We achieved this by replacing the second aldehyde in the Maitland–Japp reaction sequence with the dimethyl acetal of a N,N-dimethyl amide (Table 1).17|
|
||||
|---|---|---|---|---|
| DHP (10) | R1 | R2 | R3 | Yield (%) |
| a | Ph | Me | Me | 60 |
| b | Et | Me | Me | 71 |
| c | Pr | Me | Me | 70 |
| d | i-Pr | Me | Me | 70 |
| e | 2-Furyl | Me | Me | 40 |
| f | CH2OBn | Me | Me | 61 |
| g | CH2CH2OBn | Me | Me | 72 |
| h | Ph | i-Pr | Me | 62 |
| i | Cy-hex | i-Pr | Me | 81 |
| j | Pr | i-Pr | Me | 78 |
| k | CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH3 CHCH3 |
i-Pr | Me | 69 |
| l | Pr | i-Pr | Ph | 68 |
| m | CHCHCH3 |
i-Pr | Ph | 56 |
| n | i-Pr | i-Pr | Ph | 53 |
| o | CH2CH2OBn | Me | Ph | 41 |
| p | Cy-hex | Me | Ph | 33 |
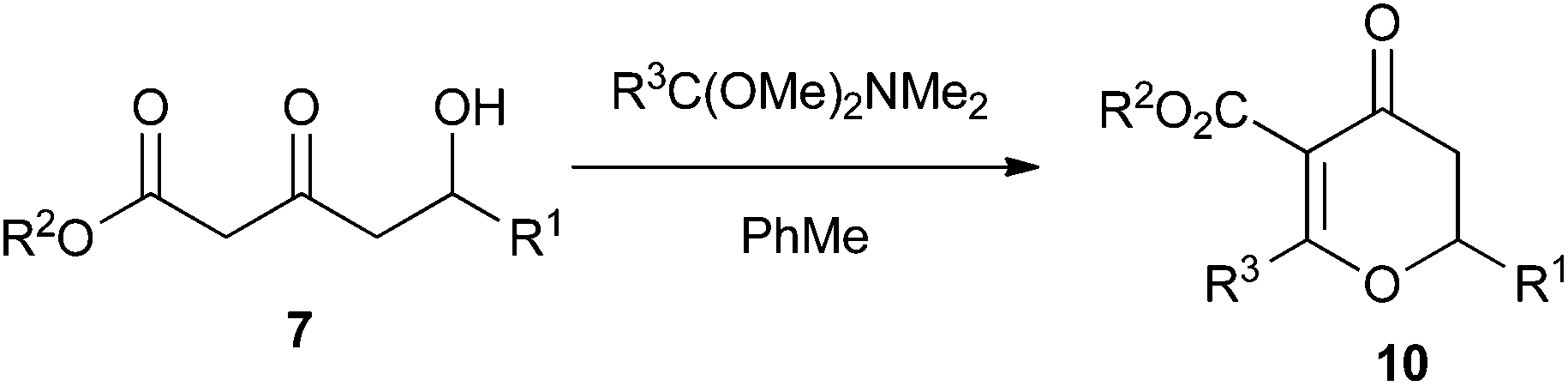
As can be seen from Table 1, a wide range of δ-hydroxy-β-ketoesters 7 can be reacted with the dimethyl acetals of N,N-dimethyl acetamide or benzamide to generate dihydropyran-4-ones 10 in good to excellent yields. However, the scope of this approach is limited by the commercial availability and synthetic accessibility of such orthoamides. While the dimethyl acetal of N,N-dimethyl acetamide was commercially available, the corresponding dimethyl acetal of N,N-dimethyl benzamide required a two-step synthesis. This involved first reacting the N,N-dimethyl benzamide with dimethyl sulfate and then treating the resulting product with NaOMe in methanol.18 Thus, while unfunctionalised alkyl and aryl dimethyl acetals of N,N-dimethyl amides can be formed, this procedure cannot be used for any amides containing either Lewis acid or base sensitive functional groups.
In order to overcome this problem, we studied the use of orthoesters, which are more easily accessible than their orthoamide counterparts. We selected two commercially available orthoesters to study: trimethyl orthoacetate and trimethyl orthovalerate (Table 2). However, it is worth noting that functionalised orthoesters can be synthesised in two steps from the appropriate nitrile.19
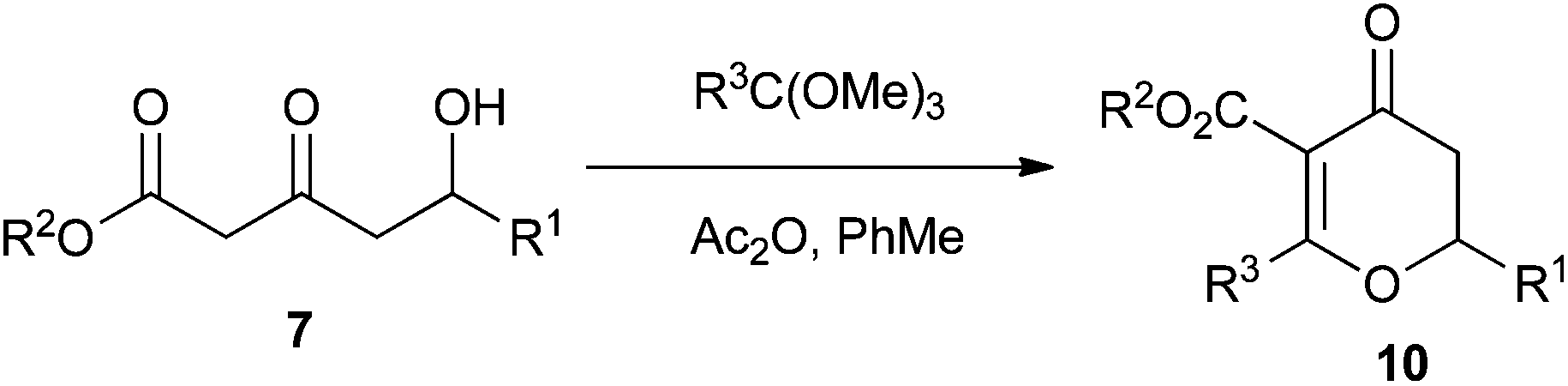
We found that these orthoester Maitland–Japp reactions required heating under reflux, the presence of acetic anhydride as a dehydrating agent and a large excess of orthoester in order to achieve completion. However, the large excess of orthoester caused problems in the isolation of the dihydropyran-4-one products 10. We therefore investigated the use of microwave heating,20 which enabled us to reduce the amount of orthoester to only 2 equiv. and still maintain reasonable yields. Microwave heating also reduced the reaction time from hours to a matter of minutes.
Conversion of dihydropyran-4-ones to 2,6-cis-tetrahydropyran-4-ones
Having developed the Maitland–Japp cyclisation to form dihydropyran-4-ones 10, we turned our attention to reduction of the double bond to form tetrahydropyran-4-ones 11. We predicted that addition of a hydride to the double bond would occur from a pseudo-axial trajectory, thus generating the 2,6-cis-tetrahydropyran-4-one stereoselectively. A number of reducing agents were investigated, with L-Selectride® proving to be the best. Treatment of dihydropyran-4-ones 10 with L-Selectride® delivered tetrahydropyran-4-ones 11 as the sole products as mixtures of ketone and enol tautomers with excellent 2,6-cis-diastereoselectivity (Table 3).17|
|
|||||
|---|---|---|---|---|---|
| THP (11) | R1 | R2 | R3 | Keto![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enol :enol |
Yield (%) |
| a | Ph | Me | Me | 1:0.34 |
69 |
| b | Et | Me | Me | 1:0.10 |
54 |
| c | Pr | Me | Me | 1:0.13 |
61 |
| d | i-Pr | Me | Me | 1:0.13 |
67 |
| e | 2-Furyl | Me | Me | 1:0.20 |
84 |
| f | CH2OBn | Me | Me | 1:0.15 |
62 |
| g | CH2CH2OBn | Me | Me | 1:0.19 |
67 |
| h | Ph | i-Pr | Me | 1:0 |
79 |
| k | CHCHCH3 |
i-Pr | Me | 1:0.15 |
51 |
| q | Pr | Me | Bu | 1:0 |
65 |
| r | i-Pr | Me | Bu | 1:0 |
60 |
| l | Pr | i-Pr | Ph | 0:1 |
63 |
| m | CHCHCH3 |
i-Pr | Ph | 0:1 |
73 |
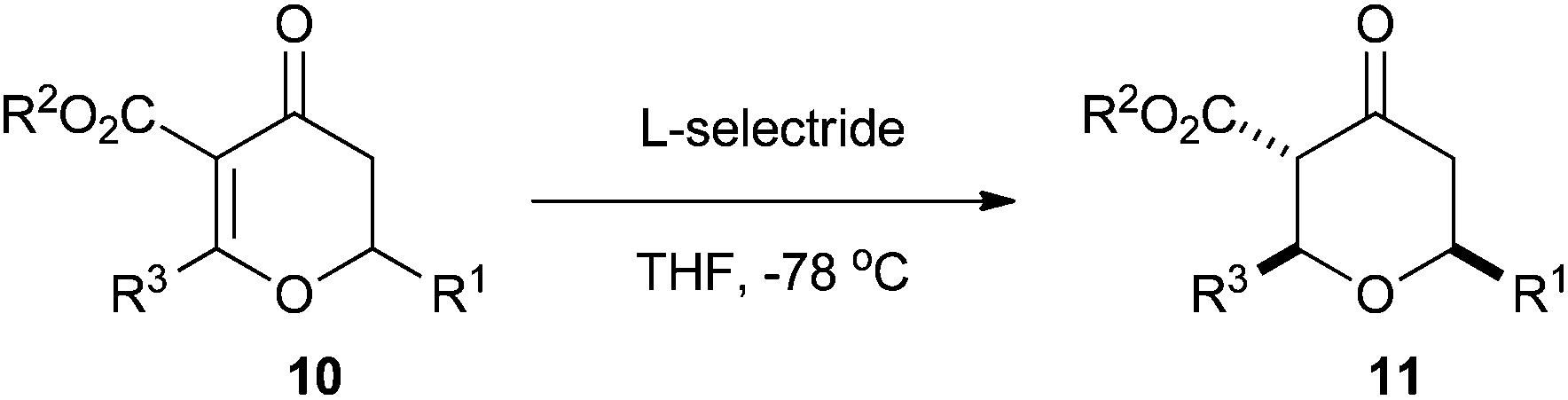
In the case of the 2-methyl tetrahydropyran-4-ones 11a–k a trace amount of the 2,6-trans-tetrahydropyran-4-one was formed, although this could be separated from the major 2,6-cis-product by flash column chromatography using cyclohexane–ethyl acetate mixtures. We believe that the 2,6-trans products arose from a retro-Michael/Michael equilibration, rather than from pseudo-equatorial addition of a hydride. Indeed, we have seen this equilibration in these tetrahydropyran-4-ones previously, especially under Lewis or Brønsted acid conditions.13 With larger C2 substituents, the 2,6-trans-tetrahydropyran-4-ones were not observed; the 2-butyl tetrahydropyran-4-ones 11q and 11r were formed solely as the ketone tautomer. Interestingly, the 2-phenyl tetrahydropyran-4-ones 11l and 11m were formed exclusively as the enol tautomer.
The structures of the 2,6-cis ketone tautomers were elucidated by analysis of the coupling constants in 1H NMR and nOe studies. Coupling constants of about 10 Hz were observed between H2/H3 and H5ax/H6, indicating that the two pairs had trans-diaxial relationships and thus all of the protons occupied axial positions. Positive nOe correlations between H2 and H6 of 1.7–2.6% confirmed the 2,6-cis relationship. The 2,6-cis enol stereochemistry was also confirmed by positive nOe correlations between H2 and H6 of around 1.0–1.5%.
Synthesis of a constituent of Civet cat secretion
Having developed methods for the synthesis of dihydropyran-4-ones 10 and for their conversion to 2,6-cis-tetrahydropyran-4-ones 11, we looked to apply them to the synthesis of the small 2,6-cis-tetrahydropyran natural product that is found in the glandular secretions of the Civet cat (Viverra civetta) and is used in the fragrance industry.21Our synthesis began with the Maitland–Japp formation of dihydropyran-4-one 10g in 72% yield using the orthoamide procedure. This was then treated with L-Selectride® to furnish the 2,6-cis-tetrahydropyran-4-one 11g in 67% yield in a 1:0.19 ratio of ketone and enol tautomers. Microwave mediated decarboxylation in wet DMF provided 2,6-cis-tetrahydropyran-4-one 12 quantitatively. Tetrahydropyran-4-one 12 was converted into tetrahydropyran 13, quantitatively, by formation of the dithiolane and removal of the benzyl group with BCl3·SMe2 in CH2Cl2. Reduction of the dithiolane with RANEY® Ni and H2 gave alcohol 14 in 68% yield. Alcohol 14 was then oxidized with Jones reagent to give the carboxylic acid in 63% yield, thus completing the total synthesis of the Civet cat secretion natural product 15 in 7 steps (Scheme 4).
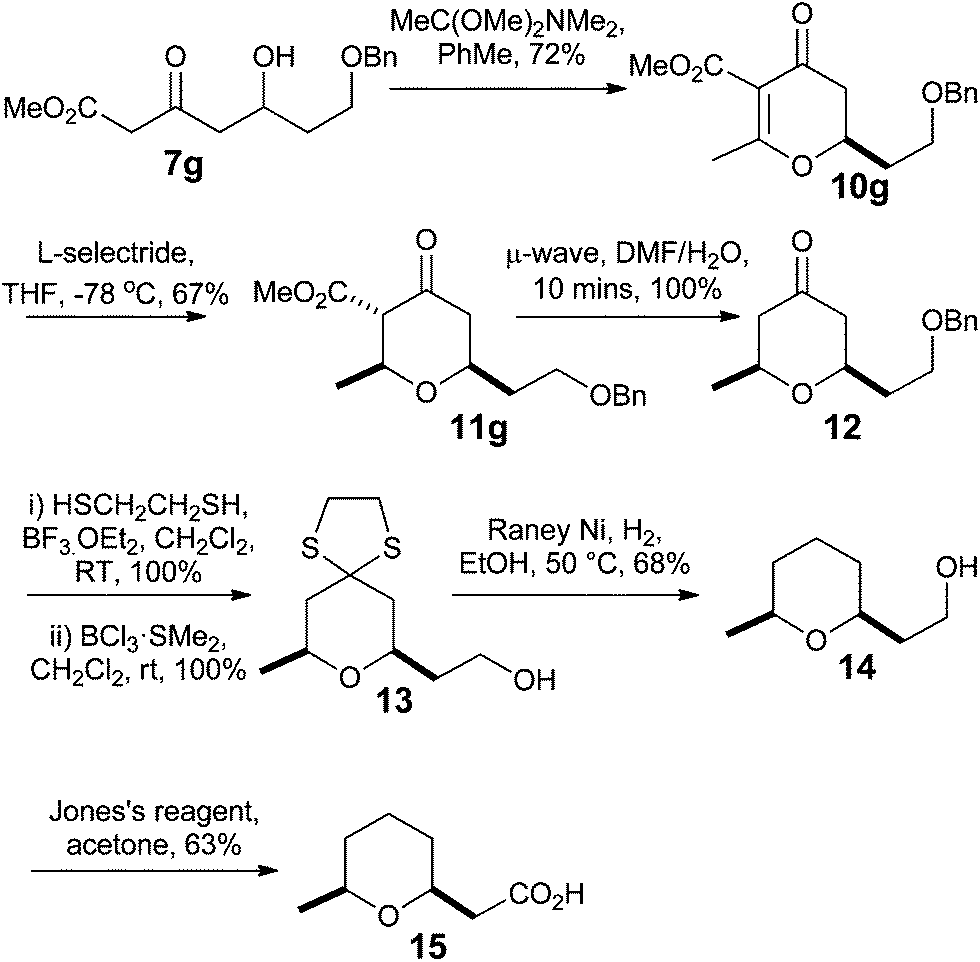 | ||
| Scheme 4 Synthesis of Civet. | ||
Synthesis of tetrahydropyran-4-ones with quaternary stereocentres
As the addition of L-Selectride® to dihydropyran-4-ones 10 generated an enolate, we wondered whether it would be possible to trap the enolate with a carbon electrophile. We envisaged that the enolate trapping should occur anti to the incoming hydride nucleophile; thus if MeI were used as an electrophile this should lead to structures containing the substitution found on the A-ring of (−)-lasonolide A, specifically the quaternary stereocentre. L-Selectride® was added to a solution of the dihydropyran-4-ones 10 in THF at −78 °C and after an hour MeI was introduced and the reaction was warmed to room temperature.The desired 2,6-cis-tetrahydropyran-4-ones 16 were formed in moderate to good yields with alkylation at C3 and with the methyl substituent in an axial position (Table 4). The exceptions to this were dihydropyran-4-ones 10l and 10m where R3 was a phenyl group. In these cases alkylation occurred on the C4 oxygen to give enol ethers 17l and 17m. The 2,6-cis stereochemistry was again confirmed by trans-diaxial couplings between H5ax and H6 of around 11.0–12.0 Hz and positive nOe correlations between H2 and H6 of 2.8%. Positive nOe correlations between H5ax and the C3 methyl substituent of 1.2% showed that the methyl quench occurred from the expected pseudo-axial trajectory, anti to the addition of a hydride.
|
|
|||||
|---|---|---|---|---|---|
| THP (16/17) | R1 | R2 | R3 | Ratio (16:17) |
Yield (%) |
| a | Ph | Me | Me | 1:0 |
61 |
| b | Pr | Me | Me | 1:0 |
35 |
| d | i-Pr | Me | Me | 1:0 |
47 |
| e | 2-Furyl | Me | Me | 1:0 |
49 |
| f | CH2OBn | Me | Me | 1:0 |
61 |
| j | Pr | i-Pr | Me | 1:0 |
62 |
| r | i-Pr | Me | Bu | 1:0 |
41 |
| t | CH2OBn | Me | Bu | 1:0 |
57 |
| l | Pr | i-Pr | Ph | 0:1 |
72 |
| m | CHCHCH3 |
i-Pr | Ph | 0:1 |
46 |
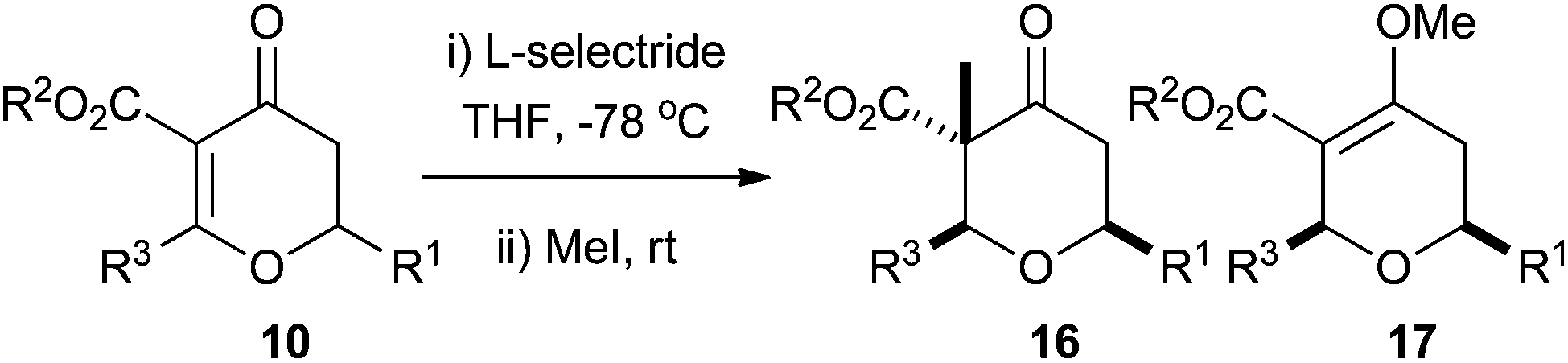
Synthesis of the tetrahydropyran A-ring of lasonolide A
With a procedure in place for the alkylation of the C3 position we could focus on completing a synthesis of a model A-ring of lasonolide A (Scheme 5).22 Tetrahydropyran 16t with the quaternary stereocentre at C3 was treated with an excess of LiAlH4 in THF to reduce both the ketone and ester functional groups. This furnished diol 18, where a hydride had been delivered to the ketone in a pseudo-axial manner to generate the equatorial alcohol. The stereochemistry of the new alcohol was confirmed by analysis of the coupling constants that H4 had with both H5ax and H5eq. The coupling constant between H4 and H5ax was 12.0 Hz, indicating a trans-diaxial relationship, while that between H4 and H5eq was only 4.8 Hz (Fig. 2). Reduction of 16t with L-Selectride® in THF resulted in the formation of 19, where delivery of a hydride occurred from the pseudo-equatorial trajectory placing the hydroxyl group in an axial position. Once again, 1H NMR coupling constants confirmed the stereochemistry. Now H4 had a coupling constant of 2.7 Hz to H5ax and 5.7 Hz to H5eq, indicating that H4 was indeed equatorial (Fig. 2). Treatment of 19 with LiAlH4 reduced the ester to the primary alcohol, thus generating tetrahydropyranol 20, which has the substitution and relative configuration present in the A-ring of lasonolide A.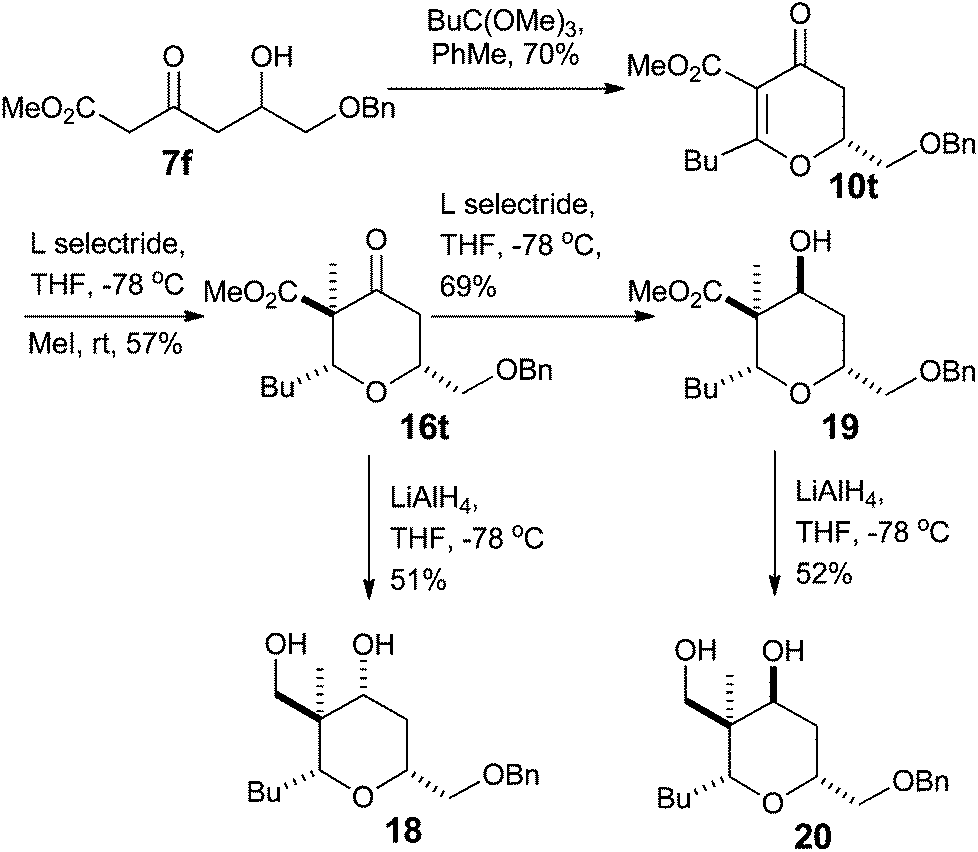 | ||
| Scheme 5 Synthesis of a model A-ring of lasonolide A. | ||
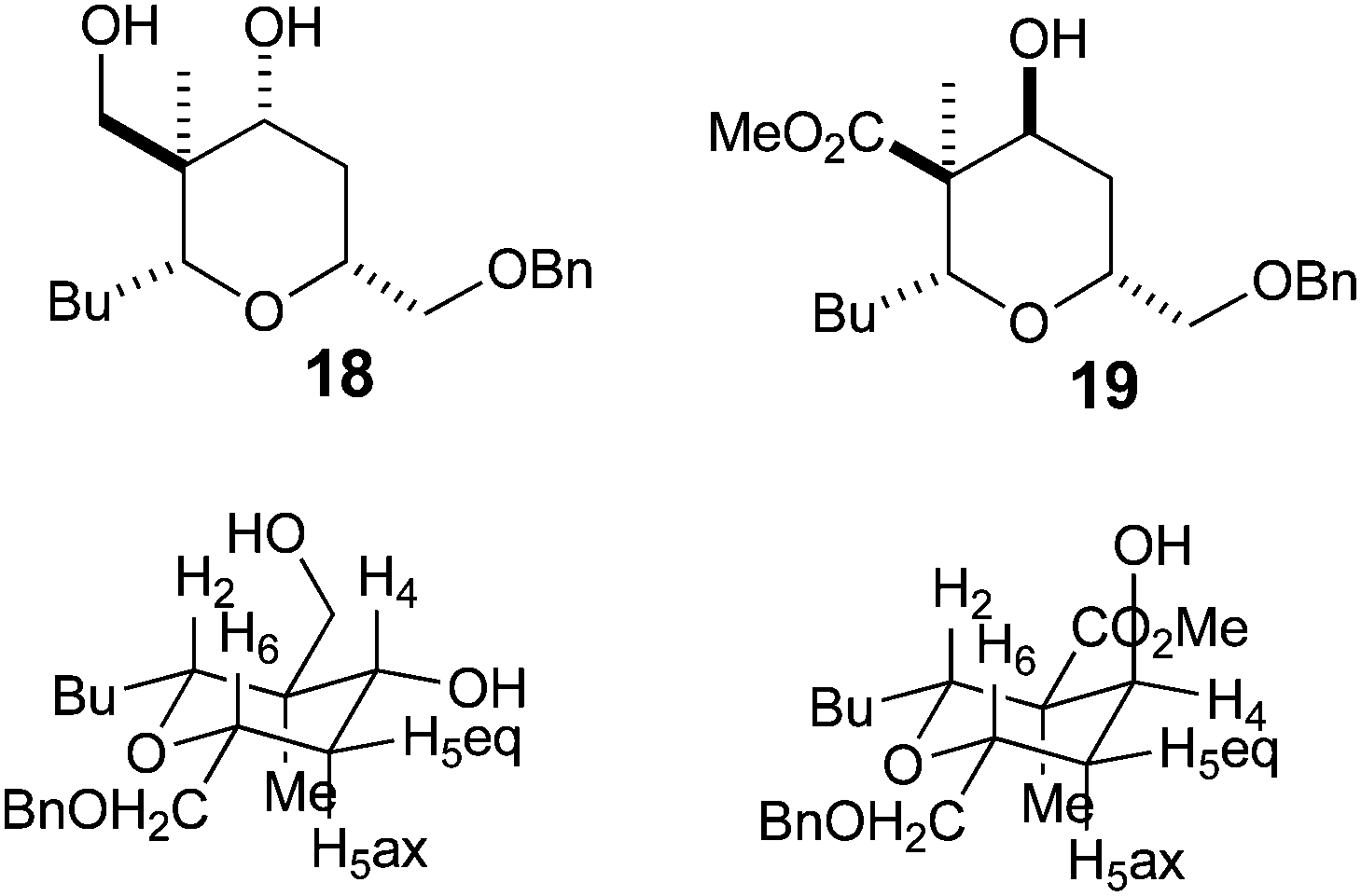 | ||
| Fig. 2 Conformations and stereochemistry of 18 and 19. | ||
Synthesis of 2,2,6-substituted tetrahydropyran-4-ones from dihydropyran-4-ones
We next turned our attention to extending the scope of the nucleophile we could employ in the conjugate addition reaction. Gilman cuprates had been previously reported in the conjugate addition reaction to dihydropyran-4-ones.23 In addition to these we extended the scope of the investigation to other nucleophiles (Table 5).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Nucleophile | Additive | Yield (%) | |||
| 10 | 21 | 22 | 23 | |||
| 1 | MeMgBr | None | 33 | 17 | 15 | 0 |
| 2 | MeMgBr | CuBr2·SMe2 | 0 | 68 | 9 | 20 |
| 3 | Me2CuLi | None | 0 | 49 | 0 | 14 |
| 4 | Me2CuLi | TMSCl | 0 | 70 | 4 | 8 |
| 5 | (2-Th)Cu(CN)MeLi2 | None | 0 | 44 | 32 | 5 |
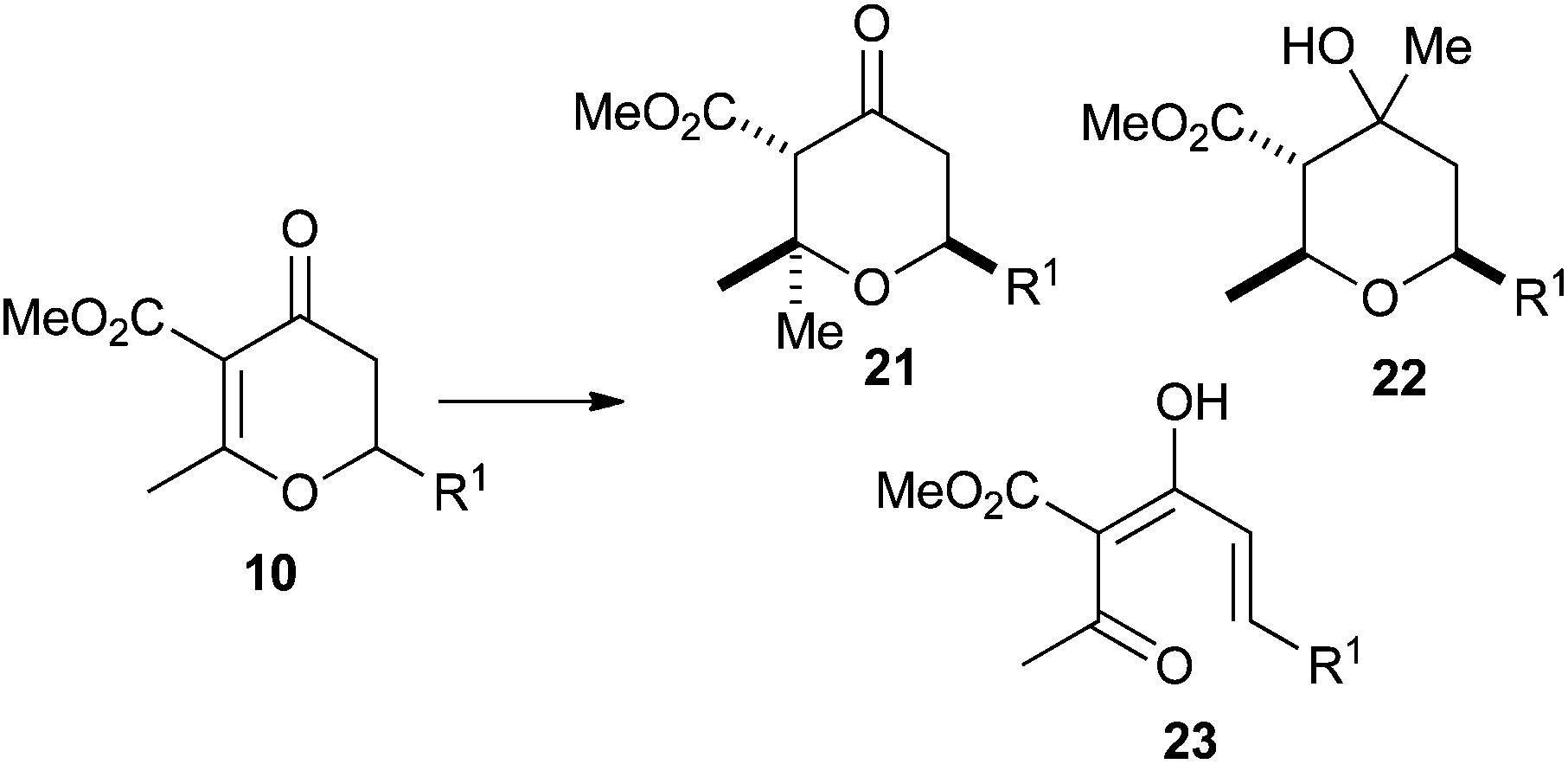
As can be seen from Table 5, when MeMgBr was used, the reaction generated essentially equal amounts of the 1,4- and 1,2-addition products 21 and 22. The inclusion of a CuBr2·SMe2 additive did bias this in favour of the 1,4-addition product 21, but also resulted in the formation of 23 which presumably arose from an elimination reaction. Gilman cuprate (Me2CuLi) also resulted in both 1,4-addition and elimination products. However, when TMSCl was added to the reaction,24 an increase in rate and selectivity for the 1,4-addition product 21 was seen. Finally, the use of a higher order cuprate was investigated but this did not lead to any further improvements and actually gave a sizable amount of the 1,2-addition product 22. As a result of these studies we opted for the use of Gilman cuprates.
We chose to investigate the reactions of Me2CuLi, Bu2CuLi, (H2CCH)2CuLi and Ph2CuLi with a representative number of dihydropyran-4-ones 10 (Table 6).
|
|
|||||
|---|---|---|---|---|---|
| THP | R1 | R2 | Ratio 21:24:25 |
Overall yield (%) | Yield (%) 26 |
| a | Ph | Me | 3.3:1.0:6.7 |
75 | 89 |
| b | Pr | Me | 2.4:1.0:3.0 |
65 | 74 |
| c | i-Pr | Me | 2.1:1.0:3.0 |
60 | 59 |
| d | 2-Furyl | Me | 2.4:1.0:3.7 |
27 | 73 |
| e | CH2OBn | Me | 2.3:1.0:2.3 |
69 | 92 |
| f | CH2CH2OBn | Me | 2.4:1.0:4.2 |
58 | 86 |
| g | Ph | Bu | 0.9:1.0:6.3 |
67 | 85 |
| h | Pr | Bu | 2.0:1.0:5.9 |
75 | 63 |
| i | Ph | CHCH2 |
0.1:0.1:1.0 |
59 | 90 |
| j | Pr | CHCH2 |
0.1:0.1:1.0 |
52 | 77 |
| k | i-Pr | CHCH2 |
0.1:0.1:1.0 |
59 | 69 |
| l | 2-Furyl | CHCH2 |
0.1:0.1:1.0 |
60 | 90 |
| m | CH2OBn | CHCH2 |
0.1:0.1:1.0 |
76 | 85 |
| n | CH2CH2OBn | CHCH2 |
0.1:0.1:1.0 |
47 | 100 |
| o | Ph | Ph | 0.0:0.0:1.0 |
85 | — |
| p | Pr | Ph | 0.0:0.0:1.0 |
67 | — |
| q | i-Pr | Ph | 0.0:0.0:1.0 |
67 | — |
| r | 2-Furyl | Ph | 0.0:0.0:1.0 |
61 | — |
| s | CH2OBn | Ph | 0.0:0.0:1.0 |
75 | — |
The Gilman cuprates all added from a pseudo-axial trajectory to form products with a 2,6-cis relationship between the new C2 substituent and H6, which was shown by positive nOe correlations of 4% in the case of the butyl, vinyl and phenyl substituents. Interestingly the tetrahydropyran-4-ones were actually formed as mixtures of three tautomers: the enol tautomer 25 and two ketone tautomers 21 and 24 which resulted from protonation of the intermediate enolate from either face. The product of the pseudo-axial protonation 21 had a positive nOe correlation of 1.6% between H3 and H5ax, confirming the stereochemistry, whilst in the product of pseudo-equatorial protonation, H5ax was shifted about 0.5 ppm downfield in 1H NMR due to an interaction with the nearby axial ester substituent (Fig. 3). When Ph2CuLi was used as the nucleophile the enol-tautomer 25 was the only product. Where mixtures of tautomers occurred they could be converted into single enol acetate products 26 in good yields by treatment with acetic anhydride in pyridine at 40 °C. This conversion provided further support for our assignment of these 1,4-addition products as compounds 21, 24 and 25.
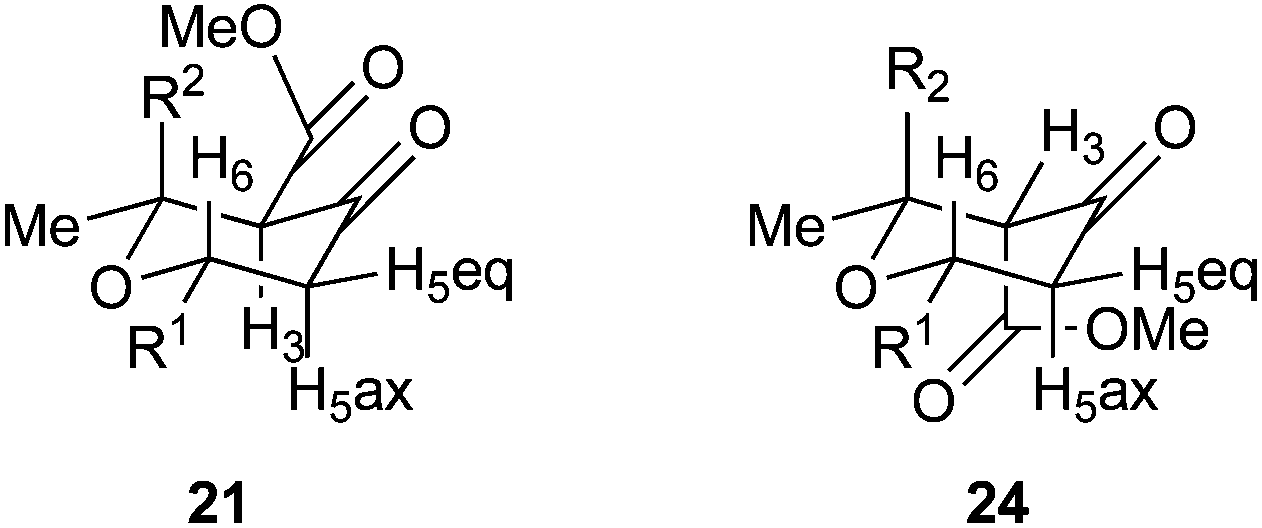 | ||
| Fig. 3 Conformation and stereochemistry of 21 and 24. | ||
Conclusions
We have developed a new modification of the Maitland–Japp reaction using orthoamides and orthoesters, which provides access to a range of dihydropyran-4-ones 10 in good yields. These dihydropyran-4-ones 10 can be converted to 2,6-cis tetrahydropyran-4-ones 11 by the stereoselective addition of L-Selectride®. The intermediate enolate resulting from this addition can be trapped stereoselectively with either a proton or MeI to form tetrahydropyran-4-ones with a quaternary stereocentre at C3 16. The utility of these procedures was demonstrated by their use in the total synthesis of a constituent of the Civet cat secretion and for the synthesis of a model A-ring of lasonolide A. Treatment of the dihydropyran-4-ones 10 with a Gilman cuprate has led to the development of a procedure for the stereoselective formation of tetrahydropyran-4-ones 25 that are doubly substituted at the C2 position. Hence, we have overcome the difficulties inherent in the functionalisation of 2,6-cis-tetrahydropyran-4-one products of the Maitland–Japp reaction 1, and provided a route to the stereoselective construction of highly functionalised tetrahydropyran rings.Experimental
General methods
For general experimental details, including information on solvent purification and the spectrometers used in this research, as well as for procedures and spectroscopic and crystallographic data not reported below, see ESI.†General procedure for the synthesis of 2-methyl dihydropyrans
N,N-Dimethylacetamide dimethyl acetal (0.16 mL, 1.08 mmol) was added to a stirred solution of δ-hydroxy-β-ketoester (0.54 mmol) in dry toluene (4 mL) at room temperature. The solution was stirred at room temperature and monitored by TLC. Upon completion of the reaction, the solvent was removed in vacuo. Purification by flash column chromatography (petroleum ether–ethyl acetate) afforded the product.General procedure for L-Selectride® reduction of dihydropyran-4-ones with methyl iodide quench
A 1.0 M solution of L-Selectride® in THF (0.04 mL, 0.04 mmol) was added to a stirred solution of DHP (0.04 mmol) in THF (1 mL) at −78 °C. The mixture was stirred for 1 hour, after which time iodomethane (0.4 mmol) was added. The reaction mixture was stirred at room temperature until completion, when it was partitioned between Et2O (10 mL) and sat. aq. NHCl4 (10 mL). The aqueous layer was washed with Et2O (10 mL) and the combined organic extracts were washed with brine (20 mL), dried over MgSO4 and concentrated in vacuo. Purification by flash column chromatography (petroleum ether–ethyl acetate) afforded the product.General procedure for Gilman cuprate addition to dihydropyran-4-ones
An organolithium solution (0.41 mmol) was added to a suspension of copper iodide (38.7 mg, 0.20 mmol) in THF (1.17 mL) at 0 °C. The mixture was stirred at this temperature for 20 minutes and then cooled to −78 °C. Addition of TMSCl (0.08 mL, 0.64 mmol) was followed by addition of DHP (0.13 mmol) in THF (1.17 mL) at −78 °C. The reaction mixture was stirred at this temperature for 4 hours, then quenched with sat. aq. NH4Cl (1 mL) and allowed to warm to rt with vigorous stirring. The mixture was diluted with sat. aq. NH4Cl (10 mL) and extracted with EtOAc (4 × 15 mL). The combined organic extracts were washed with H2O (15 mL) and brine (15 mL), then dried over MgSO4 and concentrated in vacuo. Flash column chromatography (petroleum ether–ethyl acetate) afforded the products.:enol:ketoax 3.3:6.7:1); νmax (film) 3016, 2985, 2930, 2889, 1723, 1692, 1619, 1582, 1418, 1356, 1317, 1257, 1200, 1111, 1048 cm−1; δH (400 MHz, C6D6) 13.27 (1H, s), 7.80–7.00 (5H, m), 7.80–7.00 (5H, m, ketoeq), 7.80–7.00 (5H, m, ketoax), 4.64–4.59 (1H, m, ketoeq), 4.64–4.59 (1H, m, ketoax), 4.57 (1H, dd, J = 10.7, 2.9 Hz), 3.47 (1H, s, ketoeq), 3.39 (3H, m, ketoeq), 3.38 (1H, m, ketoax), 3.27 (1H, s, ketoax), 3.26 (3H, s), 3.19 (3H, s, ketoax), 2.46 (1H, dd, J = 17.4, 10.7 Hz), 2.44 (1H, m, ketoax), 2.35–2.30 (1H, m), 2.35–2.30 (1H, m, ketoeq), 2.02 (1H, dd, J = 13.7, 10.7 Hz, ketoeq), 1.62 (3H, s), 1.47 (3H, s), 1.45 (3H, s, ketoeq), 1.35 (3H, s, ketoeq), 1.23 (3H, s, ketoax) and 0.90 (3H, s, ketoax) ppm; δC (100 MHz, C6D6) 201.9 (ketoax), 200.8 (ketoeq), 172.3, 171.8, 168.1 (ketoeq), 168.0 (ketoax), 142.1, 141.6, 128.7, 128.6, 128.5, 127.9, 127.7, 126.6, 126.2, 126.0, 105.1, 77.6, 76.4, 74.0, 73.4, 72.7 (ketoeq), 68.6, 67.0 (ketoeq), 66.0 (ketoax), 53.2 (ketoax), 51.5 (ketoeq), 51.0, 48.8 (ketoeq), 47.3 (ketoax), 37.6, 29.8, 29.3 (ketoeq), 27.7 (ketoax), 25.8, 24.7 (ketoax) and 21.5 (ketoeq) ppm; m/z (ESI+) 285 (M + Na)+ (Found 285.1092 (M + Na)+. C15H18NaO4 requires 285.1097).
General procedure for acylation of tetrahydropyrans
The THP mixture (0.03 mmol), acetic anhydride (0.1 mL, 0.1 mmol) and DMAP (cat.) were stirred in pyridine (0.47 mL) at 40 °C for 40 minutes. The mixture was cooled to rt, concentrated in vacuo, and then partitioned between Et2O (30 mL) and H2O (10 mL). The organic layer was washed with H2O (10 mL) and brine (10 mL), then dried over MgSO4 and concentrated in vacuo. Flash column chromatography (petroleum ether–diethyl ether) gave the product.Acknowledgements
We thank the University of York (P.B.S.) and the Malaysian Ministry of Higher Education (N.M.N.) for funding.Notes and references
- (a) C. A. C. Araujo, L. V. Alegrio and L. L. Leon, Phytochemistry, 1998, 49, 751 CrossRef CAS; (b) C. A. C. Araujo, L. V. Alegrio, D. C. F. Gomes, M. E. F. Lima, L. Gomes-Cardoso and L. L. Leon, Mem. Inst. Oswaldo. Cruz., 1999, 94, 791 CrossRef CAS PubMed.
- For the phorboxazoles see: (a) P. A. Searle and T. F. Molinski, J. Am. Chem. Soc., 1995, 117, 8126 CrossRef CAS; (b) P. A. Searle, T. F. Molinski and L. J. Brzezinski, J. Am. Chem. Soc., 1996, 118, 9422 CrossRef CAS; (c) T. F. Molinski, Tetrahedron Lett., 1996, 37, 7879 CrossRef CAS.
- For lasonolide A see: P. A. Horton, F. E. Koehn, R. E. Longley and O. J. McConnell, J. Am. Chem. Soc., 1994, 116, 6015 CrossRef CAS.
- For a recent review see: (a) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC; (b) M. A. Perry, S. D. Rychnovsky and N. Sizemore, Synthesis of Saturated Oxygenated Heterocycles I, Topics in Heterocyclic Chemistry 35, ed. J. Cossy, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 43–95 Search PubMed.
- For a review on the Prins reaction and its application to tetrahydropyran synthesis see: C. Olier, M. Kaafarani, S. Gastaldi and M. P. Bertrand, Tetrahedron, 2010, 66, 413 CrossRef CAS PubMed.
- (a) K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 3558 CrossRef; (b) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS PubMed; (c) K. Gademann, D. E. Chavez and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 3059 CrossRef CAS.
- P. A. Clarke and K. Ermanis, Curr. Org. Chem., 2013, 17, 2025 CrossRef CAS.
- P. A. Clarke and W. H. C. Martin, Tetrahedron, 2005, 61, 5433 CrossRef CAS PubMed.
- P. A. Clarke, S. Santos, N. Mistry, L. Burroughs and A. C. Humphries, Org. Lett., 2011, 13, 624 CrossRef CAS PubMed.
- P. A. Clarke and K. Ermanis, Org. Lett., 2012, 14, 5550 CrossRef CAS PubMed.
- F. R. Japp and W. Maitland, J. Chem. Soc., 1904, 85, 1473 RSC.
- P. A. Clarke and W. H. C. Martin, Org. Lett., 2002, 4, 315 CrossRef PubMed.
- (a) P. A. Clarke, W. H. C. Martin, J. M. Hargreaves, C. Wilson and A. J. Blake, Chem. Commun., 2005, 1061 RSC; (b) P. A. Clarke, W. H. C. Martin, J. M. Hargreaves, C. Wilson and A. J. Blake, Org. Biomol. Chem., 2005, 3, 3551 RSC.
- P. A. Clarke, S. Santos and W. H. C. Martin, Green Chem., 2007, 9, 438 RSC.
- M. Iqbal, N. Mistry and P. A. Clarke, Tetrahedron, 2011, 67, 4960 CrossRef CAS PubMed.
- P. A. Clarke, J. M. Hargreaves, D. J. Woollaston and R. M. Rodríguez Sarmiento, Tetrahedron Lett., 2010, 51, 4731 CrossRef CAS PubMed.
- P. A. Clarke, P. B. Sellars and N. Mistry, Tetrahedron Lett., 2011, 52, 3564 Search PubMed.
- S. Hanessian and E. Moralioglu, Can. J. Chem., 1972, 50, 233 CrossRef CAS.
- H. Ueno, A. Maruyama, M. Miyake, E. Nakao, K. Nakao, K. Umezu and I. Nitta, J. Med. Chem., 1991, 34, 2468 CrossRef CAS.
- CEM Discover microwave oven.
- For other recent syntheses of Civet cat secretion see: (a) S. Sultana, K. Indukuri, M. J. Deka and A. K. Saikia, J. Org. Chem., 2013, 78, 12181 CrossRef PubMed; (b) O. Karblubikova, M. Babjak and T. Gracza, Tetrahedron, 2011, 67, 4980 CrossRef PubMed; (c) F. K. Chio, J. Warne, D. Gough, M. Penny, S. Green, S. J. Coles, M. B. Hursthouse, P. Jones, L. Hassell, T. M. McGuire and A. P. Dobbs, Tetrahedron, 2011, 67, 5107 CrossRef CAS PubMed; (d) H. Zhou and T.-P. Loh, Tetrahedron Lett., 2009, 50, 4368 CrossRef CAS PubMed.
- For syntheses of lasonolide A see: (a) B. M. Trost, C. E. Stivala, K. L. Hull, A. Huang and D. R. Fandrick, J. Am. Chem. Soc., 2014, 136, 88 CrossRef CAS PubMed; (b) A. K. Ghosh and G. Gong, Chem. – Asian J., 2008, 3, 1811 CrossRef CAS PubMed; (c) T. Yoshimura, F. Yakushiji, S. Kondo, X. Wu, M. Shindo and K. Shishido, Org. Lett., 2006, 8, 475 CrossRef CAS PubMed; (d) H. Y. Song, J. M. Joo, J. W. Kang, D.-S. Kim, C.-K. Jung, H. S. Kwak, J. H. Park, E. Lee, C. Y. Hong, S. W. Jeong, K. Jeon and J. H. Park, J. Org. Chem., 2003, 68, 8080 CrossRef PubMed.
- (a) D. S. Reddy, D. V. Veld and J. Aube, J. Org. Chem., 2004, 69, 1716 CrossRef CAS PubMed; (b) J. L. Romine, Z. Yang, G. Wang, N. Nguyen, J. A. Bender, D. R. St. Laurent and M. Belema, International Patent, WO 2014/065791 A1, 2014 Search PubMed; (c) K. M. Cottrell, J. Maxwell, Q. Tang, A.-L. Grillot, A. Le Tiran and E. Perola, US Patent, US 7,964,624 B1, 2011 Search PubMed.
- E. J. Corey and N. W. Boaz, Tetrahedron Lett., 1985, 26, 6015 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures and copies of spectroscopic data. See DOI: 10.1039/c5ob00292c |
| This journal is © The Royal Society of Chemistry 2015 |