
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chirality extension of an oxazine building block en route to total syntheses of (+)-hyacinthacine A2 and sphingofungin B†
Seok-Hwi
Park‡
a,
Xiangdan
Jin‡
a,
Jong-Cheol
Kang
a,
Changyoung
Jung
a,
Seong-Soo
Kim
a,
Sung-Soo
Kim
a,
Kee-Young
Lee
b and
Won-Hun
Ham
*a
aSchool of Pharmacy, Sungkyunkwan University, Suwon 440-746, Republic of Korea. E-mail: whham@skku.edu; Fax: +82 31 292 8800; Tel: +82 31 290 7706
bYonsung Fine Chemicals Co., Ltd, Hwaseong 445-944, Republic of Korea. E-mail: uncleduly@skku.edu; Fax: +82 31 351 6624; Tel: +82 31 351 6622
First published on 3rd March 2015
Abstract
Concise and stereocontrolled syntheses of (+)-hyacinthacine A2 and sphingofungin B were achieved via a diastereomerically enriched oxazine intermediate. The key strategies include palladium(0)-catalyzed intramolecular oxazine formation and diastereoselective nucleophilic addition to an aldehyde. (+)-Hyacinthacine A2 was synthesized in 13 steps and 10.2% overall yield and the synthesis of sphingofungin B proceeded in a linear sequence over 15 steps and 6.9% overall yield from (R)-methyl 2-benzamido-3-((tert-butyldimethylsilyl)oxy)propanoate.
Introduction
As part of an ongoing research program aimed at developing total syntheses of biologically active compounds, we recently described a facile strategy towards the preparation of syn,syn-, syn,anti-,1 and anti,syn-oxazines2via a stereoselective palladium(0)-catalyzed reaction (Scheme 1). The diastereoselectivity of syn,syn- and syn,anti- cyclizations can be critically dependent upon whether the reaction temperature results in kinetic or thermodynamic control of the products. Meanwhile, the diastereoselectivity of anti,syn-oxazine ring formation is predominantly controlled by the bulky OTBS group. Based on the oxazine library, piperidine alkaloid D-fagomine,2d pyrrolidine alkaloid DAB-1,2d indolizidine alkaloid (−)-lentiginosine,2b phytosphingosines,1a,2c and other natural products bearing three contiguous chiral centers were successfully synthesized.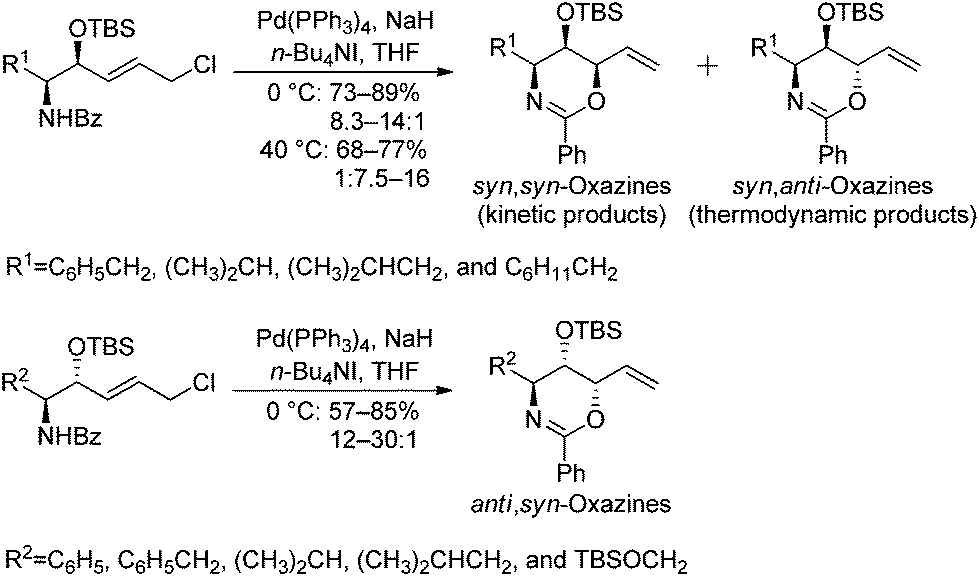 | ||
| Scheme 1 Oxazine library generated via a stereoselective palladium(0)-catalyzed reaction. | ||
Polyhydroxylated alkaloids (or aminopolyols) isolated from plants and microorganisms should be thoroughly investigated because they can act as glycosidase inhibitors by mimicking natural monosaccharide substrates, which may allow for the development of new antiviral, antidiabetic, and anticancer agents.3 More than two hundred of these naturally-occurring and water-soluble compounds have been isolated and classified structurally as piperidines, pyrrolidines, pyrrolizidines, indolizidines, and nortropanes. (+)-Hyacinthacine A2 (1), which is a representative pyrrolizidine alkaloid, was first isolated from Muscari armeniacum bulbs and is a good inhibitor of both amyloglucosidase and lactase (Fig. 1).4
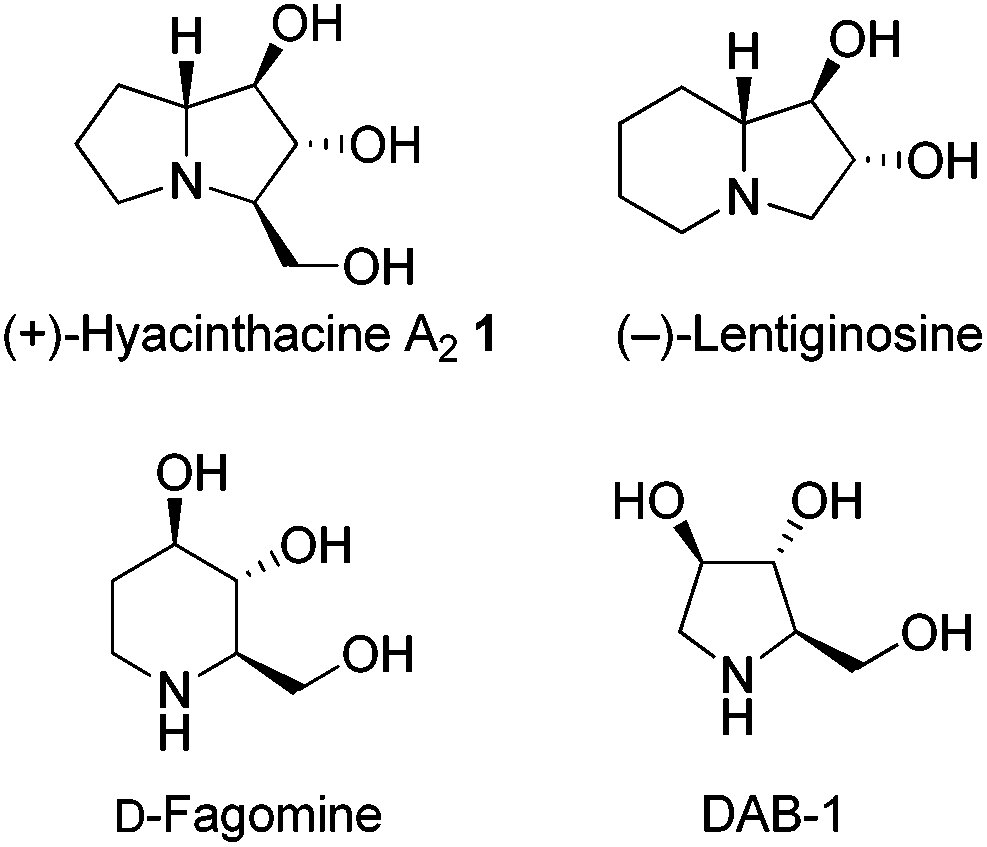 | ||
| Fig. 1 The structures of several polyhydroxylated alkaloids. | ||
Its promising biological properties and the existence of four contiguous stereogenic centers in its structure have prompted the development of numerous synthetic approaches.5,6 For example, in 2011, Fox et al. described the concise synthesis of (+)-hyacinthacine A2, which relied on a novel transannular hydroamination of 5-aza-cyclooctene.5a Zheng and Huang's approach for the synthesis of 1 was SmI2-mediated radical coupling reaction of an activated amide from (R,R)-tartarimide.5b Bonaccini et al. reported a stereocontrolled cyclic nitrone cycloaddition for the synthesis of 1.5c Another previous result by Goti and Merino started from a nitrone, which is readily prepared from D-arabinose.5e Marco et al. demonstrated a stereoselective synthesis of 1via a double cyclization with the one-pot formation of two C–N bonds.5f
Sphingofungins isolated from fungi are also of significant interest owing to their biological activities as immunosuppressants and potential antifungal agents via the inhibition of serine palmitoyl-CoA transferase (SPT).7 In addition to sphingofungins, congeners such as myriocin, sulfamisterin, and mycestericins exhibit similar functions.7 Sphingofungin B (2), having a polar polyhydroxy amino acid head group and a long lipid chain containing an (R)-hydroxy group at C-14, was isolated from the fermentation broth of Aspergillus fumigatus and its structure was elucidated by Merck group in 1992 (Fig. 2).8
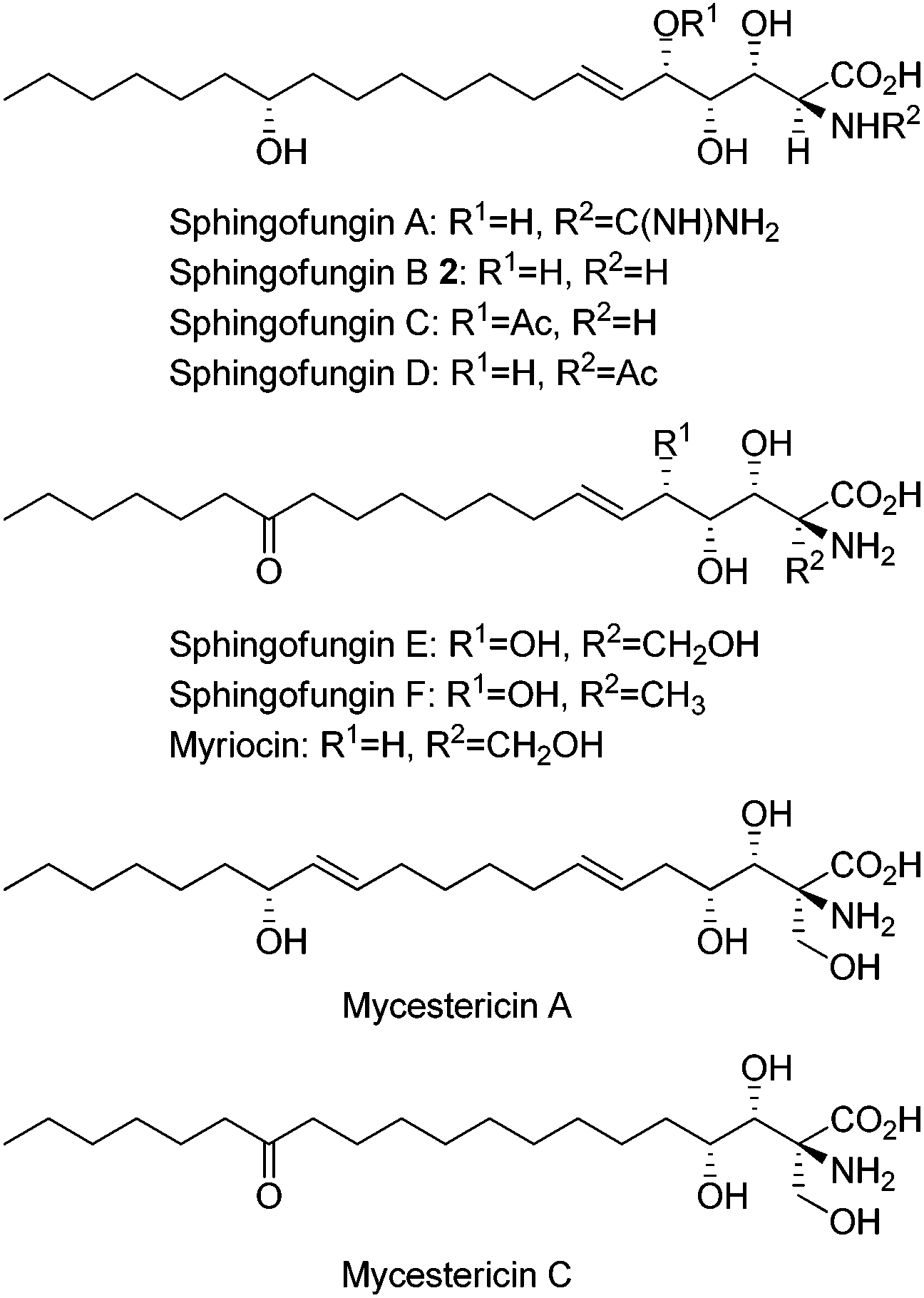 | ||
| Fig. 2 The structures of several sphingolipids. | ||
The promising biological activities and more than three contiguous chiral centers present in these compounds also have prompted the development of numerous synthetic approaches.9–15 For example, the first total synthesis of sphingofungin B was successfully achieved by Kobayashi et al. in 1996 making use of catalytic asymmetric aldol reactions.9 Meanwhile, the first total synthesis of sphingofungin D was reported by Mori et al. in 1994 using a polar building block derived from N-acetyl-D-mannoside.10a,c Shortly thereafter, Chida et al. accomplished the synthesis of sphingofungin D starting from myo-inositol.10b
Herein we report the highly stereocontrolled total syntheses of (+)-hyacinthacine A2 and sphingofungin B via straightforward procedures that rely on a chiral anti,syn-oxazine building block.
Results and discussion
Chirality extension of a chiral anti,syn-oxazine
The first step in the syntheses, the preparation of the known chiral 1,3-oxazine 4 beginning with (R)-methyl 2-benzamido-3-((tert-butyldimethylsilyl)oxy)propanoate 3, proceeded in 56.8% yield after five steps (Scheme 2).2b,d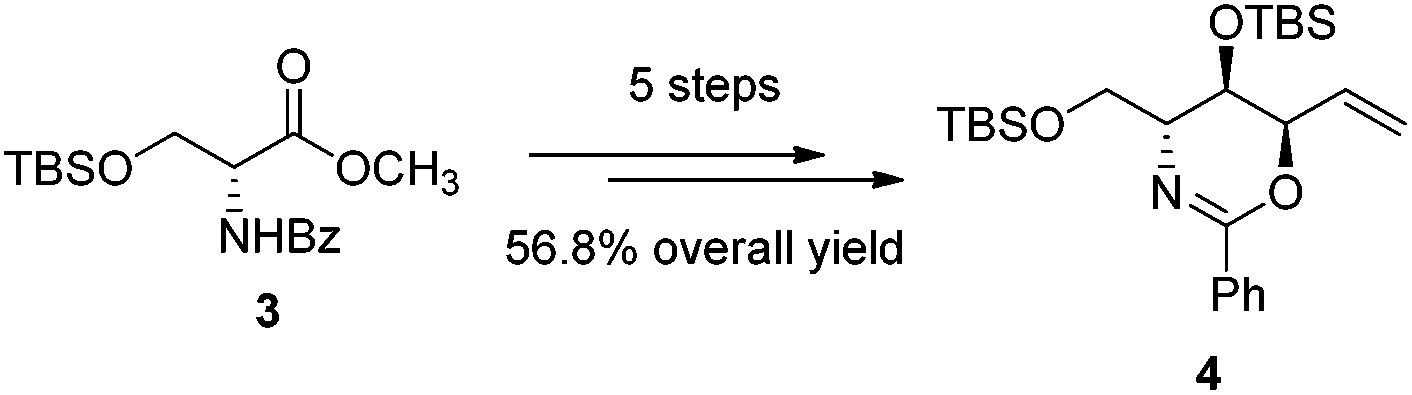 | ||
| Scheme 2 Synthesis of anti,syn-oxazine 4. | ||
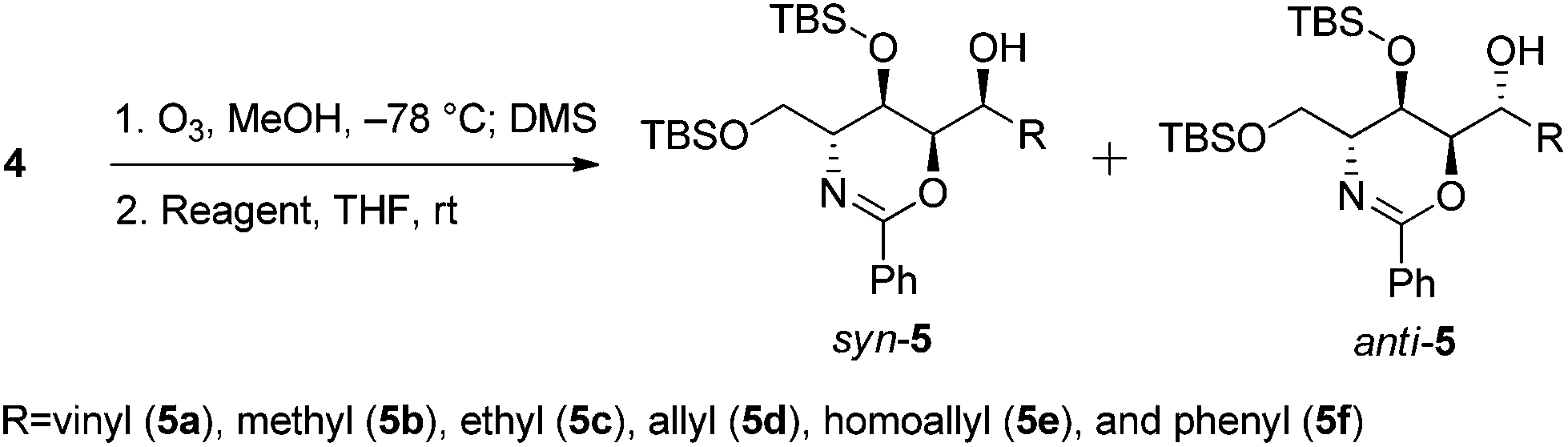
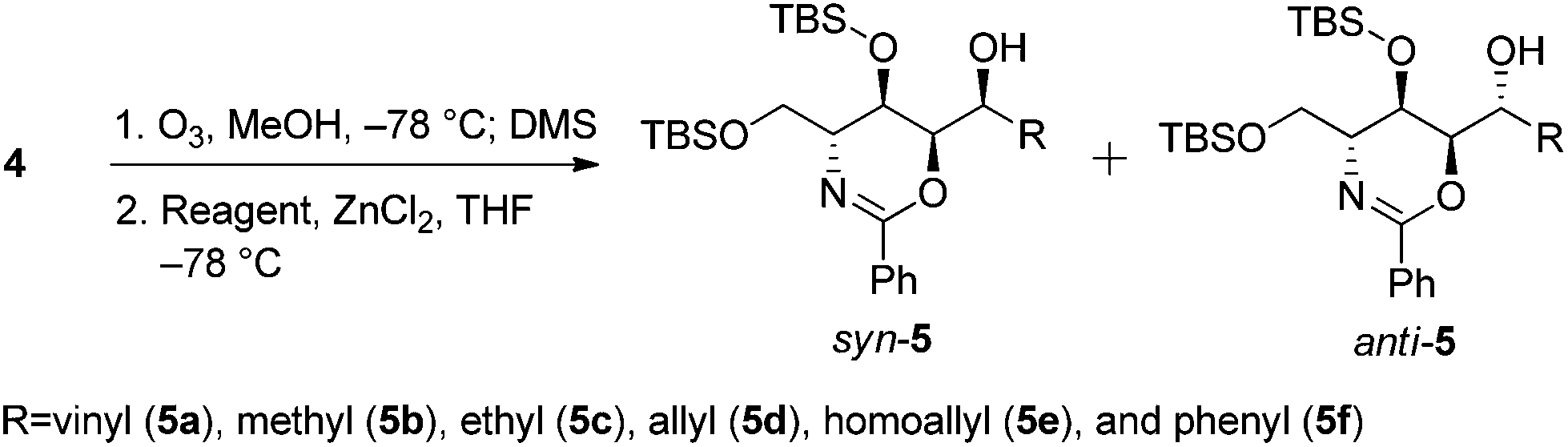
We envisioned that a stereocontrolled Grignard reaction or Grignard-like nucleophilic addition16 to the oxazine-derived aldehyde could generate the desired fourth contiguous stereocenter. In most cases, the chelation-control product of a Grignard reaction is produced via the chelation of organomagnesium nucleophiles rather than bisligation of Lewis acids; this is why ethers, which are the universal solvents of Grignard reactions, coordinate to Lewis acids to form octahedral complexes.16e,17 Therefore, it is uncertain whether Lewis acid additives such as ZnCl2,18,19 ZnBr2,18–21 ZnI2,22,23 MgBr2·OEt2,20,23,24 CdCl2,18 and Ti(OiPr)420 act as bidentate ligands, although stereoselective addition should be possible via a chelated transition state.25 Hence, ZnCl2 was tested in the following reaction to optimize the diastereomeric enrichment.
The results of the introduction of Grignard reagents are shown in Table 1. Unfortunately, in the absence of additives, organomagnesiums cannot chelate. Meanwhile, Table 2 summarizes the results of reactions in which ZnCl2 was added to the reaction mixture; in each case, 1.1 equivalents of the Lewis acid were added dropwise followed by the addition of 3.0 equivalents of each Grignard reagent. Surprisingly, reactions favored the syn-product, indicating the occurrence of α-chelation with the oxygen inside the 1,3-oxazine moiety.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Time (h) | Product | Ratioa (syn/anti) | Yieldb (%) |
| a Ratios were determined by 1H NMR peak intensities. b Yields refer to isolated yields over the two steps. | |||||
| 1 | CH2CHMgBr | 0.5 | 5a | 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
78 |
| 2 | MeMgBr | 0.5 | 5b | 1:1.5 |
71 |
| 3 | EtMgBr | 0.5 | 5c | 1.5:1 |
70 |
| 4 | CH2CHCH2MgBr | 0.5 | 5d | 2.0:1 |
70 |
| 5 | CH2CH(CH2)2MgBr | 0.5 | 5e | 1.5:1 |
70 |
| 6 | PhMgBr | 0.5 | 5f | 4:1 |
70 |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Time (h) | Product | Ratioa (syn/anti) | Yieldb (%) |
| a Ratios were determined by 1H NMR peak intensities. b Yields refer to isolated yields over the two steps. | |||||
| 1 | CH2CHMgBr | 1 | 5a | 10:1 |
62 |
| 2 | MeMgBr | 1 | 5b | 2.2:1 |
59 |
| 3 | EtMgBr | 1 | 5c | 14:1 |
60 |
| 4 | CH2CHCH2MgBr | 1 | 5d | >20:1 |
60 |
| 5 | CH2CH(CH2)2MgBr | 1 | 5e | >20:1 |
62 |
| 6 | PhMgBr | 1 | 5f | 8:1 |
65 |
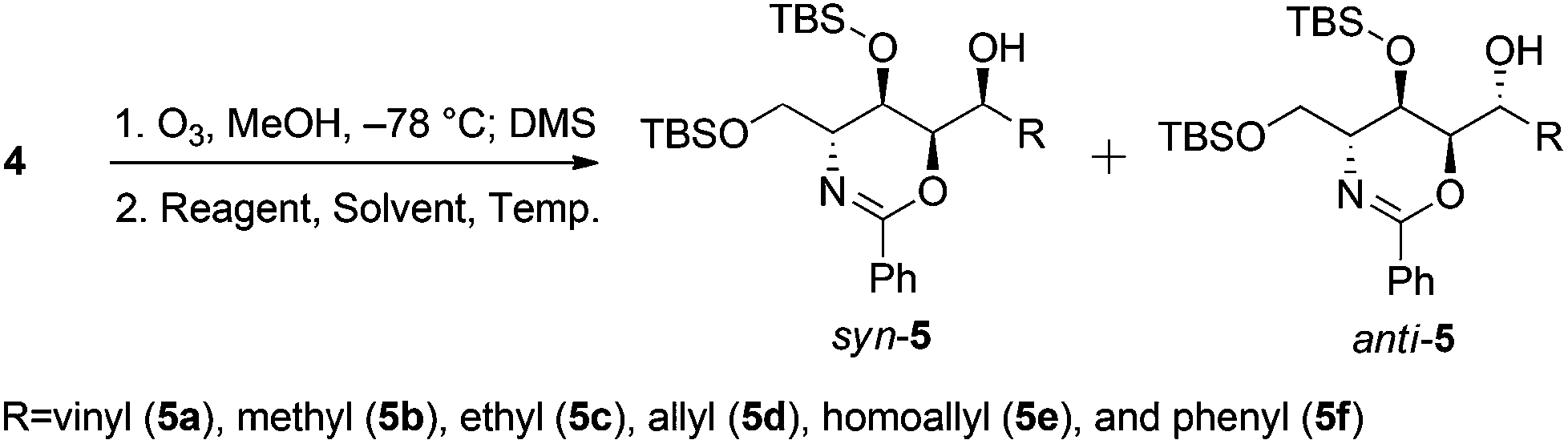
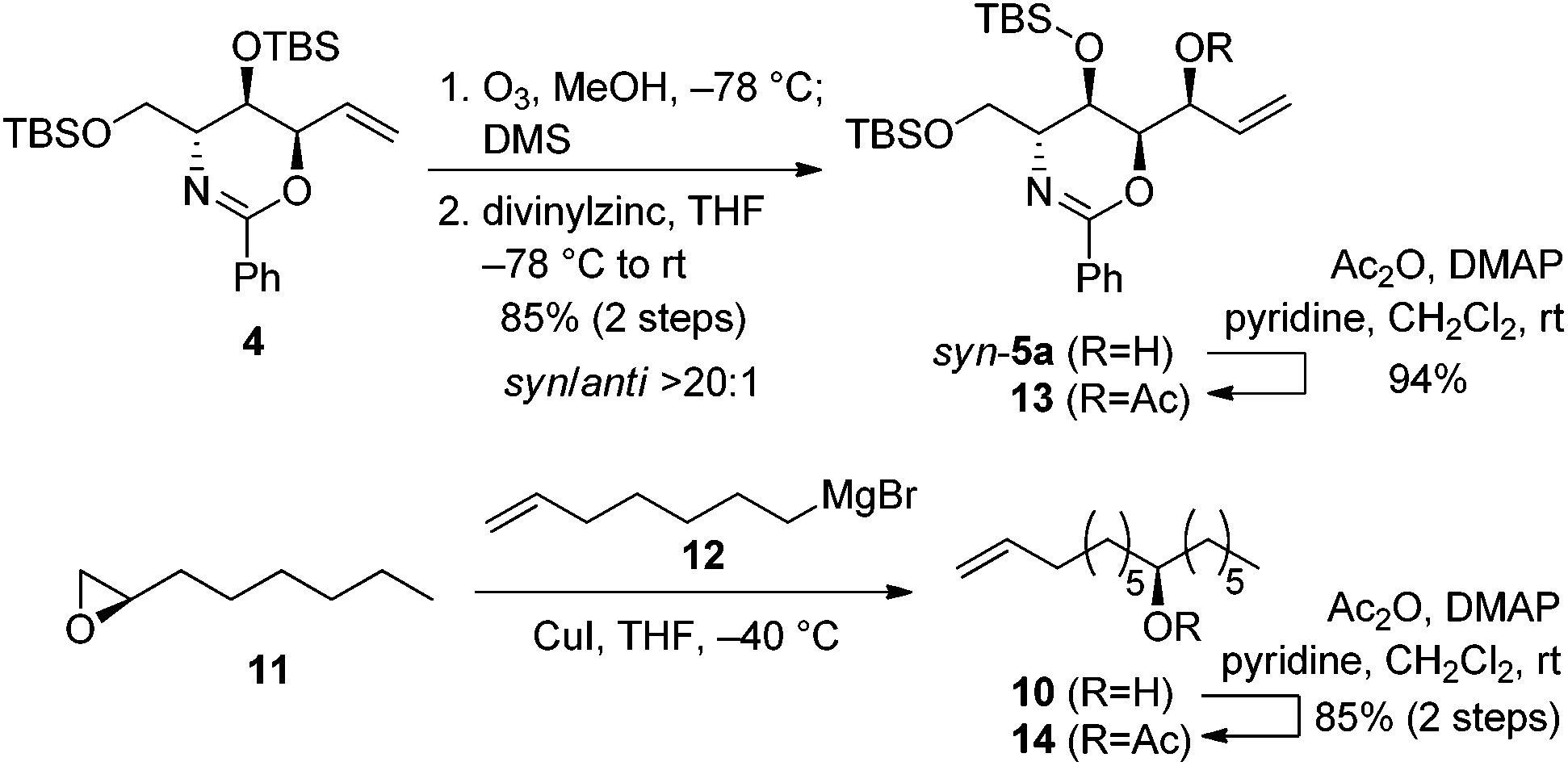
Further experiments for using other organometallic compounds were conducted (Table 3). Replacing magnesium with lithium, which is less prone to chelation, shifted the product composition slightly to favor the Felkin–Anh variant (entry 1). Vinylation using vinylzinc chloride and divinylzinc, which were prepared in situ from zinc chloride and vinylmagnesium bromide,26 generated the syn-alcohol exclusively (entries 2–4). Unfortunately, these conditions have limited applications; reactions using Me2Zn, Et2Zn, and other reagents proceeded sluggishly (entries 5–8), while that involving Ph2Zn proceeded much more quickly (entry 9).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Temp. | Time (h) | Product | Ratioa (syn/anti) | Yieldb (%) |
| a Ratios were determined by 1H NMR peak intensities. b Yields refer to isolated yields over the two steps. | |||||||
| 1 | CH2CHLi | Et2O | −78 °C | 1 | 5a | 1:2.0 |
58 |
| 2 | CH2CHZnCl | THF | −78 °C to rt | 3 | 5a | >20:1 |
75 |
| 3 | (CH2CH)2Zn | THF | −78 °C to rt | 3 | 5a | >20:1 |
85 |
| 4 | (CH2CH)2Zn/ZnCl2 | THF | −78 °C to rt | 3 | 5a | >20:1 |
80 |
| 5 | Me2Zn | THF | −78 °C to rt | 3 | 5b | trace | |
| 6 | Et2Zn | THF | −78 °C to rt | 3 | 5c | trace | |
| 7 | (CH2CHCH2)2Zn | THF | −78 °C to rt | 3 | 5d | trace | |
| 8 | (CH2CH(CH2)2)2Zn | THF | −78 °C to rt | 3 | 5e | trace | |
| 9 | Ph2Zn | THF | −78 °C to rt | 3 | 5f | >20:1 |
85 |
To determine the relative stereochemistry of syn-5a, diastereoisomer anti-5a was prepared (Table 1, entry 1). Oxazine rings of syn-5a and anti-5a were cleaved by hydrogenolysis. Two secondary alcohols gave the corresponding acetals, respectively. Unfortunately, the coupling constants between H4 and H5 of trans-6 and cis-6 were not in good agreement with theoretical values [J4,5(trans-6) = 8.0 Hz and J4,5(cis-6) = 5.4 Hz]. Therefore, NOESY spectra of trans-6 and cis-6, representing the correlations between H4 or H5 and acetal methyl protons, confirm the identification of the diastereoisomers (Scheme 3).
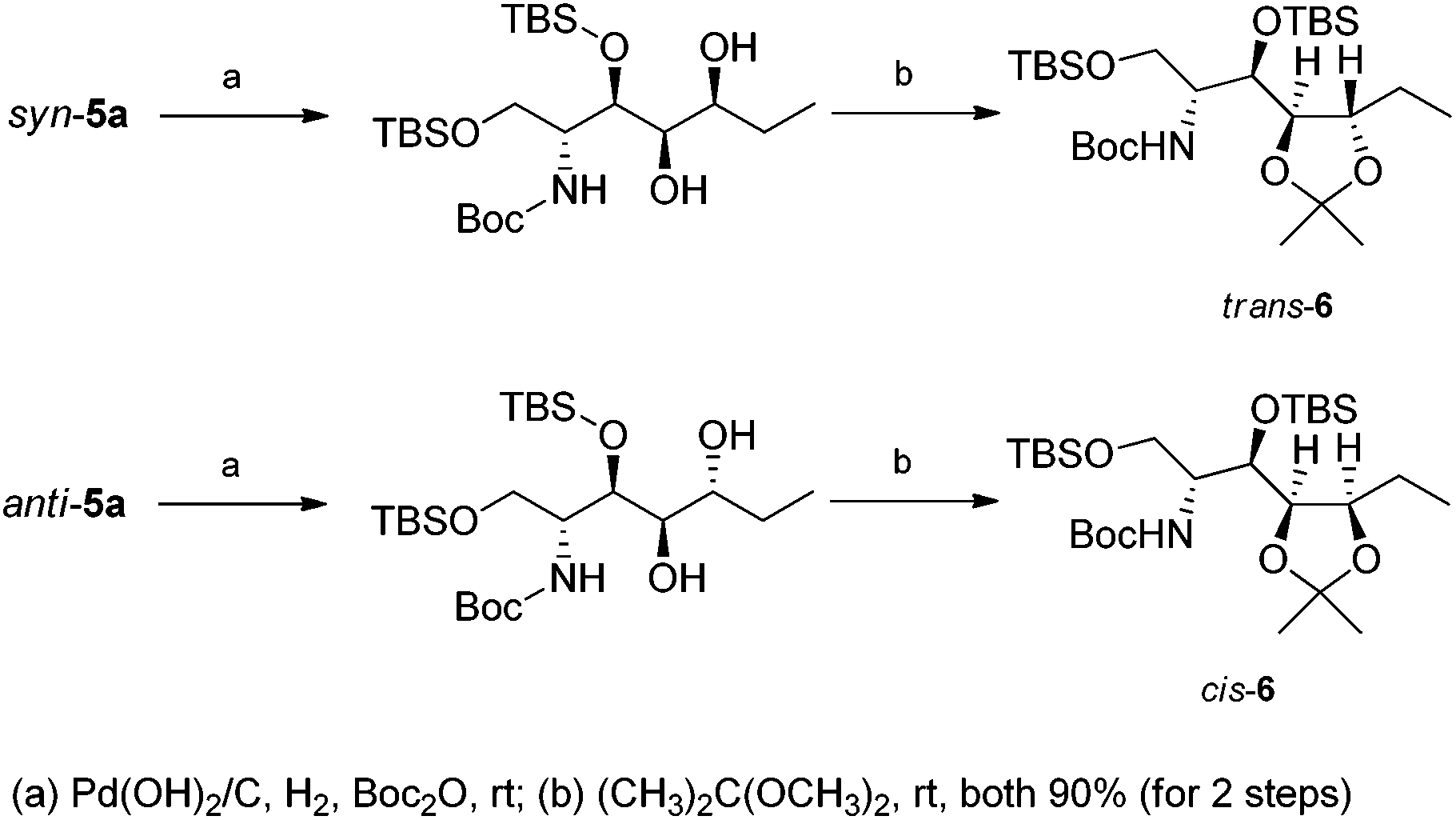 | ||
| Scheme 3 Determination of the relative stereochemistry of trans-6 and cis-6. | ||
Total synthesis of (+)-hyacinthacine A2
Our retrosynthesis (Scheme 4) suggests that the bicyclic structure of 1 could be synthesized from tertiary amine 7, which could in turn be prepared from secondary mesylate 8via intramolecular SN2 substitution. Conveniently, mesylate 8 could be obtained from the pentenyl alcohol syn-5e. The synthesis of syn-5e could be accomplished by nucleophilic addition to the corresponding aldehyde, which could be derived from conversion of the exomethylene moiety of anti,syn-oxazine 4.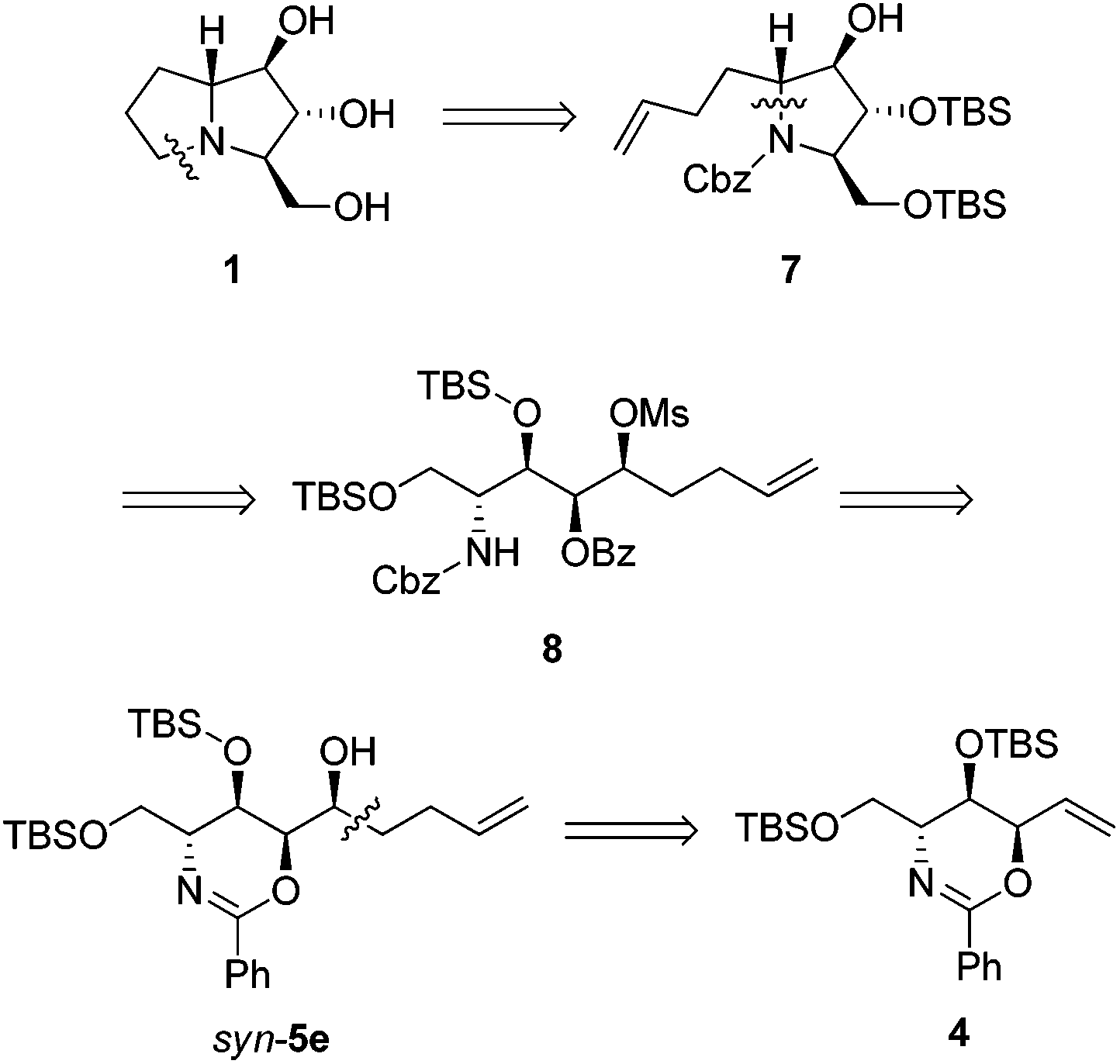 | ||
| Scheme 4 Retrosynthesis of 1. | ||
Pentenyl alcohol syn-5e was consecutively treated with MsCl and benzyl chloroformate under biphasic conditions to afford carbamate 8 in 69% yield over 2 steps. The room temperature exposure of 8 to sodium hydride led to the formation of tertiary amine 7 (mixture of rotamers) in 83% yield via intramolecular cyclization as well as O-benzoyl hydrolysis. Next, ozonolysis and hydrogenolysis of 7 gave protected 9 in 72% yield over 2 steps. Finally, removal of TBS-protecting groups by treatment of 9 with concentrated acid yielded the 1·HCl salt, which was neutralized with an ion-exchange resin to give 1 in 70% yield (Scheme 5). Our [α]D +15.3 (c 0.1, H2O) compared to the reported [α]D +20.1 (c 0.44, H2O),4 [α]D +12 (c 0.40, H2O),5a [α]D +10.6 (c 1.64, H2O),5b and [α]D +10.5 (c 0.6, H2O)5i confirms the identity of the absolute configuration. We could also confirm the relative stereochemistry of chromatographically separable syn-5e after comparing spectroscopic data of 1 with those of 7a-epimer, 7-deoxyalexine.27
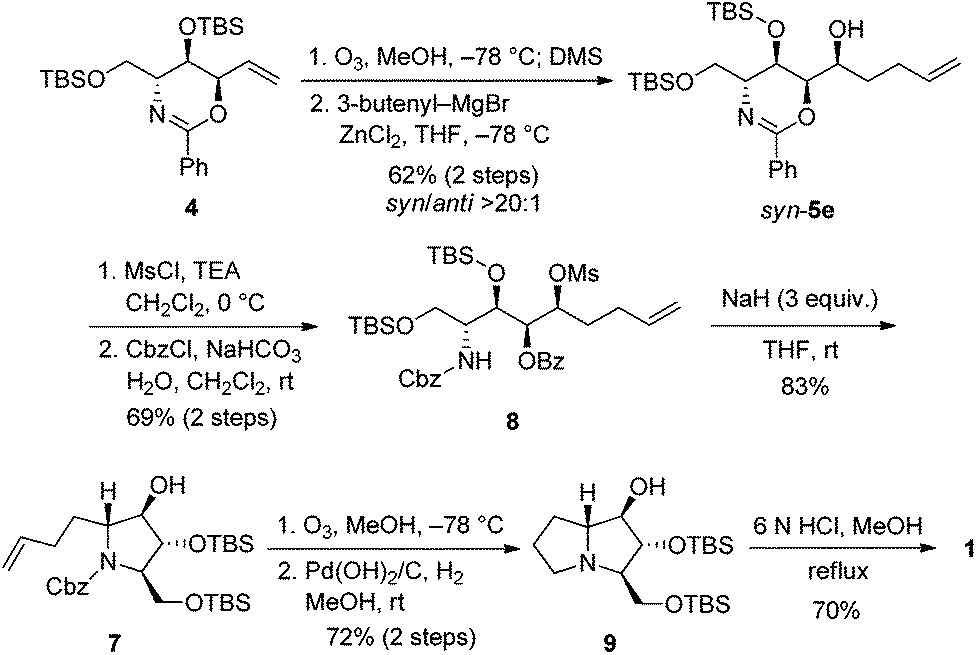 | ||
| Scheme 5 Synthesis of 1. | ||
Total synthesis of sphingofungin B
Our retrosynthesis (Scheme 6) suggested that 2 could be generated from allylic alcohol syn-5a and lipid chain 10 by an olefin cross-metathesis reaction. Chiral alcohol 10 could be obtained from (R)-epoxyoctane 11 and 6-heptenylmagnesium bromide 12.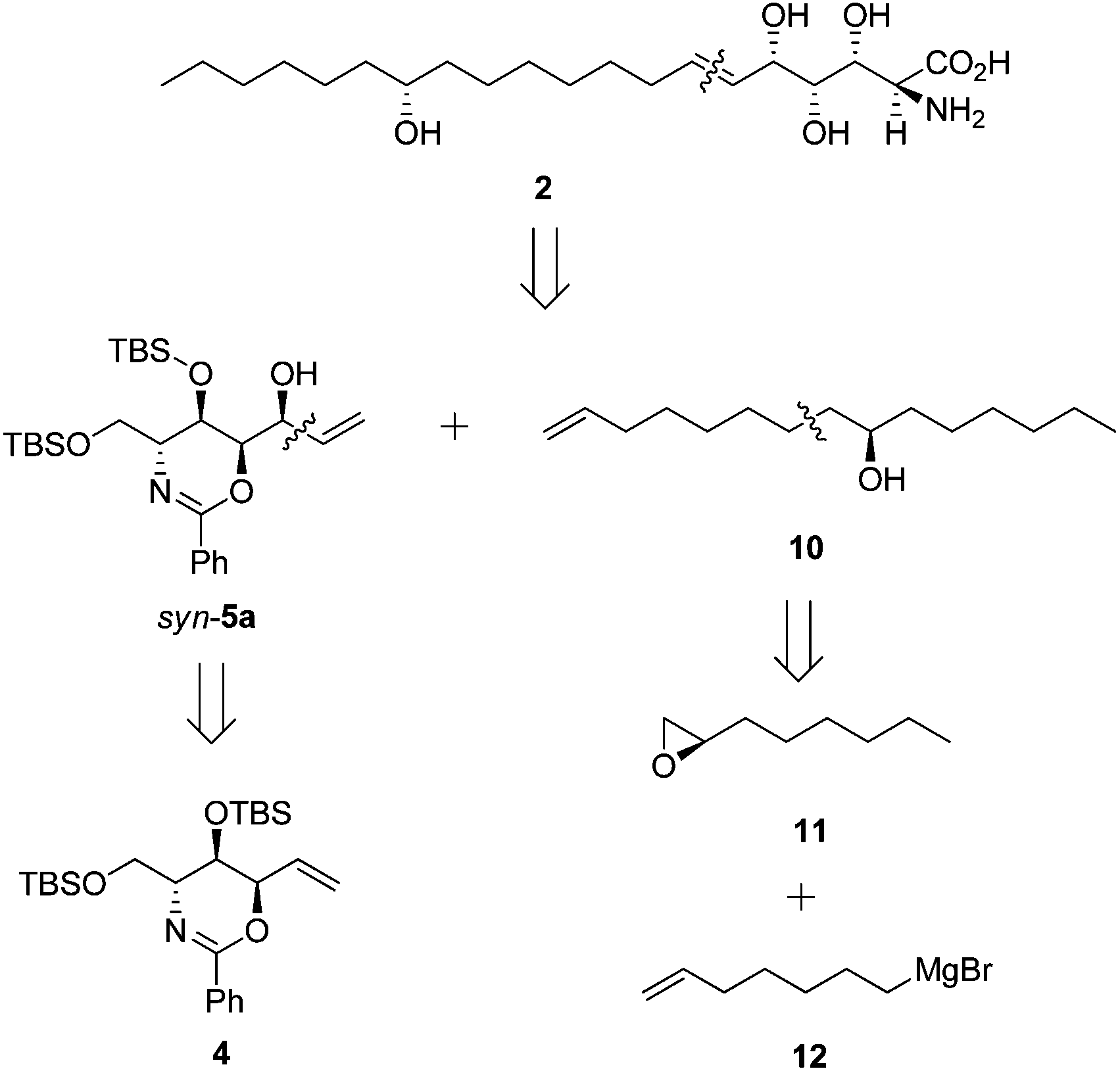 | ||
| Scheme 6 Retrosynthesis of 2. | ||
Subsequent acetylation of the hydroxyl group of syn-5a yielded secondary allylic acetate 13 in 94% yield. Meanwhile, the nucleophilic addition reaction between (R)-epoxyoctane 11 and 6-heptenylmagnesium bromide 12, which are both commercially available, and the subsequent acetylation provided the lipid chain subunit 14 in 85% yield over two steps (Scheme 7).
 | ||
| Scheme 7 Syntheses of subunits 13 and 14. | ||
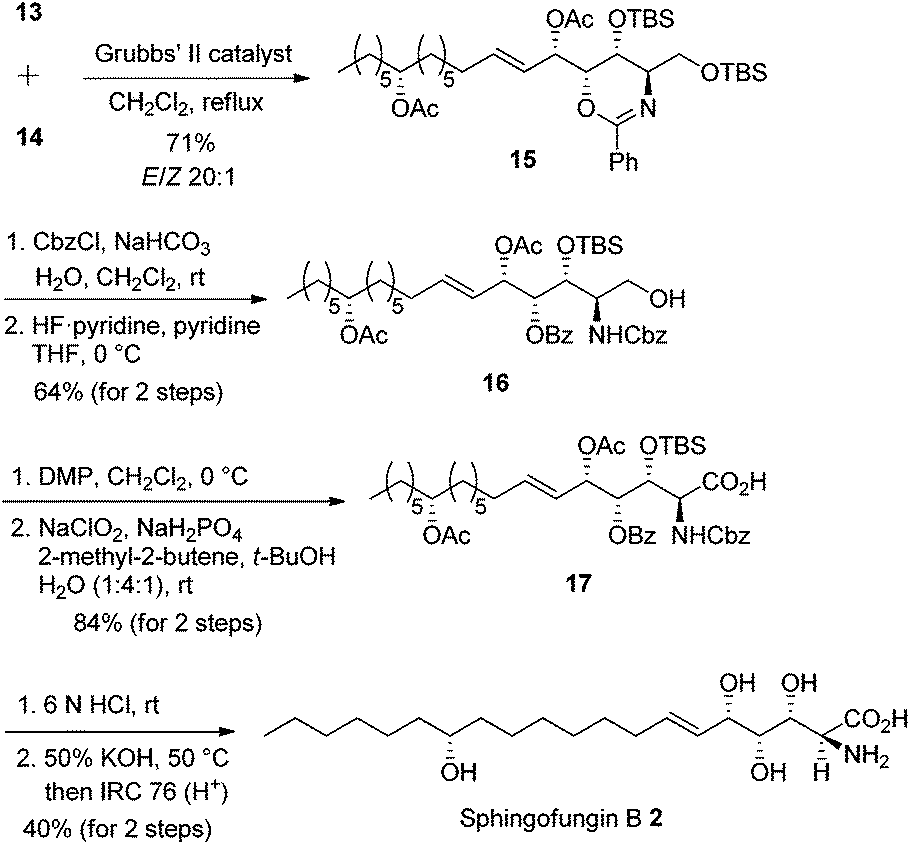
Intermolecular olefin cross-metathesis between 13 and 14 resulted in compound 15 in 71% yield (E/Z > 20:1 ratio). 1,3-Oxazine 15 was treated with benzyl chloroformate under biphasic conditions followed by primary OTBS deprotection affording carbamate 16 in 64% yield over two steps. Primary alcohol 16 was oxidized to carboxylic acid 17 in 84% yield over two steps. Finally, acid- and base-promoted hydrolysis cleaved all protecting groups, and subsequent neutralization afforded sphingofungin B 2 in 40% yield over two steps (Scheme 8). The synthetic compound was spectroscopically in good agreement with the reported sphingofungin B.9
 | ||
| Scheme 8 Synthesis of 2. | ||
Conclusions
We described new procedures for stereoselective nucleophilic addition to anti,syn-oxazine. The diastereoselectivity of the Grignard reaction was predominantly controlled by zinc chloride. Furthermore, the reactions that used divinylzinc and diphenylzinc also favored the syn-adducts. We took advantage of these reactions and various other available transformations to synthesize (+)-hyacinthacine A2 (1) and sphingofungin B (2) from a common source via chiral alcohol syn-5e and syn-5a. In addition to the diastereoselective reactions, these syntheses rely on a palladium(0)-catalyzed intramolecular oxazine formation reaction. Starting from readily available 3, 1 was obtained in 13 steps and 10.2% overall yield, whereas starting from oxazine chiral building block 4, 1 was afforded in 8 steps and 17.9% overall yield. Meanwhile, the synthesis of 2 proceeded in a linear sequence beginning from 3 over 15 steps and 6.9% overall yield and from the chiral oxazine 4 over 10 steps and 12.2% overall yield. The main advantage of this strategy is its high versatility, which allows the synthesis of not only 1 and 2, but also a range of structural analogs. Using this protocol, we are in the process of synthesizing castanospermines, australine, broussonetines, and other natural products; the results will be reported in due course.Experimental
General methods
Flash chromatography was executed using a Merck Kieselgel 60 (230–400 mesh) stationary phase and mixtures of ethyl acetate and hexanes as the eluents. Ethyl acetate and hexanes were dried and purified by distillation prior to use. Tetrahydrofuran and diethyl ether were distilled over sodium and benzophenone (indicator). Dichloromethane was mixed with concentrated sulfuric acid (Caution), dried over potassium carbonate, and distilled. Commercially available compounds were used without further purification. 1H and 13C NMR spectra were recorded at the Center for Cooperative Research Facility at Sungkyunkwan University on FT-NMR 500 and 700 MHz spectrometers. Chemical shifts are reported as δ values in ppm relative to the CHCl3 residual peak (7.26 ppm in CDCl3). IR spectra were recorded on a Bruker FT-IR spectrometer. Optical rotation was measured on a Jasco Dip 1020 digital polarimeter. Mass spectral data were recorded at the Korea Basic Science Institute (Daegu) on a Jeol JMS 700 high resolution mass spectrometer.:1; white solid; mp 115–132 °C; Rf = 0.50 (1:6 ethyl acetate/hexanes); [α]25D +40.1 (c 1.0, CHCl3); IR (neat) 698, 777, 836, 937, 1110, 1161, 1256, 1279, 1638, 2855, 2928, 2955, 3387 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.02–0.03 (m, 3H), 0.06–0.07 (m, 3H), 0.12–0.15 (m, 6H), 0.86–0.88 (m, 18H), 3.04 (d, J = 2.1 Hz, 1H), 3.45 (dd, J = 10.8, 8.2 Hz, 1H), 3.71 (m, 1H), 3.97 (dd, J = 10.3, 3.9 Hz, 1H), 4.05 (dd, J = 5.6, 2.2 Hz, 1H), 4.30 (t, J = 2.0 Hz, 1H), 4.57 (d, J = 1.7 Hz, 1H), 5.36 (dt, J = 10.6, 1.3 Hz, 1H), 5.53 (dt, J = 17.2, 1.3 Hz, 1H), 5.97–6.02 (m, 1H), 7.38 (t, J = 7.5 Hz, 2H), 7.42–7.45 (m, 1H), 7.91–7.93 (m, 2H); 13C NMR (175 MHz, CDCl3) δ –5.5, –5.4, –4.5, –4.1, 18.0, 18.2, 25.7, 25.7, 25.8, 25.9, 61.1, 64.7, 65.5, 72.6, 76.1, 118.0, 127.3, 128.1, 130.5, 133.6, 135.4, 155.5; HRMS (FAB) m/z: [M + H]+ calcd for C26H46O4NSi2 492.2965, found 492.2962.
:1; white solid; mp 106–110 °C; Rf = 0.53 (1:6 ethyl acetate–hexanes); [α]25D +21.6 (c 0.6, CHCl3); IR (neat) 699, 778, 836, 937, 1033, 1118, 1256, 1280, 1651, 2934, 2951, 3358 cm−1; 1H NMR (CDCl3, 500 MHz) δ 0.03 (s, 3H), 0.07 (s, 3H), 0.16 (s, 3H), 0.17 (s, 3H), 0.87 (s, 9H), 0.89 (s, 9H), 2.74 (d, J = 5.0 Hz, 1H), 3.58 (dd, J = 10.5, 7.0 Hz, 1H), 3.67–3.70 (m, 1H), 3.96 (dd, J = 10.5, 3.5 Hz, 1H), 4.05 (dd, J = 8.0, 2.5 Hz, 1H), 4.47 (t, J = 2.5 Hz, 1H), 4.51–4.56 (m, 1H), 5.33 (dt, J = 12.0, 1.5 Hz, 1H), 5.45 (dt, J = 19.0, 1.5 Hz, 1H), 7.23–7.43 (m, 3H), 7.90–7.92 (m, 2H); 13C NMR (125 MHz, CDCl3) δ –5.2, –4.3, –4.2, 18.2, 18.4, 26.0, 26.1, 60.8, 64.6, 71.6, 75.1, 75.4, 77.4, 117.1, 127.6, 128.2, 130.7, 133.7, 137.8, 155.4; HRMS (FAB) m/z: [M + H]+ calcd for C26H46O4NSi2 492.2965, found 492.2963.
:1; colorless liquid; Rf = 0.4 (1:6 ethyl acetate–hexanes); [α]25D +3.7 (c 1.0, CHCl3); IR (neat) 667, 777, 837, 1071, 1111, 1141, 1257, 1653, 2858, 2930, 2955, 3385 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.05 (s, 3H), 0.08 (s, 3H), 0.13 (s, 6H), 0.81–0.94 (m, 18H), 2.94 (s, 1H), 3.44 (dd, J = 10.5, 8.4 Hz, 1H), 3.70 (m, 1H), 3.92 (dd, J = 6.0, 0.9 Hz, 1H), 3.98 (dd, J = 10.5, 4.0 Hz, 1H), 4.20 (t, J = 6.2 Hz, 1H), 4.28 (t, J = 1.6 Hz, 1H), 7.36–7.39 (m, 2H), 7.41–7.43 (m, 1H), 7.91–7.93 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.3, –5.2, –3.9, 18.2, 18.3, 18.4, 61.4, 65.1, 65.8, 67.9, 127.5, 128.2, 130.7, 134.0, 155.8; HRMS (FAB) m/z: [M + H]+ calcd for C25H46O4NSi2 480.2965, found 480.2964.
:1; white solid; mp 122–124 °C; Rf = 0.45 (1:6 ethyl acetate–hexanes); [α]25D +2.8 (c 1.0, CHCl3); IR (neat) 702, 777, 838, 1103, 1140, 1363, 1648, 2856, 2929, 2978, 3193 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.04 (s, 3H), 0.09 (s, 3H), 0.17 (s, 6H), 0.87–0.90 (m, 18H), 1.40 (d, J = 6.2 Hz, 3H), 2.53 (s, 1H), 3.63–3.69 (m, 2H), 3.92 (dd, J = 8.1, 2.5 Hz, 1H), 3.97 (dd, J = 10.1, 3.0 Hz, 1H), 4.15–4.18 (m, 1H), 4.63 (dd, J = 3.2, 2.9 Hz, 1H), 7.33–7.38 (m, 2H), 7.40–7.43 (m, 1H), 7.91–7.93 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.1, –4.4, –4.3, 18.2, 18.5, 20.6, 26.0, 60.6, 64.5, 66.6, 127.6, 128.2, 130.6, 133.8, 155.2; HRMS (FAB) m/z: [M + H]+ calcd for C25H46O4NSi2 480.2965, found 480.2961.
:1; colorless liquid; Rf = 0.5 (1:6 ethyl acetate–hexanes); [α]25D +3.9 (c 1.0, CHCl3); IR (neat) 698, 777, 836, 1112, 1257, 1653, 2858, 2930, 2956, 3385 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.04–0.15 (m, 12H), 0.81–0.94 (m, 18H), 1.07 (t, J = 7.4 Hz, 3H), 1.63–1.68 (m, 1H), 1.71–1.74 (m, 1H), 3.02 (s, 1H), 3.47 (dd, J = 10.5, 8.3 Hz, 1H), 3.68–3.71 (m, 1H), 3.93–3.99 (m, 2H), 4.04 (dd, J = 5.2, 0.4 Hz, 1H), 4.30 (t, J = 1.7 Hz, 1H), 7.35–7.38 (m, 2H), 7.41–7.43 (m, 1H), 7.91–7.93 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.3, –5.2, –4.7, –3.9, 9.9, 10.1, 18.2, 25.5, 26.1, 61.5, 65.1, 66.3, 73.1, 75.6, 127.5, 128.2, 130.7, 134.0, 155.9; HRMS (FAB) m/z: [M + H]+ calcd for C26H48O4NSi2 494.3122, found 494.3120.
:1; colorless liquid; Rf = 0.55 (1:6 ethyl acetate–hexanes); [α]25D +3.2 (c 1.0, CHCl3); IR (neat) 697, 778, 836, 1140, 1256, 1655, 2858, 2930, 2956, 3385 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.04 (s, 3H), 0.08 (s, 3H), 0.16–0.17 (m, 6H), 0.87–0.90 (m, 18H), 1.07 (t, J = 7.4 Hz, 3H), 1.60–1.64 (m, 1H), 1.87–1.91 (m, 1H), 2.53 (s, 1H), 3.63–3.65 (m, 1H), 3.67–3.69 (m, 1H), 3.96–3.98 (m, 2H), 4.00–4.03 (m, 1H), 4.45 (dd, J = 3.3, 2.6 Hz, 1H), 7.35–7.38 (m, 2H), 7.40–7.42 (m, 1H), 7.90–7.93 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.1, –4.3, –4.2, 9.5, 18.2, 18.5, 64.6, 71.4, 75.3, 127.6, 128.2, 130.6, 133.8, 155.3; HRMS (FAB) m/z: [M + H]+ calcd for C26H48O4NSi2 494.3122, found 494.3120.
:1; colorless liquid; Rf = 0.5 (1:6 ethyl acetate–hexanes); [α]25D +4.6 (c 1.0, CHCl3); IR (neat) 777, 837, 1007, 1033, 1057, 1110, 1257, 1655, 2859, 2930, 2955, 3419 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.03–0.08 (m, 6H), 0.15 (s, 6H), 0.80–0.95 (m, 18H), 2.42–2.45 (m, 1H), 2.50–2.53 (m, 1H), 3.12 (s, 1H), 3.47 (dd, J = 10.4, 8.2 Hz, 1H), 3.69–3.71 (m, 1H), 3.97 (dd, J = 10.5, 3.8 Hz, 1H), 4.08–4.13 (m, 2H), 4.32 (s, 1H), 5.14–5.19 (m, 1H), 5.21 (d, J = 1.61 Hz, 1H), 5.92–6.00 (m, 1H), 7.35–7.39 (m, 2H), 7.41–7.44 (m, 1H), 7.92–7.95 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.3, –5.2, –4.4, –3.9, 18.2, 18.4, 25.9, 26.1, 37.3, 61.3, 66.5, 71.4, 74.8, 118.2, 127.6, 128.2, 128.4, 130.7, 134.0, 134.3, 155.9; HRMS (FAB) m/z: [M + H]+ calcd for C27H48O4NSi2 506.3122, found 506.3121.
:1; colorless liquid; Rf = 0.55 (1:6 ethyl acetate–hexanes); [α]25D +4.0 (c 1.0, CHCl3); IR (neat) 697, 778, 837, 1072, 1119, 1257, 1656, 2858, 2929, 2955, 3356 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.04 (s, 3H), 0.08 (s, 3H), 0.15 (s, 6H), 0.80–0.98 (m, 18H), 2.37–2.41 (m, 2H), 2.67–2.69 (m, 1H), 3.60 (dd, J = 10.4, 7.1 Hz, 1H), 3.67–3.90 (m, 1H), 3.97 (dd, J = 10.4, 3.4 Hz, 1H), 4.01 (dd, J = 8.3, 1.9 Hz, 1H), 4.05–4.08 (m, 1H), 4.45 (t, J = 2.4 Hz, 1H), 5.20–5.26 (m, 2H), 5.90–6.00 (m, 1H), 7.35–7.39 (m, 2H), 7.40–7.43 (m, 1H), 7.91–7.93 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.2, –4.4, –4.3, 9.7, 13.9, 18.2, 18.4, 26.2, 38.4, 61.0, 64.4, 64.7, 68.7, 75.2, 119.0, 127.5, 128.2, 130.6, 133.9, 134.3, 155.2; HRMS (FAB) m/z: [M + H]+ calcd for C27H48O4NSi2 506.3122, found 506.3120.
:1; colorless liquid; Rf = 0.5 (1:6 ethyl acetate–hexanes); [α]25D +10.0 (c 1.0, CHCl3); IR (neat) 698, 778, 837, 1071, 1111, 1257, 1655, 2858, 2930, 2954, 3423 cm−1; 1H NMR (CDCl3, 700 MHz) δ –0.07 (s, 2H), –0.03 (s, 2H), 0.00 (s, 2H), 0.67 (d, J = 5.2 Hz, 6H), 0.79 (s, 9H), 0.81 (s, 9H), 1.65–1.67 (m, 2H), 2.14–2.17 (m, 1H), 2.27–2.31 (m, 1H), 3.02 (s, 1H), 3.62–3.63 (m, 1H), 3.90 (dd, J = 10.5, 3.9 Hz, 1H), 3.97 (s, 2H), 4.23 (s, 1H), 4.94 (d, J = 11.3 Hz, 1H), 5.02 (d, J = 20.4 Hz, 1H), 5.79–5.84 (m, 1H), 7.30 (t, J = 9.0 Hz, 2H), 7.36 (t, J = 8.0 Hz, 1H), 7.85 (d, J = 7.1 Hz, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.5, –5.4, –4.7, –4.1, 17.9, 18.2, 25.6, 25.7, 29.6, 31.7, 61.1, 64.8, 66.1, 71.1, 75.5, 115.0, 127.3, 128.1, 130.5, 133.7, 138.2, 155.7; HRMS (FAB) m/z: [M + H]+ calcd for C28H50O4NSi2 520.3278, found 520.3275.
:1; colorless liquid; Rf = 0.55 (1:6 ethyl acetate–hexanes); [α]25D +3.8 (c 1.0, CHCl3); IR (neat) 698, 778, 836, 1117, 1256, 1654, 2858, 2930, 2954, 3384 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.04 (s, 3H), 0.08 (s, 3H), 0.16 (s, 6H), 0.80–0.94 (s, 18H), 1.65–1.71 (m, 1H), 1.95–1.99 (m, 1H), 2.22–2.27 (m, 1H), 2.34–2.38 (m, 1H), 2.51 (s, 1H), 3.62 (dd, J = 10.3, 6.8 Hz, 1H), 3.68 (6, J = 3.4 Hz, 1H), 3.97 (dd, J = 10.3, 3.4 Hz, 1H), 3.99–4.01 (m, 1H), 4.01–4.06 (m, 1H), 4.45 (s, 1H), 5.00 (dd, J = 10.1, 1.8 Hz, 1H), 5.08 (dd, J = 17.2, 1.8 Hz, 1H), 5.88–5.91 (m, 1H), 7.35–7.37 (m, 2H), 7.37–7.40 (m, 1H), 7.90–7.93 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.2, –5.1, –4.4, –4.3, 18.2, 18.5, 26.0, 29.6, 33.1, 60.7, 64.6, 69.7, 75.7, 115.2, 127.6, 128.2, 130.7, 133.8, 138.7, 155.3; HRMS (FAB) m/z: [M + H]+ calcd for C28H50O4NSi2 520.3278, found 520.3282.
:1; colorless liquid; Rf = 0.57 (1:6 ethyl acetate–hexanes); [α]25D +5.5 (c 1.0, CHCl3); IR (neat) 699, 778, 837, 1112, 1134, 1256, 1472, 1648, 2857, 2929, 2955, 3284 cm−1; 1H NMR (CDCl3, 700 MHz) δ –0.07 (s, 3H), 0.00 (s, 3H), 0.12 (s, 6H), 0.80 (s, 9H), 0.90 (s, 9H), 3.37–3.41 (m, 2H), 3.73 (dd, J = 8.1, 3.7 Hz, 1H), 3.93 (dd, J = 10.4, 3.9 Hz, 1H), 4.16 (d, J = 1.3 Hz, 1H), 4.28 (d, J = 5.4 Hz, 1H), 5.11 (d, J = 5.5 Hz, 1H), 7.33–7.44 (m, 6H), 7.50–7.53 (m, 2H), 7.88–7.92 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.7, –5.5, –4.4, –3.9, 18.1, 25.8, 61.1, 64.7, 65.9, 74.3, 126.0, 127.4, 128.1, 128.3, 128.4, 128.5, 130.5, 133.7, 139.7, 155.7; HRMS (FAB) m/z: [M + H]+ calcd for C30H48O4NSi2 542.3122, found 542.3121.
:1; white solid; mp 145–155 °C; Rf = 0.48 (1:6 ethyl acetate–hexanes); [α]25D +2.5 (c 1.0, CHCl3); IR (neat) 699, 775, 830, 1016, 1033, 1057, 2858, 2925, 2950, 3211 cm−1; 1H NMR (CDCl3, 700 MHz) δ –0.06 (s, 3H), 0.00 (s, 3H), 0.10–0.25 (m, 6H), 0.70–0.95 (m, 18H), 3.09 (s, 1H), 3.53 (dd, J = 10.2, 6.9 Hz, 1H), 3.65–3.69 (m, 1H), 3.90 (dd, J = 10.4, 3.4 Hz, 1H), 4.17 (dd, J = 7.9, 2.1 Hz, 1H), 4.44 (t, J = 2.7 Hz, 1H), 5.00 (dd, J = 7.9, 2.8 Hz, 1H), 7.20–7.40 (m, 8H), 7.61–7.65 (m, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.3, –5.0, –4.5, –4.3, 18.5, 26.0, 60.8, 64.6, 64.8, 73.00, 76.4, 127.1, 127.4, 127.6, 127.7, 127.9, 128.1, 128.2, 128.5, 128.6, 130.5, 133.5, 141.6, 155.3; HRMS (FAB) m/z: [M + H]+ calcd for C30H48O4NSi2 542.3122, found 542.3124.
:6 ethyl acetate–hexanes); [α]25D +19.3 (c 1.0, CHCl3); IR (neat) 699, 778, 837, 926, 1110, 1178, 1256, 1359, 1658, 2858, 2930, 2954, 3423 cm−1; 1H NMR (CDCl3, 700 MHz) δ –0.07 (s, 2H), –0.04 (s, 2H), 0.00 (s, 2H), 0.04 (d, J = 5.2 Hz, 6H), 0.78 (s, 9H), 0.80 (s, 9H), 1.71–1.74 (m, 1H), 1.88–1.92 (m, 1H), 2.21–2.25 (m, 2H), 2.98 (s, 3H), 3.42 (dd, J = 10.5, 7.9 Hz, 1H), 3.63–3.65 (m, 1H), 3.90 (dd, J = 10.6, 3.8 Hz, 1H), 4.17 (s, 1H), 4.26 (dd, J = 8.5, 0.7 Hz, 1H), 4.96 (d, J = 10.2 Hz, 1H), 5.00–5.04 (m, 2H), 5.75–5.79 (m, 1H), 7.30 (t, J = 7.9 Hz, 2H), 7.35 (t, J = 8.0 Hz, 1H), 7.79 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.5, –5.4, –4.6, –3.9, 18.0, 18.1, 25.6, 25.8, 28.4, 29.6, 38.8, 60.6, 64.0, 64.5, 74.9, 82.4, 115.7, 127.4, 128.2, 130.5, 133.8, 136.8, 155.9; HRMS (FAB) m/z: [M + H]+ calcd for C29H52O6NSi2S 598.3054, found 598.3055. A solution of NaHCO3 (0.6 M in distilled water, 1.5 mL, 0.892 mmol) and benzyl chloroformate (0.127 mL, 0.892 mmol) was successively added to a solution of this mesylate (133 mg, 0.223 mmol) in dichloromethane (1.5 mL). The reaction mixture was stirred at rt for 48 h. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layer was washed with distilled water and brine, dried with MgSO4, and filtered. The filtrate was concentrated in vacuo. Purification using silica gel column chromatography gave 8 (125 mg, 0.167 mmol); yield 69% for two steps; rotamer ratio >20:1; colorless liquid; Rf = 0.35 (1:6 ethyl acetate–hexanes); [α]25D –6.3 (c 1.0, CHCl3); IR (neat) 712, 778, 836, 914, 1033, 1056, 1099, 1175, 1258, 1345, 1722, 2858, 2931, 2954, 3453 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.00 (s, 3H), 0.03 (d, J = 6.8 Hz, 3H), 0.10 (d, J = 15.5 Hz, 6H), 0.82 (s, 9H), 0.87 (s, 9H), 1.88–1.90 (m, 1H), 2.00–2.02 (m, 1H), 2.26–2.29 (m, 2H), 2.89 (s, 3H), 3.66 (dd, J = 10.3, 5.8 Hz, 1H), 3.80–3.83 (m, 1H), 3.89–3.91 (m, 1H), 4.23–4.25 (m, 1H), 5.01 (d, J = 10.4 Hz, 1H), 5.05 (d, J = 12.2 Hz, 1H), 5.09–5.12 (m, 2H), 5.45 (t, J = 5.6 Hz, 1H), 5.81–5.83 (m, 1H), 7.30–7.37 (m, 6H), 7.54–7.56 (m, 2H), 8.07 (d, J = 7.4 Hz, 2H); 13C NMR (CDCl3, 175 MHz) δ –5.7, –5.5, –4.8, –4.3, 18.0, 18.2, 25.9, 29.1, 31.2, 38.7, 54.6, 61.4, 66.8, 69.4, 74.1, 80.6, 115.7, 127.9, 128.4, 128.6, 129.9, 133.4, 136.5, 136.9, 155.9, 165.4; HRMS (FAB) m/z: [M + H]+ calcd for C37H60O9NSi2S 750.3527, found 750.3528.
:1; colorless liquid; Rf = 0.5 (1:6 ethyl acetate–hexanes); [α]25D +2.8 (c 0.1, CHCl3); IR (neat) 474, 1012, 1032, 1055, 1656, 2825, 2936, 2953, 3420 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.04–0.10 (m, 12H), 0.84–0.90 (m, 18H), 1.85–1.88 (m, 2H), 2.00–2.15 (m, 2H), 3.61 (dd, J = 10.6, 1.3 Hz, 1H), 3.70–3.85 (m, 2H), 3.86 (d, J = 2.1 Hz, 1H), 4.05–4.12 (m, 2H), 4.49 (dd, J = 10.6, 3.0 Hz, 0.75H), 4.57 (d, J = 11.3 Hz, 0.25H), 4.87 (dd, J = 11.9 Hz, 0.75H), 4.90–4.92 (m, 0.25H), 4.97 (d, J = 15.9 Hz, 0.75H), 5.07 (d, J = 15.9 Hz, 0.25H), 5.10–5.16 (m, 2H), 5.65–5.75 (m, 0.75H), 5.80–5.88 (m, 0.25H), 7.30–7.38 (m, 5H); 13C NMR (CDCl3, 175 MHz) δ –5.8, –5.0, 17.9, 18.3, 25.7, 30.7, 31.1, 61.4, 62.8, 66.7, 68.5, 68.7, 114.7, 127.9, 128.2, 128.3, 128.5, 128.6, 136.5, 136.7, 137.9, 138.0, 154.2, 154.7; HRMS (FAB) m/z: [M + H]+ C29H52O5NSi2 calcd for 550.3384, found 550.3383.
:2 ethyl acetate–hexanes); [α]25D +12.5 (c 1.0, CHCl3); IR (neat) 663, 672, 775, 837, 1016, 1058, 1089, 2855, 2926, 2957 cm−1; 1H NMR (CDCl3, 700 MHz) δ 0.05–0.12 (m, 12H), 0.88–0.95 (m, 18H), 1.68–1.73 (m, 2H), 1.82–1.84 (m, 1H), 1.95–2.10 (m, 1H), 2.30–2.40 (br, s, 1H), 2.66–2.74 (m, 1H), 2.80–2.86 (m, 1H), 3.00–3.08 (m, 1H), 3.28–3.32 (m, 1H), 3.60–3.63 (m, 1H), 3.70–3.92 (m, 2H), 3.91 (t, J = 4.8 Hz, 1H); 13C NMR (CDCl3, 175 MHz) δ –5.2, –5.1, –4.7, –4.1, 18.1, 18.6, 25.4, 25.9, 26.2, 30.3, 56.3, 65.9, 70.9, 74.0, 80.7, 82.5; HRMS (FAB) m/z: [M + H]+ calcd for C20H44O3NSi2 402.2860, found 402.2861.
:1 methanol–chloroform); [α]25D +10.4 (c 0.1, CH3OH) and +15.3 (c 0.1, H2O); IR (neat) 1033, 1391, 1642, 2923, 3385 cm−1; 1H NMR (D2O, 700 MHz) δ 1.85–1.93 (m, 2H), 1.95–2.00 (m, 1H), 2.00–2.06 (m, 1H), 2.88–2.96 (m, 2H), 3.05–3.10 (m, 1H), 3.36–3.40 (m, 1H), 3.71–3.75 (m, 1H), 3.80–3.88 (m, 3H); 13C NMR (D2O, 175 MHz) δ 24.4, 29.4, 55.0, 61.2, 66.9, 69.3, 76.3, 79.4; HRMS (FAB) m/z: [M + H]+ calcd for C8H16O3N 174.1130, found 174.1128.
:15 ethyl acetate–hexanes) gave the carbamate intermediate; colorless oil; [α]25D +6.1 (c 1.0, CHCl3); 1H NMR (700 MHz, CDCl3) δ 0.05–0.07 (m, 12H), 0.87–0.88 (m, 21H), 1.20–1.26 (m, 16H), 1.48–1.50 (m, 4H), 1.94 (s, 5H), 2.03 (s, 3H), 3.70–3.75 (m, 1H), 3.75–3.80 (m, 1H), 3.90–4.00 (m, 1H), 4.25 (s, 1H), 4.84 (m, 1H), 5.02–5.10 (m, 3H), 5.42 (dd, J = 15.4, 7.2 Hz, 1H), 5.48 (dd, J = 6.5, 4.2 Hz, 1H), 5.76 (t, J = 6.9 Hz, 1H), 5.87 (m, 1H), 7.26–7.31 (m, 5H), 7.35–7.40 (m, 2H), 7.50–7.60 (m, 1H), 8.02 (d, J = 8.1 Hz, 2H); 13C NMR (175 MHz, CDCl3) δ –5.6, –4.6, –4.5, 14.0, 18.0, 18.1, 21.0, 21.2, 22.5, 25.2, 25.7, 25.8, 28.6, 29.0, 29.1, 31.7, 32.3, 34.0, 54.6, 61.2, 66.4, 69.8, 72.8, 74.2, 74.3, 74.6, 124.0, 124.1, 127.8, 128.3, 128.4, 129.6, 129.7, 129.9, 133.0, 136.6, 137.2, 137.3, 155.7, 165.6, 169.9, 170.8. Pyridine (1.7 mL) buffered HF-pyridine (0.5 mL, 0.497 mmol) was added to a solution of carbamate (0.24 g, 0.257 mmol) in THF (5 mL) at 0 °C and then stirred at rt. After 3 h, the reaction mixture was quenched by saturated NaHCO3. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with saturated CuSO4 and brine, dried with MgSO4, and the filtrate was concentrated in vacuo. The resulting substance purified by silica gel column chromatography (1:15 ethyl acetate–hexanes) gave primary alcohol 16 (0.173 g, 0.213 mmol); yield 64% (for 2 steps); rotamer ratio >20:1; colorless oil; [α]25D +8.9 (c 1.0, CHCl3); IR (neat) 713, 778, 838, 1026, 1068, 1108, 1243, 1727, 2857, 2930, 3451 cm−1; 1H NMR (700 MHz, CDCl3) δ –0.06 (s, 3H), 0.00 (s, 3H), 0.75 (s, 9H), 0.82 (m, 3H), 1.12–1.24 (m, 16H), 1.35–1.50 (m, 4H), 1.85–1.95 (m, 2H), 1.96–2.05 (m, 6H), 3.69 (dd, J = 11.6, 3.7 Hz, 1H), 3.73 (d, J = 3.7 Hz, 1H), 3.96 (d, J = 3.0 Hz, 1H), 4.23 (br, s, 1H), 4.73–4.82 (m, 1H), 4.99 (d, J = 12.2 Hz, 1H), 5.05 (d, J = 12.1 Hz, 1H), 5.30–5.40 (m, 2H), 5.46 (d, J = 6.9 Hz, 1H), 5.51–5.59 (m, 1H), 5.72–5.80 (m, 1H), 7.25–7.29 (m, 5H), 7.32–7.38 (m, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.97 (d, J = 7.2 Hz, 2H); 13C NMR (175 MHz, CDCl3) δ –4.6, 0.0, 8.3, 14.1, 18.0, 21.1, 21.3, 22.6, 25.2, 25.3, 25.8, 28.6, 29.0, 29.2, 29.3, 31.8, 32.3, 34.1, 53.7, 58.9, 60.4, 62.2, 66.8, 72.2, 72.6, 74.4, 74.8, 123.7, 128.1, 128.5, 129.8, 133.3, 136.4, 137.3, 156.0, 165.8, 170.0, 171.0; HRMS (FAB) m/z: [M + H]+ calcd for C45H70O10NSi 812.4769, found 812.4773.
:4:1). After 2 h, the mixture was diluted with H2O and extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, and the filtrate was concentrated in vacuo. The resulting substance purified by silica gel column chromatography (1:20 methanol–chloroform) gave carboxylic acid 17 (32 mg, 0.039 mmol); yield 84% (for two steps); rotamer ratio >20:1; colorless oil; Rf = 0.2 (1:20 methanol–chloroform); [α]25D +4.7 (c 1.0, CH3OH); IR (neat) 713, 778, 838, 1055, 1067, 1119, 1230, 1248, 1728, 2857, 2930, 3434 cm−1; 1H NMR (700 MHz, CDCl3) δ –0.16 (s, 3H), 0.00 (s, 3H), 0.67–0.74 (m, 9H), 0.86–0.88 (m, 3H), 1.14–1.28 (m, 18H), 1.40–1.50 (m, 4H), 1.90 (br, s, 2H), 2.00–2.10 (m, 3H), 2.12 (s, 3H), 4.41 (s, 1H), 4.52 (br, s, 1H), 4.80–4.91 (m, 1H), 5.00–5.10 (m, 3H), 5.13 (d, J = 11.6 Hz, 1H), 5.41 (br, s, 1H), 5.69–5.78 (m, 4H), 7.25–7.31 (m, 5H), 7.33–7.45 (m, 2H), 7.50–7.60 (m, 1H), 8.02 (d, J = 7.4 Hz, 2H); 13C NMR (175 MHz, CDCl3) δ –4.6, –4.5, 0.0, 14.1, 18.0, 21.3, 22.6, 25.1, 25.3, 25.7, 28.6, 28.8, 29.2, 29.7, 31.8, 32.1, 34.1, 56.7, 66.8, 72.6, 72.9, 74.8, 75.5, 124.2, 128.1, 128.4, 129.9, 130.2, 133.1, 136.0, 136.5, 155.3, 165.9, 170.8, 171.7; HRMS (FAB) m/z: [M + Na]+ calcd for C45H67O11NSiNa 848.4381, found 848.4386.
Acknowledgements
This research was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science and Technology (2010-0022900, 2011-0029199) and by the Yonsung Fine Chemicals Corporation. S.H.P. also gratefully acknowledges the Global Ph. D. Fellowship Grant.Notes and references
- (a) Y. Mu, T. Jin, G.-W. Kim, J.-S. Kim, S.-S. Kim, Y.-S. Tian, C.-Y. Oh and W.-H. Ham, Eur. J. Org. Chem., 2012, 2614–2620 CrossRef CAS; (b) J.-E. Joo, V.-T. Pham, Y.-S. Tian, Y.-S. Chung, C.-Y. Oh, K.-Y. Lee and W.-H. Ham, Org. Biomol. Chem., 2008, 6, 1498–1501 RSC; (c) J.-E. Joo, K.-Y. Lee, V.-T. Pham and W.-H. Ham, Eur. J. Org. Chem., 2007, 1586–1593 CrossRef CAS; (d) J.-E. Joo, K.-Y. Lee, V.-T. Pham, Y.-S. Tian and W.-H. Ham, Org. Lett., 2007, 9, 3627–3630 CrossRef CAS PubMed.
- (a) T. Jin, J.-S. Kim, Y. Mu, S.-H. Park, X. Jin, J.-C. Kang, C.-Y. Oh and W.-H. Ham, Tetrahedron, 2014, 70, 2570–2575 CrossRef CAS PubMed; (b) G.-W. Kim, T. Jin, J.-S. Kim, S.-H. Park, K.-H. Lee, S.-S. Kim, I.-S. Myeong and W.-H. Ham, Tetrahedron: Asymmetry, 2014, 25, 87–91 CrossRef CAS PubMed; (c) Y. Mu, J.-Y. Kim, X. Jin, S.-H. Park, J.-E. Joo and W.-H. Ham, Synthesis, 2012, 2340–2346 CAS; (d) J.-Y. Kim, Y. Mu, X. Jin, S.-H. Park, V.-T. Pham, D.-K. Song, K.-Y. Lee and W.-H. Ham, Tetrahedron, 2011, 67, 9426–9432 CrossRef CAS PubMed; (e) V.-T. Pham, J.-E. Joo, K.-Y. Lee, T.-W. Kim, Y. Mu and W.-H. Ham, Tetrahedron, 2010, 66, 2123–2131 CrossRef CAS PubMed.
- For recent reviews, see: (a) P. Compain and O. R. Martin, Iminosugar: From Synthesis to Therapeutic Applications, Wiley, Chichester, 2007 Search PubMed; (b) W. Zou, Curr. Top. Med. Chem., 2005, 5, 1363–1391 CrossRef CAS; (c) Y. Nishimura, Curr. Top. Med. Chem., 2003, 3, 575–591 CrossRef CAS; (d) P. Compain and O. R. Martin, Curr. Top. Med. Chem., 2003, 3, 541–560 CrossRef CAS; (e) T. M. Wrodnigg, Monatsh. Chem., 2002, 133, 393–426 CrossRef CAS; (f) A. A. Watson, G. W. J. Fleet, N. Asano, R. J. Molyneux and R. J. Nash, Phytochemistry, 2001, 56, 265–295 CrossRef CAS; (g) A. E. Stütz, Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond, Wiley-VCH, Weinheim, 1999 Search PubMed.
- N. Asano, H. Kuroi, K. Ikeda, H. Kizu, Y. Kameda, A. Kato, I. Adachi, A. A. Watson, R. J. Nash and G. W. J. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1–8 CrossRef CAS.
- For previous syntheses of (+)-hyacinthacine A2, see: (a) M. Royzen, M. T. Taylor, A. DeAngelis and J. M. Fox, Chem. Sci., 2011, 2, 2162–2165 RSC; (b) X.-K. Liu, S. Qiu, Y.-G. Xiang, Y.-P. Ruan, X. Zheng and P.-Q. Huang, J. Org. Chem., 2011, 76, 4952–4963 CrossRef CAS PubMed; (c) C. Bonaccini, M. Chioccioli, C. Parmeggiani, F. Cardona, D. Lo Re, G. Soldaini, P. Vogel, C. Bello, A. Goti and P. Gratteri, Eur. J. Org. Chem., 2010, 5574–5585 CrossRef CAS; (d) W.-J. Liu, J.-L. Ye and P.-Q. Huang, Org. Biomol. Chem., 2010, 8, 2085–2091 RSC; (e) I. Delso, T. Tejero, A. Goti and P. Merino, Tetrahedron, 2010, 66, 1220–1227 CrossRef CAS PubMed; (f) C. Ribes, E. Falomir, M. Carda and J. A. Marco, Tetrahedron, 2009, 65, 6965–6971 CrossRef CAS PubMed; (g) P. Dewi-Wülfing and S. Blechert, Eur. J. Org. Chem., 2006, 1852–1856 CrossRef; (h) S. Desvergnes, S. Py and Y. Vallée, J. Org. Chem., 2005, 70, 1459–1462 CrossRef CAS PubMed; (i) I. Izquierdo, M. T. Plaza and F. Franco, Tetrahedron: Asymmetry, 2003, 14, 3933–3935 CrossRef CAS PubMed; (j) F. Cardona, E. Faggi, F. Liguori, M. Cacciarini and A. Goti, Tetrahedron Lett., 2003, 44, 2315–2318 CrossRef CAS; (k) L. Rambaud, P. Compain and O. R. Martin, Tetrahedron: Asymmetry, 2001, 12, 1807–1809 CrossRef CAS.
- For previous syntheses of (−)-hyacinthacine A2, see: (a) E. A. Brock, S. G. Davies, J. A. Lee, P. M. Roberts and J. E. Thomson, Org. Biomol. Chem., 2013, 11, 3187–3202 RSC; (b) J. Calveras, J. Casas, T. Parella, J. Joglar and P. Clapés, Adv. Synth. Catal., 2007, 349, 1661–1666 CrossRef CAS.
- For recent reviews, see: (a) H.-S. Byun, X. Lu and R. Bittman, Synthesis, 2006, 2447–2474 CAS; (b) J. Liao, J. Tao, G. Lin and D. Liu, Tetrahedron, 2005, 61, 4715–4733 CrossRef CAS PubMed; (c) S. H. Kang, S. Y. Kang, H.-S. Lee and A. J. Buglass, Chem. Rev., 2005, 105, 4537–4558 CrossRef CAS PubMed; (d) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127–5143 CrossRef CAS; (e) M. Brunner and A. M. P. Koskinen, Curr. Org. Chem., 2004, 8, 1629–1645 CrossRef CAS.
- (a) F. VanMiddlesworth, R. A. Giacobbe, M. Lopez, G. Garrity, J. A. Bland, K. Bartizal, R. A. Fromtling, J. Polishook, M. Zweerink, A. M. Edison, W. Rozdilsky, K. E. Wilson and R. L. Monaghan, J. Antibiot., 1992, 45, 861 CrossRef CAS; (b) F. VanMiddlesworth, C. Dufresne, F. E. Wincott, R. T. Mosley and K. E. Wilson, Tetrahedron Lett., 1992, 33, 297–300 CrossRef CAS.
- For previous syntheses of sphingofungin B, see: (a) S. Kobayashi and T. Furuta, Tetrahedron, 1998, 54, 10275–10294 CrossRef CAS; (b) S. Kobayashi, T. Furuta, T. Hayashi, M. Nishijima and K. Hanada, J. Am. Chem. Soc., 1998, 120, 908–919 CrossRef CAS; (c) S. Kobayashi, T. Hayashi, S. Iwamoto, T. Furuta and M. Matsumura, Synlett, 1996, 672–674 CrossRef CAS PubMed.
- For previous syntheses of sphingofungin D, see: (a) K. Otaka and K. Mori, Eur. J. Org. Chem., 1999, 1795–1802 CrossRef CAS; (b) N. Chida, H. Ikemoto, A. Noguchi, S. Amano and S. Ogawa, Nat. Prod. Lett., 1995, 6, 295–302 CrossRef CAS; (c) K. Mori and K. Otaka, Tetrahedron Lett., 1994, 35, 9207–9210 CrossRef CAS.
- For previous syntheses of sphingofungin E, see: (a) K. Ikeuchi, M. Hayashi, T. Yamamoto, M. Inai, T. Asakawa, Y. Hamashima and T. Kan, Eur. J. Org. Chem., 2013, 6789–6792 CrossRef CAS; (b) M. Martinková, J. Gonda, J. Š. Raschmanová, M. Slaninková and J. Kuchár, Carbohydr. Res., 2010, 345, 2427–2437 CrossRef PubMed; (c) B. Wang and G.-Q. Lin, Eur. J. Org. Chem., 2009, 5038–5046 CrossRef CAS; (d) C. J. Hayes, D. M. Bradley and N. M. Thomson, J. Org. Chem., 2006, 71, 2661–2665 CrossRef CAS PubMed; (e) T. Oishi, K. Ando, K. Inomiya, H. Sato, M. Iida and N. Chida, Bull. Chem. Soc. Jpn., 2002, 75, 1927–1947 CrossRef CAS; (f) T. Oishi, K. Ando, K. Inomiya, H. Sato, M. Iida and N. Chida, Org. Lett., 2002, 4, 151–154 CrossRef CAS PubMed; (g) T. Nakamura and M. Shiozaki, Tetrahedron, 2002, 58, 8779–8791 CrossRef CAS; (h) B. M. Trost and C. Lee, J. Am. Chem. Soc., 2001, 123, 12191–12201 CrossRef CAS PubMed; (i) T. Nakamura and M. Shiozaki, Tetrahedron Lett., 2001, 42, 2701–2704 CrossRef CAS; (j) B. Wang, X.-M. Yu and G.-Q. Lin, Synlett, 2001, 904–906 CrossRef CAS.
- For previous syntheses of sphingofungin F, see: (a) Y.-C. Luo, H.-H. Zhang, Y. Wang and P.-F. Xu, Acc. Chem. Res., 2010, 43, 1317–1330 CrossRef CAS PubMed; (b) F.-F. Gan, S.-B. Yang, Y.-C. Luo, W.-B. Yang and P.-F. Xu, J. Org. Chem., 2010, 75, 2737–2740 CrossRef CAS PubMed; (c) See ref. 11c; (d) M. Li and A. Wu, Synlett, 2006, 2985–2988 CrossRef CAS PubMed; (e) K.-Y. Lee, C.-Y. Oh and W.-H. Ham, Org. Lett., 2002, 4, 4403–4405 CrossRef CAS PubMed; (f) See ref. 11h; (g) D.-G. Liu, B. Wang and G.-Q. Lin, J. Org. Chem., 2000, 65, 9114–9119 CrossRef CAS PubMed; (h) See ref. 9a; (i) B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 1998, 120, 6818–6819 CrossRef CAS; (j) S. Kobayashi, M. Matsumura, T. Furuta, T. Hayashi and S. Iwamoto, Synlett, 1997, 301–303 CrossRef CAS PubMed.
- For previous syntheses of myriocin, see: (a) M. Inai, T. Goto, T. Furuta, T. Wakimoto and T. Kan, Tetrahedron: Asymmetry, 2008, 19, 2771–2773 CrossRef CAS PubMed; (b) M. C. Jones and S. P. Marsden, Org. Lett., 2008, 10, 4125–4128 CrossRef CAS PubMed; (c) K.-Y. Lee, C.-Y. Oh, Y.-H. Kim, J.-E. Joo and W.-H. Ham, Tetrahedron Lett., 2002, 43, 9361–9363 CrossRef CAS and references cited therein; (d) See ref. 11e; (e) T. Oishi, K. Ando and N. Chida, Chem. Commun., 2001, 1932–1933 RSC.
- For previous syntheses of mycestericin A, see: (a) H. Yamanaka, K. Sato, H. Sato, M. Iida, T. Oishi and N. Chida, Tetrahedron, 2009, 65, 9188–9201 CrossRef CAS PubMed; (b) H. Sato, K. Sato, M. Iida, H. Yamanaka, T. Oishi and N. Chida, Tetrahedron Lett., 2008, 49, 1943–1947 CrossRef CAS PubMed.
- For previous synthesis of mycestericin C, see: S. Sakamoto, N. Kazumi, Y. Kobayashi, C. Tsukano and Y. Takemoto, Org. Lett., 2014, 16, 4758–4761 CrossRef CAS PubMed.
- For reviews, see: (a) A. Mengel and O. Reiser, Chem. Rev., 1999, 99, 1191–1223 CrossRef CAS PubMed; (b) M. T. Reetz, Chem. Rev., 1999, 99, 1121–1162 CrossRef CAS PubMed; (c) M. T. Reetz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1531–1546 CrossRef; (d) J. Jurczak and A. Gołębiowski, Chem. Rev., 1989, 89, 149–164 CrossRef CAS; (e) M. T. Reetz, Angew. Chem., Int. Ed. Engl., 1984, 23, 556–569 CrossRef.
- R. S. Coleman and A. J. Carpenter, Tetrahedron Lett., 1992, 33, 1697–1700 CrossRef CAS.
- T. Fujisawa, Y. Ukaji, M. Funabora, M. Yamashita and T. Sato, Bull. Chem. Soc. Jpn., 1990, 63, 1894–1897 CrossRef CAS.
- G. Boireau, A. Deberly and D. Abenhaïm, Tetrahedron, 1989, 45, 5837–5844 CrossRef CAS.
- S. Kobayashi, P. Das, G. X. Wang, T. Mita, M. J. Lear and M. Hirama, Chem. Lett., 2002, 300–301 CrossRef CAS.
- (a) M. Asami and R. Kimura, Chem. Lett., 1985, 1221–1222 CrossRef CAS; (b) M. Asami and T. Mukaiyama, Chem. Lett., 1983, 93–96 CrossRef CAS.
- R. Bloch and L. Gilbert, Tetrahedron Lett., 1987, 28, 423–426 CrossRef CAS.
- G. E. Keck, M. B. Andrus and D. R. Romer, J. Org. Chem., 1991, 56, 417–420 CrossRef CAS.
- J. Mulzer and C. Pietschmann, J. Org. Chem., 1997, 62, 3938–3943 CrossRef CAS.
- “Although the actual role of ZnBr2 is not clear at present” by Asami (ref. 21a, p. 1222), “its generality is unknown” by Reetz (ref. 16e, p. 562), “although the actual stereochemical course is still an open question” by Fujisawa (ref. 18, p. 1895), “argues further against simple chelation model” by Coleman (ref. 17, p. 1698), and “the actual role of Ti(OiPr)4 in our case… in THF is still a matter of debate” by Hirama (ref. 20, p. 301).
- (a) J. L. von dem Bussche-Hünnefeld and D. Seebach, Tetrahedron, 1992, 48, 5719–5730 CrossRef; (b) D. Seebach, L. Behrendt and D. Felix, Angew. Chem. Int., Ed. Engl., 1991, 30, 1008–1009 CrossRef.
- I. Izquierdo, M. T. Plaza, R. Robles and F. Franco, Tetrahedron: Asymmetry, 2001, 12, 2481–2487 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of 1, 2, and all new compounds; and NOESY spectra of trans-6 and cis-6. See DOI: 10.1039/c5ob00251f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |