
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and photosensitivity of isoxazolin-5-one glycosides†
Tobias
Becker
a,
Prashant
Kartikeya
b,
Christian
Paetz
a,
Stephan H.
von Reuß
a and
Wilhelm
Boland
*a
aMax Planck Institute for Chemical Ecology, Beutenberg Campus, Hans-Knoell-Straße 8, D-07745 Jena, Germany. E-mail: boland@ice.mpg.de
bDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721 302, India
First published on 23rd February 2015
Abstract
A novel procedure for the synthesis of isoxazolin-5-one glycosides starting from unprotected carbohydrates is described. The substrate scope of the one-pot synthetic protocol was explored using D-configured glucose, xylose, maltose, fructose, ribose and 2-deoxyribose. Naturally occurring 2-(β-D-glucopyranosyl)-3-isoxazolin-5-one and four novel isoxazolin-5-one glycosides derived from xylose, maltose and fructose were synthesized and purified by flash chromatography. The compounds were characterized in terms of chemical structure, photophysical properties as well as pH stability. The photohydrolysis rates of the synthesized glycosides were compared with uridine as a standard to determine the quantum yields for the photoreactions in water.
Introduction
Isoxazolin-5-one derivatives of glucose, β-aminopropionitrile and amino acids occur as secondary metabolites in different plant- and insect families.1–14 2-(β-D-Glucopyranosyl)-3-isoxazolin-5-one (1) is one of the major components of the defensive secretions of diverse leaf beetle species (Chrysomelina).7,10–13 Seedlings of a variety of plants within the legume family (Fabaceae) contain high amounts of this compound during development and growth.1–5 It is described that 3-unsubstituted isoxazolin-5-one derivatives show rapid hydrolysis under neutral conditions upon exposure to low intense UV light.1,3,8 Consequently, UV irradiation corresponding to the absorption band of isoxazolin-5-one glucoside 1 results in the release of free D-glucose (Scheme 1).1,3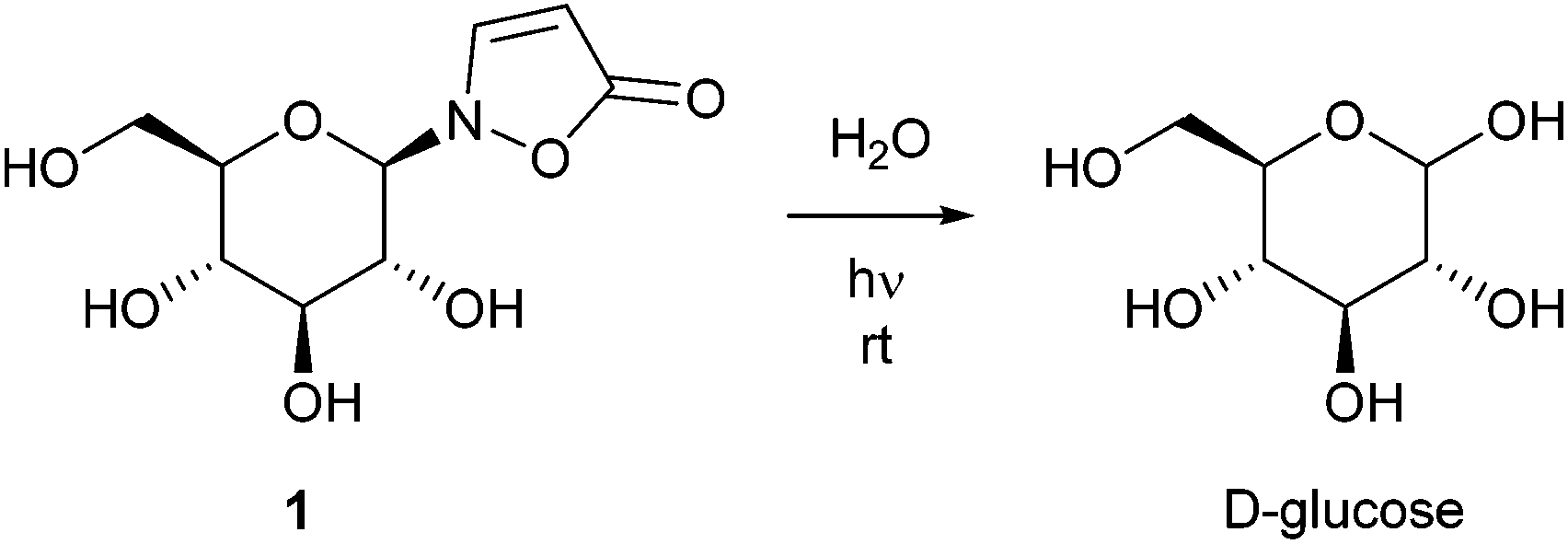 | ||
| Scheme 1 Photoactivity of naturally occurring isoxazolin-5-one glucoside 1 in water. | ||
The mechanism and the efficiency of the isoxazolin-5-one decomposition in aqueous solution was studied in case of plant metabolites that derive from amino acids as well as β-aminopropionitrile.8 Due to a lack of synthetic access, the quantum yield of the decomposition process in glucosides and the efficiency of the sugar release have not been determined so far. Furthermore, the synthesis of other isoxazolin-5-one glycosides has not been described.
Different approaches for the synthesis of glucoside 1 based on the Koenigs–Knorr substitution and a cascade reaction have previously been reported.3,15 Both methods depend on protection and deprotection of the hydroxyl functions in the sugar moiety using acetate esters or benzyl ethers.
In order to study the efficiency of its photoactivity and to explore its biological activity we required significant amounts of compound 1 and have developed an alternative direct synthetic route starting from D-glucose. Applying our novel synthetic approach we introduced the isoxazolin-5-one moiety into common carbohydrates to study its suitability as a general photoactive group for the release of sugars from anomeric-protected precursors.
Results and discussion
Synthesis
The novel synthetic protocol is based on a two-step one-pot strategy starting with the reaction of an unprotected carbohydrate with a free anomeric position and hydroxylamine that is freshly produced from the hydrochloride.16 Then, the solvent is removed and water is added. Propynoic acid and DCC, both in MeCN, are added simultaneously at rt to the aqueous solution (Scheme 2).17 This counterintuitive procedure results in quantitative chemoselective acylation of the N-hydroxy function without the need for an additional esterification catalyst. Furthermore, it allows a simple and complete separation of the sugar derivatives from the water insoluble side product 1,3-dicyclohexylurea (DCU) via aqueous extraction of the dried crude mixture.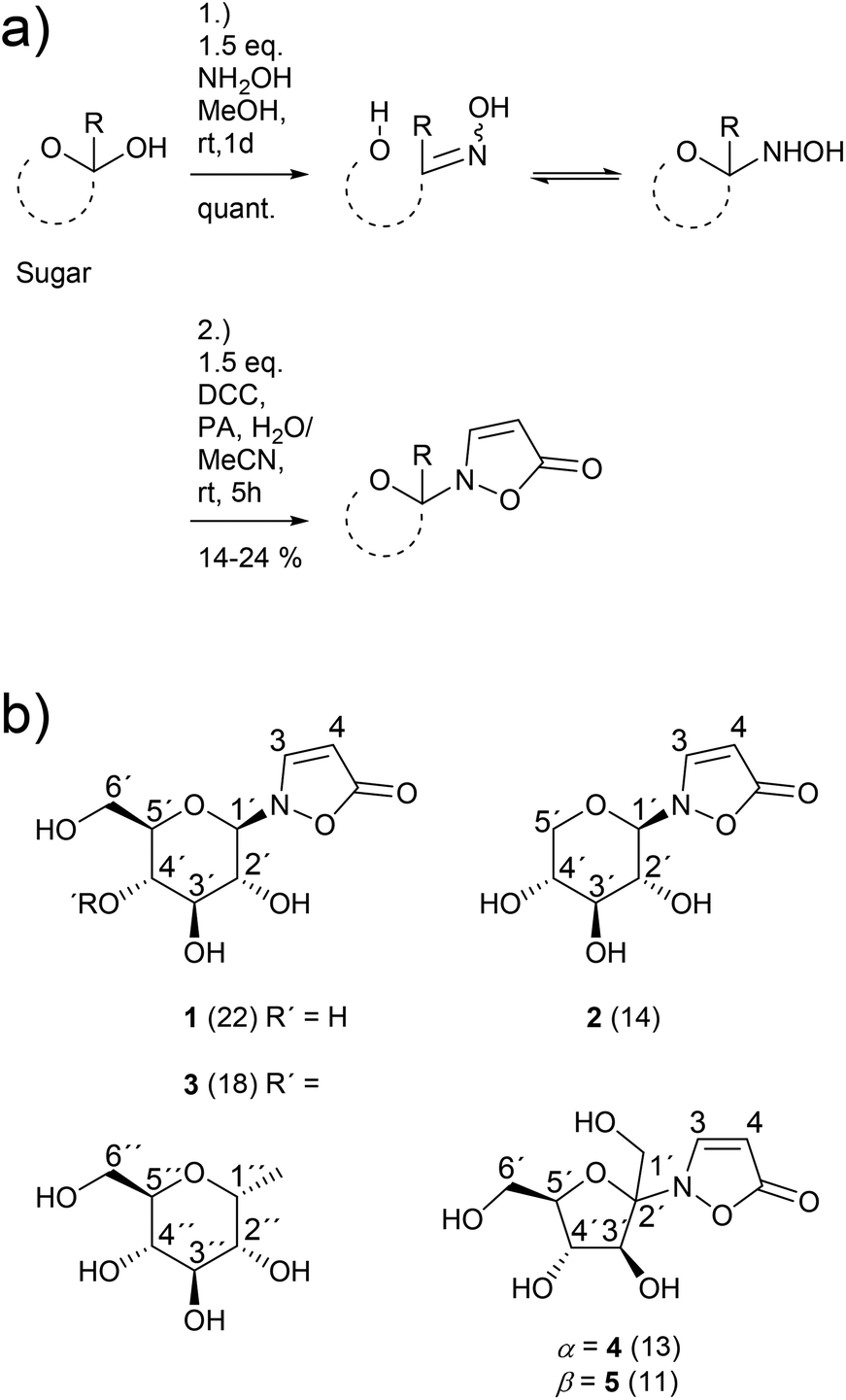 | ||
| Scheme 2 (a) Conditions of the synthetic protocol; sugar: D-glucose, D-xylose, D-maltose or D-fructose; quant. = quantitative; R = H, CH2OH; DCC = dicyclohexylcarbodiimide; PA = propynoic acid; (b) structures of the isolated products 1–5, yields are given in brackets. | ||
The 1H NMR spectrum of a typical reaction mixture using D-glucose (Glc) reveals the formation of the free sugar as the main product upon hydrolysis in the aqueous reaction medium (Fig. 1). The signals corresponding to the isoxazolin-5-one glucoside display the second highest intensity and show that the β-isomer is formed selectively (β/α is ca. 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The total isolated yield of 2-(β-D-glucopyranosyl)-3-isoxazolin-5-one (1) is in the range of previous reported syntheses (here: 22%, lit.15: 21%). The time that is needed for the synthesis and isolation of the isoxazolin-5-one glucoside is reduced from several days to hours.3,4,15
:1). The total isolated yield of 2-(β-D-glucopyranosyl)-3-isoxazolin-5-one (1) is in the range of previous reported syntheses (here: 22%, lit.15: 21%). The time that is needed for the synthesis and isolation of the isoxazolin-5-one glucoside is reduced from several days to hours.3,4,15
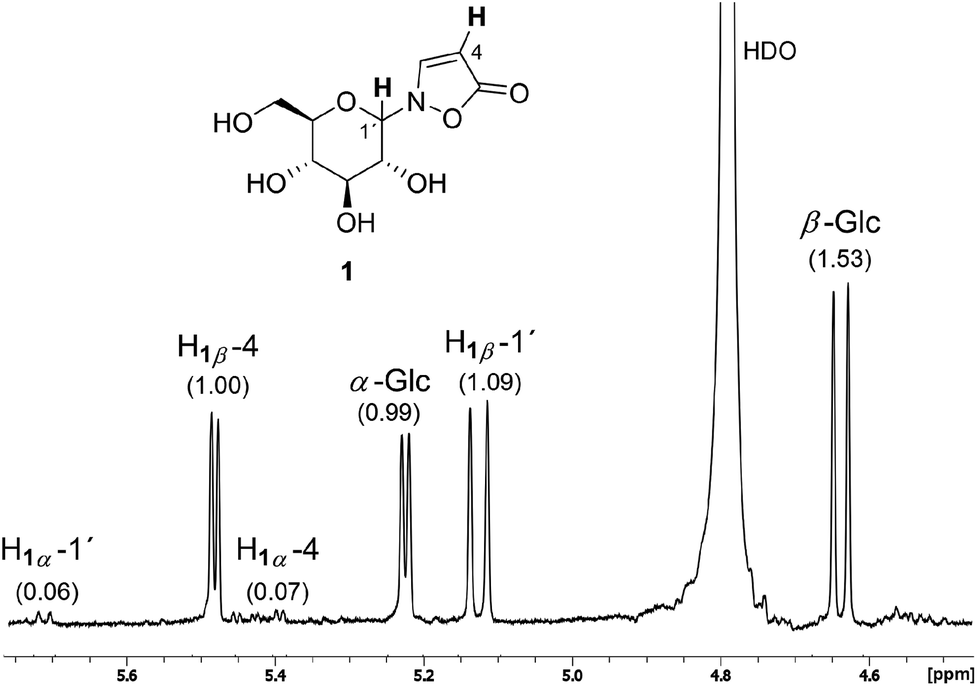 | ||
| Fig. 1 Typical 1H NMR spectrum at 400 MHz of a D2O extract of a reaction mixture after applying the described protocol to glucose showing the anomeric protons (H-1′) as well as one of the isoxazolin-5-one protons (H-4). Integral values are given in brackets. | ||
The β/α-selectivity for the formation of analogous isoxazolin-5-one xylosides 2 is 4:1. For the corresponding maltosides 3 a ratio of 12:1 was determined. In case of fructose an anomeric mixture of the furanosides 4 and 5 in a ratio of ca. 1:1 was obtained. The isoxazolin-5-one ribosides and (2-deoxy)ribosides were not isolated due to product yields of less than 5% as shown by 1H NMR measurements.
The product scope and observed β-selectivities can be understood considering the result of the condensation reaction with hydroxylamine (first step) as well as kinetic effects. In case of glucose and maltose the formation of the corresponding open chain (methanol soluble) aldoxime is the first step, followed by the crystallization of the β-N-pyranosylhydroxylamine in quantitative yield.16,18 This observation explains the high selectivity of the β- over the α-anomer in case of isoxazolin-5-one glycosides derived from glucose and maltose (Scheme 3, right side).
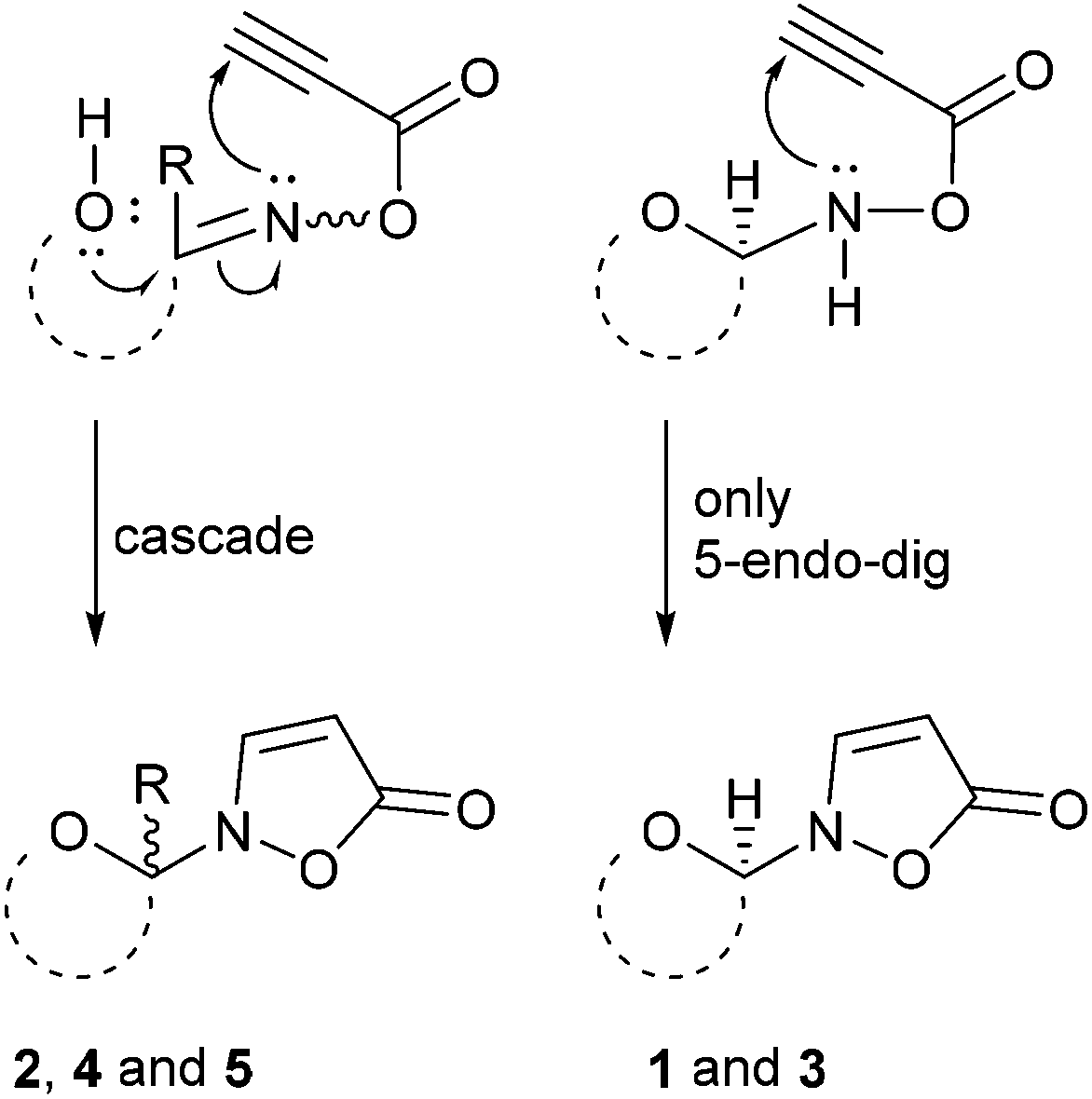 | ||
| Scheme 3 Proposed mechanisms for the formation of the isoxazolin-5-one moiety in glycosides 1–5 after acylation of the condensation products with propynoic acid. | ||
In contrast, xylose and fructose form open chain oximes after addition of hydroxylamine in methanol almost exclusively.16 Thus, a low stereoselectivity in the formation of isoxazolin-5-one glycosides derived from fructose and xylose is observed (Scheme 3, left side).
In case of ribose the 1H NMR spectra show that the crystalline reaction product of the condensation reaction mainly consists of the open chain oximes, too. 1H NMR analysis of the reaction mixtures of ribose and 2-deoxyribose show that after acylation with propynoic acid the hydrolysis reaction is predominant (Scheme 4).
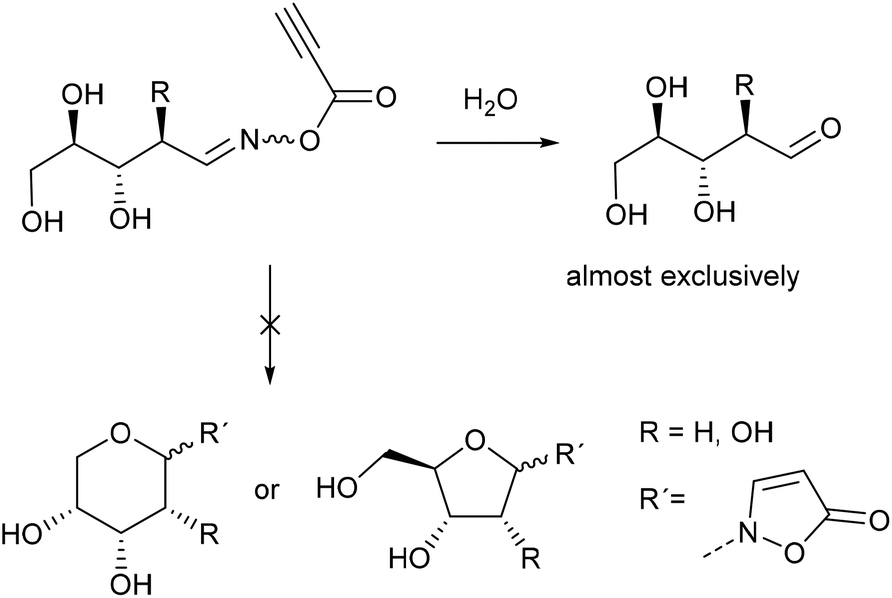 | ||
| Scheme 4 Hydrolysis of acylated ribose- and 2-deoxyribose oxime. | ||
Due to the cis-configuration of the substituents in ribopyranosides and -furanosides we conclude that the formation of isoxazolin-5-one ribosides is kinetically disfavored against the hydrolysis reaction in the aqueous medium. This holds true for the formation of 2-deoxyribopyranosides. However, the analogous 2-deoxyribofuranosides were also not observed in adequate yields.
The purification of the isoxazolin-5-one glycosides was accomplished by low pressure flash chromatography19 using MeCN–H2O eluents. Due to the low solubility of the glycosides in the applied solvent mixtures, the crude mixture was extracted with water. The extract was concentrated after addition of acetonitrile onto a small amount of dry silica. The dry silica adsorbed mixture was then applied to the column and eluted.20 The isolated products were analyzed by NMR-, HRMS-, IR-, UV- as well as optical rotation measurements.
The IR spectra of compounds 1–5 show absorptions centered at 1718–1696 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1536–1553 cm−1 (νCC) and 1038–1069 cm−1 (νC–O) being characteristic for isoxazolin-5-one glycosides.3,15 The 1H NMR spectra of compounds 1–5 show doublet signals with typical chemical shifts and coupling constants for H-3 (δ 8.45 − 8.56) and H-4 (δ 5.39 − 5.50 ppm; 3J3,4 = 3.7 Hz). The signals of H-1′ in compounds 1–3 appear at δ 5.05–5.13 ppm showing coupling constants 3J1′,2′ in the range of 8.6–9.2 Hz. The 13C NMR spectra of compounds 1–5 show chemical shifts corresponding to the isoxazolin-5-one ring in narrow ranges between δ 174.4–175.3 (C-5), 153.5–155.1 (C-3) and 89.1–91.7 (C-4) ppm. The anomeric carbon atoms (C-1′) in compounds 1–3 show absorption at δ 88.6–89.4 ppm. These data are in excellent agreement with literature values.3,4,9,151H NMR spectra of the fructosides 4 and 5 provide chemical shifts of 4.61 and 4.36 ppm that correspond to the H-3′position. The coupling constants 3J3′,4′ are 5.0 (α-anomer) and 8.4 Hz (β-anomer) respectively. The chemical shifts of HB-6′ equal 3.73 (3J5′,6′ = 4.6 Hz, comp. 4) and 3.75 ppm (3J5′,6′ = 6 Hz, comp. 5). The 13C NMR spectra of compounds 4 and 5 show signals at δ 100.3 (C-2′, α-anomer 4) and 97.5 ppm (C-2′, β-anomer 5). The signals at 61.1 (comp. 4) and δ 61.5 ppm (comp. 5) correspond to the C-6′-position. Moreover, the optical rotations are 68.4 (α-anomer 4) and −27.8° (β-anomer 5). All of these data are in excellent agreement with previous reported results for N-fructofuranosides.21–23
O), 1536–1553 cm−1 (νCC) and 1038–1069 cm−1 (νC–O) being characteristic for isoxazolin-5-one glycosides.3,15 The 1H NMR spectra of compounds 1–5 show doublet signals with typical chemical shifts and coupling constants for H-3 (δ 8.45 − 8.56) and H-4 (δ 5.39 − 5.50 ppm; 3J3,4 = 3.7 Hz). The signals of H-1′ in compounds 1–3 appear at δ 5.05–5.13 ppm showing coupling constants 3J1′,2′ in the range of 8.6–9.2 Hz. The 13C NMR spectra of compounds 1–5 show chemical shifts corresponding to the isoxazolin-5-one ring in narrow ranges between δ 174.4–175.3 (C-5), 153.5–155.1 (C-3) and 89.1–91.7 (C-4) ppm. The anomeric carbon atoms (C-1′) in compounds 1–3 show absorption at δ 88.6–89.4 ppm. These data are in excellent agreement with literature values.3,4,9,151H NMR spectra of the fructosides 4 and 5 provide chemical shifts of 4.61 and 4.36 ppm that correspond to the H-3′position. The coupling constants 3J3′,4′ are 5.0 (α-anomer) and 8.4 Hz (β-anomer) respectively. The chemical shifts of HB-6′ equal 3.73 (3J5′,6′ = 4.6 Hz, comp. 4) and 3.75 ppm (3J5′,6′ = 6 Hz, comp. 5). The 13C NMR spectra of compounds 4 and 5 show signals at δ 100.3 (C-2′, α-anomer 4) and 97.5 ppm (C-2′, β-anomer 5). The signals at 61.1 (comp. 4) and δ 61.5 ppm (comp. 5) correspond to the C-6′-position. Moreover, the optical rotations are 68.4 (α-anomer 4) and −27.8° (β-anomer 5). All of these data are in excellent agreement with previous reported results for N-fructofuranosides.21–23
Having access to isoxazolin-5-one glycosides we studied their pH dependent stability1,3,4 as well as the stability of compound 1 against β-glucosidase. 1H NMR and UV measurements of solutions of compound 1 in D2O showed that the N-glycosidic moiety is inert in a range of 7 ≥ pH ≥ 0 at rt, which is untypical for many kinds of glycosidic bonds. The π-conjugation in the aromatic isoxazolin-5-one ring reduces the basicity of the free electron pair of the nitrogen atom and thus the stability against electrophilic attack is increased. Consequently, β-glucosidase from almonds could not cleave the N-glycosidic bond in compound 1 at pH = 5 and rt due to its catalytic mechanism.24 In contrast, the absorption band disappears rapidly at pH > 7. The decay rates increase with ascending pH (see ESI, Fig. S2†). 1H NMR spectra in D2O show that the N-glycosidic bond is inert at pH > 7 while the double bond signals H-3 and H-4 disappear due to a β-addition of an hydroxyl anion to the α,β-unsaturated carbonyl structure. After re-acidification the N-glycosidic bond is cleaved and the free sugar is formed as expected due to the increased basicity of the lone electron pair of the nitrogen atom.
Photophysical properties
All of the novel glycosides show molar extinction coefficients around 10800 M−1 cm−1 at wavelengths of maximum extinction between 260 and 266 nm (Table 1). Upon irradiation with a low intense UV lamp (λmax = 254 nm) 1H NMR studies in D2O show a quantitative decay of the glycosides releasing the sugar from which they were derived. The quantum yield of this photohydrolysis reaction was determined by comparison of the decomposition kinetics of the isolated compounds 1–5 with uridine (U) as a standard. Equally concentrated solutions of the glycosides were placed in a cuvette and irradiated with a weak UV lamp. Simultaneously the absorbances of the solutions have been measured over the time. The intensity of the light source was determined (Fig. 2).
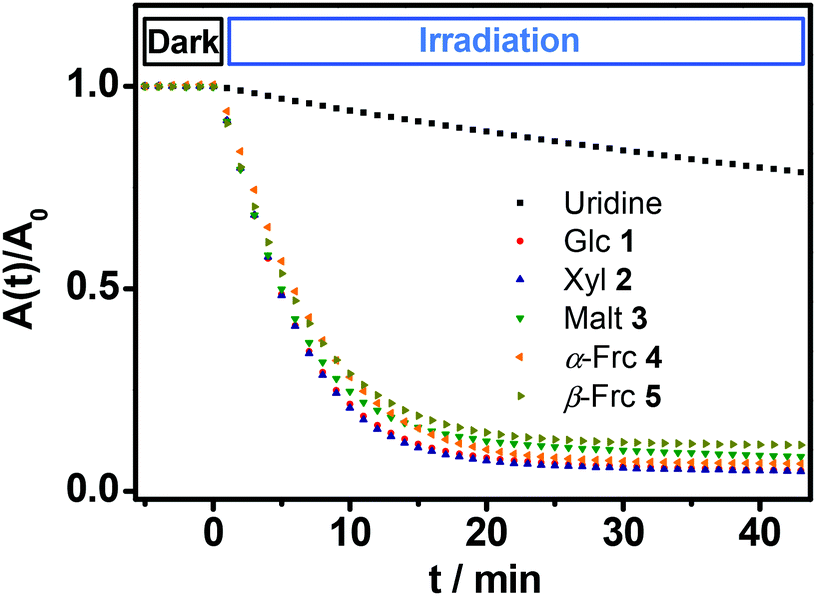 | ||
| Fig. 2 Decay kinetics for the photohydrolysis of the glycosides; black: uridine, red: β-glucoside 1, blue: β-xyloside 2, green: β-maltoside 3, orange: α-fructofuranoside 4, yellow: β-fructofuranoside 5; λ = 261 nm; c0 = 3.5 × 10−5 M; I261 nm = 0.18 mW cm−2; dlamp = 5 cm; stirred; over the first five minutes absorbances were measured without illumination (dark). | ||
| S | λ max/nm | ε/103 M−1 cm−1 | Φ |
|---|---|---|---|
| U | 260 | 9.7 | 0.019 ± 0.00125 |
| 1 | 260 | 10.8 | 0.324 ± 0.018 |
| 2 | 260 | 10.8 | 0.327 ± 0.019 |
| 3 | 261 | 10.8 | 0.299 ± 0.017 |
| 4 | 266 | 10.8 | 0.284 ± 0.016 |
| 5 | 265 | 10.8 | 0.275 ± 0.016 |
The first five minutes without illumination show a constant absorption with no significant change in case of all glycosides. After starting the irradiation with the low intense UV lamp all of the curves show a decrease in absorbance that is significantly faster compared to the decay of uridine (U) under the same conditions. The slopes of the curves at the beginning of the irradiation were determined. Via comparison with the standard the quantum yields of the photoreactions were calculated (see Experimental section). The determined quantum yields lie in a range of 0.275 to 0.327 (Table 1).
These values are in the same order of magnitude as those reported for quantum yields of naturally occurring amino acid derivatives of isoxazolin-5-one (0.5).8 Thus, the described compounds 1–5 show a rapid and very efficient release of their corresponding free sugars at room temperature upon irradiation with very weak UV light.
Conclusion
A novel synthetic protocol has been developed for the synthesis of isoxazolin-5-one glycosides that does not depend on the use of protecting groups. The reaction was successfully applied to glucose, xylose, maltose and fructose to afford the corresponding glycosides 1–5. Although this one-pot strategy gives only low yields, it can be used to provide rapidly a significant amount of the described glycosides. The product scope and selectivity of the reaction have been discussed. The main products were isolated and characterized in terms of their chemical structure, stability and photophysical properties. High quantum yields of the photohydrolysis reactions have been observed. As a consequence of the described photolability as well as the stability against low pH and enzymatic hydrolysis, we conclude that the isoxazolin-5-one moiety provides potential for the use as a photoactive and bioorthogonal protecting group for the anomeric position in glycosides.Experimental section
General
Melting points were determined with a capillary melting point apparatus. Infrared spectra were measured with an IR spectrometer in a range of 700–4000 cm−1 in transmission mode with a spectral resolution of 6 cm−1. Optical rotations were measured at 589 nm in water (temperatures are given). NMR spectra were measured using a spectrometer operating at 400 MHz (1H) and 100 MHz (13C). Chemical shifts (δ) are quoted in parts per million (ppm) and are referenced to the signal of residual protonated solvent (HDO at δ 4.79 ppm). Acetonitrile was added as a reference for 13C NMR spectra in D2O. Assignment of peaks was carried out using 2D NMR experiments (COSY, HSQC, and HMBC). The multiplicities are given as follows: d, doublet; dd, doublet of doublets; m, multiplet. High-resolution mass spectra were recorded on a UHR-qTOF mass spectrometer.The columns for preparative chromatography were packed by pouring a suspension of silica gel (0.03–0.063 mm) in the eluent into the column containing 40 ml of the eluent. When the sedimentation process was completed the column was opened and additional pressure was slowly increased up to 1.2 bar. The column was washed with 300 ml of solvent before adding the silica-adsorbed mixture. The separations were carried out at 1.1 bar additional pressure.
Thin-layer chromatography was performed on TLC silica gel 60 F254 aluminum sheets. Compounds were visualized using an UV lamp with a maximum emission at 254 nm. All reagents and solvents were purchased in the highest purity that was commercially available and used without further purification.
Photophysical measurements
Absorptions were recorded on a UV spectrophotometer with a spectral resolution of 2 nm at 261 nm and room temperature. Quantum yields and photon flux were determined by using commercially available uridine (99%) as a chemical actinometer according to eqn (1).8,25 The accuracies were estimated using eqn (2). The intensity of the UV lamp was determined applying eqn (3).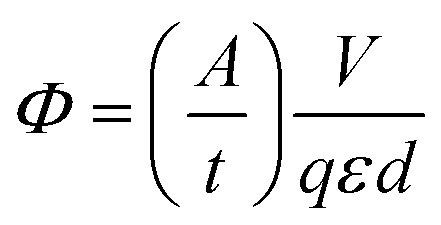 | (1) |
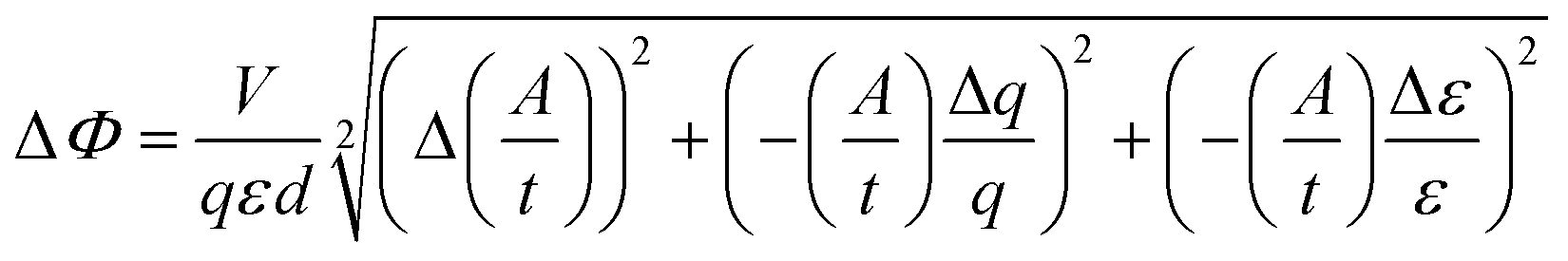 | (2) |
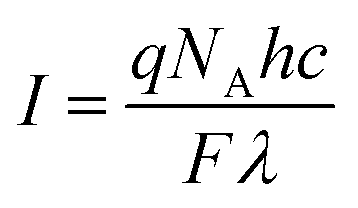 | (3) |
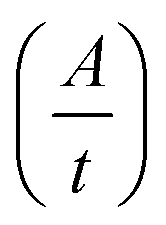 = slope of the change in absorption over short time periods in s−1, V = volume of the irradiated solution in l, q = photon flux in Einstein s−1, ε = molar extinction coefficient in l mol−1 cm−1, d = length of the cuvette in cm, Na = Avogadro constant in mol−1, h = Planck constant in J s, c = speed of light in ms−1, F = area of irradiation (1 cm2), λ = wavelength (261 nm), I = intensity in W cm−2 and ΔX = the standard deviation of X. The substrates were dissolved in a buffer (Na2HPO4–NaH2PO4; 50 mM) at pH 7 in a concentration of (3.5 ± 0.2) × 10−5 M. The total volume of the solution was (1.63 ± 0.03) ml. A UV lamp (λmax = 254 nm, 6 W) was used for the illumination. The cuvette (d = 1 cm) was irradiated vertically to the measurement geometry at a distance (dlamp) of 5 cm. The solution was stirred via magnetic induction. The absorption at a wavelength of 261 nm was measured time dependently and simultaneously to irradiation and stirring. The observed molar extinction coefficients are in excellent agreement with the literature results.1–4,6,8,15,26
= slope of the change in absorption over short time periods in s−1, V = volume of the irradiated solution in l, q = photon flux in Einstein s−1, ε = molar extinction coefficient in l mol−1 cm−1, d = length of the cuvette in cm, Na = Avogadro constant in mol−1, h = Planck constant in J s, c = speed of light in ms−1, F = area of irradiation (1 cm2), λ = wavelength (261 nm), I = intensity in W cm−2 and ΔX = the standard deviation of X. The substrates were dissolved in a buffer (Na2HPO4–NaH2PO4; 50 mM) at pH 7 in a concentration of (3.5 ± 0.2) × 10−5 M. The total volume of the solution was (1.63 ± 0.03) ml. A UV lamp (λmax = 254 nm, 6 W) was used for the illumination. The cuvette (d = 1 cm) was irradiated vertically to the measurement geometry at a distance (dlamp) of 5 cm. The solution was stirred via magnetic induction. The absorption at a wavelength of 261 nm was measured time dependently and simultaneously to irradiation and stirring. The observed molar extinction coefficients are in excellent agreement with the literature results.1–4,6,8,15,26
General synthetic procedure
To a stirred solution of 209 mg (3 mmol, 1.5 eq.) hydroxylamine hydrochloride in 2 ml dry methanol 314 mg (2.8 mmol, 1.4 eq.) potassium tert-butoxide were added in portions at 0 °C under stirring. After 1 h at rt the solution was filtered under vacuum, washed with 1 ml of dry methanol and 2 mmol sugar were added. After 1 d of stirring at rt the solvent was removed under reduced pressure at 40 °C. The dry residue was dissolved in 1 ml of water. Under stirring 0.5 ml of a freshly prepared solution of DCC in MeCN (c = 0.4 M, 0.1 eq.) was added at once. Then further 7 ml of DCC in MeCN (c = 0.4 M, 1.4 eq.) and 7 ml of propynoic acid in MeCN (c = 0.42 M, 1.47 eq.) were added simultaneously at rt over 5 h. After 20 h of stirring at rt the solvents were removed at 25 °C under reduced pressure. The mixture was taken up in 10 ml of water and applied to an ultrasound bath for 1 h at 22–27 °C. The suspension was filtrated and washed with water (3 × 5 ml). To the filtrate 150 ml of MeCN were added and the solvents were removed at 25 °C and 75 mbar. Then 1.25 g of dry silica and 150 ml MeCN were added. The solvents were removed again at 25 °C and 75 mbar to yield a dry crude mixture.Column chromatography and analytical data of 2-(β-D-glucopyranosyl)-3-isoxazolin-5-one (1): The dry mixture was applied to a column and eluted (MeCN–H2O 55:1, silica). The product fractions were combined and concentrated to yield 109 mg (0.44 mmol, 22%) of 1 as a colorless powder.
[α]24D +14.6 (c 0.81, H2O); Rf(MeCN–H2O 55:1) = 0.15; 1H NMR (400 MHz, D2O) δ 8.47 (d, 3J3,4 = 3.7 Hz, 1H, H-3), 5.47 (d, 3J3,4 = 3.7 Hz, 1H, H-4), 5.12 (d, 3J1′,2′ = 9.2 Hz, 1H, H-1′), 3.90–3.85 (m, 2H, HA-6′ and H-2′), 3.71 (dd, 2J6′A,6′B = 12.5 Hz, 3J5′,6′B = 6.9 Hz, 1H, HB-6′), 3.62− 3.54 (m, 2H, H-3′ and H-5′), 3.46 (dd, 3J3′,4′ = 3J4′,5′ = 9.5 Hz, 1H, H-4′); 13C NMR (100 MHz, D2O) δ 174.8 (C-5), 154.9 (C-3), 91.2 (C-4), 88.9 (C-1′), 78.8 (C-5′), 76.8 (C-3′), 70.0 (C-2′), 69.6 (C-4′), 61.0 (C-6′); HRMS (ESI-TOF) m/z calcd for C9H14NO7 248.0765 [M + H]+, found 248.0758 (Δm/z 2.8 ppm); IR (thin film, cm−1) 3382 (br, s), 2922 (m), 1718 (s, br), 1546 (s), 1069 (br, s); UV (H2O) λmax/nm (ε/L mol−1 cm−1) 260 (10800 ± 200); mp 147–149 °C (decomp.).
Column chromatography and analytical data of 2-(β-D-xylopyranosyl)-3-isoxazolin-5-one (2): MeCN–H2O 85:1; yield: 58 mg (0.28 mmol, 14%); colorless oil.
[α]24D −53.2 (c 0.84, H2O); Rf(MeCN–H2O 85:1) = 0.22; 1H NMR (400 MHz, D2O) δ 8.45 (d, 3J3,4 = 3.7 Hz, 1H, H-3), 5.49 (d, 3J3,4 = 3.7 Hz, 1H, H-4), 5.05 (d, 3J1′,2′ = 9.2 Hz, 1H, H-1′), 3.99 (dd, 2J5′A,5′B = 11.6 Hz, 3J4′,5′A = 5.5 Hz, 1H, HA-5′), 3.87 (dd, 3J2′,3′ = 3J1′,2′ = 9.2 Hz, 1H, H-2′), 3.67 (m, 1H, H-4′), 3.55 (dd, 3J2′,3′ = 3J3′,4′ = 9.3 Hz, 1H, H-3′), 3.42 (dd, 2J5′A,5′B = 3J4′,5′B = 11.0 Hz, 1H, HB-5′); 13C NMR (100 MHz, D2O) δ 174.7 (C-5), 155.1 (C-3), 91.7 (C-4), 89.4 (C-1′), 76.8 (C-3′), 69.9 (C-2′), 69.2 (C-4′), 67.9 (C-5′); HRMS (ESI-TOF) m/z calcd for C8H12NO6 218.0659 [M + H]+, found 218.0658 (Δm/z 0.5 ppm); IR (thin film, cm−1) 3374 (br, s), 1717 (s, br), 1547 (s), 1055 (s); UV (H2O) λmax/nm (ε/L mol−1 cm−1) 260 (10800 ± 200).
Column chromatography and analytical data of 2-(β-D-maltopyranosyl)-3-isoxazolin-5-one (3): MeCN–H2O 7:1; yield: 148 mg (0.36 mmol, 18%); colorless powder.
[α]25D +105.4 (c 0.61, H2O); Rf(MeCN–H2O 7:1) = 0.26; 1H NMR (400 MHz, D2O) δ 8.46 (d, 3J3,4 = 3.7 Hz, 1H, H-3), 5.48 (d, 3J3,4 = 3.7 Hz, 1H, H-4), 5.43 (d, 3J1′′,2′′ = 3.9 Hz, 1H, H-1′′), 5.13 (d, 3J1′,2′ = 8.6 Hz, 1H, H-1′), 3.94–3.82 (m, 4H), 3.78 − 3.65 (m, 6H), 3.58 (dd, 3J2′′,3′′ = 9.9 Hz, 3J1′′,2′′ = 3.9 Hz, 1H, H-2′′), 3.41 (dd, 3J5′,6A′ = 2J6A′,6B′ = 9.4 Hz, 1H, HA-6′); 13C NMR (100 MHz, D2O) δ 174.8 (C-5), 154.8 (C-3), 100.2 (C-1′′), 91.3 (C-4), 88.6 (C-1′), 77.4, 77.2, 76.4, 73.4, 73.4, 72.3 (C-2′′), 69.9, 69.8, 61.1, 61.0; HRMS (ESI-TOF) m/z calcd for C15H24NO12 410.1293 [M + H]+, found 410.1280 (Δm/z 3.2 ppm); IR (thin film, cm−1) 3367 (br, s), 1717 (s, br), 1594 (s), 1553 (s), 1038 (br, s); UV (H2O) λmax/nm (ε/L mol−1 cm−1) 261 (10760 ± 200); mp 195–200 °C (decomp.).
Column chromatography and analytical data of 2-(α-D-fructofuranosyl)-3-isoxazolin-5-one (4): DCM–MeOH 5:1 and MeCN–H2O 35:1; yield: 65 mg (0.26 mmol, 13%); colorless oil.
[α]25D +68.4 (c 0.66, H2O); Rf(MeOH–DCM 1:5) = 0.30; 1H NMR (400 MHz, D2O) δ 8.56 (d, 3J3,4 = 3.7 Hz, 1H, H-3), 5.43 (d, 3J3,4 = 3.5 Hz, 1H, H-4), 4.61 (d, 3J3′,4′ = 5.0 Hz, 1H, H-3′), 4.15 − 4.09 (m, 2H, H-4′ and H-5′), 3.93–3.83 (m, 3H, H-1′ and HA-6′), 3.73 (dd, 2J6′A,6′B = 12.7 Hz, 3J5′,6′B = 4.6 Hz, 1H, HB-6′); 13C NMR (100 MHz, D2O) δ 175.3 (C-5), 153.7 (C-3), 100.3 (C-2′), 89.6 (C-4), 83.8 (C-4′ or C-5′), 79.9 (C-3′), 76.3 (C-4′ or C-5′), 61.5 (C-1′), 61.1 (C-6′); HRMS (ESI-TOF) m/z calcd for C9H14NO7 248.0765 [M + H]+, found 248.0755 (Δm/z 4.0 ppm); IR (thin film, cm−1) 3366 (br, s), 1717 (s, br), 1541 (s), 1058 (br, s); UV (H2O) λmax/nm (ε/L mol−1 cm−1) 266 (10820 ± 200).
Column chromatography and analytical data of 2-(β-D-fructofuranosyl)-3-isoxazolin-5-one (5): DCM–MeOH 5:1 and MeCN–H2O 35:1; yield: 55 mg (0.22 mmol, 11%); colorless oil.
[α]25D −27.8 (c 0.85, H2O); Rf(MeOH–DCM 1:5) = 0.20; 1H NMR (400 MHz, D2O) δ 8.56 (d, 3J3,4 = 3.7 Hz, 1H, H-3), 5.39 (d, 3J3,4 = 3.7 Hz, 1H, H-4), 4.36 (d, 3J3′,4′ = 8.4 Hz, 1H, H-3′), 4.26 (dd, 3J3′,4′ = 3J4′,5′ = 8.4 Hz, 1H, H-4′), 4.06 (d, 2J1A′,1B′ = 12.7 Hz, 1H, HA-1′), 3.97 (d, 2J1A′,1B′ = 12.7 Hz, 1H, HB-1′), 3.92–3.86 (m, 2H, H-5′ and HA-6′), 3.75 (dd, 2J6′A,6′B = 12.9 Hz, 3J5′,6′B = 6.0 Hz, 1H, HB-6′); 13C NMR (100 MHz, D2O) δ 175.0 (C-5), 153.5 (C-3), 97.5 (C-2′), 89.1 (C-4), 82.1 (C-5′), 78.4 (C-3′), 73.8 (C-4′), 62.6 (C-1′), 61.5 (C-6′); HRMS (ESI-TOF) m/z calcd for C9H14NO7 248.0765 [M + H]+, found 248.0756 (Δm/z 3.6 ppm); IR (thin film, cm−1) 3360 (br, s), 1696 (s, br), 1536 (s), 1046 (s); UV (H2O) λmax/nm (ε/L mol−1 cm−1) 265 (10780 ± 200).
Author information
TB and WB planned the experiments. TB and PK synthesized the compounds. TB performed the photophysical characterizations. TB, SHvR and CP performed NMR analyses. TB, SHvR and WB wrote the manuscript.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We thank Kerstin Ploss for the HRMS measurements. This work was financed by the Max Planck Society.Notes and references
- F. Lambein and R. Van Parijs, Biochem. Biophys. Res. Commun., 1970, 40, 557 CrossRef CAS.
- L. Van Rompuy, N. Schamp, N. De Kimpe and R. Van Parijs, J. Chem. Soc., Perkin Trans. 1, 1973, 2503 RSC.
- L. Van Rompuy, F. Lambein, R. De Gussem and R. Van Parijs, Biochem. Biophys. Res. Commun., 1974, 56, 199 CrossRef CAS.
- F. Lambein, L. Van Rompuy, A. De Bruyn and R. Van Parijs, Arch. Physiol. Biochem., 1974, 82, 187 CAS.
- F. Ikegami, F. Lambein, Y.-H. Kuo and I. Murakoshi, Phytochemistry, 1984, 23, 1567 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington and D. J. Birch, Tetrahedron Lett., 1985, 26, 5931 CrossRef CAS.
- T. Randoux, J. C. Braekman, D. Daloze and J. M. Pasteels, Naturwissenschaften, 1991, 78, 313 CrossRef CAS.
- A. De Bruyn, G. Verhegge and F. Lambein, Planta Med., 1992, 58, 159 CrossRef CAS PubMed.
- W. Sugeno and K. Matsuda, Appl. Entomol. Zool., 2002, 37, 191 CrossRef CAS.
- J. M. Pasteels, J. C. Braekman, D. Daloze and R. Ottinger, Tetrahedron, 1982, 38, 1891 CrossRef CAS.
- M. Rowell-Rahier and J. M. Pasteels, J. Chem. Ecol., 1986, 12, 1189 CrossRef CAS PubMed.
- J. M. Pasteels, D. Daloze and M. Rowellrahier, Physiol. Entomol., 1986, 11, 29 CrossRef CAS PubMed.
- P. Laurent, J.-C. Braekman and D. Daloze, Top. Curr. Chem., 2005, 240, 167 CAS.
- J. E. Baldwin, R. M. Adlington and L. C. Mellor, Tetrahedron, 1994, 50, 5049 CrossRef CAS.
- T. Becker, H. Görls, G. Pauls, R. Wedekind, M. Kai, S. H. von Reuß and W. Boland, J. Org. Chem., 2013, 78, 12779 CrossRef CAS PubMed.
- J. Brand, T. Huhn, U. Groth and J. C. Jochims, Chem. – Eur. J., 2006, 12, 499 CrossRef PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522 CrossRef.
- C. Pöhner, V. Ullmann, R. Hilpert, E. Samain and C. Unverzagt, Tetrahedron Lett., 2014, 55, 2197 CrossRef PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- J. Jindrich, P. Josef and B. Lindberg, Carbohydr. Res., 1995, 275, 1 CrossRef CAS.
- W. C. Kett, M. Batley and J. W. Redmond, Carbohydr. Res., 1997, 299, 129 CrossRef CAS.
- A. Bouali, D. F. Ewing and G. Mackenzie, Nucleosides Nucleotides, 1994, 13, 491 CAS.
- A. Grouiller, G. Mackenzie, B. Najib, G. Shaw and D. Ewing, J. Chem. Soc., Chem. Commun., 1988, 671 RSC.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 1999, 33, 11 CrossRef PubMed.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 2004, 76, 2105 CrossRef CAS.
- F. De Sarlo, G. Dini and P. Lacrimini, J. Chem. Soc. C, 1971, 86 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. 1H and 13C NMR spectra of compounds 1–5, pH and beta-glucosidase stability plots of compound 1, decay kinetics of uridine and compounds 1–5. See DOI: 10.1039/c5ob00244c |
| This journal is © The Royal Society of Chemistry 2015 |