
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concise total syntheses of (±)-mesembrane and (±)-crinane†
Mrinal Kanti
Das‡
,
Subhadip
De‡
,
Shubhashish
and
Alakesh
Bisai
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal, MP-462066, India. E-mail: alakesh@iiserb.ac.in
First published on 10th February 2015
Abstract
A straightforward and unified strategy to access Amaryllidaceae alkaloids comprising a cis-3a-aryloctahydroindole scaffold has been developed. The strategy features Eschenmoser–Claisen rearrangement of allylalcohol as a key step for the installation of all-carbon quaternary stereocenters present in these alkaloids. The consequent iodolactonization–reduction–oxidation sequence beautifully assembles the advanced intermediate keto-aldehyde 10a, b in synthetically viable yields. The methodology has been successfully applied in the efficient syntheses of (±)-mesembrane (1a) and (±)-crinane (2a).
The cis-3a-aryloctahydroindole alkaloids possessing an all-carbon quaternary stereocenter1 constitute the core structure of many alkaloids with impressive diversity of biological activity.2 Their biological potential is significantly manifested by their anti-viral, anti-tumor, anti-cholinergic and anti-HIV properties.3 These activities together with their intriguing structures have brought a major impetus for synthetic exploration in this direction from organic chemists across the globe.
In particular, the cis-3a-aryloctahydroindole alkaloids 1 and 2 (Fig. 1) are found in plants of the Amaryllidaceae family4 and elicit continuing interest in the synthetic research community due to their intriguing physiological activities.5,6 Biogenetically, crinane (2a) and related alkaloids are closely related to other major Amaryllidaceae family natural products, lycorane- and galanthamine-type alkaloids as they all are derived from the same precursor norbelladine.7 These cis-3a-aryloctahydroindole alkaloids8 display vicinal quaternary and tertiary carbon stereocenters with a fused pyrrolidine ring whose stereochemical incorporation is indeed a challenge.9 We envisaged a unified strategy to access all of these alkaloids having the cis-3a-aryloctahydroindole skeleton (Fig. 1) with a sterically congested quaternary carbon center located at the hydroindolone bridgehead (C-3a) position as a common structural feature. Herein, we report the development of a powerful strategy involving Eschenmoser–Claisen rearrangement followed by iodolactonization which would permit the late stage, divergent introduction of a range of functionality to address the total synthesis of several congeners of this family.
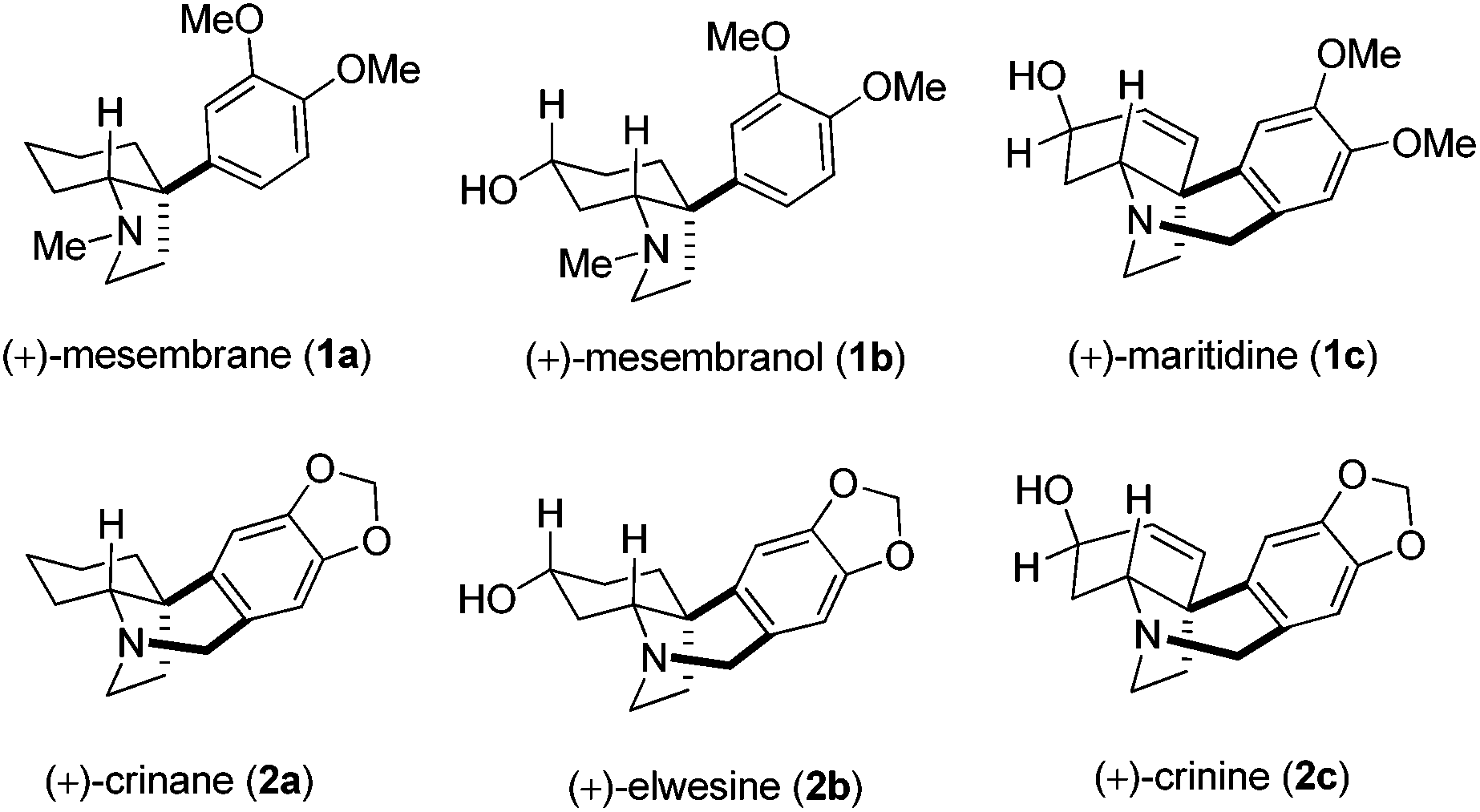 | ||
| Fig. 1 The Amaryllidaceae alkaloids. | ||
Retrosynthetically, we envisioned that the advanced intermediate ketoaldehydes 10a, b would lead to a unified pathway to access both mesembrane (1a) and crinane (2a). The dimethylamides 4a, b (Scheme 1) would afford 3a, b,10via iodolactonization, which in turn can be synthesized from allylalcohols 5a, b following Eschenmoser–Claisen rearrangement.11 Allylalcohols 5a, b can be accessed from 3-aryl-2-cyclohexenones 7a, b (Scheme 2), and the latter could easily be obtained directly from vinylogous ester 6via a well-known Stork–Danheiser sequence.12
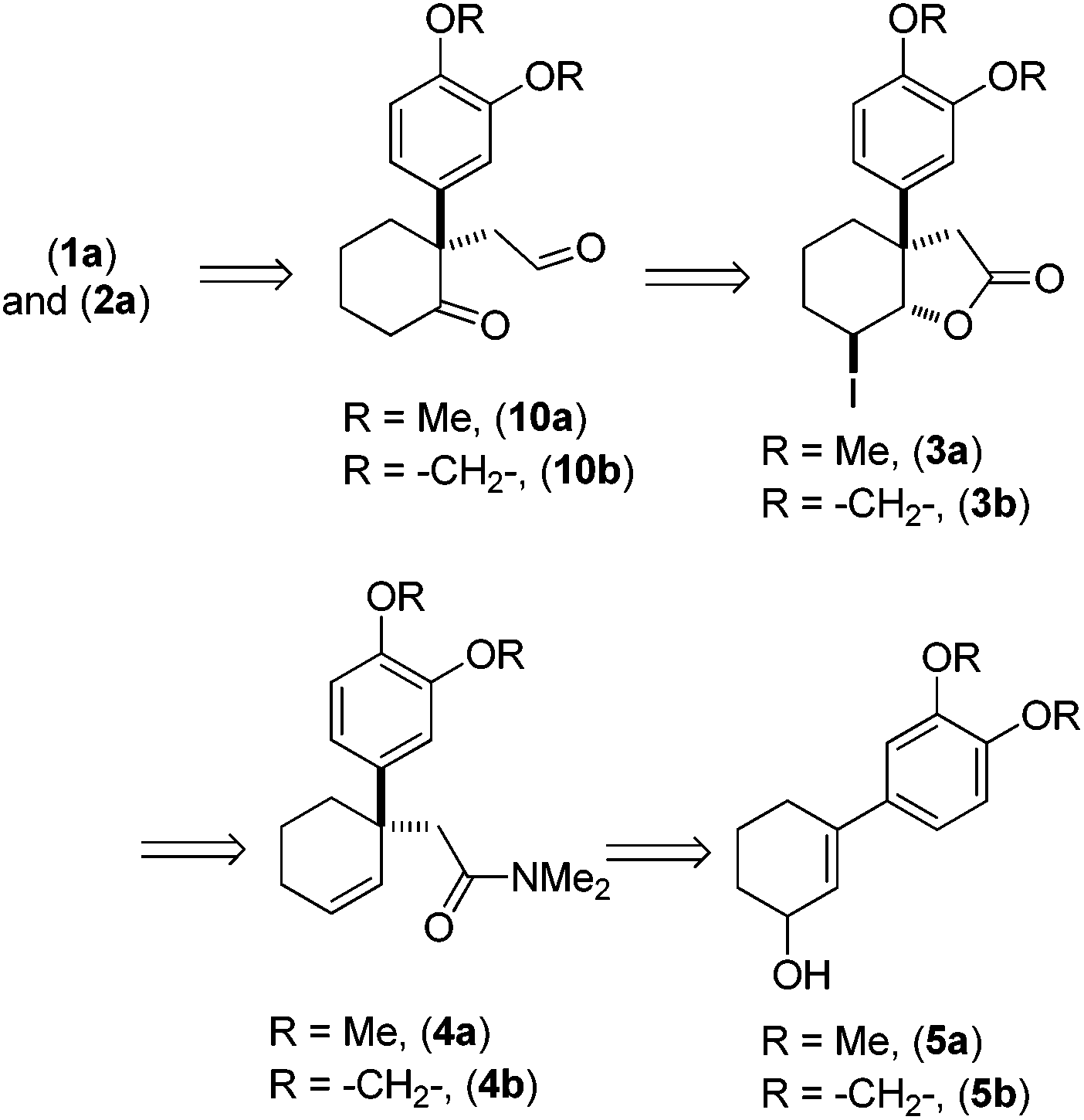 | ||
| Scheme 1 Retrosynthetic analysis of (±)-1a and (±)-2a. | ||
 | ||
| Scheme 2 Synthesis of 3-aryl-cyclohexenols (±)-5a, b. | ||
Moving forward with our proposed strategy, we performed the Stork–Danheiser sequence on compound 6 using arylmagnesium bromides to afford 3-aryl-2-cyclohexenones 7a, b in 73–85% yields (Scheme 2). The latter were then reduced under Luche reduction13 to access allylalcohols 5a, b in 92–96% yields. With allyl alcohols 5a, b in hand, we sought after conditions to effect Eschenmoser–Claisen rearrangement for the synthesis of 1-alkyl-1-aryl-2-cyclohexenes 4a, b (Scheme 3) having an all-carbon quaternary stereocenter.
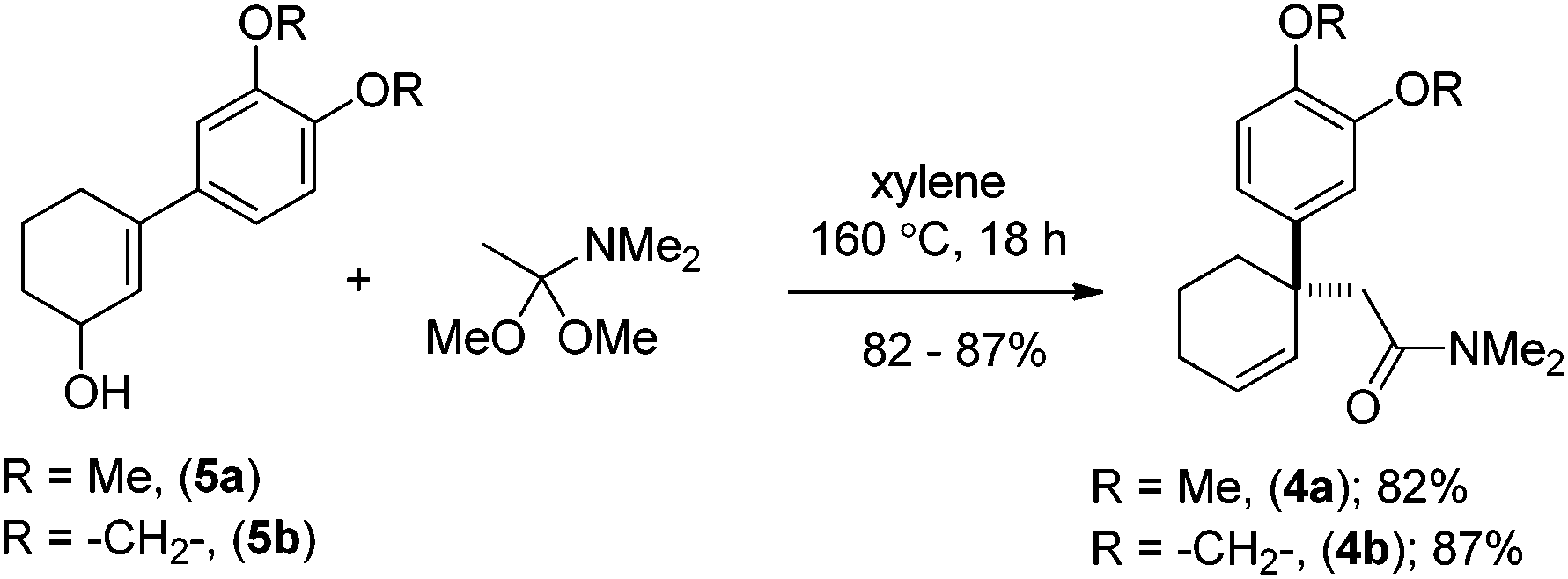 | ||
| Scheme 3 Eschenmoser–Claisen rearrangement of (±)-5a, b. | ||
Preliminary studies indicate that 2–6 equiv. of dimethylacetal of N,N-dimethylacetamide (DMA–DMA) in different solvents furnished product 4a only in 26–53% yields. After exhaustive optimization, it was found that 7 equiv. of DMA–DMA under heating at 160 °C led to the formation of the desired product in 82% yield (Scheme 3). Under optimized conditions, 5b afforded product 4b in 87% isolated yield (Scheme 3). We then turned our attention to functionalize the 2-position of the cyclohexene ring. Iodolactonization of 1-alkyl-1-aryl substituted cyclohexenes 4a, b in the presence of iodine in the THF and water mixture provided iodolactone intermediates 3a, b in 85–86% yield (Scheme 4). The iodolactones 3a, b upon treatment with DBU furnished alkenes 9a, b in excellent yields, which can in turn be utilized as advanced intermediates for the synthesis of various Amaryllidaceae alkaloids. However, for total synthesis of mesembrane (1a) and crinane (2a) we required γ-keto aldehydes 10a, b to be further charged under reductive amination conditions to afford cis-3a-aryloctahydroindole. To synthesize γ-keto aldehydes 10a, b, we reduced 3a, b in the presence of lithium aluminum hydride to afford 1,4-diols 8a, b in quantitative yield (Scheme 4). Among the various oxidation procedures tried to synthesize γ-keto aldehydes 10a, b, we found that the Swern oxidation14 afforded 10a, b in 89–92% yields (Scheme 4).
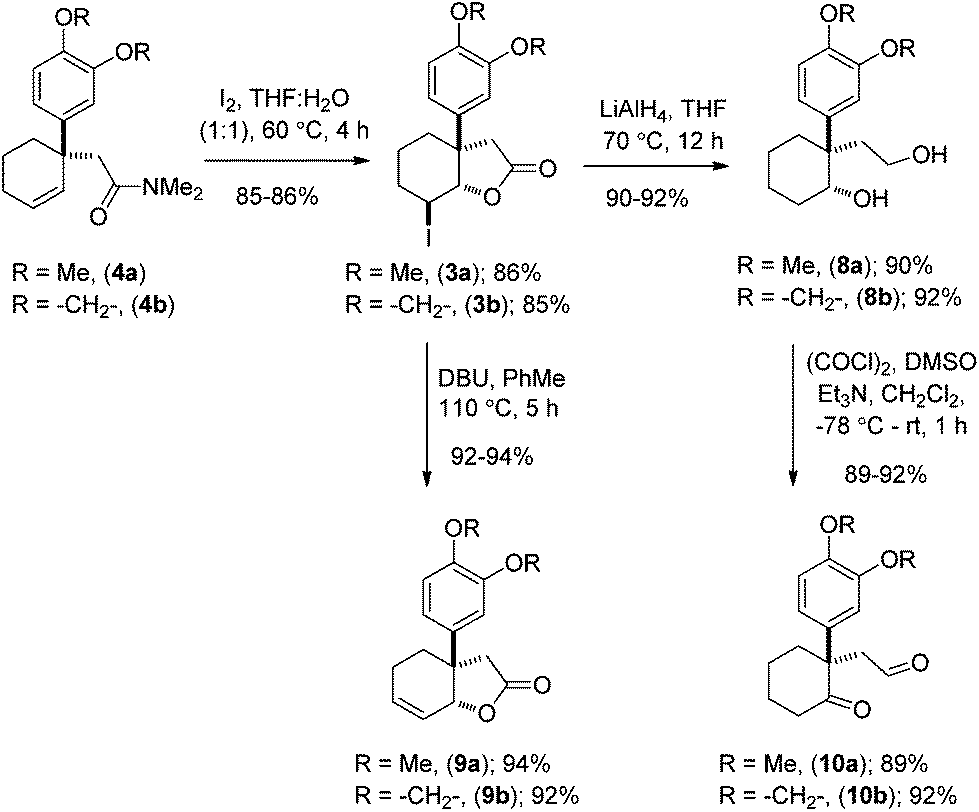 | ||
| Scheme 4 Synthesis of ketoaldehydes (±)-10a, b. | ||
Optimization studies were further conducted to achieve reductive amination of compound 10a in order to complete the total synthesis of mesembrane (1a) (Table 1). Initially, we carried out reductive amination of 10a in the presence of 2 equiv. of ammonium acetate and 4 equiv. of sodium cyano borohydride in different solvents such as MeOH, EtOH, and THF in the presence of 1 equiv. of trifluoroacetic acid and acetic acid. To our delight, we found that cis-3a-aryloctahydroindole 11a could be obtained in 32–89% isolated yields (entries 1–6, Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid | Solvent | Temp. | Time | Yielda,b (%) |
| a 2.0 equiv. of NH4OAc and 4.0 equiv. NaBH3CN were used in each case and all the reactions were performed on a 0.20 mmol of (±)-10a in 2 mL of the solvent under an inert atmosphere. b Isolated yields after column chromatography. | |||||
| 1 | TFA (1 equiv.) | MeOH | 0–25 °C | 12 h | 72% |
| 2 | AcOH (1 equiv.) | MeOH | 0–25 °C | 12 h | 75% |
| 3 | TFA (1 equiv.) | EtOH | 0–25 °C | 10 h | 89% |
| 4 | AcOH (1 equiv.) | EtOH | 0–25 °C | 10 h | 88% |
| 5 | TFA (1 equiv.) | THF | 0–25 °C | 18 h | 35% |
| 6 | AcOH (1 equiv.) | THF | 0–25 °C | 18 h | 32% |
| 7 | TFA (10 mol%) | EtOH | 0–25 °C | 16 h | 83% |
| 8 | AcOH (10 mol%) | EtOH | 0–25 °C | 16 h | 85% |
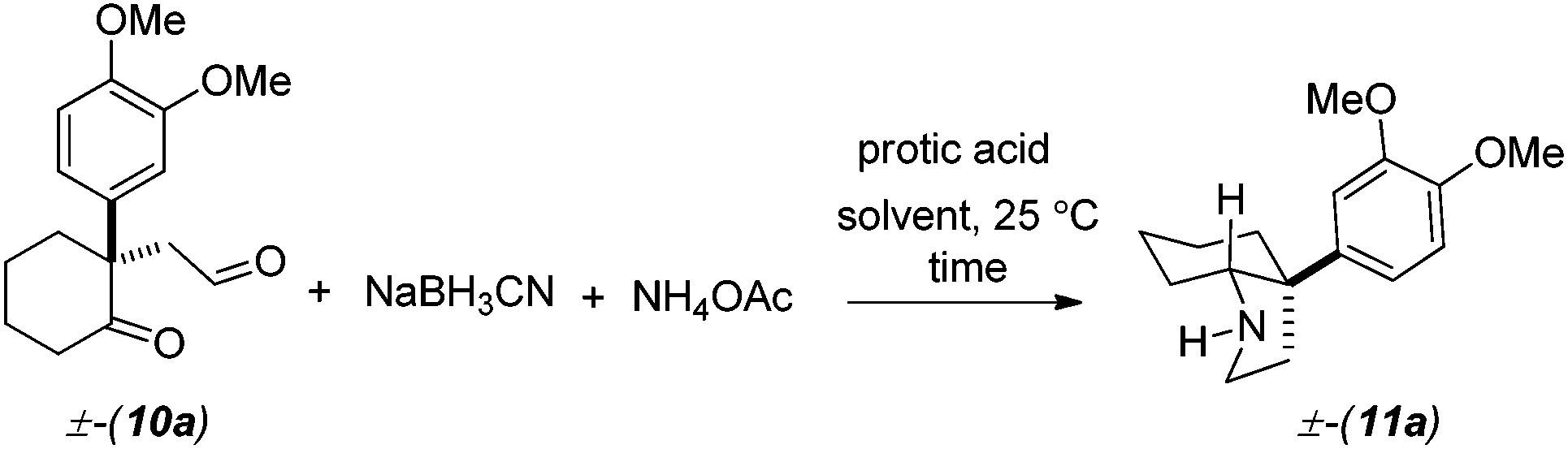
Following further optimization, we were pleased to find that secondary amine 11a could be obtained in 83–85% yields when reductive amination was carried out in the presence of only 10 mol% of trifluoroacetic acid and acetic acid, respectively (entries 7 and 8, Table 1). Further, we synthesized carbamates 12a, b in 82–85% yields from 11a by treatment with chloromethylformate and benzyl chloroformate in the presence of NaHCO3 (Scheme 5). In fact, we strongly feel that 12a, b could serve as potential precursors for the synthesis of a tricyclic core with additional amide functionality (see red arrows) related to many Amaryllidaceae alkaloids (see, 2a, c, Fig. 1) via a Bischler–Napieralski type process.15
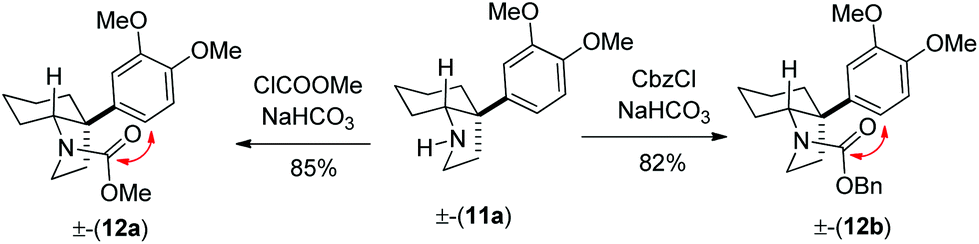 | ||
| Scheme 5 Synthesis of cis-3a-aryloctahydroindole derivatives (12a, b). | ||
For total synthesis of (±)-mesembrane 1a, we then carried out reductive amination using methylamine under optimized conditions, which in turn provided 1a in 86–88% yields (Scheme 6). Along similar lines, we have also synthesized 13 in 87–91% isolated yields (Scheme 6).
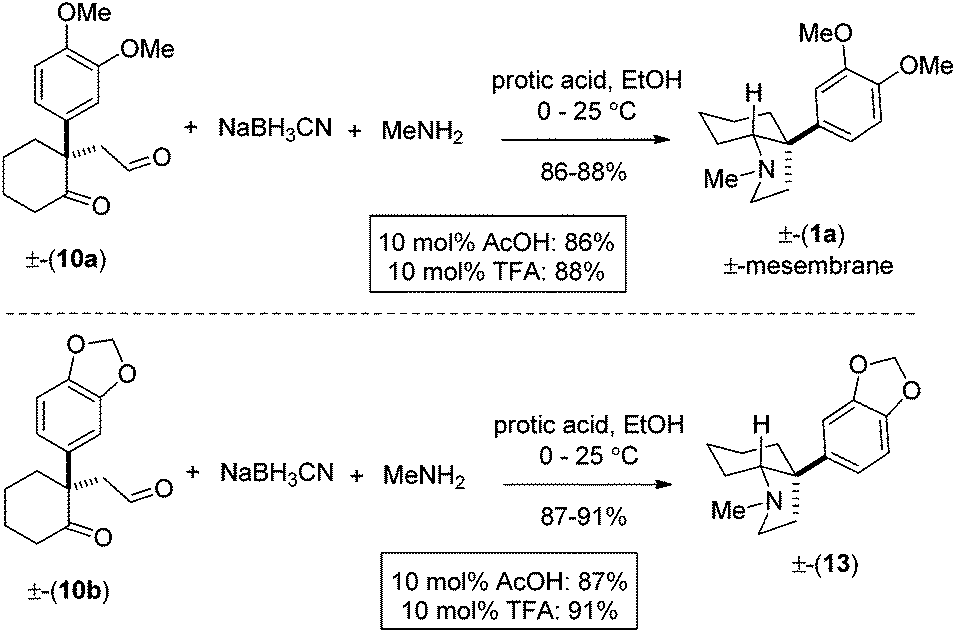 | ||
| Scheme 6 Total synthesis of (±)-mesembrane (1a). | ||
Next, we shifted our attention for a concise total synthesis of crinane (2a). Towards this end, we carried out the reductive amination of γ-keto aldehyde 10b, affording cis-3a-aryloctahydroindole 11b in 84–88% isolated yields (Scheme 7). Finally, 11b was treated with Eschenmoser's salt,9d to complete the total synthesis of (±)-crinane (2a). Following our optimized conditions shown in Scheme 7, we have also synthesized 14a, b in 74–83% yields.
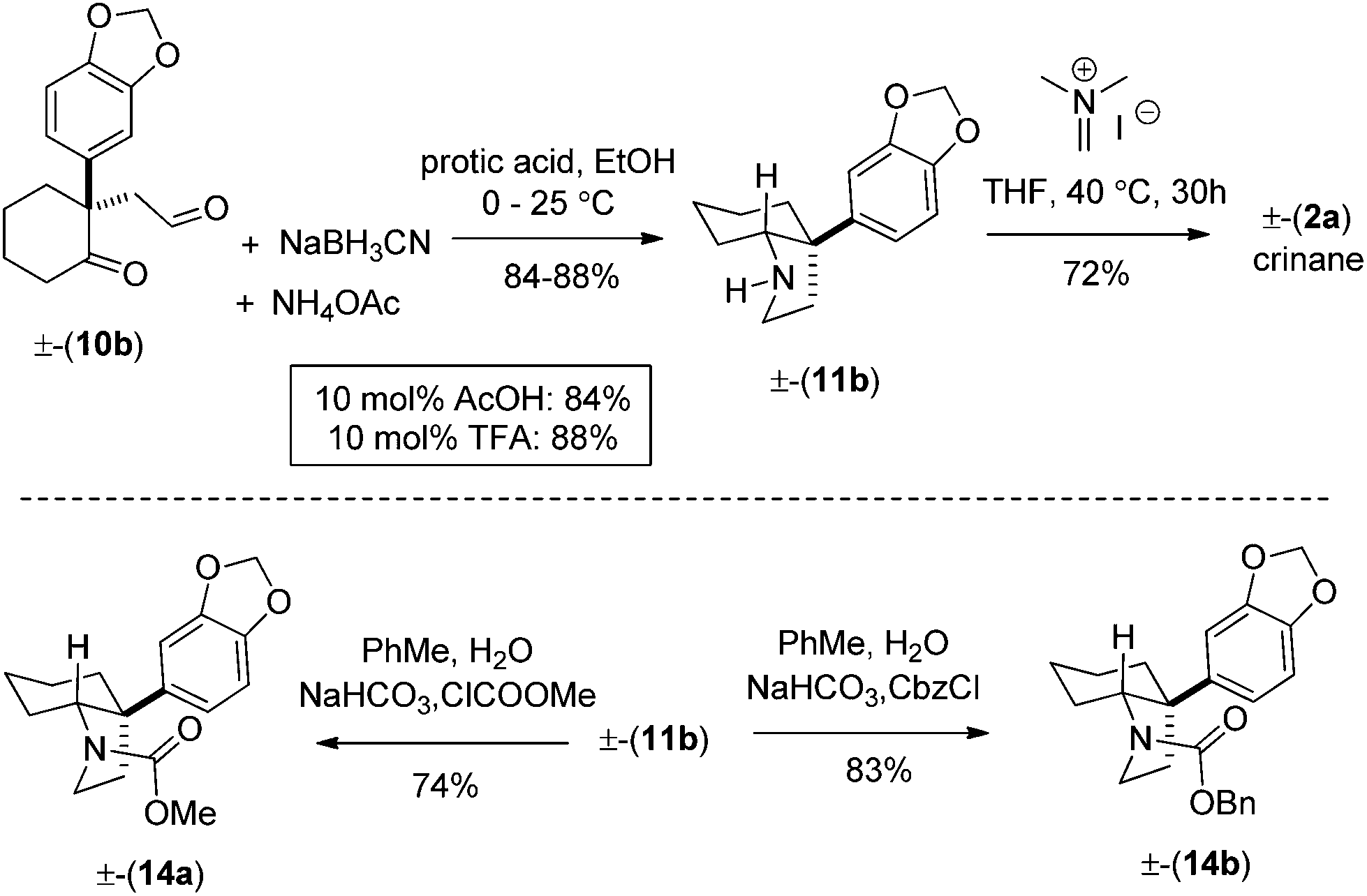 | ||
| Scheme 7 Total synthesis of (±)-crinane (2a). | ||
Conclusions
In conclusion, total synthesis of the Amaryllidaceae alkaloids mesembrane (1a) and crinane (2a) has been demonstrated. The strategy features the Eschenmoser–Claisen rearrangement as the key step to install an all carbon quaternary stereocenter. As allylic alcohols of the type 5a, b could easily be accessed in an enantioenriched form either using resolution or employing CBS reduction,16 our strategy could be nicely adopted to an enantioselective version as well.Acknowledgements
A.B. thanks the DST, SERB, India, for a research grant through FAST-TRACK scheme (SB/FT/CS-54/2011). M.K.D. and S.D. thank the Council of Scientific and Industrial Research (CSIR), New Delhi, for predoctoral fellowships. We thank the Department of Chemistry, IISER Bhopal for the infrastructure.Notes and references
- (a) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (b) For numerous examples, see: K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis, Wiley-VCH, New York, 1st edn, 1996 Search PubMed; (c) K. C. Nicolaou and S. A. Snyder, Classics in Total Synthesis II, Wiley-VCH, Weinheim, 1st edn, 2003 Search PubMed. For reviews, see: (d) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef; (e) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS; (f) B. M. Trost and C. H. Jiang, Synthesis, 2006, 369 CrossRef CAS PubMed; (g) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473 CrossRef CAS.
- (a) P. M. Jeffs, in The Alkaloids, ed. R. G. A. Rodrigo, Academic Press, New York, 1981, vol. 19, pp. 1–80 Search PubMed; (b) S. F. Martin, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1987, vol. 30, pp. 251–376 Search PubMed; (c) P. M. Jeffs, in MTP International Review of Science, Alkaloids, Organic Chemistry, Series One, ed. D. H. Hey and K. F. Wiesner, Butterworth, London, 1973, vol. 9, pp. 273–318 Search PubMed.
- L.-Z. Lin, S.-F. Hu, H.-B. Chai, T. Pengsuparp, J. M. Pezzuto, G. A. Cordell and N. Ruangrungsi, Phytochemistry, 1995, 40, 1295 CrossRef CAS.
- For reviews on Amaryllidaceae alkaloids, see: (a) Z. Jin, Nat. Prod. Rep., 2009, 26, 363 RSC and references cited therein; (b) J. R. Lewis, Nat. Prod. Rep., 2002, 19, 223 RSC; (c) Z. Jin, Z. Li and R. Huang, Nat. Prod. Rep., 2002, 19, 454 RSC.
- (a) R. Lebeuf, F. Robert, K. Schenk and Y. Landais, Org. Lett., 2006, 8, 4755 CrossRef CAS PubMed; (b) F.-M. Zhang, Y.-Q. Tu, J.-D. Liu, X.-H. Fan, L. Shi, X.-D. Hu, S.-H. Wang and Y.-Q. Zhang, Tetrahedron, 2006, 62, 9446 CrossRef CAS PubMed; (c) W. H. Pearson and F. E. Lovering, J. Org. Chem., 1998, 63, 3607 CrossRef CAS; (d) S. F. Martin and C. L. Campbell, J. Org. Chem., 1988, 53, 3184 CrossRef CAS; (e) L. E. Overman and L. T. Mendelson, J. Am. Chem. Soc., 1981, 103, 5579 CrossRef CAS.
- J. McNulty, J. J. Nair, C. Codina, J. Bastida, S. Pandey, J. Gerasimoff and C. Griffin, Phytochemistry, 2007, 68, 1068 CrossRef CAS PubMed.
- P. W. Jeffs, H. F. Campbell, D. S. Farrier, G. Ganguli, N. H. Martin and G. Molina, Phytochemistry, 1974, 13, 933 CrossRef CAS.
- Approaches to cis-3a-aryloctahydroindole alkaloids, see: (a) (±)-Martidine (1c): G. Pandey, N. R. Gupta and T. M. Pimpalpalle, Org. Lett., 2009, 11, 2547 CrossRef CAS PubMed; (b) (−)-Mesembranol (ent-1b): N. Chida, K. Sugihara, S. Amano and S. Ogawa, J. Chem. Soc., Perkin Trans. 1, 1997, 275 RSC; (c) M. Asaoka, N. Fuji and H. Takei, Chem. Lett., 1988, 17, 1655 CrossRef; (d) J. J. Nieuwenhius, H. F. Strauss and A. Wiechers, J. Chem. Soc., Perkin Trans. 1, 1981, 284 RSC; (e) H. F. Strauss and A. Wiechers, Tetrahedron Lett., 1979, 20, 4495 CrossRef; (f) K. Psotta and A. Wiechers, Tetrahedron, 1979, 35, 255 CrossRef CAS; (g) H. F. Strauss and A. Wiechers, Tetrahedron, 1978, 34, 127 CrossRef CAS; (h) G. Schwenker and G. Metz, Arch. Pharm., 1968, 301, 592 CrossRef CAS.
- For total synthesis of crinane (2a), see: (a) T. Kano, Y. Hayashi and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 7134 CrossRef CAS PubMed; (b) A. Padwa, M. A. Brodney, M. Dimitroff, B. Liu and T. H. Wu, J. Org. Chem., 2001, 66, 3119 CrossRef CAS PubMed; (c) J. M. Schkeryantz and W. H. Pearson, Tetrahedron, 1996, 52, 3107 CrossRef CAS; (d) G. E. Keck and R. R. Webb, J. Am. Chem. Soc., 1981, 103, 3173 CrossRef CAS; (e) G. E. Keck and R. R. Webb II, J. Org. Chem., 1982, 47, 1302 CrossRef CAS. For total synthesis of mesembrane (1a), see: (f) D. F. Taber and T. D. Neubert, J. Org. Chem., 2001, 66, 143 CrossRef CAS; (g) J. H. Rigby and W. Dong, Org. Lett., 2000, 2, 1673 CrossRef CAS PubMed; (h) K. Ogasawara and O. Tamada, Tetrahedron Lett., 1998, 39, 7747 CrossRef; (i) M. Mori, S. Kuroda, C. Zhang and Y. Sato, J. Org. Chem., 1997, 62, 3263 CrossRef CAS PubMed; (j) S. E. Denmark and L. R. Marcin, J. Org. Chem., 1997, 62, 1675 CrossRef CAS; (k) S. F. Martin, T. A. Puckette and J. A. Colapret, J. Org. Chem., 1979, 44, 3391 CrossRef CAS.
- Iodolactonization of Eschenmoser–Claisen products, see: (a) V. Bisai and R. Sarpong, Org. Lett., 2010, 12, 2551 CrossRef CAS PubMed; (b) L. Zhu, J. Luo and R. Hong, Org. Lett., 2014, 16, 2162 CrossRef CAS PubMed.
- (a) K. Hayashi, H. Tanimoto, H. Zhang, T. Morimoto, Y. Nishiyama and K. Kakiuchi, Org. Lett., 2012, 14, 5728 CrossRef CAS PubMed; (b) M. Asaoka, N. Fuji and H. Takei, Chem. Lett., 1988, 17, 1655 CrossRef; (c) V. H. Bruderer and K. Bernauer, Helv. Chim. Acta., 1983, 66, 570 CrossRef; (d) A. E. Wick, D. Felix, K. Steen and A. Eschenmoser, Helv. Chim. Acta, 1964, 47, 2425 CrossRef CAS; (e) For a review, see: A. M. M. Castro, Chem. Rev., 2004, 104, 2939 CrossRef CAS PubMed.
- (a) G. Stork and R. L. Danheiser, J. Org. Chem., 1973, 38, 1775 CrossRef CAS. For recent examples, see: (b) A. Bisai, S. P. West and R. Sarpong, J. Am. Chem. Soc., 2008, 130, 7222 CrossRef CAS PubMed; (c) N. B. Bennett, A. Y. Hong, A. M. Harned and B. M. Stoltz, Org. Biomol. Chem., 2012, 10, 56 RSC; (d) B. N. Kakde, S. Bhunia and A. Bisai, Tetrahedron Lett., 2013, 54, 1436 CrossRef CAS PubMed.
- (a) J. L. Luche, J. Am. Chem. Soc., 1978, 100, 2226 CrossRef CAS; (b) G. A. Molander, Chem. Rev., 1992, 92, 29 CrossRef CAS; (c) S. P. West, A. Bisai, A. D. Lim, R. R. Narayan and R. Sarpong, J. Am. Chem. Soc., 2009, 131, 11187 CrossRef CAS PubMed.
- (a) K. Omura and D. Swern, Tetrahedron, 1978, 36, 1651 CrossRef; (b) G. Tojo and M. I. Fernandez, Oxidation of Alcohols to Aldehyde and Ketones: A Guide to Current Common Practice, Springer, New York, 2006 Search PubMed.
- For a Bischler–Napieralski reaction of carbamate for a synthesis of trans-dihydronarciclasine, see: N. T. Tam and C.-G. Cho, Org. Lett., 2008, 10, 601 CrossRef CAS PubMed.
- (a) E. J. Corey, R. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 7925 CrossRef CAS; (b) E. J. Corey and J. O. Link, J. Am. Chem. Soc., 1992, 114, 1906 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra. See DOI: 10.1039/c5ob00183h |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |