
Design, synthesis, and kinetic analysis of potent protein N-terminal methyltransferase 1 inhibitors†
Gang
Zhang
,
Stacie Lynn
Richardson
,
Yunfei
Mao
and
Rong
Huang
*
Department of Medicinal Chemistry, and the Institute of Structural Biology & Drug Discovery, Virginia Commonwealth University, Richmond, VA, USA. E-mail: rhuang@vcu.edu
First published on 17th February 2015
Abstract
The protein N-terminal methyltransferase 1 (NTMT1) methylates the α-N-terminal amines of proteins. NTMT1 is upregulated in a variety of cancers and knockdown of NTMT1 results in cell mitotic defects. Therefore, NTMT1 inhibitors could be potential anticancer therapeutics. This study describes the design and synthesis of the first inhibitor targeting NTMT1. A novel bisubstrate analogue (NAM-TZ-SPKRIA) was shown to be a potent inhibitor (Ki = 0.20 μM) for NTMT1 and was selective versus protein lysine methyltransferase G9a and arginine methyltransferase 1. NAM-TZ-SPKRIA was found to exhibit a competitive inhibition pattern for both substrates, and mass spectrometry experiments revealed that the inhibitor substantially suppressed the methylation progression. Our results demonstrate the feasibility of using a triazole group to link an S-adenosyl-L-methionine analog with a peptide substrate to construct bisubstrate analogues as NTMT1 potent and selective inhibitors. This study lays a foundation to further discover small molecule NTMT1 inhibitors to interrogate its biological functions, and suggests a general strategy for the development of selective protein methyltransferase inhibitors.
Protein methylation is an important epigenetic modification that regulates celluar signal pathways. Protein methyltransferases utilize S-adenosyl-L-methionine (SAM) as the methyl donor and catalyze the post-translational protein methylation. Dysregulation of the methyltransferases leads to diverse diseases including cancer, inflammation, and cardiovascular diseases.1–3 Hence, specific inhibitors that modulate protein methylation are valuable probes to investigate methylation-mediated biological processes, and even act as therapeutic agents.4 While the importance of lysine and arginine methylation have received significant attention in the past decades, a growing number of studies has reported the important roles of α-N-terminal methylation.5–8 α-N-terminal methylation is essential to stabilize the interactions between regulator of chromosome condensation 1 (RCC1) and chromatin during mitosis, and to localize and enhance the interaction of centromere proteins with centromeric chromatin.6–9 N-terminal methylation of damaged DNA-binding protein 2 facilitates its nuclear location for DNA repair.5 Protein N-terminal methyltransferases (NTMTs) are the enzymes that are responsible for methylating the α-amines of proteins.10 NTMT1, one of the two known NTMTs, recognizes a canonical motif X–P–K (X: A, P, or S).11,12 NTMT1 is overexpressed in a variety of cancers including melanoma, gastrointestinal and brain cancers.11 It is important for cell proliferation, and knockdown of NTMT1 leads to mitotic defects and reduces nuclear localization.5,9,11 Therefore, NTMT1 specific inhibitors may have therapeutic potential. Currently, there is no chemical inhibitor available for NTMT1, which has impeded efforts to characterize its biological functions.
Protein methyltransferases have two binding pockets: a protein substrate binding pocket and a methyl donor SAM binding site. Our group has piloted studies to characterize the kinetic mechanism and found that NTMT1 catalysis involves a ternary complex formation.13 On the basis of this mechanism, we focused on designing and synthesizing bisubstrate analogues that covalently link a SAM analogue with a peptide substrate moiety in order to develop potent and specific inhibitors for NTMT1. The rationale is that bisubstrate analogues have the potential to afford a more potent and selective pharmacologic agent by combining the free binding energies of the individual interactions without suffering the entropic loss.14,15 There have been precedent attempts to generate selective bisubstrate inhibitors of protein arginine methyltransferases (PRMTs). Ward et al. attached guanidine functionality to the N-adenosyl-L-methionine (NAM) via a variable linker to yield potent PRMT1 inhibitors with IC50s of 3–6 μM.16 Although those compounds demonstrated around 20-fold selectivity for PRMT1 over protein lysine methyltransferase SET7, the inhibitory activity was far from ideal. It may be due to the fact that these prototype PRMT1 bisubstrate inhibitors only contain a guanidine group instead of a peptide substrate recognition moiety. Martin et al. recently reported that adenosine–guanidine conjugates containing three-carbon spacers showed micromolar to submicromolar inhibitory activities to PRMT1, 4, and 6.17 Thompson et al. reported the in situ synthesis of a bisubstrate analogue inhibitor of PRMT1 which linked link NAM with an histone 4 peptide through an ethylene group to yield a PRMT1 bisubstrate inhibitor with an IC50 of 350 μM.18 However, there has been no chemical synthesis available to link a SAM analogue with a peptide substrate portion to prepare bisubstrate analogues for protein methyltransferases to test their inhibitory abilities.
Here, we report the design, synthesis, and kinetic characterization of the first NTMT1 inhibitor that potently and specifically targets NTMT1. A novel bisubstrate analogue (NAM-TZ-SPKRIA) was shown to be a potent inhibitor for NTMT1 with an IC50 of 0.81 ± 0.13 μM. This first NTMT1 inhibitor was more than 60-fold selective versus other representative protein methyltransferases such as lysine methyltransferase G9a and arginine methyltransferase 1. NAM-TZ-SPKRIA was found to exhibit a competitive inhibition pattern for both the peptide substrate and SAM, and mass spectrometry experiments revealed that the inhibitor substantially suppressed the methylation progression. This study is significant because it not only generates the first potent and selective inhibitor for NTMT1, but also provides a new and simple method to synthesize SAM-peptide conjugates that can be leveraged to develop bisubstrate inhibitors for any SAM-utilizing protein methyltransferases.
We focused on designing bisubstrate analogues that covalently link a SAM analogue with a peptide substrate moiety via a triazole linker. Since the sulfonium center of SAM is very reactive, the sulfur was replaced with a nitrogen to yield the NAM as a stable analogue of SAM.19 The sequence of the peptide part is derived from the N-terminus of RCC1. For initial efforts, we incorporated a hexapeptide (SPKRIA) into the bisubstrate analogue in order to retain the substrate recognition (Fig. 1A). There is no crystal structure available for the NTMT1-peptide complex. Docking the SPKRIA to the crystal structure of NTMT1 with SAH (PDB ID 2EX4) suggested that the distance between the structure α amino group and the S atom of the SAM is 3.6 Å.11 Considering the distance and size, we hypothesized that a triazole linker could be used to couple both substrate portions to construct a bisubstrate analogue. To support our hypothesis, we carried out docking studies using Gold 5.2 (Table S1†). Our results suggested NAM-TZ-SPKRIA can fit into the NTMT1 binding sites and the triazole linker can be accommdated (Fig. 1B and C). The NAM part superimposes well with the SAH and retains the similar interactions with NTMT1. The Pro, Arg, and Ala of the peptide part exhibit interactions with Asn169, Tyr216, and Asp179 of NTMT1, and side chains of Lys and Arg interact with Gly32 and Glu214. Hence, the clicked NAM-peptide conjugate was designed and synthesized as the NTMT1 bisubstrate inhibitor.
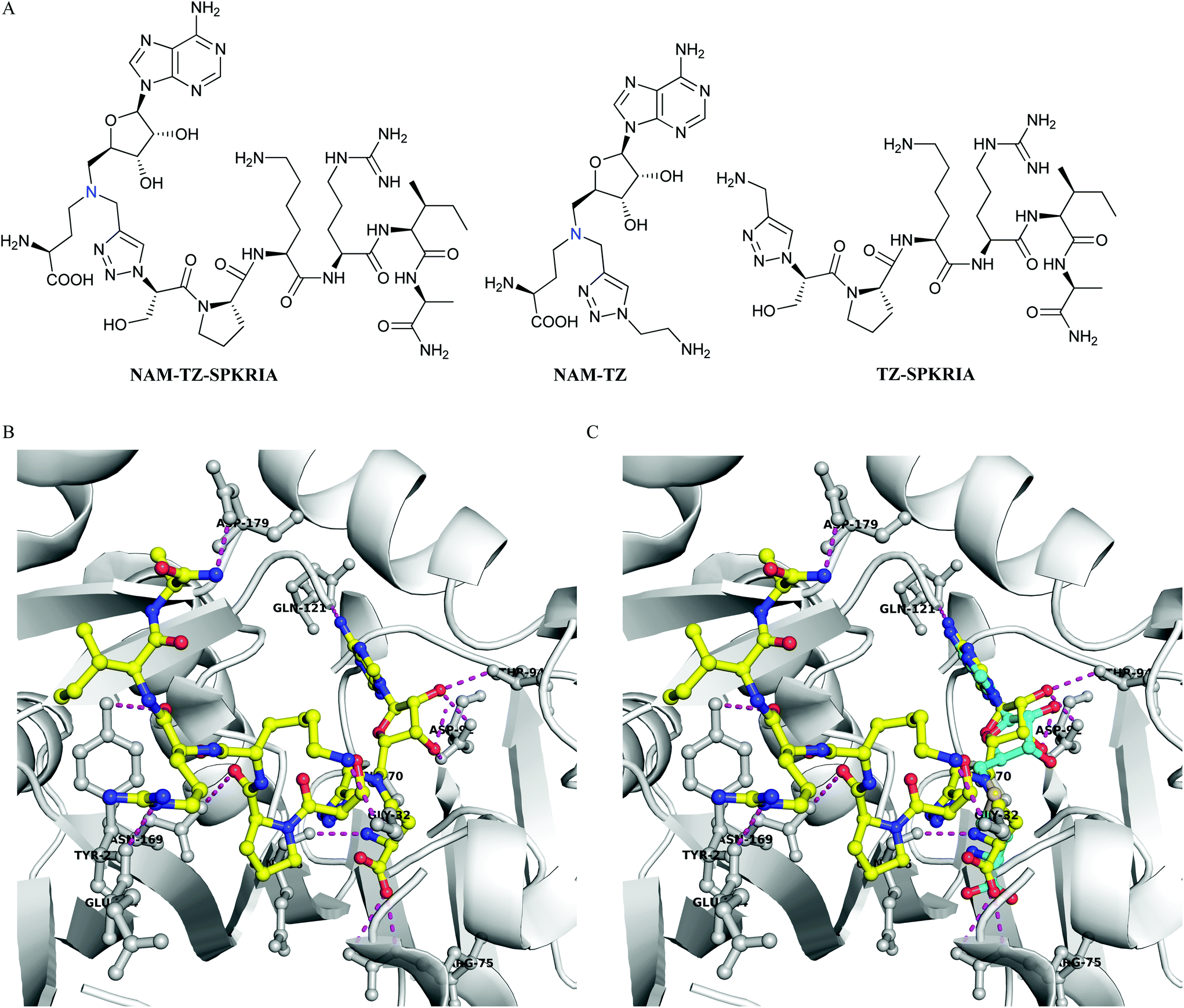 | ||
| Fig. 1 Inhibitor design. (A) Structures of NAM-TZ-SPKRIA, NAM-TZ, and TZ-SPKRIA. Nitrogen atom (blue) replaces the sulfur atom of SAH. (B) Docking study of NAM-TZ-SPKRIA (yellow) to crystal structure of NTMT1 complexed with SAH (PDB: 2EX4). (C) Superimposed structure of NAM-TZ-SPKRIA (yellow) with SAH (cyan) in the complex. Purple line indicates the hydrogen bonding between NAM-TZ-SPKRIA and NTMT1. | ||
The synthesis of the bisubstrate analogue is illustrated in Scheme 1. Briefly, the synthesis started from the commercially available adenosine, of which the 2′- and 3′-hydroxyl groups were selectively protected by the isopropylidene group to quantitatively yield 1.16,20 Compound 1 was converted to the azide in the presence of diphenylphosphoryl azide (dppa) and sodium azide, followed by hydrogenation to provide 2.21 Subsequent reductive amination with Boc-protected aspartic aldehyde provided 3 and treatment with propargyl bromide produced 4.16,22,23 The N-terminal free amino group of SPKRIA peptide was converted to the azide group on resin by treated with a mixture of triflyl azide, potassium carbonate and copper(II) sulfate pentahydrate.24 The azide was then reacted with 4 to form the NAM-peptide conjugate through a triazole linker via click chemistry on resin, followed by a standard peptide cleavage condition to provide the bisubstrate analogue NAM-TZ-SPKRIA.24
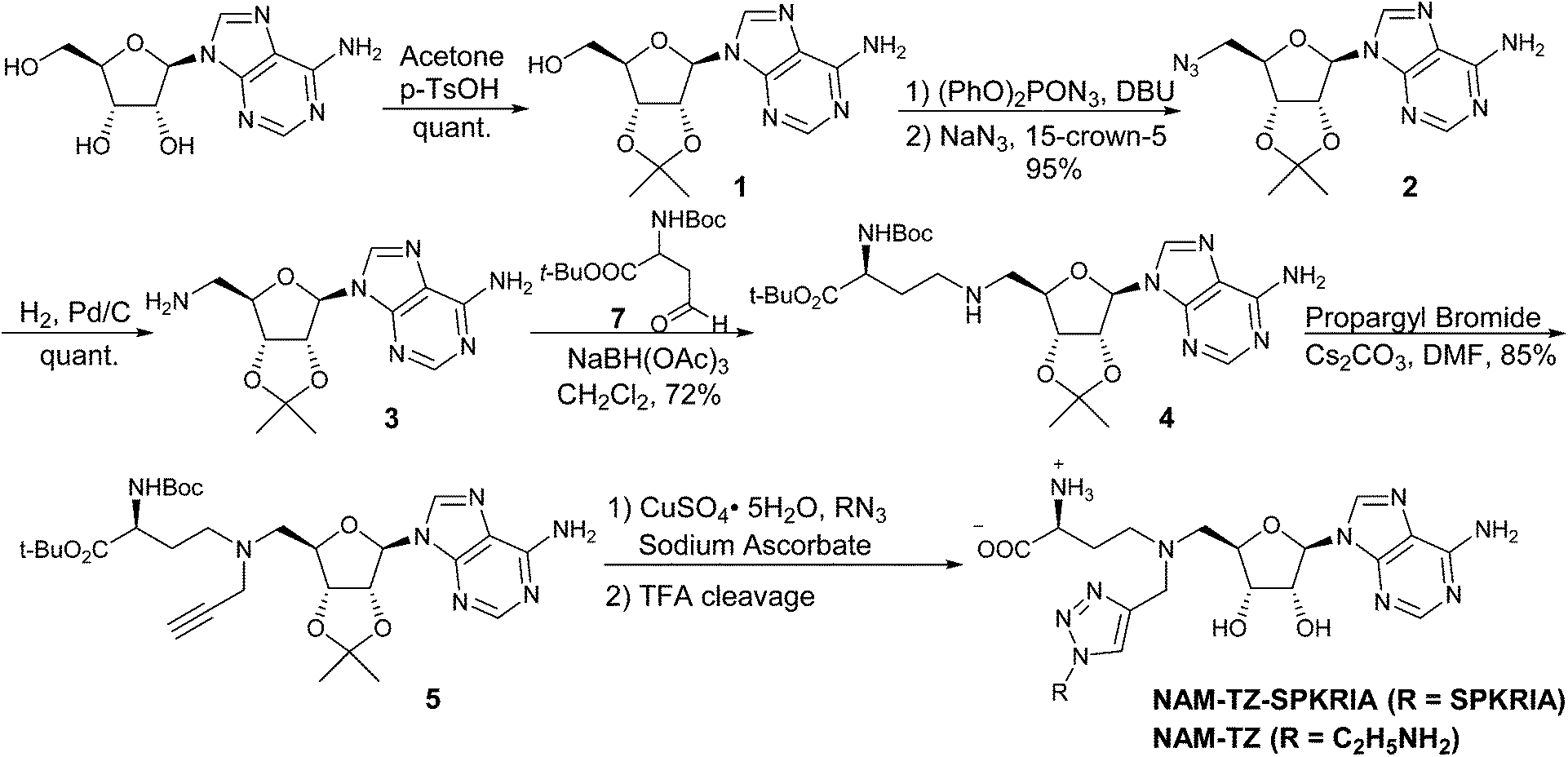 | ||
| Scheme 1 Synthesis of Bisubstrate Analog NAM-TZ-SPKRIA. | ||
To assess how both substrate portions contribute to the activities of the bisubstrate analogue, we also designed and prepared two control compounds NAM-TZ and TZ-SPKRIA (Fig. 1). NAM-TZ only contains an NAM moiety and a triazole linker, while TZ-SPKRIA contains a triazole linker and the peptide portion. TZ-SPKRIA was synthesized in a similar manner as illustrated in Scheme 1 by reacting azidohexapeptide with propargylamine.25 The synthesis of NAM-TZ started by converting 2-bromoethylamine to 2-azidoethanamine in the presence of sodium azide, followed by reacting with 4via click chemistry and acid deprotection.26
We first evaluated the ability of NAM-TZ-SPKRIA, NAM-TZ, and TZ-SPKRIA to inhibit NTMT1 through a fluorescence assay.13 The results of IC50 values revealed that NAM-TZ-SPKRIA is a highly potent NTMT1 inhibitor (IC50 = 0.81 ± 0.13 μM), while both NAM-TZ and TZ-SPKRIA did not show any detectable inhibition at 100 μM (Fig. 2A). This corroborated our hypothesis that linking both substrate portions of NTMT1 through a triazole linker can provide a potent inhibitor for NTMT1. To our knowledge, NAM-TZ-SPKRIA is the first inhibitor for NTMT1.
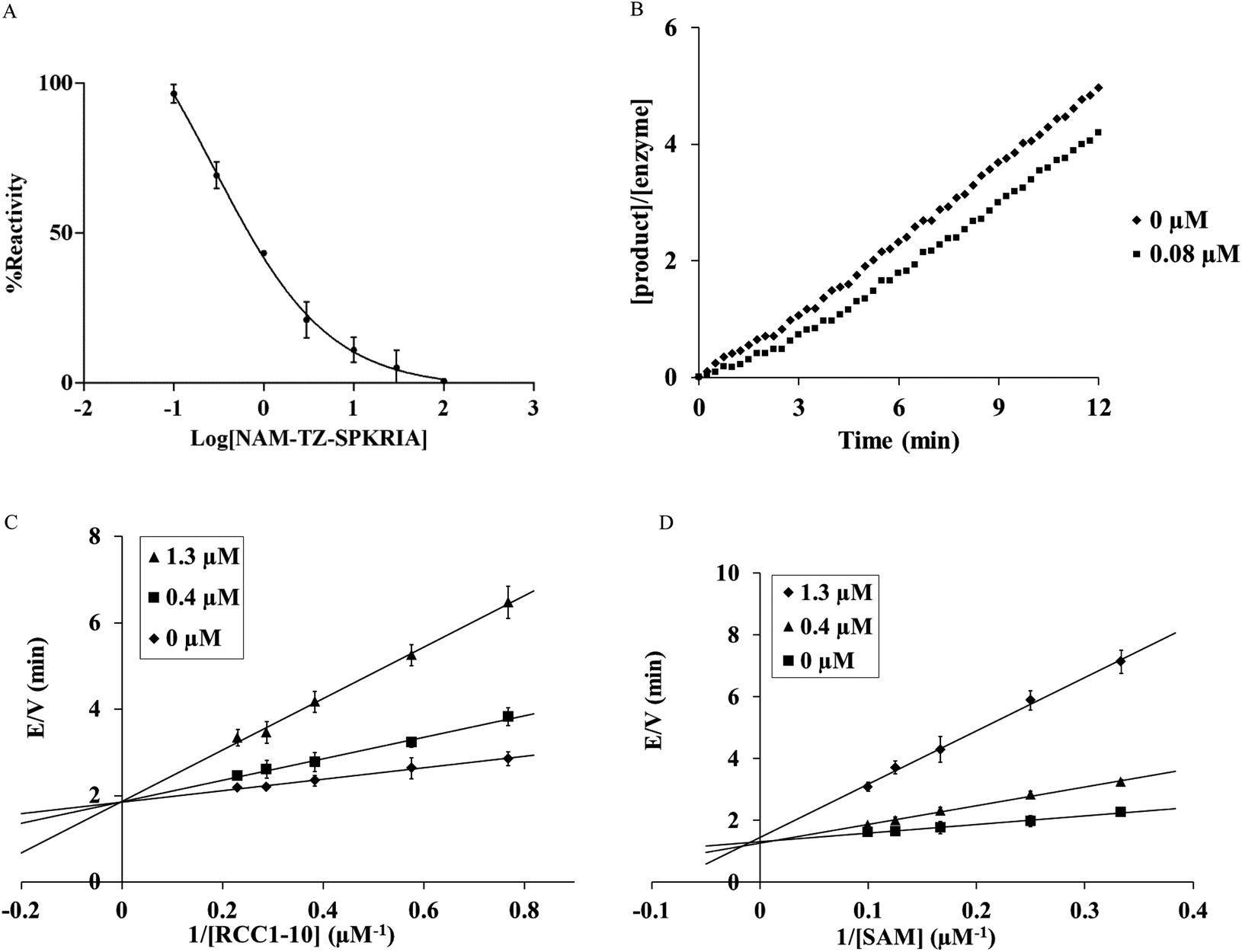 | ||
| Fig. 2 Kinetic analysis of the inhibition of NTMT1 by bisubstrate analog NAM-TZ-SPKRIA. (A) Steady-state inhibition of NTMT1 by NAM-TZ-SPKRIA ranging from 0 to 100 μM. (B) Recovery activity of NTMT1 after a rapid dilution of the enzyme-inhibitor (NAM-TZ-SPKRIA) complex. (C) E/V versus 1/RCC1 in the presence of varying concentration of the inhibitor NAM-TZ-SPKRIA. (D) E/V versus 1/SAM in the presence of varying concentration of the inhibitor NAM-TZ-SPKRIA. | ||
Given that all protein methyltransferases share a SAM binding site, we examined the selectivity of NAM-TZ-SPKRIA for NTMT1 over other protein methyltransferases. We assessed its ability to inhibit two representative protein methyltransferases: protein lysine methyltransferase G9a and protein arginine methyltransferase 1 (PRMT1) in a fluorescence assay.27,28 Our results exhibited that NAM-TZ-SPKRIA showed less than 50% inhibitory effect on G9a and less than 15% inhibition effect on PRMT1 at 50 μM, respectively. These data revealed the selectivity of NAM-TZ-SPKRIA for NTMT1 is more than 60-fold over G9a and PRMT1. Since NAM-TZ-SPKRIA showed the highest potency and selectivity, we focused on this bisubstrate analogue for further studies. To gain further insight into the nature of the inhibition of the NTMT1 by NAM-TZ-SPKRIA, we performed a kinetic analysis designed to characterize the inhibitory properties of this bisubstrate analogue. The mixture with 20 μM NTMT1 and 8 μM NAM-TZ-SPKRIA was preincubated for 30 min and diluted 100-fold into assay buffer containing 5 μM RCC1-10.15,29 The recovery of activity was monitored for 12 min, and the SAH product was quantified as described previously. Product formation was linear with no apparent lag in the recovery of NTMT1 activity at 0.08 μM, which is typical for a reversible inhibitor (Fig. 2B).
We then characterized the mechanism of inhibition of NAM-TZ-SPKRIA for NTMT1 by determining the inhibition pattern for SAM in the absence and presence of various concentrations of NAM-TZ-SPKRIA at a fixed concentration of RCC1-10. The results of this analysis revealed that NAM-TZ-SPKRIA acts as a competitive inhibitor when the concentration of SAM is varied from 3 to 10 μM and RCC1-10 substrate peptide is at a fixed concentration at 3 μM. We also determined the inhibition pattern of NAM-TZ-SPKRIA for RCC1-10 substrate peptide in the absence and presence of various concentrations of NAM-TZ-SPKRIA at a fixed concentration of SAM. The results indicated that NAM-TZ-SPKRIA also acts as a competitive inhibitor when the concentration of RCC1-10 is varied from 1.3 to 4.3 μM and SAM is at a fixed concentration at 4 μM. These inhibition patterns suggest that NAM-TZ-SPKRIA competes with both SAM and RCC1-10 to bind NTMT1. As bisubstrate analogues are designed to retain binding to both substrate binding sites, they are competitive inhibitors to either substrate. Hence, competitive inhibition patterns of NAM-TZ-SPKRIA to both substrates further validate that NAM-TZ-SPKRIA is a bisubstrate analogue, as well as support our hypothesis of covalently attaching NAM and peptide portions through a triazole linker to build bisubstrate analogues for NTMT1.
To confirm the NTMT1 inhibition fluorescence assay results obtained with NAM-TZ-SPKRIA, we applied MALDI mass spectrometry to directly and quantitatively assess the effects on N-α-amine methylation.30 The IC50 value for NAM-TZ-SPKRIA was determined to be 1.3 ± 0.1 μM, which corroborates the result obtained from fluorescence assay (Fig. S1†). We also chose to use NAM-TZ-SPKRIA at concentrations of its IC50 value and a 5-fold of IC50 to evaluate how it affects the progression of N-α-amine methylation. Triplicate samples of RCC1-10 peptide along with NAM-TZ-SPKRIA were subjected to NTMT1 methylation assays. Following these assays, samples were analyzed at 20 min to monitor the methylation progression (Fig. 3). At both concentrations, dimethylation and trimethylation of RCC1-10 were completely abolished. Monomethylated RCC1 was substantially reduced to 10% at 5.30 μM.
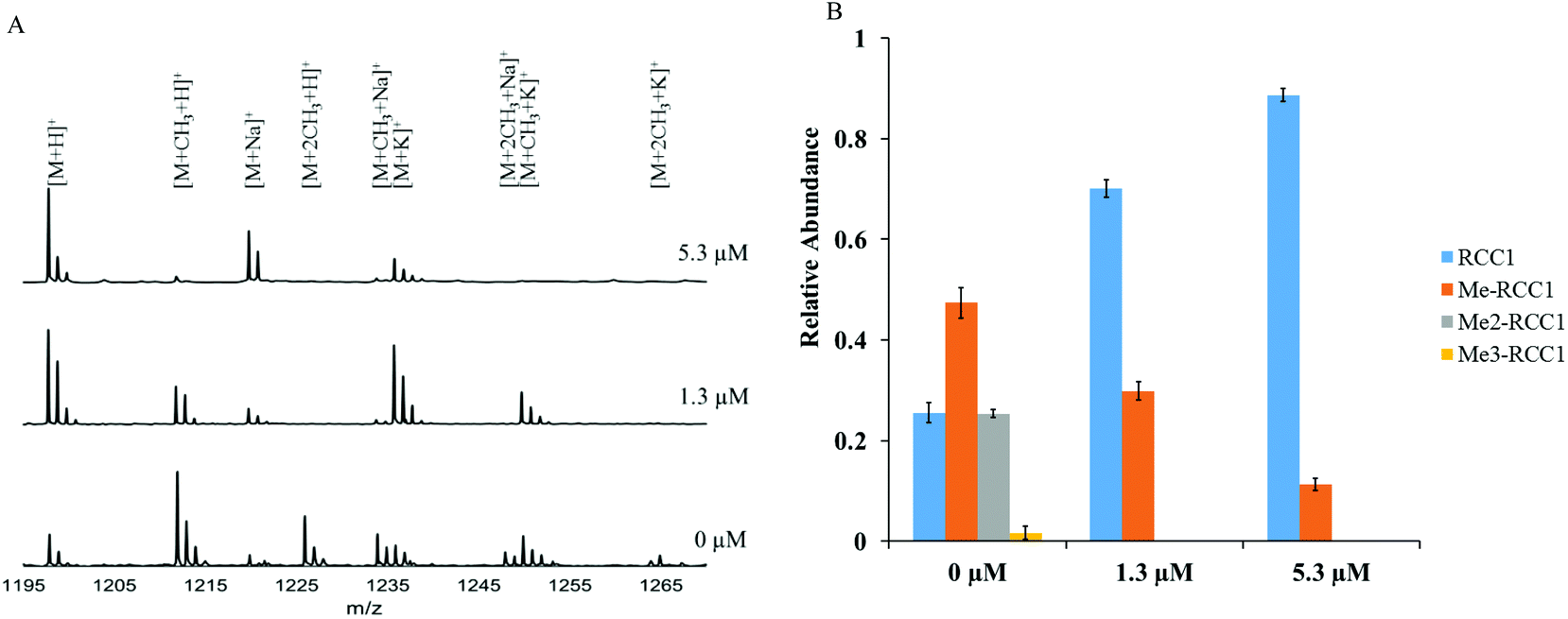 | ||
| Fig. 3 Inhibitory effects on methylation states of RCC1-10 with NAM-TZ-SPKRIA. (A) Stacked plot of MALDI-MS of RCC1-10 methylation progression as a result of NTMT1 inhibition by NAM-TZ-SPKRIA at 20 minutes. (B) Quantification of methylation states of RCC1-10 as a result of NTMT1 inhibition by NAM-TZ-SPKRIA at 20 min. | ||
In summary, NAM-TZ-SPKRIA (Ki = 0.20 μM) represents the first NTMT1 inhibitor that selectively targets NTMT1. The availability of NTMT1 inhibitors is critical to advance our understanding of the function of NTMT1. More importantly, we demonstrated that a triazole group can be applied to couple a SAM analogue and a peptide to build bisubstrate analogues for protein methyltransferase. As most protein methyltransferases catalysis involves a ternary complex, it seems plausible to predict that the bisubstrate analogue could be mimicked by compounds that contain a SAM moiety attached to a less specific peptide portion to provide a selective and potent inhibitor for any SAM-containing protein methyltransferases. While we prepared this manuscript, Martin et al. reported a PRMT1 inhibitor that using a triazole group to attach two small molecules to bind two binding sites,17 which further support our hypothesis of using NAM-peptide conjugates to design protein methyltransferases inhibitors. Therefore, herein we have provided a convenient synthetic route to construct a potent inhibitor for NTMT1 through a triazole linker, which can be adapted to prepare any NAM-peptide conjugates. Considering the emerging importance of protein methylation in epigenetics, our synthetic strategy offers a general approach that has potential to generate specific and potent probes for each methyltransferase.
Acknowledgements
We thank Dr Raymond Trievel for the SAHH plasmid, and Dr Yujun Zheng for the PRMT1 plasmid. We appreciate Dr Darrel L. Peterson for purifying NTMT1 and G9a. This work was supported by the VCU Presidential Research Quest Fund (R.H.) and VCU Massey Cancer Center Pilot Project (R.H.).Notes and references
- R. A. Copeland, E. J. Olhava and M. P. Scott, Curr. Opin. Chem. Biol., 2010, 14, 505 CrossRef CAS PubMed.
- P. N. Alpoim, L. P. Sousa, A. P. Mota, D. R. Rios and L. M. Dusse, Clin. Chim. Acta, 2014, 440C, 36 Search PubMed.
- N. Kang, P. Chen, Y. Chen, H. Zeng, X. He and Y. Zhu, Int. Immunopharmacol., 2014, 24, 95 CrossRef PubMed.
- T. K. Kelly, D. D. De Carvalho and P. A. Jones, Nat. Biotechnol., 2010, 28, 1069 CrossRef CAS PubMed.
- Q. Cai, L. Fu, Z. Wang, N. Gan, X. Dai and Y. Wang, J. Biol. Chem., 2014, 289, 16046 CrossRef CAS PubMed.
- A. O. Bailey, T. Panchenko, K. M. Sathyan, J. J. Petkowski, P. J. Pai, D. L. Bai, D. H. Russell, I. G. Macara, J. Shabanowitz, D. F. Hunt, B. E. Black and D. R. Foltz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11827 CrossRef CAS PubMed.
- X. Dai, K. Otake, C. You, Q. Cai, Z. Wang, H. Masumoto and Y. Wang, J. Proteome Res., 2013, 12, 4167 CrossRef CAS PubMed.
- E. Hitakomate, F. E. Hood, H. S. Sanderson and P. R. Clarke, BMC Cell Biol., 2010, 11, 43 CrossRef PubMed.
- T. Chen, T. L. Muratore, C. E. Schaner-Tooley, J. Shabanowitz, D. F. Hunt and I. G. Macara, Nat. Cell Biol., 2007, 9, 596 CrossRef CAS PubMed.
- A. Villar-Garea, I. Forne, I. Vetter, E. Kremmer, A. Thomae and A. Imhof, Nucleic Acids Res., 2012, 40, 1536 CrossRef CAS PubMed.
- C. E. Tooley, J. J. Petkowski, T. L. Muratore-Schroeder, J. L. Balsbaugh, J. Shabanowitz, M. Sabat, W. Minor, D. F. Hunt and I. G. Macara, Nature, 2010, 466, 1125 CrossRef PubMed.
- A. Stock, S. Clarke, C. Clarke and J. Stock, FEBS Lett., 1987, 220, 8 CrossRef CAS.
- S. L. Richardson, Y. Mao, G. Zhang, P. Hanjra, D. L. Peterson and R. Huang, J. Biol. Chem., 2015 Search PubMed , submitted.
- R. Huang, I. Martinez-Ferrando and P. A. Cole, Nat. Struct. Mol. Biol., 2010, 17, 646 CAS.
- K. Parang, J. H. Till, A. J. Ablooglu, R. A. Kohanski, S. R. Hubbard and P. A. Cole, Nat. Struct. Biol., 2001, 8, 37 CrossRef CAS PubMed.
- J. Dowden, W. Hong, R. V. Parry, R. A. Pike and S. G. Ward, Bioorg. Med. Chem. Lett., 2010, 20, 2103 CrossRef CAS PubMed.
- M. van Haren, L. Q. van Ufford, E. E. Moret and N. I. Martin, Org. Biomol. Chem., 2015, 13, 549 CAS.
- T. Osborne, R. L. Roska, S. R. Rajski and P. R. Thompson, J. Am. Chem. Soc., 2008, 130, 4574 CrossRef CAS PubMed.
- J. F. Couture, G. Hauk, M. J. Thompson, G. M. Blackburn and R. C. Trievel, J. Biol. Chem., 2006, 281, 19280 CrossRef CAS PubMed.
- A. Hampton, J. C. Fratantoni, P. M. Carroll and S. Wang, J. Am. Chem. Soc., 1965, 87, 5481 CrossRef CAS.
- F. Liu and D. J. Austin, J. Org. Chem., 2001, 66, 8643 CrossRef CAS.
- A. P. Townsend, S. Roth, H. E. Williams, E. Stylianou and N. R. Thomas, Org. Lett., 2009, 11, 2976 CrossRef CAS PubMed.
- C. Marian, R. Huang and R. F. Borch, Tetrahedron, 2011, 67, 10216 CrossRef CAS PubMed.
- J. T. Lundquist 4th and J. C. Pelletier, Org. Lett., 2001, 3, 781 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed.
- T. Mayer and M. E. Maier, Eur. J. Org. Chem., 2007, 4711 CrossRef CAS.
- F. Liu, X. Chen, A. Allali-Hassani, A. M. Quinn, T. J. Wigle, G. A. Wasney, A. Dong, G. Senisterra, I. Chau, A. Siarheyeva, J. L. Norris, D. B. Kireev, A. Jadhav, J. M. Herold, W. P. Janzen, C. H. Arrowsmith, S. V. Frye, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2010, 53, 5844 CrossRef CAS PubMed.
- Y. Feng, N. Xie, M. Jin, M. R. Stahley, J. T. Stivers and Y. G. Zheng, Biochemistry, 2011, 50, 7033 CrossRef CAS PubMed.
- C. M. Kim and P. A. Cole, J. Med. Chem., 2001, 44, 2479 CrossRef CAS PubMed.
- L. P. Blair, N. L. Avaritt, R. Huang, P. A. Cole, S. D. Taverna and A. J. Tackett, Epigenetics, 2011, 6, 490 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Organic synthesis, characterization, assay procedure, and docking study. See DOI: 10.1039/c5ob00120j |
| This journal is © The Royal Society of Chemistry 2015 |