
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
γ-(S)-Trifluoromethyl proline: evaluation as a structural substitute of proline for solid state 19F-NMR peptide studies†
Vladimir
Kubyshkin
ab,
Sergii
Afonin
c,
Sezgin
Kara
a,
Nediljko
Budisa
b,
Pavel K.
Mykhailiuk
*de and
Anne S.
Ulrich
*ac
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: anne.ulrich@kit.edu
bInstitute of Chemistry, Technical University of Berlin, Müller-Breslau-Str. 10, 10623 Berlin, Germany
cInstitute of Biological Interfaces (IBG-2), KIT, POB 3640, 76021 Karlsruhe, Germany
dFaculty of Chemistry, Taras Shevchenko National University of Kyiv, vul. Volodymyrska 62a, 01601 Kyiv, Ukraine. E-mail: pavel.mykhailiuk@gmail.com; pavel.mykhailiuk@mail.enamine.net
eEnamine Ltd., vyl. Chervonotkatska 78, 02660 Kyiv, Ukraine
First published on 13th February 2015
Abstract
γ-(4S)-Trifluoromethyl proline was synthesised according to a modified literature protocol with improved yield on a multigram scale. Conformational properties of the amide bond formed by the amino acid were characterised using N-acetyl methyl ester model. The amide populations (s-trans vs. s-cis) and thermodynamic parameters of the isomerization were found to be similar to the corresponding values for intact proline. Therefore, the γ-trifluoromethyl proline was suggested as a structurally low-disturbing proline substitution in peptides for their structural studies by 19F-NMR. Indeed, the exchange of native proline for γ-trifluoromethyl proline in the peptide antibiotic gramicidin S was shown to preserve the overall amphipathic peptide structure. The utility of the amino acid as a selective 19F-NMR label was demonstrated by observing the re-alignment of the labelled gramicidin S in oriented lipid bilayers.
Introduction
Proline (Pro) is the only amino acid with a secondary amine function among the canonical α-amino acids. Locally, in the polypeptide backbone Pro forms unique tertiary X-Pro amide bonds which endows a stable s-cis conformation (Fig. 1A).1 The Pro residue is conformationally restricted by the side chain to backbone cyclization, and is therefore the only canonical amino acid with the rigidly constrained phi angle. Due to these features, prolyl residues often play determining roles in the definition of the three-dimensional structures of proteins and limit the folding kinetics.2 Consequently, proline-rich regions in proteins are predominantly located within the solvent-exposed sections with enigmatic secondary structures such as intra-domain connections, loops, intrinsically disordered segments. In addition, prolines are ample in the folded domains which constitute the inter-molecular interfaces of the protein–protein and protein–drug recognition sites.3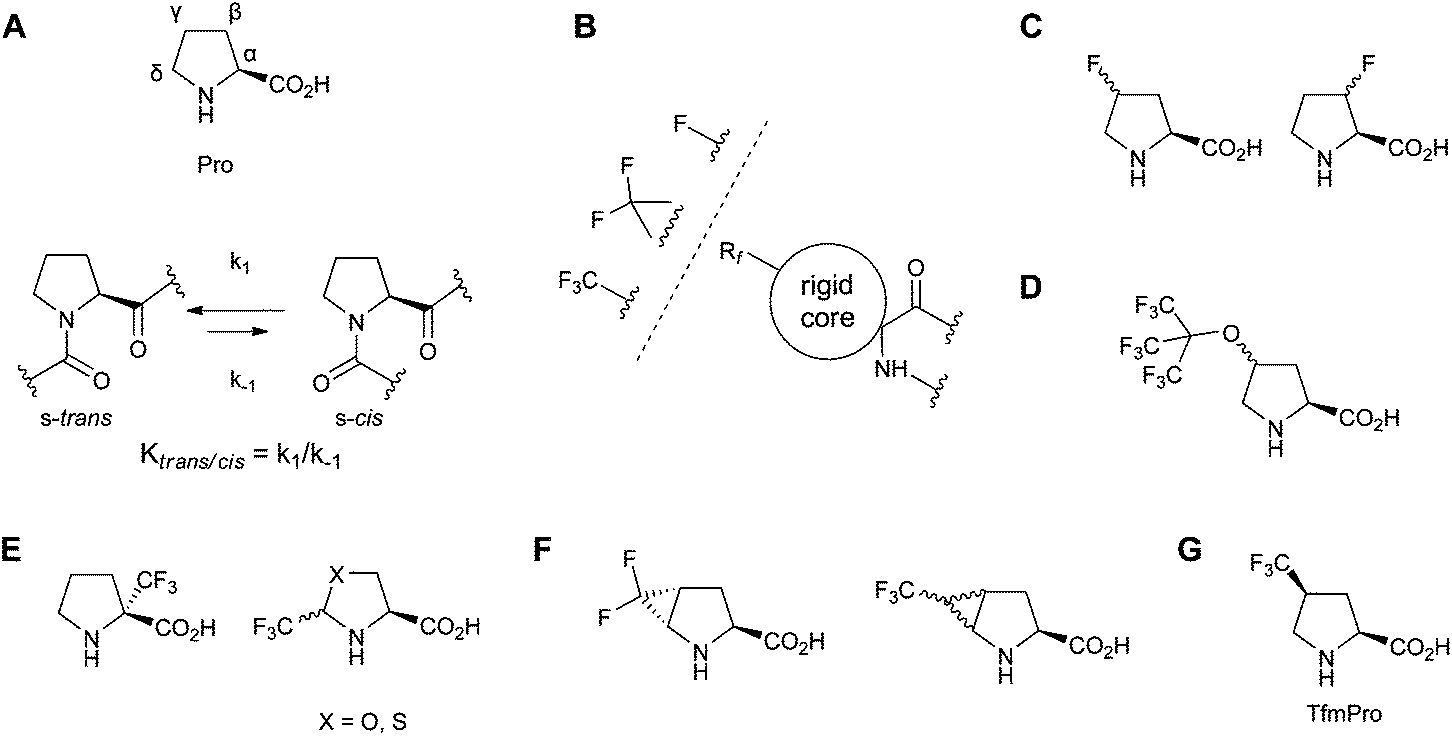 | ||
| Fig. 1 (A) Structure of proline (Pro) and the trans–cis equilibrium in a peptidyl-prolyl fragment; (B) attachment of a conformationally constrained 19F-NMR reporter to the polypeptide backbone; (C–G) known fluorine-substituted α-amino acids which could be considered as Pro-replacing 19F-NMR labels: (C) monofluorine-substituted γ- and β-fluoroprolines; (D) perfluoro-tert-butyl-γ-hydroxyprolines;22 (E) carriers of the CF3-group: α-CF3-proline and δ-CF3-pseudo and thiopseudoprolines; (F) methanoproline-based difluoro-trans-γ,δ-methanoproline and CF3-γ,δ-methanoprolines; (G) γ-CF3-(S)-proline (TfmPro) explored in this study. | ||
The absence of NH not only precludes participation in the secondary structure-stabilizing hydrogen bonds, but also makes Pro sites invisible for many routine NMR methods of the polypeptide structure analysis. Yet, the NMR visibility of Pro can be restored by selective isotope labelling. The labelling per se serves enhancing the NMR sensitivity, where among conventional isotope labels, 2H, 13C, 15N, and 19F, the latter is by far the most effective nucleus.4 However, introduction of the 19F isotope, an unnatural element in the proteinogenic amino acids, requires chemical modification of the side chain, i.e. a mutation. While for large proteins a single mutation does not necessarily interfere with the native fold, for small peptides such a modification may affect significant percentage of the sequence. For 19F-NMR labelling which aims determination of the structure of small peptides, in particular employing anisotropic NMR parameters (solid state NMR), the introduction of a fluorine-containing motif is further complicated. In an ideal label, not only should the physico-chemical nature of the side chain be preserved, but simultaneously a rigid molecular connectivity between the NMR-reporter group and the aminocarboxylate must be present.5,6 This challenge is solved in 19F-NMR by designing the residue type-specific substitutive amino acids. These have to be close steric surrogates, which possess conformationally rigid side chains alongside with the NMR-reporter groups - a single fluorine atom,7,8 a CF2-,9 or the CF3-group10,11 (Fig. 1B). In the case of the intrinsically constricted Pro side chain, design of such analogues is particularly difficult, because the residue steric size always increases, the side chain conformation, unique amide bond isomerization equilibrium and consequently the preferred near-proline backbone conformational angles are strongly influenced by the substituents in the pyrrolidine ring.12,13
There are several potential candidates worth considering for the proline substitutive 19F-labelling in polypeptides. For instance, β-14 and γ-mono-fluoroprolines,15–18 (Fig. 1C) that are among most explored proline modifications. The utility of these amino acids is toughened by their compatibility with the natural ribosomal protein expression machinery.19,20 However, mono-fluoroprolines are particularly known for the over-stabilization of their preferred ring puckers and both significantly deviate in their trans–cis amide bond equilibria from Pro.
Another beneficial possibility is a straight incorporation of the CF3-group. The latter carries three times more 19F spins per residue and allows utilization of the strong homonuclear F–F dipolar coupling. In the solid state 19F-NMR of peptides, the F–F coupling is a structural restraint which is superior to the chemical shift anisotropy.6,7,10 However, the trifluoromethylation not only impose significant steric demand and potentially modulate electronic configuration of the pyrrolidine ring, but is also known to render template structures becoming more lipophilic,21 which directly affects function of the membrane-active peptides, for instance. The steric demand and the increase in lipophilicity will be very large when multiple CF3-groups are present, e.g. like in the perfluoro-tert-butyl-γ-hydroxyproline (Fig. 1D) as was recently proposed for 19F-NMR labelling.22 Furthermore, in the solid state a strongly coupled homonuclear 9-spin system will produce NMR spectra with a very complex multiplicity, which might obscure the data analysis and cancel the initial sensitivity advantage. Therefore an isolated CF3-group should be preferred in the solid state 19F-NMR studies.
Synthetically, a sole CF3-group can be introduced into proline or the pseudoproline template in different positions. In particular, it has been described that α-CF3 proline23 and δ-CF3 oxazolidine-based pseudoprolines24 (Fig. 1E) could be incorporated in peptides. Still, in both Pro surrogates with the CF3-substituent adjoining the aminocarboxylate, conformational freedom within the adjacent backbone swerves the parent Pro structure. Besides, incorporation of these analogues into a peptide chain is still a challenge as the amino acid reactivity in the peptide synthesis is predictably lowered.25 Poor compatibility with the standard solid-phase peptide synthesis (SPPS) and sophisticated synthesis are the major utilization problem also for the recently described difluoro-26 and trifluoromethyl-γ,δ-methanoprolines11c (Fig. 1F). In contrast, simple γ-CF3-prolines (Fig. 1G) are chemically stable and carry the CF3-group in the most remote position relative to both amino acid functionalities and correspondingly from the future polypeptide backbone. Therefore they may be the most balanced candidates for an intact proline substitution.
The conformational impact of a large number of γ-substituents has been addressed within ample mutagenesis investigations. Curiously, such studies for a single CF3-group in the γ-position have been not described so far. We fill this knowledge gap and determine herein conformational and thermodynamic parameters of the γ-(S)-CF3-proline (TfmPro) amide bond in a model compound. Judging by these parameters, we demonstrate TfmPro being close to proline, and further test the amino acid utility as a structural NMR label, in particular in the solid state 19F-NMR study of the membrane-active peptide gramicidin S.
Results and discussion
Synthesis of TfmPro
The synthesis of TfmPro was described in several publications about a decade ago.27–30 Among these approaches we choose the simplest and the most economic (to our opinion) synthesis of the N-Boc derivative 1 reported by Qiu & Qing,29 which starts from hydroxyproline 2.Instead of using the benzyl ester which was obtained in the original study in a poor 45% yield we employed methyl-ester protection and got the protected hydroxyproline 3 in 96% yield (Scheme 1). In the following, hydroxyl group was converted to the keto-function (leading to 4) with 76% yield (7 gram) using chromium(VI) oxide–pyridine (Collins reagent). However, for the scaled up synthesis we altered to the Parikh–Doering oxidation and increased the yield of 4 to 90% (50 gram). Installation of the CF3-group and subsequent water abstraction worked well using the original protocols. This way we obtained 5 in a 94% yield. The compound 5 heated with thionyl chloride in pyridine for 20 min under reflux gave 20 gram of the alkene 6 in a satisfactory 60% yield. The second stereocenter was conventionally generated by hydrogenation of the double bond. Resulting TfmPro derivative 7 was obtained in 94% yield. The carboxyl-group was then deprotected by saponification giving 1 (86% yield, 9 gram). The overall yield of 1 was therefore 39%, almost twice higher than the 21% reported by Qiu & Qing.
 | ||
| Scheme 1 The synthesis of γ-(S)-trifluoromethylproline, as N-Boc (1), its derivative Ac-TfmPro-OMe (8) used for the conformational analysis, and the gramicidin S analogues used for solid state 19F-NMR. The X-ray crystal structure of 8 (carbon – blue, oxygen – red, fluorine – green, nitrogen – purple). | ||
For the following conformational study the model compound Ac-TfmPro-OMe (8) was prepared (vide infra). In addition, the Boc-protection in 1 was exchanged to Fmoc (9) with quantitative yield. The Fmoc amino acid was later used for the SPPS.
NMR spectra, in particular 19F-NMR of compounds 1, 7, 8, 9 were consistent with only one diastereomeric form of TfmPro in our synthesis. Furthermore, the [α]D value of −77° determined for 1 was in full agreement with the −77.6° reported by Qiu & Qing. We therefore found that in our modification the induction of the second stereo center under hydrogenation of 6 yields TfmPro of the correct cis relative stereochemistry despite the induction step was not coupled with the Bn-ester hydrogenolysis as described in the initial report.
We also obtained crystals of 8 which were then taken for X-ray analysis. To our surprise, the substance crystallized as racemic mixture in the orthorhombic cell containing four (2S,4S) and four (2R,4R) enantiomeric molecules. The main part of the substance 8 remained liquid and did not crystallize. The fact that the substance was diastereomerically pure strongly indicates that the partial epimerization of the proline α-(S)-chirality indeed occurred before the induction of the second stereo center. We presume that partial epimerization happened at the water elimination step (5 to 6). Finally by derivatisation with N-(2,4-dinitrophenyl)proline and subsequent 19F-NMR we determined the enantiomeric excess of 1 being 92%.
Conformation analysis of the Ac-TfmPro-OMe model
For detailed conformational characterization of TfmPro in a quasi-peptide context, we studied the properties of its tertiary amide bond and the pucker in a small derivative. We selected Ac-TfmPro-OMe model as a well characterised framework in which many Pro analogues were studied.1,15It is generally known that proline prefers s-trans amide rotamer, and the amide bond trans–cis interconversion could be conveniently characterized by the Ktrans/cis constant or by corresponding free energy difference. For instance, for Ac-Pro-OMe model in aqueous solution, the ΔG300 K was reported being −3.90 kJ mol−1, which corresponds to the “native” Pro Ktrans/cis of 4.8.19 It is also known that substituents in the γ-position influence this ratio mainly through stabilization of particular pyrrolidine ring γ-puckers. Comparative studies12 suggest that generally, an electron withdrawing substituent in the position γ-cis shifts the equilibrium towards the s-cis configuration; whereas an electron-donating group alleviates the s-trans rotameric form due to the gauche-effect. At the same time, a sterically demanding moiety in the γ-cis configuration should also lead to the s-trans stabilization. Thus, the electronegative31 and sterically demanding trifluoromethyl group might exhibit two counteracting effects: an influence on the pyrrolidine ring conformation and on the amide bond configuration in TfmPro. Should these effects be mutually compensated, TfmPro residue may be rendered as an almost intact proline analogue, i.e. to possess backbone conformational propensities very close to the ones in Pro.
The electronegative substituent effect was already seen in the pKa value for TfmPro which we determined to be 8.5 ± 0.1 close to that of γ-(S)-fluoroproline (9.232). In order to test the conformational impact, we performed first the van't Hoff type of analysis for 8 in aqueous medium employing 19F-NMR, which experimentally gives more accurate results than could be obtained from the crowded 1H-NMR spectra. The chemical shift difference between the two amide bond rotamers in the 19F spectra was 0.35 ppm at 25 °C and changed to 0.22 ppm at 90 °C. In agreement with our hypothesis, we found (Table 1) the equilibrium constant Ktrans/cis for Ac-TfmPro-OMe being equal 4.0. The γ-(S)-CF3-group thus only marginally shifts the overall equilibrium towards the cis-amide bond in the contrast to the large perturbation a single fluorine atom causes at the same ring position (Ktrans/cis = 2.5 for γ-(S)-fluoroproline). In addition, we have determined kinetic parameters of the isomerisation process. In particular, the activation energy of the cis-to-trans rotation process (81.8 kJ mol−1) was slightly lower but overall similar to that of proline (84.5 kJ mol−1). This effect is potentially related to the higher acidity of the imonium function likewise in γ,γ-difluoroproline, where corresponding activation barrier value was even lower, 80.8 kJ mol−1.19 We also observed cis–trans and trans–cis isomerization processes to be both driven enthalpically (amide conjugation), the same way as this is known for proline.
| Equilibrium | K trans/cis 300 K | ΔH, kJ mol−1 | ΔS, J mol−1 K | ΔG300 K, kJ mol−1 |
|---|---|---|---|---|
| Ac-TfmPro-OMe | 4.0 | −4.86 ± 0.11 | −4.72 ± 0.33 | −3.44 ± 0.21 |
| Ac-Pro-OMe19 | 4.8 | −5.04 ± 0.05 | −3.82 ± 0.16 | −3.90 ± 0.10 |
| Ac-(R)-Flp-OMe19 | 6.8 | −7.73 ± 0.26 | −9.81 ± 0.81 | −4.78 ± 0.50 |
| Ac-(S)-Flp-OMe19 | 2.5 | −3.04 ± 0.03 | −2.47 ± 0.11 | −2.30 ± 0.07 |
| cis-to-trans | k 300 K, s−1 | ΔH≠, kJ mol−1 | ΔS≠, J mol−1 K | E a 300 K, kJ mol−1 |
| Ac-TfmPro-OMe | 0.034 | 78.3 ± 0.3 | −11.9 ± 1.0 | 81.8 ± 0.6 |
| Ac-Pro-OMe19 | 0.012 | 87.2 | 9.00 | 84.5 |
| Ac-(R)-Flp-OMe19 | 0.026 | 84.2 | 5.37 | 82.6 |
| Ac-(S)-Flp-OMe19 | 0.015 | 84.7 | 2.42 | 84.0 |
| trans-to-cis | k 300 K, s−1 | ΔH≠, kJ mol−1 | ΔS≠, J mol−1 K | E a 300 K, kJ mol−1 |
| Ac-TfmPro-OMe | 0.009 | 81.4 ± 0.5 | −12.6 ± 1.6 | 85.2 ± 1.0 |
| Ac-Pro-OMe19 | 0.003 | 92.3 | 12.8 | 88.4 |
| Ac-(R)-Flp-OMe19 | 0.004 | 91.8 | 14.8 | 87.4 |
| Ac-(S)-Flp-OMe19 | 0.006 | 87.5 | 4.17 | 86.3 |
The 1D 1H-NMR spectra of Ac-TfmPro-OMe have also been analysed to assess the preferred conformation of the pyrrolidine ring. Since in our model, the α-CH couples to both protons of the adjacent β-CH2-group, two characteristic patterns are expected. For the exo-puckers the two 3Jαβ couplings should be in the range of 7–11 Hz, whereas for the endo-configuration one coupling should be in the range of 6–10 and the other of 2–3 Hz.33 For the major s-trans isomer of Ac-TfmPro-OMe we observed two equal 3Jαβ values of 8.3 Hz, thus confirming its preference for the exo-pucker. Remarkably, the same α-CH triplet shape for the major s-trans amide rotamer has been reported for s-trans rotamer of glycosylated γ-(S)-hydroxyproline which exhibits the exo-pucker.34 The splitting of 8.3 Hz persisted in water, chloroform and DMSO solutions of 8. The exo-pucker configuration for the s-trans was also confirmed in the solid state (X-ray structure, Scheme 1). In contrast, the minor s-cis form exhibited two distinct 3Jαβ values of 3.8 and 9.8 Hz (in water), having therefore no qualitative pucker preference. Following the same NMR criteria, proline itself should be classified as preferring an endo-pucker in the s-cis form and a mixed pucker in the s-trans form.35
TfmPro represents therefore a proline substitute which in summary has the pucker favouring the exo-conformer, whereas its amide bond thermodynamic preference is slightly shifted towards s-cis. From the structural labelling perspective, despite differences in the pucker preferences, the two amino acids can be considered mutual analogues since the overall amide bond configuration in TfmPro is very close to the one in proline.
TfmPro in gramicidin S
To observe TfmPro in a real peptide and to later evaluate the amino acid as an NMR label for peptide studies we incorporated TfmPro in the cyclic antimicrobial peptide gramicidin S (cyclo[Pro-Val-Orn-Leu-DPhe]2, where Orn stands for ornithine; GS). The two possible peptides 1TfmPro-GS (one TfmPro to Pro substitution, mono-substituted) and 2TfmPro-GS (two TfmPro to Pro substitutions, doubly-substituted) were produced by conventional Fmoc-SPPS as reported earlier.9,36 The peptides were synthesised in two steps: first a linear sequence was constructed on the solid support, followed by the cyclisation in solution. Since the linear precursors have to be pre-organized for the cyclisation, success of the reaction per se indirectly confirms the compatibility of TfmPro with a turn conformation of the native Pro-containing segment. The peptides were standardly purified using TFA-free RP-HPLC,7 performing which we observed an expected stepwise increase in the retention times (compared against GS) as hydrophobicity rose upon successive introduction the CF3-groups (Fig. 2). In the view of GS being the membrane-active peptide, this suggests a potential modulation of the peptide functional activity, but may not necessarily be a sign of the structural deviation.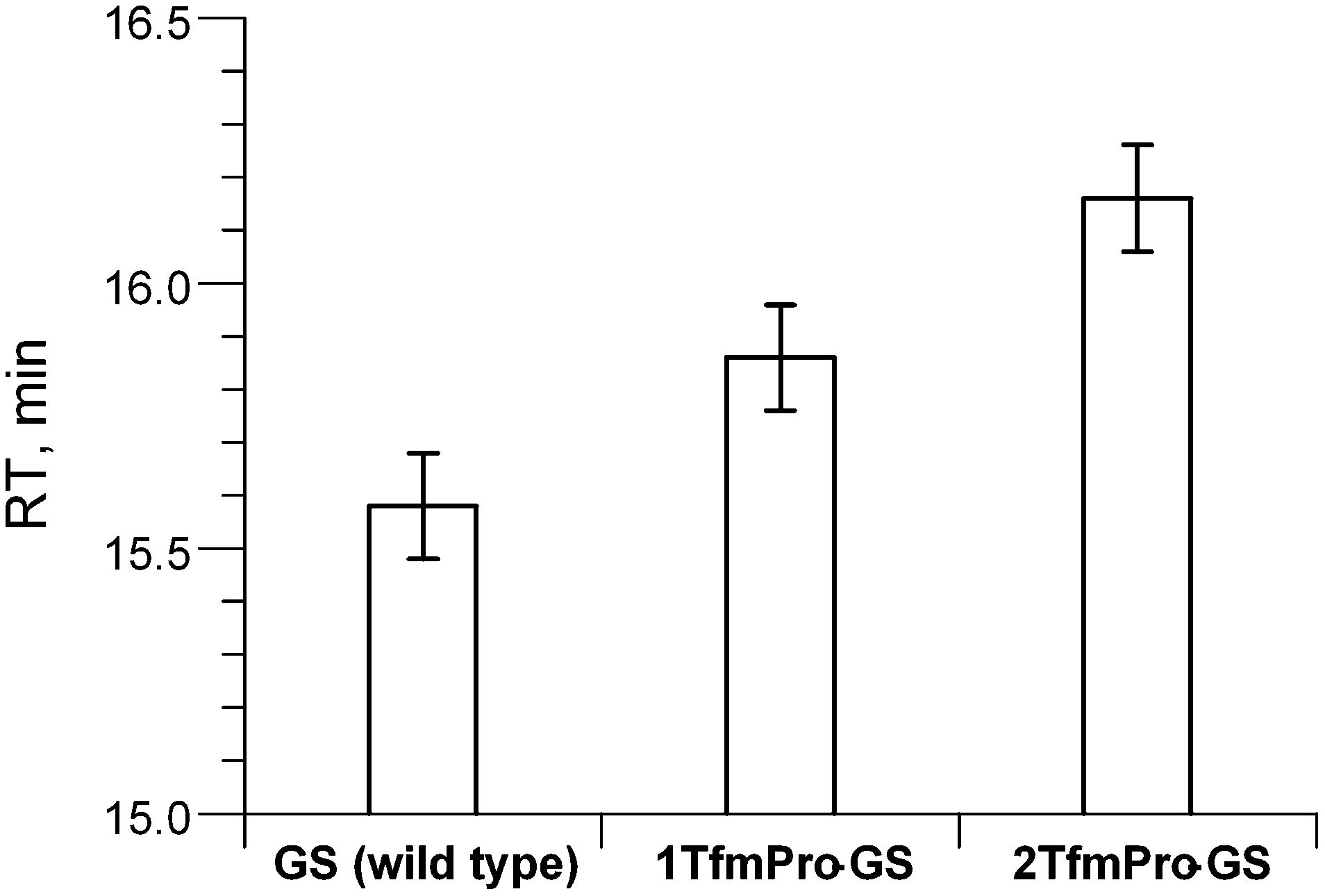 | ||
| Fig. 2 RP-HPLC retention times of GS and its analogues containing TfmPro. The error bars represent deviations of the values measured in triplicate. | ||
Since we address only structural aspect, in the following we only focused on the studies of potentially more perturbed doubly-substituted analogue 2TfmPro-GS.
As it is usually done in the label-validation studies to verify global conformational impact of a substitute, the new analogue was compared against GS by circular dichroism spectroscopy (CD) in a membrane-mimicking environment (Fig. 3). Practically the same spectral shapes for both molecules suggest convergence of the modified backbone conformation to the parent GS structure. Formally, since we have demonstrated TfmPro possesses proline-like intrinsic amide conformational preferences and not likely to influence overall geometry of the GS skeleton, we therefore could call TfmPro a reasonable 19F-label to substitute proline.6,11c
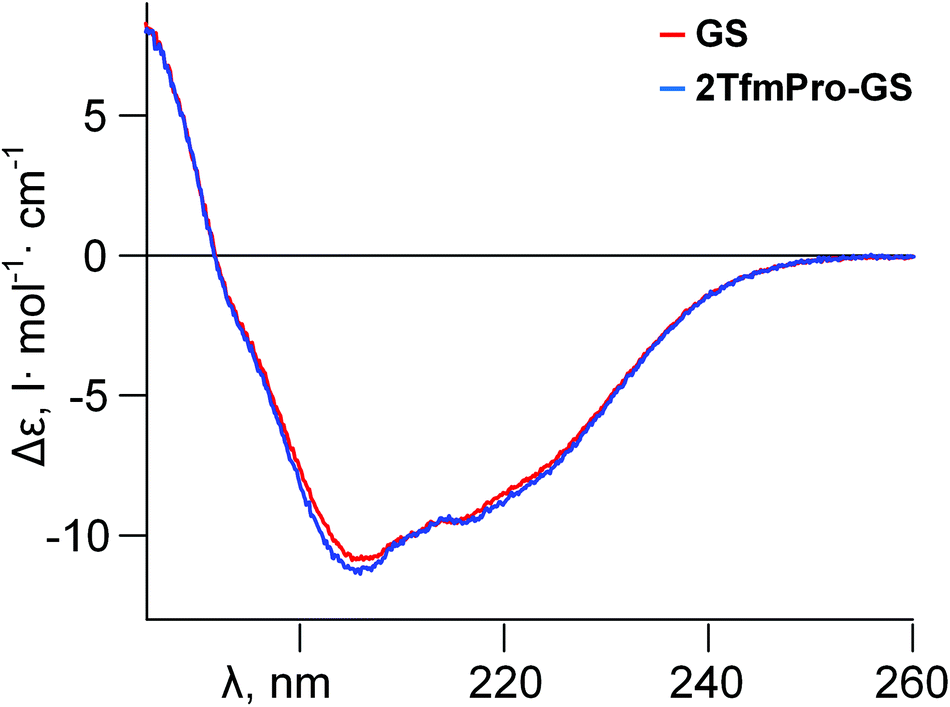 | ||
| Fig. 3 Circular dichroism spectra of GS and its analogue 2TfmPro-GS in aqueous buffer–trifluoroethanol mixture (phosphate buffer (PB), 10 mM, pH 7.4; TFE − 30%) at 25 °C. | ||
To validate the use of TfmPro in the solid state NMR structural studies of peptides, we used the GS system, solid state NMR behaviour of which is comprehensively studied. In particular, we benefited from the knowledge of the conditions in which the peptide changes its membrane-bound state. Using conventional 19F-labels it was earlier shown that in the oriented lipid bilayers molecules of GS change their alignment from the surfacially-bound basic state (“S-state”, Fig. 4A) to an upright orientation (“I-state”), the latter being attributed to the formation of homooligomeric pores relevant to the biological activity.9,37
 | ||
Fig. 4 (A) Schematic representation of GS in its membrane-bound S-state viewed from the bilayer plane; definition of the membrane normal (n), and rotational axis of C2 symmetry in GS molecule. Backbone structure as determined by Xu et al.41 The side-chains are taken as statistically most probable rotamers, protons are not shown for clarity, the prolines are highlighted in red. (B) Definition of the angle Θ (pucker is arbitrarily selected) which can be determined from solid state 19F-NMR from the dipolar splitting F–F according to the relation F − Fobs = F − Fmax × Smol·(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2Θ − 1)/2. (C) Temperature-dependent re-alignment of 2TfmPro-GS in DLPC as followed by solid state 19F-NMR. The sample is oriented with the membrane normal parallel to the magnetic field, temperatures and characteristic F−F values are indicated. cos2Θ − 1)/2. (C) Temperature-dependent re-alignment of 2TfmPro-GS in DLPC as followed by solid state 19F-NMR. The sample is oriented with the membrane normal parallel to the magnetic field, temperatures and characteristic F−F values are indicated. | ||
We knew that the amount of molecules in the I-state increases: (i) with the increase of the concentration, (ii) is more pronounced at temperatures close to the gel-to-fluid phase transition (Tm) of the bilayers and (iii) in the short-chain saturated glycerophospholipids.38 To satisfy these conditions we prepared 2TfmPro-GS-containing oriented proteobilayers using 1,2-didodecanoyl-sn-glycero-3-phosphocholine (DLPC) as a lipid matrix and 1/40 as a peptide/lipid ratio. From these samples a series of solid state 19F-NMR spectra were collected as a function of temperature. The temperature range was chosen to include the Tm of DLPC. Notably, the gel-to-fluid transition in DLPC is a complex process, characterised by conversions of several polymorphic mesophases and is rather broad (spans between −2 °C and +7 °C).39 As can be seen in the Fig. 4C, the oriented peptide in fluid lipids revealed single 19F-NMR signal (F–F dipolar coupling of +2.3 kHz), which was no longer resolved in the more rigid gel-phase bilayers. Such behaviour was expected and in both cases the peptide molecules should reside in the same orientation (the S-state), but should have different mobilities. The re-orientation to the I-state was indeed observed upon cooling at around Tm (the triplet with ca. −6.0 kHz splitting). Thus we reproduced temperature-dependent re-alignment of GS and demonstrated a qualitative application of TfmPro to monitor this process, but also showed this process for the first time by using F–F dipolar couplings, not the chemical shift.37,40
Based upon the above confirmation that 2TfmPro-GS has the same membrane alignment as GS, we attempted to validate the membrane-bound structure of the molecule. Intrinsically symmetric GS molecules in the S-state (Fig. 4A) should have both Pro residues identically oriented with respect to the membrane normal. We indeed observed only one signal, thus confirmed the C2 symmetry of the 2TfmPro-GS in the lipid-bound situation. The angle Θ (Fig. 4B) between the 19F-reporter group (the vector which connects the Cγ and the carbon of the CF3) and the external magnetic field (B0) can be directly determined from the value of the orientation-dependent F–F dipolar coupling.11 The observed F–F splitting (+2.3 kHz), considering the order parameter Smol of 0.3440 and the maximum F–F of the rotating CF3 group of 15.8 kHz, gives Θ of ∼37°. When we docked the TfmPro crystal geometry (exo-pucker, s-trans) onto NMR-determined backbone structure of GS41 and align the model in the S-state as it was done in the earlier studies,37,38 to our dissatisfaction, we obtain Θ of 45–50°. However, when we do the same using one of the reported crystal GS structures,42Θ assumes the range of 25–30°. Remarkably, our experimental value of ∼37° lays in-between these two values, but this appears to be a pure coincidence as clearly the major uncertainty in membrane-bound structure determination is the assumed backbone conformation of GS.
The uncertainty in the peptide backbone urged us to re-evaluate the degree of conformational perturbance TfmPro provides on the GS backbone. From CD analysis above we have concluded 2TfmPro-GS does not interfere with the GS backbone in the rough approximation only. To have a more detailed conformational insight we have additionally characterized the structure of 2TfmPro-GS by NMR. The 19F-NMR solution spectra of the 2TfmPro-GS showed single resonances in DMSO, water-trifluoroethanol and aqueous SDS micelles at −69.5, −71.4 and −70.8 ppm, respectively, confirming the overall C2 symmetry in solution. Conformation of backbone amides was examined by 1H15N single bond correlation NMR in 30% trifluoroethanol (Fig. 5A), i.e. exactly at conditions of CD (Fig. 3). In addition we inspected 2TfmPro-GS in DMSO environment (Fig. 5B), which is a better membrane mimic,43 but a condition inaccessible to CD spectroscopy. In the first case the DPhe NH signals of were affected only in the 1H dimension implying a different solvent exposure, which may come from the deviations within the β-turn atom arrangements. The Val NH, which is a part of the Val-TfmPro peptide bond, exhibited in contrast a prominent shift in the 15N dimension which persisted in both solvents. This is a potential indication of a local conformational effect of the γ-CF3-substituent in the proline analogue. Alternatively, an electrophilic influence of the CF3-substituent to the proline carbonyl could cause such a shift. Nevertheless, the rest of the signals remained in the modified peptide at same or close positions as they were in the wild type peptide. NMR data therefore corroborates the CD spectra-based conclusion that the TfmPro is compatible with the global secondary structure of GS and could affect only the local arrangement. The former finding is critically important to conclude solid state 19F-NMR data consistency with conditions-dependent states of the native peptide in the membrane environment. Whereas the correspondence of the local atomic arrangements is a technical prerequisite for correct conversion of the experimental anisotropic parameters (F–F dipolar couplings) into alignment angles for the wild type peptide molecule.
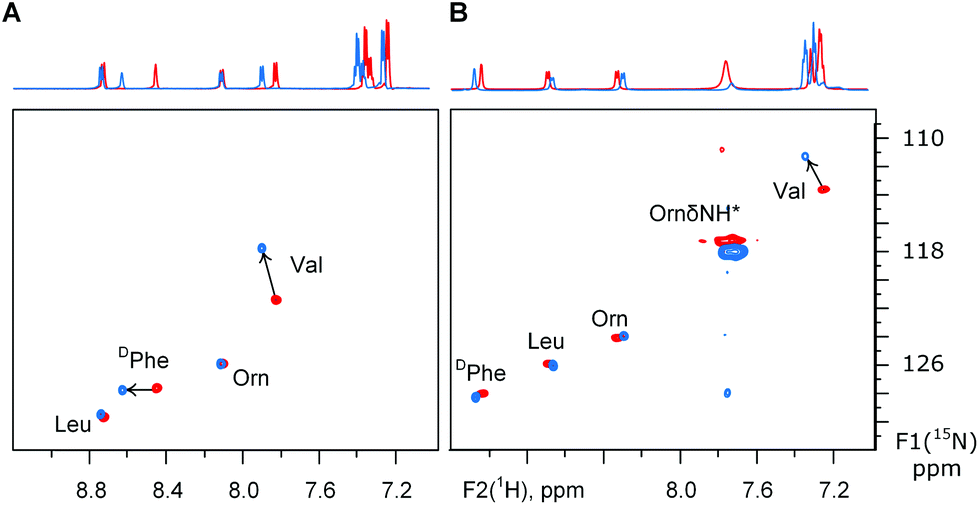 | ||
| Fig. 5 1H15N sofastHMQC spectra on the GS and its analogue 2TfmPro-GS in: (A) aqueous buffer-trifluoroethanol and (B) DMSO. The ornithine δ-NH resonance is a folded signal. | ||
In the DMSO solution we found also, that the Pro ring conformation in GS (α-CH as d, J = 7.2 Hz) was different to the TfmPro containing analogue (α-CH as dd, J = 9.8 and 4.5 Hz). In 2TfmPro-GS, the α-CH multiplicity of the imino acid is no longer consistent with the preference for the exo-pucker seen in the trans-amide of the Ac-TfmPro-OMe. This observation alludes additional reason to the uncertainties in the 2TfmPro-GS structure analysis from the solid state NMR (vide supra). Besides, it demonstrates that the substituted prolines may not necessarily maintain their “free” conformations (Ac-TfmPro-OMe model) in the restrained context of a structured polypeptide. Just as in the recent reports for γ-mono-fluoroprolines in the thiredoxin fold44 and γ-hydroxyprolines in the cyclotide kalata B1.45
Conclusions
In summary we describe herein the properties of a peptide bond formed by the γ-(S)-CF3-proline as an analogue of proline. The CF3-group in the γ-position of the pyrrolidine ring showed minimal influence on both the trans–cis amide equilibrium ratio (slightly shifted towards cis) and the rotational barrier (minimally reduced). The energy parameters of the γ-(S)-CF3-proline amide were described in details. In the β-turn of gramicidin S, the proline surrogate did not change the overall peptide structure. Used as a solid state 19F-NMR label, γ-(S)-CF3-proline confirmed the changes of the peptide alignment in lipid membranes in a qualitative way. Straightforward synthesis, marginal perturbation of the amide bond conformation, remote position from the peptide bond, and excellent NMR properties make γ-(S)-CF3-proline a good choice for a broad range of 19F-NMR proline labelling studies in peptides and proteins.Experimental part
The chemical shifts are given in δ scale according to the conventional internal deuterium referencing. The standards are TMS (Me4Si) and Freon-11 (CCl3F).Methyl ester N-Boc hydroxyproline (3) was obtained from hydroxyproline (2, 10.5 g) according to the literature protocol (19.0 g, 96% yield).46 4-ketoproline (4) was then prepared as following. 3 (50 g) was dissolved in dichloromethane (700 ml), triethylamine (91 ml) was added and the solution was cooled down in an ice bath under argon atmosphere. Suspension of pyridine·SO3 complex (63.5 g, 2 eq.) in DMSO (250 ml) was added portionwise within 8 min. The mixture was stirred for 18 hours at ambient temperature. It was then poured in 1 kg of ice. Organic layer was separated, washed with citric acid solution (10%, 2 × 500 ml), hydrochloric acid (4%, 1 × 500 ml), sodium hydrogencarbonate (saturated, 1 × 500 ml), citric acid solution (10%, 1 × 500 ml) and sodium hydrogencarbonate (saturated, 1 × 500 ml), dried over sodium sulphate, filtered and concentrated under reduced pressure. 4 was obtained as yellowish oil (46.9 g, 90% yield). 1H NMR data was consistent with the literature.47
Compound 5 was prepared as following. 4 (44.72 g, 184 mmol) was dissolved in THF (300 ml) under argon atmosphere. The solution was cooled down in an ice bath. CF3TMS (28.5 ml, 193 mmol) was added within 5 min followed by TBAF (1 M in THF, 1 ml). The ice was allowed to melt down, and the mixture was stirred at ambient temperature for 22 hours. Saturated ammonium chloride (300 ml) was added, the mixture was stirred for 20 min, then TBAF solution (1 M in THF, 190 ml) was added. The mixture was stirred for 1 hour. Organic layer was separated, and aqueous layer was extracted with diethyl ether (2 × 150 ml). Combined organic fractions were washed with water (1 × 300 ml), brine (1 × 300 ml), dried over sodium sulphate, filtered and concentrated under reduced pressure. 5 (54.1 g, 94%) was obtained as glassy oil. 1H-NMR (CDCl3, 400 MHz), δ, ppm (two rotamers): 4.52 and 4.43 (two d, J = 4.5 Hz, 1H), 4.37 and 4.22 (two br s, 1H), 3.77 and 3.75 (two s, 3H), 3.75–3.62 (m, 2H), 2.53 (m, 1H), 2.19 (t, 1H, J = 13 Hz), 1.44 and 1.39 (two s, 9H). 19F-NMR (CDCl3, 376 MHz), δ, ppm (two rotamers 1:1): −81.1 and −81.2 (two s, CF3).
2-tert-Butyl 2-methyl (2S)-trifluoromethyl-3-pyrrolin-1,2-dicarboxylate (6)
In a 2 l reactor 5 (33.9 g, 108 mmol) and dry pyridine (1 l) were placed under argon. Thionyl chloride (100 ml) was added and the mixture was heated and refluxed. The reflux was continued for 20 min and then the mixture was allowed to cool down to ambient temperature. The mixture was poured into iced water (0.5 kg). Aqueous layer was extracted with diethyl ether (4 × 300 ml) and ethyl acetate (1 × 1 l). Combined organic fractions were concentrated under reduced pressure to have the volume of ∼300 ml. Resulting organic solution was washed with hydrochloric acid (5%, 2 × 150 ml), sodium hydrogencarbonate (saturated, 1 × 150 ml) and brine (1 × 150 ml), dried over sodium sulphate and concentrated in vacuum. The black crude material was filtered through a short silicagel (70 g) column in hexane–ethyl acetate 2:1 mixture. Final product 6 (19.2 g, 60%) was obtained as yellowish oil.
1H-NMR (CDCl3, 400 MHz), δ, ppm (two rotamers): 6.19 (td, J = 19 and 2 Hz, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 5.08 (two m, 1H, N–CH–CO2Me), 4.33 (m, 2H, N–CH2), 3.71 (two s, 3H, OCH3), 1.42 and 1.37 (two s, 9H, C(CH3)3). 13C-NMR (CDCl3, 101 MHz), δ, ppm (two rotamers): 169.2 and 168.8 (two s, CO2Me), 153.4 and 152.8 (two s, N–C(O)–O), 132.8 and 132.6 (two q, J = 36 Hz, CCH), 128.3 and 128.1 (two q, J = 5 Hz, CCH), 120.7 (two q, J = 270 Hz, CF3), 81.1 (two s, O–C(CH3)3), 66.5 and 66.3 (two s, CH–CO2Me), 52.8 and 52.6 (two s, OCH3), 51.0 and 50.8 (two s, N–CH2), 28.3 and 28.2 (two s, C(CH3)3). 19F-NMR (CDCl3, 282 MHz), δ, ppm: −65.3 (d, JF–H = 19 Hz, CF3). IR bands: 3093, 2983, 2876, 1748, 1703, 1667 rest of the peaks below 1460 cm−1. Mass-spectrum (EI), m/z: 236 [M − But]+, 195 [M − Boc]+. [α]20D = −202 (CHCl3, c = 0.55). CHN, found. C, 49.03; H, 5.30; N 4.99. C12H16F3NO4 requires C, 48.81; H, 5.46; N 4.47.
C), 5.08 (two m, 1H, N–CH–CO2Me), 4.33 (m, 2H, N–CH2), 3.71 (two s, 3H, OCH3), 1.42 and 1.37 (two s, 9H, C(CH3)3). 13C-NMR (CDCl3, 101 MHz), δ, ppm (two rotamers): 169.2 and 168.8 (two s, CO2Me), 153.4 and 152.8 (two s, N–C(O)–O), 132.8 and 132.6 (two q, J = 36 Hz, CCH), 128.3 and 128.1 (two q, J = 5 Hz, CCH), 120.7 (two q, J = 270 Hz, CF3), 81.1 (two s, O–C(CH3)3), 66.5 and 66.3 (two s, CH–CO2Me), 52.8 and 52.6 (two s, OCH3), 51.0 and 50.8 (two s, N–CH2), 28.3 and 28.2 (two s, C(CH3)3). 19F-NMR (CDCl3, 282 MHz), δ, ppm: −65.3 (d, JF–H = 19 Hz, CF3). IR bands: 3093, 2983, 2876, 1748, 1703, 1667 rest of the peaks below 1460 cm−1. Mass-spectrum (EI), m/z: 236 [M − But]+, 195 [M − Boc]+. [α]20D = −202 (CHCl3, c = 0.55). CHN, found. C, 49.03; H, 5.30; N 4.99. C12H16F3NO4 requires C, 48.81; H, 5.46; N 4.47.
Methyl (2S,4S)-N-tert-buthoxycarbonyl-4-trifluoromethylprolinate (7)
6 (13 g; 44 mmol) and Pd/C (5%, 3 g) in methanol (100 ml) were stirred under hydrogen (1 atm) for 5 hours. The mixture was filtered and concentrated under reduced pressure to give 7 (12.3 g, 94%) as white solid.
1H-NMR (CDCl3, 400 MHz), δ, ppm (two rotamers): 4.42 and 4.33 (2t, J = 8 Hz, 1H), 3.92 and 3.84 (2t, J = 10 Hz, 1H), 3.77 (s, 3H), 3.51 (t, J = 10 Hz), 2.96 (m, 1H), 2.56 (m, 1H), 2.14 (m, 1H), 1.49 and 1.43 (2s, 9H). 13C-NMR (CDCl3, 101 MHz), δ, ppm (two rotamers): 172.3 and 172.0 (2s, CO2Me), 153.8 and 153.2 (2s, N–C(O)–O), 126.0 (q, J = 277 Hz, CF3), 80.8 (s, O–C(CH3)3), 58.5 and 58.2 (2s, N–CH–CO2Me), 52.4 and 52.2 (2s, OCH3), 45.8 and 45.7 (2m, N–CH2), 41.9 and 41.1 (q, J = 30 Hz, CH–CF3), 30.1 and 29.1 (2s, CH2–CH–CO2Me), 28.3 and 28.2 (2s, C(CH3)3). 19F-NMR (CDCl3, 282 MHz), δ, ppm: −70.4 (d, JH–F = 8 Hz, CF3). IR bands: 2987, 2870, 1747, 1695, further peaks below 1481 cm−1. Mass-spectrum (EI), m/z: 297 [M]+, 240 [M − But]+, 239 [M − CO2Me]+, 224, 196 [M − Boc]+. Tmelt = 50 °C, [α]20D = −70 (CHCl3, c = 2.1). CHN, found. C, 48.59; H, 6.05; N 4.85. C12H18F3NO4 requires C, 48.48; H, 6.10; N 4.71.
The compound 1 was prepared as following. To 6 (10.88 g, 36.6 mmol) in methanol (100 ml) sodium hydroxide (1 M in methanol, 40 ml) was added. The mixture was stirred for 2 hours at the room temperature and then concentrated in vacuum (temperature in bath ≤ 40 °C). The residue was dissolved in water (200 ml) and washed with diethyl ether (2 × 40 ml) and organic fractions were discarded. The water fraction was then acidified with hydrochloric acid (13%, 17 ml) until acidic pH was reached (∼1–2). Aqueous layer was extracted with dichloromethane (4 × 60 ml), the organic fractions were dried over sodium sulphate, filtered and concentrated under reduced pressure. 1 (8.98 g, 86%) was obtained as white solid. 1H-NMR (CDCl3, 500 MHz), δ, ppm (two rotamers): 6.75 (br s, 1H), 4.43 and 4.36 (2m, 1H), 3.93 and 3.85 (2m, 1H), 3.50 (m, 1H), 2.97 (m, 1H), 2.61 and 2.54 (2m, 1H), 2.41 and 2.22 (2m, 1H), 1.49 and 1.43 (2s, 9H). Spectral data was consisted with the one published by Qiu&Qing.29 Also, the [α]D values varied in different repetitions of the full synthesis from −72 to −77 indicating different levels of racemization on the previous steps. In this particular approach described in this experimental section we obtained the product with [α]D −77 (c = 1.0, CHCl3).
(2S,4S)-N-(9-Fluorenylmethoxycarbonyl)-4-trifluoromethylproline (9)
To a solution of 1 (569 mg, 2.0 mmol) in dichloromethane (10 ml) trifluoroacetic acid (3 ml) was added, the solution was stirred at the room temperature for 2 hours. Liquids were removed under reduced pressure (temperature in bath 30 °C), then additional dichloromethane (10 ml) was added and evaporation was repeated. Water (10 ml) and sodium carbonate solution (10%, 7 ml) was added to reach pH ≈ 8–9 and acetone (5 ml) was subsequently added until clear solution was afforded. Resulting mixture was cooled down in an ice bath. Suspension of Fmoc-OSu (745 mg, 2.2 mmol) in acetone (2 ml) was added within 1 min upon stirring, and then about 10 ml of acetone was added to the reaction mixture. After 4 hours the ice bath was removed and stirring was continued for 25 hours at ambient temperature. Acetone was removed under reduced pressure (temperature in bath ≤ 31 °C), the transparent solution was poured into water (100 ml), and resulting suspension was washed by diethyl ether (4 × 30 ml). The aqueous layer was acidified by hydrogen chloride solution (1 M) until pH ≈ 1, resulting aqueous fraction was extracted with ethyl acetate (4 × 50 ml). The ethyl acetate fractions were dried over magnesium sulphate, filtered and concentrated under reduced pressure. The residue was dissolved in acetonitrile–water mixture, the solution was frozen in liquid nitrogen and lyophilized overnight to give 9 (813 mg, 100%) as beige powder (amorphous).
1H-NMR (CDCl3, 700 MHz), δ, ppm (two rotamers 3:2): 7.89 (br s, 1H, CO2H), 7.79 (major) and 7.73 (minor) (2d, J = 7 Hz, 2H, aromatic CH), 7.58 (major) and 7.54 (minor) (m, 2H, aromatic CH), 7.43 (major) and 7.37 (minor) (m, 2H, aromatic CH), 7.34 (major) and 7.30 (minor) (m, 2H, aromatic CH), 4.55–4.40 (m, 2H, CH2–O), 4.50 (major) and 4.31 (minor) (2t, J = 8, 1H, α-CH), 4.27 (major) and 4.15 (minor) (2t, J = 7 Hz, CH–CH2–O), 3.54 (m, 1H, δ-CH), 2.98 (major) and 2.94 (minor) (m, 1H, γ-CH), 2.61 (m, 1H, β-CH), 2.28 (major) and 2.21 (minor) (2m, 1H, β-CH). 13C-NMR (CDCl3, 176 MHz), δ, ppm (two rotamers): 176.1 and 175.2 (CO2H), 155.0 and 154.1 (N–C(O)–O), 143.8, 143.5, 141.4 and 141.3 (aromatic C), 127.9, 127.7, 127.2, 127.1, 125.0, 124.8, 120.04 and 120.0 (aromatic CH), 125.8 (CF3), 68.2 and 67.8 (CH2–O), 58.5 and 57.7 (α-CH), 47.13 and 47.10 (CH–CH2–O), 46.2 and 45.8 (δ-CH2), 41.9 (major) and 41.0 (minor) (q, J = 30 Hz, CH–CF3), 30.1 and 28.8 (β-CH2). 19F-NMR (CDCl3, 659 MHz), δ, ppm, (two rotamers): −70.34 (major) and −70.38 (minor) (d, J = 8 Hz). IR bands: 3600–2150 broad peak with a maximum at 2957, 1705 broad peak, further peaks below 1430 cm−1. Mass-spectrum (EI), m/z: 405 [M]+, 179 [Fmoc]+. [α]20D = −50.7 (CHCl3, c = 1.04). CHN, found. C, 62.02; H, 4.40; N 3.60. C21H18F3NO4 requires C, 62.22; H, 4.48; N 3.46.
Methyl (2S,4S)-N-acetyl-4-trifluoromethylprolinate (8)
Acetyl chloride (0.45 ml, 6.3 mmol) was mixed to dry methanol (25 ml) and resulting acidic methanol was added to 7 (1.57 g, 5.3 mmol) in methanol (75 ml). The mixture was stirred at the room temperature for 2 hours and then refluxed for the next 4 hours. The mixture was concentrated under reduced pressure. Dichloromethane (70 ml) and triethylamine (1.62 ml, 11.6 mmol) were added to the residue and the mixture was stirred for 5 min, then acetyl chloride (0.41 ml; 5.8 mmol) was added and the mixture was stirred at the room temperature for 3 days. The mixture was then concentrated under reduced pressure, the obtained residue was rinsed with diethyl ether (2 × 50 ml, 2 × 20 ml) and the organic fractions were concentrated in vacuum to give pure matter. This product was additionally purified on a silica gel column using ethyl acetate elution to give 8 (1.03 g, 82%) as colourless oil.
1H-NMR (D2O, 700 MHz), δ, ppm (two rotamers): 4.81 (dd, J = 9.7 and 3.7 Hz, minor) and 4.50 (t, J = 8.3 Hz, major, 1H, CH–CO2Me), 4.00 (dd, J = 8.7 and 9.7 Hz, 1H major, the minor rotamer resonance lays underneath, 1H, N–CHH), 3.75 (s, minor) and 3.71 (s, major, 3H, OCH3), 3.69 (t, J = 10.0 Hz, major) and 3.42 (dd, J = 12.7 and 6.3 Hz, minor, 1H, N–CHH), 3.28 (m, major) and 3.18 (m, minor, 1H, CH–CF3), 2.75 (m, minor) and 2.63 (m, major, 1H, CHH–CH–CO2Me), 2.42 (m, minor) and 2.11 (m, major, 1H, CHH–CH–CO2Me), 2.07 (s, major) and 1.98 (s, minor, 3H, CH3–CO). 13C-NMR (CDCl3, 176 MHz), δ, ppm (two rotamers): 173.62 (s, major, CO2Me), 173.60 (s, minor, N–CO), 173.3 (s, minor, CO2Me), 172.9 (s, major, N–CO), 126.7 (q, J = 277 Hz, minor) and 126.0 (q, J = 276 Hz, major, CF3), 59.7 (s, minor) and 58.6 (s, major, CH–CO2Me), 53.4 (s, minor) and 53.1 (s, major, OCH3), 47.2 (q, J = 3 Hz, major) and 45.6 (q, J = 3 Hz, minor, N–CH2), 41.3 (q, J = 29 Hz, major) and 39.4 (q, J = 29 Hz, minor, CH–CF3), 29.4 (q, J = 2 Hz, minor) and 28.3 (q, J = 3 Hz, major, CH2–CH–CO2Me), 21.2 (s, major) and 21.0 (s, minor, CH3–CO). 19F-NMR (470 MHz), δ, ppm (two rotamers): in D2O −71.0 (major, JF–H = 9 Hz) and −71.3 (minor, JF–H = 10 Hz); in DMSO-d6 −69.4 (major, JF–H = 9 Hz) and −69.9 (minor, JF–H = 10 Hz); in CDCl3 −70.7 (major, JF–H = 8 Hz) and −71.1 (minor, JF–H = 9 Hz). IR bands: 2959, 2892, 1743, 1652, further peaks below 1438 cm−1. Mass-spectrum (EI), m/z: 239 [M+], 180, 139. [α]20D = −78 (CHCl3, c = 0.56). CHN, found. C, 45.34; H, 5.30; N 6.05. C9H12F3NO3 requires C, 45.19; H, 5.06; N 5.86. X-ray crystal structure can be found in the Cambridge Crystallographic Data Center under ID CCDC 1042476.
19F-NMR parametrization of the amide rotation
The 19F-NMR spectra were measured on Bruker Avance III 500 spectrometer (470.7 MHz) equipped with a BBFO probe. The variable temperature unit was calibrated using sample with acidified glycerol. A temperature series of conventional 1-pulse spectra was run in the range 30–90 °C with solution of Ac-TfmPro-OMe (8, 75 mg) in deuterium oxide as 12 experiments with the temperature pre-equilibration delay of 5 min. Resulting spectra were baseline corrected (5th order) and integrated. Corresponding rotameric ratios (Ktrans/cis) were calculated and then converted to ΔG according to ΔG = −RTlnK. The ΔG values were plotted against the temperature, ΔH and ΔS values were then extracted using ΔG = ΔH − TΔS equation.
The amide rotation rate constants were determined in 19F cross-relaxation experiments (EXSY) with the following setup: “noesygpph” experiment from the standard Bruker library, with the mixing time of 1 s and 20 ms for referencing. 18 experiments (9 for exchange and 9 referencing) were acquired at 30–55 °C. Resulting 2D spectra were baseline corrected in both dimensions and integrated. The exchange rates (exchange rate matrices) were calculated with EXSYCalc® (Mestrec) freeware. In particular, detected at 55 °C the exchange rates were kcis–trans = 0.617 and ktrans–cis = 0.182 s−1. Standard linearisation according to the Eyring equation (in ln(k/T) − 1/T coordinates) delivered the values of ΔH and ΔS.
Peptide synthesis
Linear peptides were synthesised on a Val-preloaded 2-chlorotrityl resin. The amino acids were taken as N-Fmoc (ornithine side chain amine was Boc-protected) in 4 eq. along with 4 eq. of 6Cl-HOBt, 3.9 eq. of HCTU, 8 eq. of DIPEA premixed in 2 ml of DMF before adding to the resin for a coupling step which was performed for 2 hours. The Fmoc-removal was done with 22% piperidine in DMF for 20–30 min. The linear peptides were cleaved from the resin by treatment with 25% hexafluoroisopropanol in dichloromethane for 15 min. Cyclisation was performed under high dilution conditions in dichloromethane (0.15 mmol/1 l) with 3 eq. of HOBt and 3 eq. PyBOP and 6 eq. of DIPEA, which were pre-mixed in DMF (1.5 ml) before addition. The reaction was continued for 12 hours. The Boc-protection groups from the ornithine side chains were removed by treatment with TFA:TIS:water 92.5:5:2.5 cocktail for >15 min. The crude matters were then purified on the semi-preparative RP-HPLC C18 column (10 × 250 mm) with the water–acetonitrile gradient elution. 5 mM hydrogen chloride concentration in the eluent was used as an ion-paring agent. Analytical RP-HPLC was done as described26 on a biphenyl analytical column (4.6 × 250 mm).
:1, vol/vol, acetonitrile–water), 282 MHz), δ, ppm: −70.5 (d, JF–H = 10 Hz, CF3). Mass-spectrum (MALDI-TOF), m/z, found/calcd: 1211.2/1209.5.
:1, vol/vol, acetonitrile–water), 282 MHz), δ, ppm: −70.5 (d, JF–H = 9 Hz, CF3). Mass-spectrum (MALDI-TOF), m/z, found/calcd: 1278.3/1277.5.
Solution NMR of peptides
The spectra were recorded on Bruker Avance III 700 spectrometer (1H 700.2 MHz; 15N 71.0 MHz) equipped with a TXI probe at 25 °C. The phosphate buffer was of pH 6.0 (22 °C) and 15 mM concentration containing 10 vol% D2O for deuterium lock. The spectra in DMSO-d6 were referenced using deuterium lock signal, while the spectra in PB-TFE were referenced using TPS internal standard. Assignment of the proton spectra was done using 1H TOCSY experiments (dipsi2 spin lock of 60 ms). The 1H15N single bond correlations were detected in the sofast-HMQC experiments with the recycling delay of 100 ms.Found HN resonances (1H/15N). GS in DMSO-d6: 9.08/128.1 (DPhe), 8.72/125.8 (Leu), 8.34/124.0 (Orn), 7.24/113.3 (Val); 2TfmPro-GS in DMSO-d6: 9.12/128.0 (DPhe), 8.70/125.8 (Leu), 8.32/123.4 (Orn), 7.34/110.5 (Val); GS in PB-TFE: 8.76/129.6 (Leu), 8.48/127.5 (DPhe), 8.12/125.8 (Orn), 7.83/121.2 (Val); 2TfmPro-GS in PB-TFE: 8.77/129.4 (Leu), 8.66/127.7 (DPhe), 8.13/125.8 (Orn), 7.91/117.4 (Val).
CD spectra of the peptides
The spectra were measured on Jasco J-720 spectropolarimeter. The spectra were measured at 25 °C in the same buffer as was taken for NMR at 90 μM peptide concentrations.Solid state NMR
The solid state 19F-NMR spectra were recorded on Bruker Avance III 500 spectrometer (470.6 MHz) equipped with wide bore magnet and the home-built HF lowE flat-coil probe.An oriented sample was prepared from 0.6 mg 2TfmPro-GS and 11.7 mg of DLPC ((12:0/12:0)PC) at the peptide-to-lipid molar ratio of 1/40. Dry mixtures were co-dissolved in methanol, spread over 16 rectangular glass plates (18 × 7.5 mm; Marienfeld, Germany), were dried in vacuum (>4 hours), stacked and hydrated at 96% relative humidity (saturated potassium sulfate at 48 °C) for 26 hours. A fresh sample was wrapped in Nescofilm® and Sarogold® films for prevention of drying. Proper orientation of the lipid bilayers was checked by 31P-NMR to have at least 80% bilayer lipids being coplanar with the slide surface.
The 19F-NMR spectra in oriented samples were measured using aring composite pulse sequence (for background suppression) with proton decoupling during acquisition. Accurate temperature series (in particular, 1 °C step in the range +5 ÷ −5 °C) was performed with 10 min temperature pre-equilibration time at each temperature. The temperature was calibrated using acidified methanol sample. The spectra were processed with Lorentzian window function (LB 250 Hz).
Acknowledgements
V.K. acknowledges Dr Andi Mainz (Berlin, Germany) for provision of the sofast-HMQC setup. Enamine Ltd and Prof. Andrey Tolmachev (Kyiv, Ukraine) are acknowledged for financial support. Prof. Igor V. Komarov (Kyiv) and Dr Patrick Durkin (Berlin) are acknowledged for revision of the manuscript.References
- L. Moroder, C. Renner, J. J. Lopez, M. Mutter and G. Tuchscherer, Tailoring the cis-trans Isomerization of Amides, in cis-trans-Isomerization in Biochemistry, ed. C. Dugave, Wiley, Weinheim, 2006, pp. 225–259 Search PubMed.
- (a) K. P. Lu, G. Finn, T. H. Lee and L. K. Nicholson, Nat. Chem. Biol., 2007, 3, 619–629 CrossRef CAS PubMed; (b) A. H. Andreotti, Biochemistry, 2003, 42, 9515–9524 CrossRef CAS PubMed; (c) C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- (a) M. Srinivasan and A. K. Dunker, Int. J. Pept., 2012, 634769 Search PubMed; (b) L. J. Ball, R. Kühne, J. Schneider-Mergener and H. Oschkinat, Angew. Chem., Int. Ed., 2005, 44, 2852–2869 CrossRef CAS PubMed; (c) X. Ren and J. H. Hurley, Traffic, 2011, 12, 1282–1290 CrossRef CAS PubMed.
- E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242–1250 CrossRef CAS PubMed.
- V. S. Kubyshkin, I. V. Komarov, S. Afonin, P. K. Mykhailiuk, S. L. Grage and A. S. Ulrich, Trifluoromethyl-substituted α-amino acids as solid-state 19F NMR labels for structural studies of membrane-bound peptides in Fluorine in Pharmaceutical and medicinal chemistry, ed. V. Gouverneur and K. Müller, Imperial College Press, London, 2012, pp. 91–138 Search PubMed.
- K. Koch, S. Afonin, M. Ieronimo, M. Berditsch and A. S. Ulrich, Top. Curr. Chem., 2012, 306, 89–118 CrossRef CAS.
- S. Afonin, R. W. Glaser, M. Berditchevskaia, P. Wadhwani, K.-H. Gührs, U. Möllmann, A. Perner and A. S. Ulrich, ChemBioChem, 2003, 4, 1151–1163 CrossRef CAS PubMed.
- A. N. Tkachenko, P. K. Mykhailiuk, D. S. Radchenko, O. Babii, S. Afonin, A. S. Ulrich and I. V. Komarov, Eur. J. Org. Chem., 2014, 3584–3591 CrossRef CAS.
- V. S. Kubyshkin, P. K. Mykhailiuk, S. Afonin, S. L. Grage, I. V. Komarov and A. S. Ulrich, J. Fluorine Chem., 2013, 152, 136–143 CrossRef CAS PubMed.
- R. W. Glaser, C. Sachse, U. H. N. Dürr, P. Wadhwani and A. S. Ulrich, J. Magn. Reson., 2004, 168, 153–163 CrossRef CAS PubMed.
- (a) P. K. Mikhailiuk, S. Afonin, A. N. Chernega, E. B. Rusanov, M. O. Platonov, G. D. Dubinina, M. Berditsch, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2006, 45, 5659–5661 CrossRef CAS PubMed; (b) S. Afonin, P. K. Mykhailiuk, I. V. Komarov and A. S. Ulrich, J. Pept. Sci., 2007, 13, 614–623 CrossRef CAS PubMed; (c) P. K. Mykhailiuk, S. Afonin, G. V. Palamarchuk, O. V. Shishkin, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2008, 47, 5765–5767 CrossRef CAS PubMed; (d) D. Maisch, P. Wadhwani, S. Afonin, C. Böttcher, B. Koksch and A. S. Ulrich, J. Am. Chem. Soc., 2009, 131, 15596–15597 CrossRef CAS PubMed; (e) A. N. Tkachenko, P. K. Mykhailiuk, S. Afonin, D. S. Radchenko, V. S. Kubyshkin, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2013, 52, 1486–1489 CrossRef CAS PubMed; (f) A. N. Tkachenko, D. S. Radchenko, P. K. Mykhailiuk, S. Afonin, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2013, 52, 6504–6507 CrossRef CAS PubMed.
- A. K. Pandey, D. Naduthambi, K. M. Thomas and N. J. Zondlo, J. Am. Chem. Soc., 2013, 135, 4333–4363 CrossRef CAS PubMed.
- (a) A. Fluores-Ortega, A. I. Jiménez, C. Cativiela, R. Nussinov, C. Alemán and J. Casanovas, J. Org. Chem., 2008, 73, 3418–3427 CrossRef PubMed; (b) A. Flores-Ortega, J. Casanovas, R. Nussinov and C. Alemán, J. Phys. Chem. B, 2008, 112, 14045–14055 CrossRef CAS PubMed; (c) Y. Che and G. R. Marshall, Biopolymers, 2006, 81, 392–406 CrossRef CAS PubMed.
- C. A. Thomas, E. R. Talaty and J. G. Bann, Chem. Commun., 2009, 3366–3368 RSC.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559–564 CrossRef CAS PubMed.
- D. Naduthambi and N. J. Zondlo, J. Am. Chem. Soc., 2006, 128, 12430–12431 CrossRef CAS PubMed.
- T. Steiner, P. Hess, J. H. Bae, B. Wiltschi, L. Moroder and N. Budisa, PLoS One, 2008, 3, e1680 Search PubMed.
- C. Renner, S. Alefelder, J. H. Bae, N. Budisa, R. Huber and L. Moroder, Angew. Chem., Int. Ed., 2001, 40, 923–925 CrossRef CAS.
- W. Kim, K. I. Hardcastle and V. P. Conticello, Angew. Chem., Int. Ed., 2006, 45, 8141–8145 CrossRef CAS PubMed.
- Q. A. Huchet, B. Kuhn, B. Wagner, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, J. Fluorine Chem., 2013, 152, 119–128 CrossRef CAS PubMed.
- C. M. Tressler and N. J. Zondlo, J. Org. Chem., 2014, 79, 5880–5886 CrossRef CAS PubMed.
- G. Chaume, N. Lensen, C. Caupène and T. Brigaud, Eur. J. Org. Chem., 2009, 5717–5724 CrossRef CAS.
- G. Chaume, J. Simon, C. Chaupène, N. Lensen, E. Miclet and T. Brigaud, J. Org. Chem., 2013, 78, 10144–10153 CrossRef CAS PubMed.
- B. Koksch, D. Ullmann, H.-D. Jakubke and K. Burger, J. Fluorine Chem., 1996, 80, 53–57 CrossRef CAS.
- V. S. Kubyshkin, P. K. Mykhailiuk, S. Afonin, A. S. Ulrich and I. V. Komarov, Org. Lett., 2012, 14, 5254–5257 CrossRef CAS PubMed.
- J. R. Del Valle and M. Goodman, Angew. Chem., Int. Ed., 2002, 41, 1600–1602 CrossRef CAS.
- X.-l. Qiu and F.-l. Qing, J. Chem. Soc., Perkin Trans. 1, 2002, 2052–2057 RSC.
- X.-l. Qiu and F.-l. Qing, J. Org. Chem., 2002, 67, 7162–7164 CrossRef CAS PubMed.
- R. Nadano, Y. Iwai, T. Mori and J. Ichikawa, J. Org. Chem., 2006, 71, 8748–8754 CrossRef CAS PubMed.
- J. E. True, T. D. Thomas, R. W. Winter and G. L. Gard, Inorg. Chem., 2003, 42, 4437–4441 CrossRef CAS PubMed.
- E. S. Eberhardt, N. Panasik Jr. and R. T. Raines, J. Am. Chem. Soc., 1996, 118, 12261–12266 CrossRef CAS PubMed.
- M. Cai, Y. Huang, J. Liu and R. Krishnamoorthi, J. Biomol. NMR, 1995, 6, 123–128 CrossRef CAS.
- N. W. Owens, C. Braun, J. D. O'Neil, K. Marat and F. Schweizer, J. Am. Chem. Soc., 2007, 129, 11670–11671 CrossRef CAS PubMed.
- C. B. Braga, L. C. Ducati, C. F. Tormena and R. Rittner, J. Phys. Chem. A, 2014, 118, 1748–1758 CrossRef CAS PubMed.
- P. Wadhwani, S. Afonin, M. Ieronimo, J. Buerck and A. S. Ulrich, J. Org. Chem., 2006, 71, 55–61 CrossRef CAS PubMed.
- S. Afonin, U. H. N. Dürr, P. Wadhwani, J. Salgado and A. S. Ulrich, Top. Curr. Chem., 2008, 273, 139–154 CrossRef CAS.
- S. Afonin, R. W. Glaser, C. Sachse, J. Salgado, P. Wadhwani and A. S. Ulrich, Biochim. Biophys. Acta, 2014, 1838, 2260–2268 CrossRef CAS PubMed.
- L. Dahbi, C. Bourgaux and M. Ollivon, Prog. Colloid Polym. Sci., 2004, 126, 178–183 CAS.
- J. Salgado, S. L. Grage, L. H. Kondejewski, R. S. Hodges, R. N. McElhaney and A. S. Ulrich, J. Biomol. NMR, 2001, 21, 191–208 CrossRef CAS.
- Y. Xu, I. P. Sugár and N. R. Krishna, J. Biomol. NMR, 1995, 5, 37–48 CrossRef CAS.
- K. Yamada, M. Unno, K. Kobayashi, H. Oku, H. Yamamura, S. Araki, H. Matsumoto, R. Katakai and M. Kawai, J. Am. Chem. Soc., 2002, 124, 12684–12688 CrossRef CAS PubMed.
- D. Mihailescu and J. C. Smith, J. Phys. Chem. B, 1999, 103, 1586–1594 CrossRef CAS.
- M. Rubini, M. A. Schärer, G. Capitani and R. Glockshuber, ChemBioChem, 2013, 14, 1053–1057 CrossRef CAS PubMed.
- C. M. Taylor, S. E. Northfield, C. K. Wang and D. J. Craik, Tetrahedron, 2014, 70, 7669–7674 CrossRef CAS PubMed.
- K. K. Schumacher, J. Jiang and M. M. Joullié, Tetrahedron: Asymmetry, 1998, 9, 47–53 CrossRef CAS.
- M. Tamaki, G. Han and V. J. Hruby, J. Org. Chem., 2001, 66, 3593–3596 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1042476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00034c |
| This journal is © The Royal Society of Chemistry 2015 |