
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral nanostructuring of multivalent macrocycles in solution and on surfaces†
Marco
Caricato
a,
Arnaud
Delforge
b,
Davide
Bonifazi
bc,
Daniele
Dondi
a,
Andrea
Mazzanti
d and
Dario
Pasini
 *ae
*ae
aDepartment of Chemistry, University of Pavia, Viale Taramelli, 10, 27100, Pavia, Italy. E-mail: dario.pasini@unipv.it; Web: http://www.unipv.it/labt
bDepartment of Chemistry and Namur Research College (NARC), University of Namur (UNamur), Rue de Bruxelles 61, Namur, 5000, Belgium
cDepartment of Chemical and Pharmaceutical Sciences and INSTM UdR Trieste, University of Trieste, Piazzale Europa 1, Trieste, 34127, Italy
dDepartment of Industrial Chemistry “Toso Montanari”, School of Science, University of Bologna, V. Risorgimento 4, 40136, Bologna, Italy
eINSTM Research Unit, University of Pavia, Italy
First published on 20th January 2015
Abstract
We describe the design and synthesis of a novel functionality-rich, homochiral macrocycle, possessing the overall molecular D2 symmetry, in which multivalency is introduced into the covalent framework by means of four suitably positioned pyridine moieties. The macrocycle synthesis is carried out with functionalized, enantiopure 1,1′-binaphthyl synthons as the source of chirality by means of a room temperature esterification reaction as the cyclization procedure. Upon addition of Pd2+, coordination of the pyridine moieties occurs both intra and intermolecularly, to afford chiral ordered mono and dimeric macrocycles or multimeric aggregates depending on the solvents and conditions used. The metal binding event takes place in combination with a significant macrocyclic conformational rearrangement detected by circular dichroism spectroscopy. When in combination with a third component (C60), the macrocycle-Pd2+ hybrid undergoes surface-confined nanostructuring into chiral nanofibres.
Introduction
Organic nanotubes are currently attracting substantial interest: they are assembled nanospaces of suitable dimensions in which to carry out host–guest chemistry, reversible binding of smaller species for transport, storage or chemical transformation purposes; they have been utilized as biological ion channel mimics, or for addressing tailored material properties.1Conformationally stable macrocycles have been used with success in the realization of organic nanotubes, by means of their assembly in the third dimension by the supramolecular organization of suitable self-recognizing functionalities embedded into the cyclic skeleton. In order to form porous nanotubes, preformed covalent macrocycles do possess a number of advantages over smaller building blocks. The covalent cyclic skeleton can avoid the possibility of having interpenetrated networks, often observed in metal–organic frameworks, and can lead to a cavity of predetermined, and tunable, dimensions. Some molecular building blocks successfully adopted for this strategy include cyclic oligopeptides, macrocyclic oligophenyleneethynylenes, urea-containing macrocycles, aromatic oligoamides, calixarenes and cyclodextrins.2 Whereas early examples of cyclic oligopeptides incorporate alternating amino acid residues with opposed stereochemistries, resulting in achiral cyclic covalent molecules, more recent reports have explored similar cyclic systems with an overall net chirality.3
Several elegant examples of self-assembled helical chemical systems have been reported to date; the transmission of the chiral information often relies on “weak” molecular chirality (for example, aliphatic carbon chains with an asymmetric carbon atom) expressed far away from the assembly area (for example, π–π stacking of extended aromatics as the assembly mode).4 The introduction of a robust source of chirality into a suitable cyclic covalent skeleton can lead to innovative systems, in which helicity, and directionality, when transferred to the whole nanotube assembly, might give access to unprecedented properties on the nanoscale.5
We have recently reported the preparation of several chiral macrocycles incorporating Binol (1,1′-bi-2-naphthol) units, as a rigid, robust source of chirality induction. We have shown their applications for the chiroptical sensing of organic or ionic species.6 In this paper, we report on the design, synthesis, supramolecular and nanoscale organization of homochiral, Binol-based multivalent macrocycles using a metal-coordination strategy as the assembly mode.
Results and discussion
Design and synthesis
Our design approach to chiral nanoscale constructs from monomeric macrocycles containing binaphthyl units can be summarized by the following key points:(a) the use of rigid aromatic spacers joining two binaphthyl-based units, in order to define an internal, non-collapsible macrocyclic inner space, and of a minimum number of sp3 carbon atoms, which impart a higher flexibility and conformational mobility to the covalent structure with respect to sp or sp2 hybridized carbon atoms;
(b) the use of macrocyclization reactions able to generate chemical functionalities which could ensure both configurational and chemical stability to the resulting cyclic, covalent structure. Ester functionalities can ensure supramolecular “silence”, in terms of competition/interaction with other functionalities introduced within the macrocyclic framework, for assembly purposes;
(c) the coordination of the pyridine moieties to suitable PdCl2 complexes as the 3D assembly mode; this interaction, reported to form trans-coordinated complexes, has been previously used for the construction of complex supramolecular architectures, and it can ensure appropriate geometries in the assembly process.7,8
Fig. 1 illustrates graphically the concepts expressed above. The homochiral, D2 symmetrical macrocyclic skeleton (top left) contains four p-linked pyridine moieties, of which two are above (green) and two below (red) the mean plane of the macrocycle. In the presence of a suitable Pd2+ source, coordination of two pyridine units can form, in principle, different kinds of nanoscale structures: (i) closed monomers and dimers, with an overall D2 symmetry, which are feasible if the coordination can occur intramolecularly between pyridine moieties belonging to the two different binaphthyl units, facing each other, of the same macrocyclic molecule; (ii) irregular assemblies, or regular helical nanotubes, in which coordination involving the pyridine moieties occurs exclusively in an intermolecular fashion.
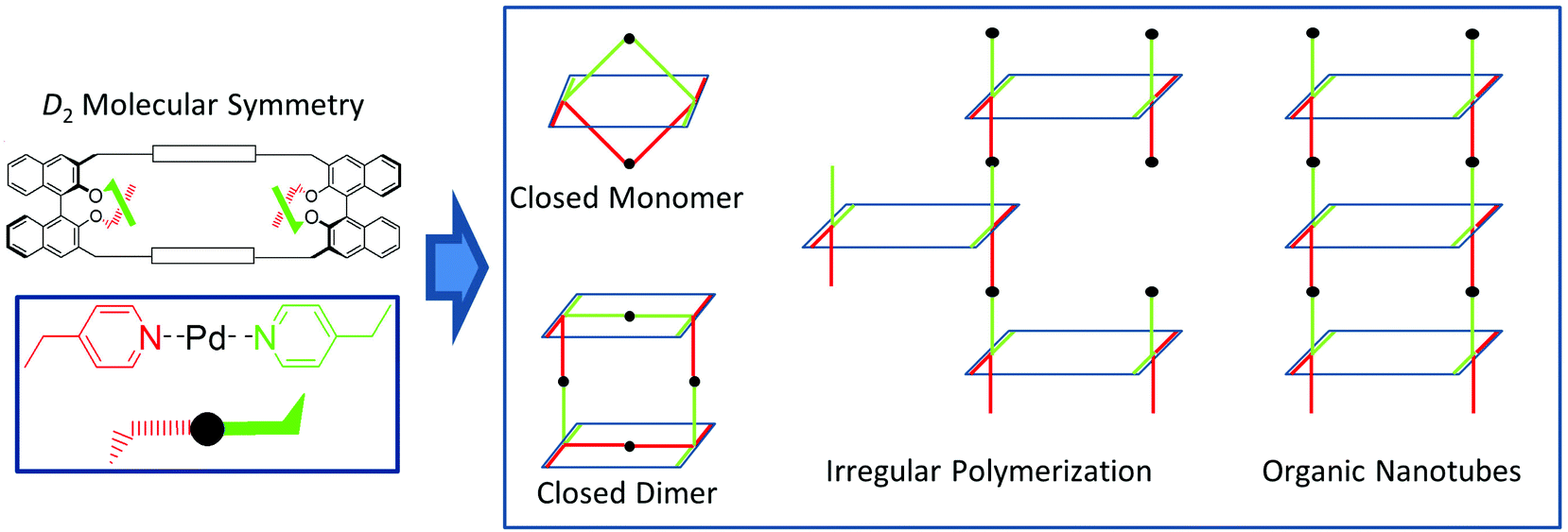 | ||
| Fig. 1 Pd2+-induced coordinative assemblies built from chiral multivalent macrocycles. | ||
The synthesis of the key macrocycle is shown in Scheme 1. We have utilized commercially available, ether bridged dicarboxylic acid 2 as the rigid spacer, deemed suitable in order to define an internal, noncollapsible cavity, and previously utilized in the construction of rigid macrocycles for nanotube assembly.2f Benzylic diol (R)-1, bearing protected methoxy functionalities in the 2,2′ positions of the binaphthyl skeleton, could be cyclized with equimolar quantities of 2 to give model macrocycle (RR)-3 in fair yields, after purification by column chromatography.
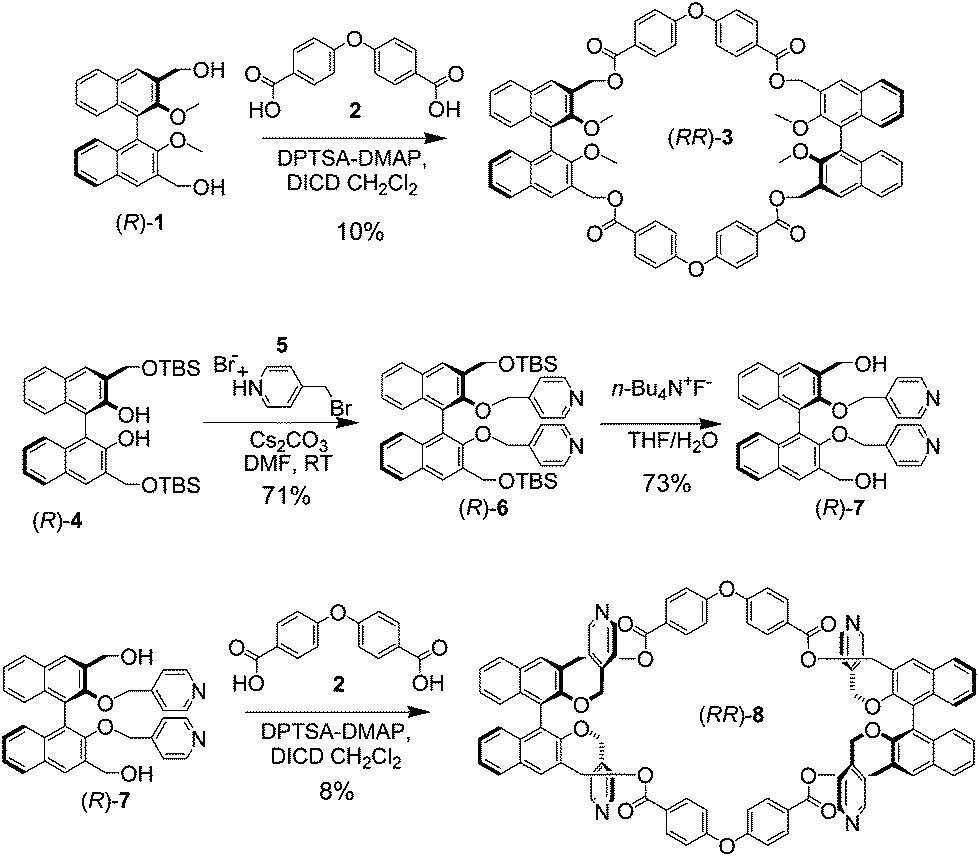 | ||
| Scheme 1 Synthesis of homochiral model macrocycle (RR)-3, and of homochiral multivalent macrocycle (RR)-8. | ||
The introduction of four pyridine functionalities within the macrocyclic framework had to be carried out convergently, since attempts to unmask the protected phenol functionalities in (RR)-3, in order to carry out postfunctionalization of the covalent structure, resulted inevitably in the degradation of the cyclic framework. The synthesis of elaborated precursor (R)-7 used our recently reported strategy6c for compound (R)-4 in excellent yields; subsequent alkylation under mildly basic conditions afforded precursor (R)-6, which could be deprotected using literature conditions to give (R)-7. The cyclization of elaborated precursor (R)-7, under the esterification conditions reported above for (R)-1, afforded macrocycle (RR)-8, after purification by column chromatography using polar eluent mixtures, in reasonable yields. In the macrocyclization reactions for the synthesis of (RR)-3 and (RR)-8, conversions of starting materials were complete; traces of higher macrocycles, and oligomeric–polymeric baseline material completed the compositions of the reaction mixtures. Precursors and macrocycles were fully characterized using NMR spectroscopy and mass spectrometric techniques. Macrocycle (RR)-8 could be stored intact for several months as a solid at 0 °C.
Spectroscopic characterization in solution
NMR, UV and CD spectroscopies were all used to characterize the adducts formed upon addition of Pd(MeCN)2Cl2 or Pd(PhCN)2Cl2, used as Pd2+ sources, to macrocycles and model compounds. Both Pd2+ salts have been previously reported for the formation of complexes in which two pyridine functionalities displace the labile nitrile ligands and leave the two chlorine ligands, to form trans-tetracoordinated, square-planar complexes.7 Both salts are fully soluble in polar organic solvents (MeCN, DMSO).1H NMR studies of macrocycle (RR)-8 in different solvents showed a complex situation which was difficult to rationalize immediately. Further complications in the interpretation of the results derived from solubility/precipitation behaviour of the adducts formed by the addition of Pd2+ by changing the concentration conditions of the samples.
When macrocycle (RR)-8 was used at relatively high concentrations (1–3 mM) in d6-DMSO by the addition of increasing amounts of Pd(MeCN)2Cl2 or Pd(PhCN)2Cl2 a completely soluble adduct was generated, and no further signal changes could be detected upon addition of more than 2 equivalents of Pd2+. Fig. 2 shows the two sets of resonances for the symmetry-related CH2 groups (bottom, two double doublets for the diastereotopic protons), becoming four different sets of resonances upon reaching two equivalents of the Pd2+ source. This splitting is in agreement with the presence of a “closed dimer” structure (Fig. 1), in which the aggregate maintains a overall D2 symmetry, but the molecular fragments (red and green in the figure) are no longer equivalent through symmetry, generating different signals in the 1H NMR spectra. To further ensure that the observed 1H NMR signals belong to the “closed dimer” proposed structure, 2D-1H-DOSY NMR spectra were recorded to obtain an estimate for the size of the aggregate and compared it to the assumed structure. The measured diffusion coefficient for the free macrocycle (D = 1.25 × 10−10 m2 s−1) was found to be essentially twice of that of the adduct formed upon addition of two equivalents of Pd(PhCN)2Cl2 (D = 6.31 × 10−11 m2 s−1) (Fig. S1, ESI†). The values can be translated by the Stokes–Einstein equation9 to a hydrodynamic radius and a dimension that is essentially double with respect to that of the macrocycle, in agreement with the calculated structures by molecular modelling (vide infra).
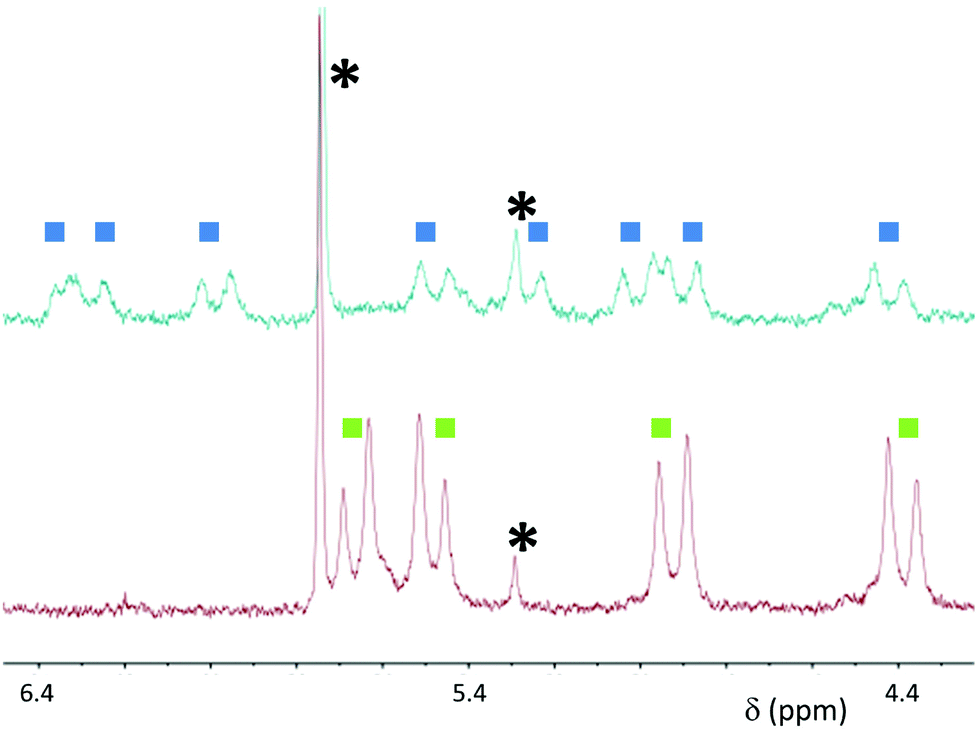 | ||
| Fig. 2 Partial 1H NMR spectra of macrocycle (RR)-8 (d6-DMSO, 3 mM, bottom) and of (RR)-8 in the presence of 2 equivalents of Pd(MeCN)2Cl2 (top). Residual solvent peaks are marked with asterisks. | ||
1H NMR studies were also performed in MeCN, in order to have a direct comparison with the titration behaviour monitored by UV and CD spectroscopies (vide infra). When macrocycle (RR)-8 was used at a relatively high concentration (1 mM) in MeCN, the addition of increasing amounts of Pd(MeCN)2Cl2 or Pd(PhCN)2Cl2 resulted in the formation of an insoluble precipitate. This observation suggested the possibility of an irregular aggregation. However, similar precipitation phenomena, at the same concentration, upon titration with Pd(MeCN)2Cl2, could be observed with control compound (R)-6. Since difunctional (R)-6 can only form linear aggregates, the precipitation suggested the possibility that the aggregate species formed in (RR)-8 could indeed be ordered, but not very soluble in the specific solvent. Even pyridine by itself in CH3CN formed, at millimolar concentrations, insoluble precipitates when Pd(MeCN)2Cl2 was added to the solution.
Control studies carried out with pyridine in MeCN at lower concentration levels (<10−4 M) showed that, upon addition of the Pd2+ source, the complex remained fully soluble; interestingly, and to our surprise, the displacement of the MeCN ligands on the Pd2+ coordinating center is not completely regiospecific. The formation of two different types of complexes, attributed to the cis and trans Pd(Py)2Cl2 square planar species, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio, respectively, is evident upon addition of more than 0.5 equivalent of Pd(MeCN)2Cl2 (Fig. S2†). In the case of difunctional compound (R)-6, under more diluted conditions (2.3 × 10−5 M), fully soluble adducts upon the addition of Pd2+ were obtained. The broadening of all proton resonances in the 1H NMR spectra supports strongly the formation of a coordination polymer with no further changes in the spectra observed when more than 1 equivalent of Pd2+ is added with respect to the ligand (R)-6. The observed stoichiometry at saturation (1:1 ligand vs. Pd2+) matches with the complete incorporation of the pyridine moieties (two per ligand) into the Pd2+ complexes (Fig. S3†).
:3 ratio, respectively, is evident upon addition of more than 0.5 equivalent of Pd(MeCN)2Cl2 (Fig. S2†). In the case of difunctional compound (R)-6, under more diluted conditions (2.3 × 10−5 M), fully soluble adducts upon the addition of Pd2+ were obtained. The broadening of all proton resonances in the 1H NMR spectra supports strongly the formation of a coordination polymer with no further changes in the spectra observed when more than 1 equivalent of Pd2+ is added with respect to the ligand (R)-6. The observed stoichiometry at saturation (1:1 ligand vs. Pd2+) matches with the complete incorporation of the pyridine moieties (two per ligand) into the Pd2+ complexes (Fig. S3†).
In the case of macrocycle (RR)-8 (Fig. 3) at a low concentration (2.8 × 10−5 M), upon addition of the Pd2+ source, no precipitation occurred and 1H NMR spectroscopy revealed the assembly of soluble aggregates; these aggregates are in part polymeric in nature, as testified by the severe broadening of the baseline. A highly symmetrical adduct is also present, which possesses the same sets of proton resonances as the monomer macrocycle.
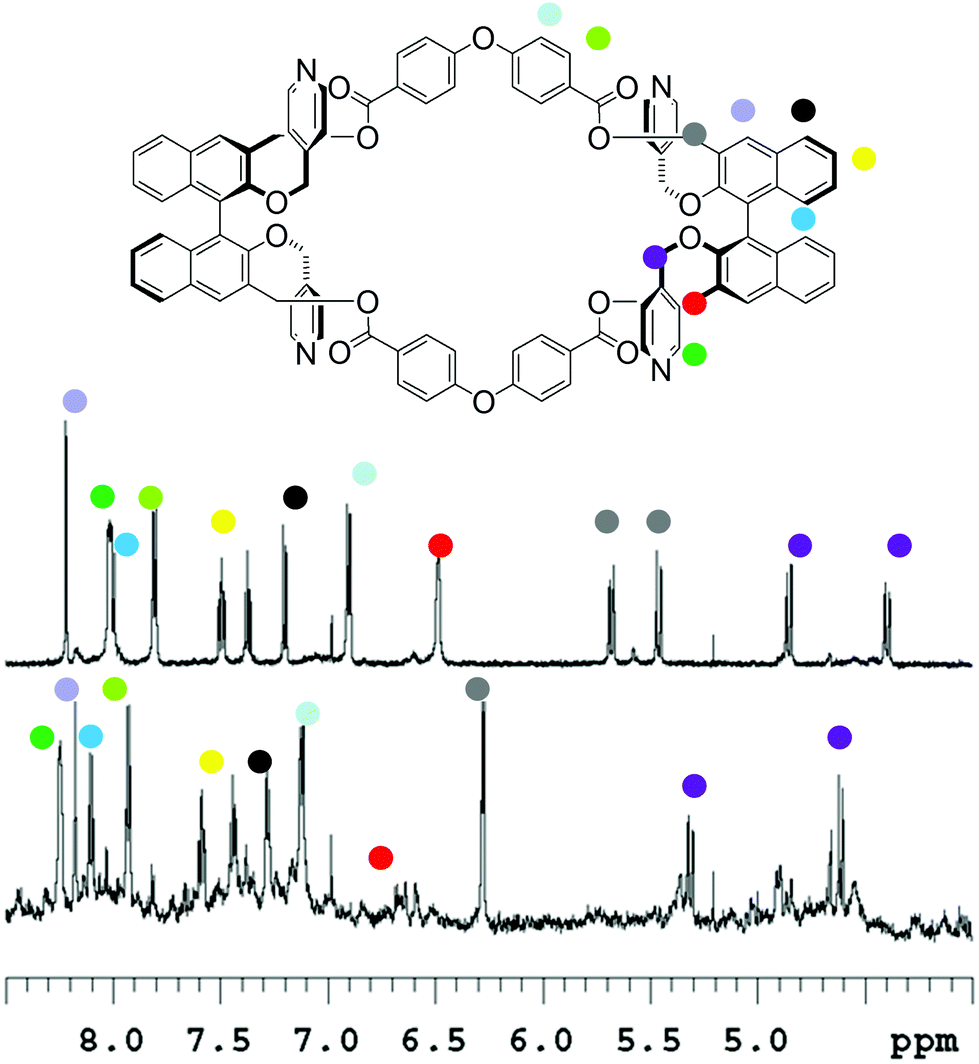 | ||
| Fig. 3 1H NMR of (in d3-MeCN at 25 °C; top) macrocycle (RR)-8 (2.8 × 10−5 M, 500 MHz); (bottom) same with 2 equiv. of Pd(MeCN)2Cl2 added. | ||
We have attributed this adduct to the “closed monomer” structure (Fig. 1), since in this structure “red” and “green” molecular fragments are equivalent by symmetry operations, therefore do not generate any splitting in the NMR pattern. Together with the downfield shifts of the α-pyridyl protons, and the broadening of the β-pyridyl protons under the baseline, which could be expected upon Pd2+ coordination, substantial variations in chemical shifts could be observed for the signals attributable to the –CH2– protons, testifying the high degree of conformational rearrangement that the macrocycle has to undertake in order to form this particular structure.
MeCN as a solvent has ideal UV cutoff properties for the study of the systems presented in this paper. In fact, three distinct chromophoric units are present, and they possess characteristic UV bands; since they are joined by sp3 carbon atoms, they are not in conjugation with each other, so that the specific electronic transitions should persist in the UV spectra of the precursors and macrocycles. The major absorption band (Fig. S4†), centered around 230 nm can be attributed to the binaphthyl chromophore, with molar absorptivities (logε between 4 and 5) fully in line with other non-conjugated binaphthyl synthons.6b,c,e The band centered at 270 nm present in macrocycles (RR)-3 and (RR)-8 is assigned to the spacer chromophore unit, given the fact it is completely absent in precursor (R)-6, and also by comparison with published UV spectra of alkyl ester derivatives of 2.10 The band of the pyridine chromophores is known to be low in intensity11 and it is probably hidden by the broad band of the binaphthyl units. Circular dichroism spectroscopy of the same compounds shows the exciton couplet typical of binaphthyl moieties (Fig. S5†), corresponding to the maximum absorption band in the UV spectra. No induced CD signal could be observed in the UV bands of the other chromophores in either macrocycles (RR)-3 and (RR)-8 or the precursors.
The UV/Vis titration was complicated by the inherent absorption of the Pd2+ source (Fig. S6†) in the absorbance region of the chromophores belonging to the macrocycle (RR)-8. Upon addition of Pd(MeCN)2Cl2 to (RR)-8, at concentrations similar to those of the 1H NMR titration, small but substantial shifts in the silent absorbance region of the added Pd2+ species (310 nm), fully saturating at 2 equivalents, could be detected (Fig. 4, top).
 | ||
| Fig. 4 Top: UV titration of macrocycle (RR)-8 (1.33 × 10−5 M) with Pd(MeCN)2Cl2 in MeCN at 25 °C. Inset: titration profile vs. Pd2+ added equivalents at 310 nm. Bottom: CD titration of macrocycle (RR)-8 (1.33 × 10−6 M) with Pd(MeCN)2Cl2 in MeCN at 25 °C. Inset: titration profile vs. Pd2+ added equivalents at 269 nm. | ||
Monitoring of the titration using CD spectroscopy was certainly more informative. An intense induced CD band appeared in correspondence with the band of the spacer chromophore unit at ca. 270 nm, and saturates upon the addition of two equivalents of Pd2+. The exciton couplet band attributable to the binaphthyl unit shows little variation in its highest energy branch at 220 nm. An intense induced CD activity could also be detected between 230 and 250 nm, and could be attributed to the chromophore band arising from the formation of the Pd2+ complex with the pyridines. This band, which is partially superimposed with the lowest energy branch of the binaphthyl couplet, and with the other band centered at 270 nm, does not saturate at 2 equivalents; a continuous shift of this band is presumably signalling a shift of the equilibrium between closed monomer/polymeric aggregates upon addition of the Pd2+ source in solution. The induced CD signal in the bands of the chromophore spacer ester and of the Pd2+ complex with the pyridine moieties indicates an overall chirality of the closed monomer/polymeric aggregates as a whole, and it stresses the high degree of conformational rearrangement occurring, upon coordination, involving all moieties of the macrocycle.
Control experiments with compound (R)-6 were carried out under identical conditions. The CD spectra revealed very little changes upon complexation, indicating how difunctional (R)-6 does not have to change its conformation in the formation of the coordination oligomeric/polymeric adduct (Fig. S7†).
Surface assembly studies
Atomic force microscopy is a powerful tool for the detection and visualization of assembled nanoscale structures.12 To further investigate and substantiate the behavior of the systems described here, AFM experiments were performed at the air/HOPG (Highly Ordered Pyrolytic Graphite) interface. AFM samples were prepared by drop-casting appropriate solutions onto freshly prepared surfaces. The samples were air-dried and imaged with an AFM apparatus in the tapping mode. In preliminary experiments, samples were prepared with two equivalents of PdCl2(CH3CN)2vs. macrocycle (RR)-8, in CHCl3 at diluted (<10−5 M) concentrations. No interesting nanoscale aggregate could be observed under these conditions. In view of the capability of similar macrocycles to form stable complexes with C60,6b,e,j solutions containing one equivalent of pristine C60vs. macrocycle (RR)-8 were realized; in this case, the solution of C60 with the macrocycle was first premixed, and the solution of PdCl2(MeCN)2 was added several hours later. Upon deposition, a nanostructured material could be observed by AFM. The images show elongated fibres together with amorphous clusters. The nanostructured materials are essentially composed of monodimensional structures with dimensions of 10–20 μm in length and 4 nm in height (Fig. S8†). The last value is higher than the one measured by molecular modelling (between 1 and 2 nm, see below). In fact, by depositing the same solutions a few days after, AFM images show different fibres. They are shorter (between 0.5 and 2 μm) and the profiles indicate heights between 0.5 and 1 nm (Fig. 5), thus suggesting a time-dependent evolutive process.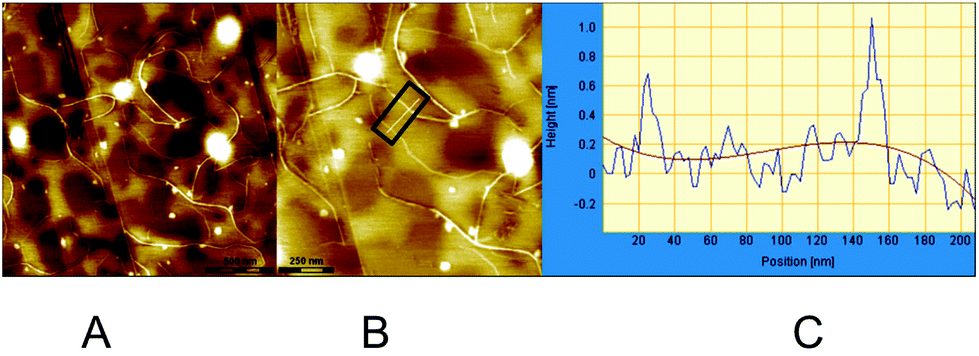 | ||
| Fig. 5 AFM images of (RR)-8, C60 and PdCl2(MeCN)2 at the HOPG interface. (A and B) Example of fibres. (C) A height profile measured along the white line (highlighted in black box) in (B). | ||
On observing the same sample after three months, we found long fibers (Fig. 6) possessing the molecular height of the single aggregates (1–1.5 nm).
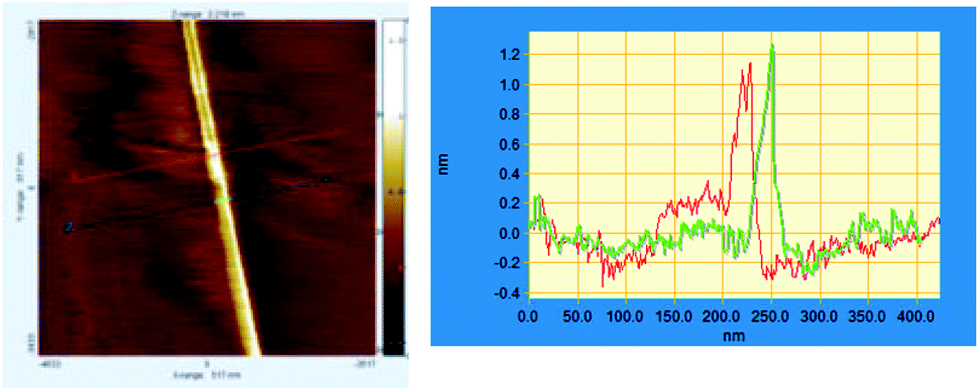 | ||
| Fig. 6 AFM images of (RR)-8, C60 and PdCl2(MeCN)2 visualized after a few months. Left: example of fibres. Right: a height profile measured along the colored lines. | ||
Control experiments of the single components and of their binary mixtures only showed amorphous materials. We also performed control experiments with difunctional compound (R)-6, again obtaining no nanostructured fibers under the same conditions as those used for macrocycle (RR)-8.
These observations are in agreement with the formation of an ordered nanostructured material, in which the nanoscale fibres are formed by single macrocyclic units piled up along a preferential dimension; the results exclude the formation of a fully crosslinked network, yet they do not support the formation of macroscopic chiral nanotubes on surfaces. Unfortunately, due to some limitations in the solubility (see above), we could not perform any studies in solution. On the other hand it is likely that the fullerene spheres are included in the macrocycle-Pd2+ coordination complexes, thus acting as templates directing the growth and the assembly of the chiral monomolecular fibres on the surface.
Molecular modelling
Calculations were initially performed on macrocycle (RR)-8 using the semi-empirical method PM3 to locate local minima. The other calculations were performed by inserting the Pd2+ atoms, the structures were then preliminarily optimized with PM3, and refined with DFT B3LYP/LANL2DZ.Initially, the trans coordination of the pyridine moieties and of the two chlorine atoms as ligands on the Pd2+ center was imposed a priori before further optimization. Even after using the preliminary PM3 screening, it appeared evident that the intramolecular positioning of two pyridine moieties belonging to two different binaphthyl units of the same macrocycle afforded a plausible and relatively stable conformer. Instead, the possible complexation of Pd2+ by two pyridines attached to the same binaphthyl unit, either in a cis or a trans complexation mode, could be thoroughly excluded. Upon optimization with DFT, two low energy conformers could be located (A and B in Fig. 7), with the form B being more stable by about 5 kcal mol−1.
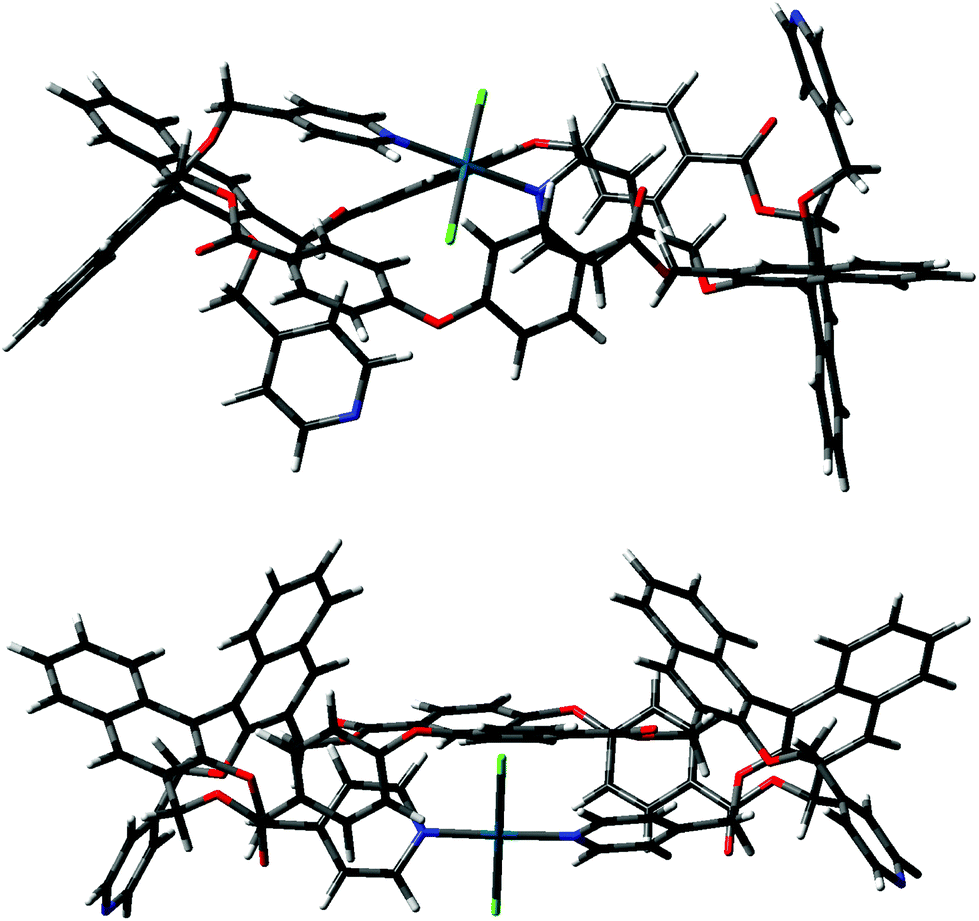 | ||
| Fig. 7 Minimized conformers A (top) and B (bottom) for (RR)-8·PdCl2. | ||
The difference between the two depends on which pyridine attached to the naphthyl unit of the facing binaphthyls is involved in the intramolecular trans complexation on the Pd2+ center. In form A, the two pyridines involved are the one above and the one below the mean plane of the macrocycle (red and green in Fig. 1), whereas in form B they are both above the mean plane of the macrocycle (both green according to Fig. 1). The molecules show similar dihedral angles for the binaphthyl units (79 and 84° for A, 86 and 82° for B) and identical bond angles for the spacer oxygen atom (PhOPh angles of 122° in all cases). The overall dimensions of the minimized structures are similar in both cases (2.6 × 1.7 nm maximum dimensions for conformer A; 2.4 × 1.3 nm maximum dimensions for conformer B). The main difference is that in form B, the two pyridine moieties not involved in the complexation are predisposed for further joining of another similar unit. In fact, conformer B can easily close to give a stable dimer (the “closed dimer” form in Fig. 1), which is shown in Fig. 8. The modelling of “closed dimer” and of conformer B was then performed independently; we did not observe any significant changes in the geometry of the intramolecularly complexed macrocycle in either monomer or dimer, as to testify that the macrocyclic structure does not have to undergo a high degree of conformational rearrangement to close to a dimeric structure.
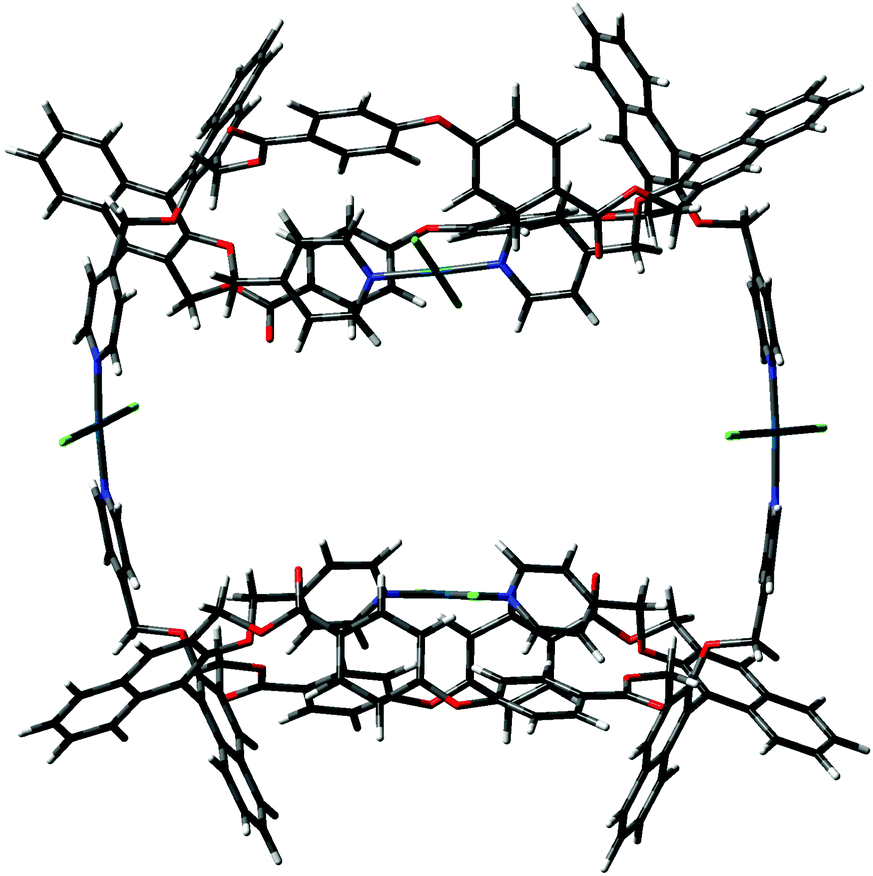 | ||
| Fig. 8 Minimized conformer for the “closed dimer”. | ||
On the other hand, it is likely that the conformer A, with two pyridine moieties pointing away from the macrocyclic structure in opposite directions, is the one involved in the formation of the nanostructured aggregates observed in solution and on surfaces.
In order to rationalize the “closed monomer” structure, which was observed under some of the conditions reported before, calculations were performed on macrocycle (RR)-8 by imposing a cis geometry for the intramolecular Pd2+ complex stabilized by two pyridine moieties of the facing binaphthyls. We have verified that this structure is plausible, both in terms of stability and symmetry, with the proposed structure shown in Fig. 1. The lowest energy conformer in this case is shown in Fig. 9.
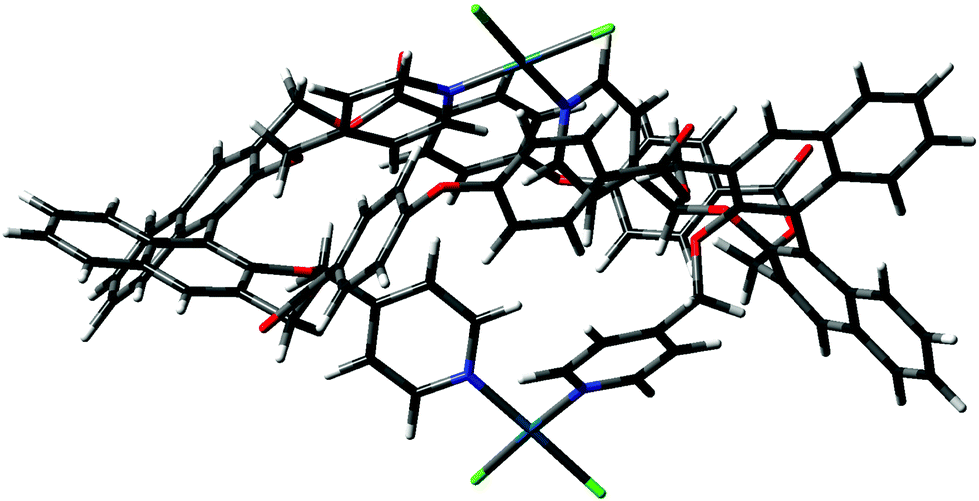 | ||
| Fig. 9 Minimized conformer for the “closed monomer”. | ||
Conclusions
The realization of chiral organic nanotubes able to organize in an helically oriented manner is of increasing scientific interest, since properties at the nanoscale, including long range directional propagation of (chiral) information, in solution and on surfaces, can be highly rewarding. Towards this target, we have studied a new multivalent homochiral macrocycle, predisposed for assembly in 3D in which chirality is robustly expressed at the molecular level. The engineered system is capable of forming nanoscale structures with a certain level of order, both in solution and on the surfaces. We are currently working towards the design of improved macrocyclic scaffolds, to be tailored and perfectioned for nanoscale transport, catalysis and sensing applications.Experimental part
General experimental
All commercially available compounds were purchased from commercial sources and used as received. Compounds 4-dimethylaminopyridinium p-toluenesulfonate (DPTSA-DMAP),13 (R)-1,14 and (R)-46c were prepared according to the literature procedures. THF (Na) and CH2Cl2 (CaH2) were dried before use. Analytical thin layer chromatography was performed on silica gel, chromophore loaded, commercially available plates. Flash chromatography was carried out using silica gel (pore size 60 Å, 230–400 mesh). 1H and 13C NMR spectra were recorded from solutions in CDCl3 on 200, 300 MHz or 500 MHz spectrometers with the solvent residual proton signal or tetramethylsilane as a standard. The UV/Vis spectroscopic studies were carried out using commercially available spectrophotometers. Mass spectra were recorded using an electrospray ionization instrument (ESI). Optical rotations were measured on a polarimeter in a 10 cm cell with a sodium lamp (λ = 589 nm) and are reported as follows: [α]rtD (c = mg(mL)−1, solvent). CD spectra were recorded at 25 °C at a scanning speed of 50 nm min−1 and were background corrected. Each spectrum is the averaged signal of four consecutive scans.Procedure for NMR, UV and CD titrations
The titration experiments were conducted as follows: to a stock solution of macrocycles or control compounds (solution A) were added several aliquots of the Pd2+ salt (solution B). Solution B is formed by the Pd2+ salt (at higher concentration) dissolved in solution A, in order to maintain one species always at the same, constant concentration.Procedure for AFM studies
Samples related to Fig. 5 and 6 were prepared as follows: a solution of macrocycle (RR)-8 (0.5 mL, 10−4 M in CH2Cl2) and a solution of C60 (0.5 mL, 10−4 M in CHCl3) were mixed and heated at 35 °C for 40 min. Then, a solution of Pd(MeCN)2Cl2 (0.5 mL, 2 × 10−4 M in CH2Cl2) was added. The following day, this solution was diluted 10× and 100× with CHCl3 to give the following concentrations: (RR)-8/Pd(MeCN)2Cl2/C60 0.33 × 10−6 M/0.66 × 10−6 M/0.33 × 10−6 M. A single drop of the solutions was added on the freshly cleaved surface of HOPG to realize the sample. Experiments were realized after total evaporation of the solvent. TM-AFM measurements on the Au substrates were carried out in air at 298 K by using Nanoscope V (model MMAFMLN, Digital Instrument Metrology Group). The tips used in all measurements were antimony-doped silicon cantilevers (T = 3.5–4.5 μm, L = 115–135 μm, fo = 271–305 kHz, k = 20–80 N m−1, Bruker) at a resonant frequency of ca. 280 kHz. The collected images were then analyzed with WsxM 5.0 software (Nanotec Electronica S. L.) to obtain the cross-sectional values and profiles of processed images.Acknowledgements
Support from the University of Pavia, MIUR (Programs of National Relevant Interest PRIN grants 2004-033354 and 2009-A5Y3N9), the CINECA Supercomputer Center (ISCRA NANOCHIR project HP10CKIGGH), and, in part, from CARIPLO Foundation (2007–2009) and INSTM-Regione Lombardia (2010–2012 and 2013–2015) is gratefully acknowledged. D.B. gratefully acknowledges the EU through the ERC Starting Grant “COLORLANDS” project, the FRS-FNRS (FRFC contract no. 2.4.550.09), the Science Policy Office of the Belgian Federal Government (BELSPO-IAP 7/05 project), the “TINTIN” ARC project (09/14-023). D.P. wishes to thank Prof. Linda Shimizu and Dr Kinkini Roy at the University of South Carolina, USA, for performing the DOSY experiments.Notes and references
- (a) M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed; (b) D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS; (c) D. Pasini and M. Ricci, Curr. Org. Synth., 2007, 4, 59–80 CrossRef CAS; (d) R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC; (e) S. Leclair, P. Baillargeon, R. Skouta, D. Gauthier, Y. Zhao and Y. L. Dory, Angew. Chem., Int. Ed., 2004, 43, 349–353 CrossRef CAS PubMed; (f) G. Dan Pantos, P. Pengo and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 194–197 CrossRef PubMed; (g) Y. Baudry, G. Bollot, V. Gorteau, S. Litvinchuk, J. Mareda, M. Nishihara, D. Pasini, F. Perret, D. Ronan, N. Sakai, M. R. Shah, A. Som, N. Sorde, P. Talukdar, D.-H. Tran and S. Matile, Adv. Funct. Mater., 2006, 16, 169–179 CrossRef CAS; (h) K. Sakaguchi, T. Kamimura, H. Uno, S. Mori, S. Ozako, H. Nobukuni, M. Ishida and F. Tani, J. Org. Chem., 2014, 79, 2980–2992 CrossRef CAS PubMed.
- (a) C. Grave and A. D. Schlüter, Eur. J. Org. Chem., 2002, 3075–3098 CrossRef CAS; (b) Y. Yamaguchi and Z.-I. Yoshida, Chem. – Eur. J., 2003, 9, 5430–5440 CrossRef CAS PubMed; (c) S. Höger, Chem. – Eur. J., 2004, 10, 1320–1329 CrossRef PubMed; (d) D. Zhao and J. S. Moore, Chem. Commun., 2003, 807–818 RSC; (e) Y. Xu, M. D. Smith, M. F. Geer, P. J. Pellechia, J. C. Brown, A. C. Wibowo and L. S. Shimizu, J. Am. Chem. Soc., 2010, 132, 5334–5335 CrossRef CAS PubMed; (f) L. S. Shimizu, A. D. Hughes, M. D. Smith, M. J. Davis, B. P. Zhang, H.-C. zur Loye and K. D. Shimizu, J. Am. Chem. Soc., 2003, 125, 14972–14973 CrossRef CAS PubMed; (g) A. J. Helsel, A. L. Brown, K. Yamato, W. Feng, L. Yuan, A. J. Clements, S. V. Harding, G. Szabo, Z. Shao and B. Gong, J. Am. Chem. Soc., 2008, 130, 15784–15785 CrossRef CAS PubMed; (h) B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed; (i) L. S. Shimizu, S. R. Salpage and A. A. Korous, Acc. Chem. Res., 2014, 47, 2116–2127 CrossRef CAS PubMed; (j) A. Ikeda and S. Shinkai, J. Chem. Soc., Chem. Commun., 1994, 2375–2376 RSC; (k) G. Gattuso, S. Menzer, S. A. Nepogodiev, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1997, 36, 1451–1454 CrossRef CAS; (l) B. H. Hong, S. C. Bae, C.-W. Lee, S. Jeong and K. S. Kim, Science, 2001, 294, 348–351 CrossRef CAS PubMed; (m) V. G. Organo, A. V. Leontiev, V. Sgarlata, H. V. R. Dias and D. R. Rudkevich, Angew. Chem., Int. Ed., 2005, 44, 3043–3047 CrossRef CAS PubMed; (n) M. Boccalon, E. Iengo and P. Tecilla, J. Am. Chem. Soc., 2012, 134, 20310–20313 CrossRef CAS PubMed; (o) P. J. Stang, D. H. Cao, S. Saito and A. M. Arif, J. Am. Chem. Soc., 1995, 117, 6273–6283 CrossRef CAS.
- (a) L. Fischer, M. Decossas, J. P. Briand, C. Didierjean and G. Guichard, Angew. Chem., Int. Ed., 2009, 48, 1625–1628 CrossRef CAS PubMed; (b) A. Hennig, L. Fischer, G. Guichard and S. Matile, J. Am. Chem. Soc., 2009, 131, 16889–16895 CrossRef CAS PubMed.
- (a) J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed; (b) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (c) D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC; (d) J. Kumaki, S. Sakurai and E. Yashima, Chem. Soc. Rev., 2009, 38, 737–746 RSC; (e) J. Clayden, Chem. Soc. Rev., 2009, 38, 817–829 RSC; (f) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed; (g) M.-Q. Zhao, Q. Zhang, G.-L. Tian and F. Wei, Nanoscale, 2014, 6, 9339–9354 RSC.
- (a) L. Pu, Chem. Rev., 1998, 98, 2405–2494 CrossRef CAS PubMed; (b) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3211 CrossRef CAS PubMed; (c) P. Kocovsky, S. Vyskocyl and M. Smrcina, Chem. Rev., 2003, 103, 3213–3245 CrossRef CAS PubMed; (d) J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef CAS PubMed; (e) A. Shockravi, A. Javadi and E. Abouzari-Lotf, RSC Adv., 2013, 3, 6717–6746 RSC; (f) M. Caricato, A. K. Sharma, C. Coluccini and D. Pasini, Nanoscale, 2014, 6, 7165–7174 RSC.
- (a) A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428–10430 RSC; (b) C. Coluccini, D. Dondi, M. Caricato, A. Taglietti, M. Boiocchi and D. Pasini, Org. Biomol. Chem., 2010, 8, 1640–1649 RSC; (c) M. Caricato, N. J. Leza, K. Roy, D. Dondi, G. Gattuso, L. S. Shimizu, D. A. Vander Griend and D. Pasini, Eur. J. Org. Chem., 2013, 6078–6083 CrossRef CAS; (d) M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS PubMed; (e) M. Caricato, C. Coluccini, D. Dondi, D. A. VanderGriend and D. Pasini, Org. Biomol. Chem., 2010, 8, 3272–3280 RSC; (f) A. Moletti, C. Coluccini, D. Pasini and A. Taglietti, Dalton Trans., 2007, 1588–1592 RSC; (g) C. Coluccini, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2010, 8, 1807–1815 RSC; (h) C. Coluccini, A. Castelluccio and D. Pasini, J. Org. Chem., 2008, 73, 4237–4240 CrossRef CAS PubMed; (i) S. Colombo, C. Coluccini, M. Caricato, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2010, 66, 4206–4211 CrossRef CAS PubMed; (j) A. Pacini, M. Caricato, S. Ferrari, D. Capsoni, A. Martínez de Ilarduya, S. Muñoz-Guerra and D. Pasini, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4790–4799 CrossRef CAS; (k) M. Caricato, S. Díez González, I. Arandia Ariño and D. Pasini, Beilstein J. Org. Chem., 2014, 10, 1308–1316 CrossRef CAS PubMed; (l) M. Caricato, N. J. Leza, C. Gargiulli, G. Gattuso, D. Dondi and D. Pasini, Beilstein J. Org. Chem., 2012, 8, 967–976 CrossRef CAS PubMed; (m) M. Agnes, A. Sorrenti, D. Pasini, K. Wurst and D. B. Amabilino, CrystEngComm, 2014, 16, 10131–10138 RSC; (n) M. Crespo Alonso, M. Arca, F. Isaia, R. Lai, V. Lippolis, S. K. Callear, M. Caricato, D. Pasini, S. J. Coles and M. C. Aragoni, CrystEngComm, 2014, 16, 8582–8590 RSC.
- For examples of pyridine moieties forming trans PdCl2 coordination complexes, see: (a) Z. Qin, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2001, 40, 6220–6228 CrossRef CAS PubMed; (b) D. Y. K. Lee, M. H. W. Lam and W.-Y. Wong, New J. Chem., 2002, 26, 330–335 RSC; (c) R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 359 Search PubMed; (d) J. Rajput, J. R. Moss, A. T. Hutton, D. T. Hendricks, C. E. Arendse and C. Imrie, J. Organomet. Chem., 2004, 689, 1553–1568 CrossRef CAS PubMed; (e) Y. M. Na, T. H. Noh, I. S. Chun, Y.-A. Lee, J. Hong and O.-S. Jung, Inorg. Chem., 2008, 47, 1391–1396 CrossRef CAS PubMed; (f) A. Bacchi, M. Carcelli, T. Chiodo and F. Mezzadri, CrystEngComm, 2008, 10, 1916–1927 RSC; (g) T. J. Burchell, D. J. Eisler and R. J. Puddephatt, Dalton Trans., 2005, 268–272 RSC; (h) R. Wang, L. Xu, J. Ji, Q. Shi, Y. Li, Z. Zhou, M. Hong and A. S. C. Chan, Eur. J. Inorg. Chem., 2005, 751–758 CrossRef CAS; (i) H. M. Lee and C.-Y. Liao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, M1447–MU932 CAS; (j) C. Coluccini, P. Metrangolo, M. Parachini, D. Pasini, G. Resnati and P. Righetti, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5202–5213 CrossRef CAS; (k) L. Pirondini, D. Bonifazi, E. Menozzi, E. Wegelius, K. Rissanen, C. Massera and E. Dalcanale, Eur. J. Org. Chem., 2001, 2311–2320 CrossRef CAS; (l) Y. S. Chong, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2001, 123, 7463–7464 CrossRef CAS; (m) M. T. Stone and J. S. Moore, J. Am. Chem. Soc., 2005, 127, 5928–5935 CrossRef CAS PubMed; (n) G. Giachi, M. Frediani, W. Oberhauser and E. Passaglia, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4708–4713 CrossRef CAS.
- For the use of Pd2+ sources in the construction of nanoscale architectures, see: (a) S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058–10059 CrossRef CAS PubMed; (b) B. A. Blight, J. A. Wisner and M. C. Jennings, Angew. Chem., Int. Ed., 2007, 46, 2835–2838 CrossRef CAS PubMed; (c) C. Gütz, R. Hovorka, G. Schnakenburg and A. Lützen, Chem. – Eur. J., 2013, 19, 10890–10894 CrossRef PubMed; (d) B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS; (e) N. L. S. Yue, D. J. Eisler, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2004, 43, 7671–7681 CrossRef CAS PubMed; (f) K. Suzuki, K. Takao, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2544–2546 CrossRef CAS PubMed; (g) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–907 CrossRef CAS PubMed; (h) B. Olenyuk and P. J. Stang, Acc. Chem. Res., 1997, 30, 502–518 CrossRef.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- A. Ulubelen, G. Topcu and N. Tan, Phytochemistry, 1995, 40, 1473–1475 CrossRef CAS.
- 4-Alkylpyridines feature absorption signals centered at 254 nm, with low molar absorptivities (ca. 2500): G. Viscardi, P. Savarino, P. Quagliotto, E. Barni and M. Botta, J. Heterocycl. Chem., 1996, 33, 1195–1200 CrossRef CAS.
- (a) A. Llanes-Pallas, K. Yoosaf, H. Traboulsi, J. Mohanraj, T. Seldrum, J. Dumont, A. Minoia, R. Lazzaroni, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2011, 133, 15412–15424 CrossRef CAS PubMed; (b) J. Kumaki, E. Yashima, G. Bollot, J. Mareda, S. Litvinchuk and S. Matile, Angew. Chem., Int. Ed., 2005, 44, 6154–6157 CrossRef CAS PubMed.
- J. S. Moore and S. I. Stupp, Macromolecules, 1990, 23, 65–70 CrossRef CAS.
- H. T. Stock and R. M. Kellogg, J. Org. Chem., 1996, 61, 3093–3105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional UV, CD and NMR spectra, and copies of NMR and mass spectra for all newly synthesized compounds. See DOI: 10.1039/c4ob02643h |
| This journal is © The Royal Society of Chemistry 2015 |