
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of the active site of β4GalT7: modifications of the aglycon of aromatic xylosides†
Anna
Siegbahn‡
a,
Karin
Thorsheim‡
a,
Jonas
Ståhle
b,
Sophie
Manner
a,
Christoffer
Hamark
b,
Andrea
Persson
c,
Emil
Tykesson
c,
Katrin
Mani
c,
Gunilla
Westergren-Thorsson
c,
Göran
Widmalm
b and
Ulf
Ellervik
*a
aCenter for Analysis and Synthesis, Center for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: ulf.ellervik@chem.lu.se
bDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden
cDepartment of Experimental Medical Science, Lund University, BMC, SE-221 00 Lund, Sweden
First published on 28th January 2015
Abstract
Proteoglycans (PGs) are macromolecules that consist of long linear polysaccharides, glycosaminoglycan (GAG) chains, covalently attached to a core protein by the carbohydrate xylose. The biosynthesis of GAG chains is initiated by xylosylation of the core protein followed by galactosylation by the galactosyltransferase β4GalT7. Some β-D-xylosides, such as 2-naphthyl β-D-xylopyranoside, can induce GAG synthesis by serving as acceptor substrates for β4GalT7 and by that also compete with the GAG synthesis on core proteins. Here we present structure–activity relationships for β4GalT7 and xylosides with modifications of the aromatic aglycon, using enzymatic assays, cell studies, and molecular docking simulations. The results show that the aglycons reside on the outside of the active site of the enzyme and that quite bulky aglycons are accepted. By separating the aromatic aglycon from the xylose moiety by linkers, a trend towards increased galactosylation with increased linker length is observed. The galactosylation is influenced by the identity and position of substituents in the aromatic framework, and generally, only xylosides with β-glycosidic linkages function as good substrates for β4GalT7. We also show that the galactosylation ability of a xyloside is increased by replacing the anomeric oxygen with sulfur, but decreased by replacing it with carbon. Finally, we propose that reaction kinetics of galactosylation by β4GalT7 is dependent on subtle differences in orientation of the xylose moiety.
1 Introduction
Proteoglycans (PGs) are highly anionic macromolecules that consist of one or several long linear polysaccharides, glycosaminoglycan chains (GAGs), covalently attached to a core protein. Two classes of GAG chains, chondroitin sulfate (CS)/dermatan sulfate (DS) and heparin/heparan sulfate (HS), are linked to the core protein by the carbohydrate xylose. The biosynthesis of these are initiated by xylosylation of the core protein by xylosyltransferases (XT). Two galactose residues are then added stepwise to the xylosylated protein by two different galactosyltransferases, β4GalT7 (GalT-I)1 and β3GalT6 (GalT-II),2 followed by addition of one glucuronic acid unit, by a glucuronyltransferase (GlcAT-I), to form a tetrasaccharide linker, i.e. GlcA(β1→3)Gal(β1→3)Gal(β1→4)Xylβ-PG (Fig. 1a). This linker is then polymerized from repeating disaccharide units. Furthermore, during and after the polymerization of the GAG chains, modifications, such as epimerization, O- and N-sulfation, and N-deacetylation, result in an extensive structural diversity of the GAG chains.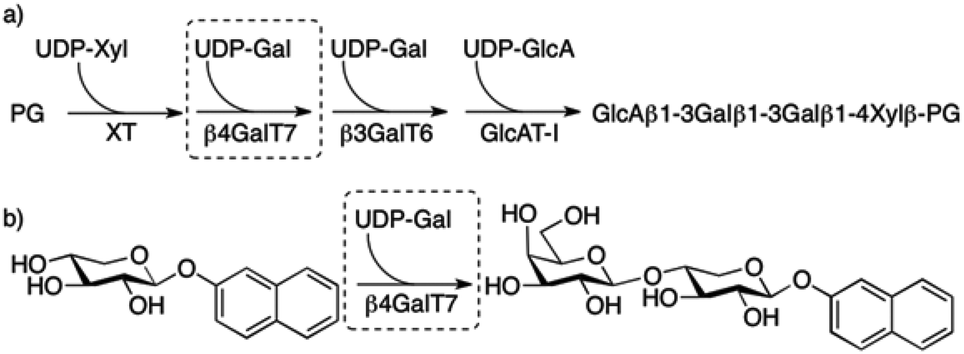 | ||
| Fig. 1 (a) Biosynthesis of the linker tetrasaccharide of HS and CS/DS PGs. (b) Galactosylation of XylNap to form GalXylNap. | ||
β-D-Xylosides, such as 2-naphthyl β-D-xylopyranoside (1, XylNap), serve as substrates for β4GalT7 and can thereby initiate formation of soluble GAG chains (Fig. 1b), and at the same time compete with GAG synthesis on core proteins forming PGs.
All enzymes that are known to be involved in the biosynthesis of the tetrasaccharide linker have been cloned,3 and we have recently reported assays for measurement of galactosylation by, and inhibition of, the enzyme β4GalT7.4 We investigated the structure–activity relationships for modifications of the xylose moiety and found that the binding pocket of β4GalT7 is narrow, with a precise set of important hydrogen bonds. We have also shown that xylose appears to be the optimal acceptor substrate for galactosylation by the enzyme and that modifications on the xylose moiety of the β-D-xylopyranosides instead can render inhibitors of galactosylation.
The biological functions of PGs are mainly due to interactions of GAG chains with different protein ligands and regulatory factors, which make them important for several normal cellular processes, but also for cancer growth, invasion, and metastasis.5 A possible therapeutic strategy may thus be to target key enzymes in the GAG biosynthesis. For example, tumor growth has been blocked by treatment of a HSPG-inhibiting xyloside in combination with an inhibitor of the formation of polyamines,6 and we have earlier shown xyloside-induced, selective inhibition of the growth of tumor cells in vitro as well as in vivo.7,8
We envision that inhibitors of β4GalT7 will be valuable tools for the exploration of GAG biosynthesis and a starting point for development of anti-tumor agents. Galactosyltransferase inhibitors are often based on donor substrate analogs, i.e. UDP-Gal.9 By instead using inhibitors based on the acceptor substrate, in this case xylose, an increased selectivity can be obtained. To broaden the knowledge of the structural requirements for β4GalT7, we now present data for modifications of the aglycon according to Chart 1.
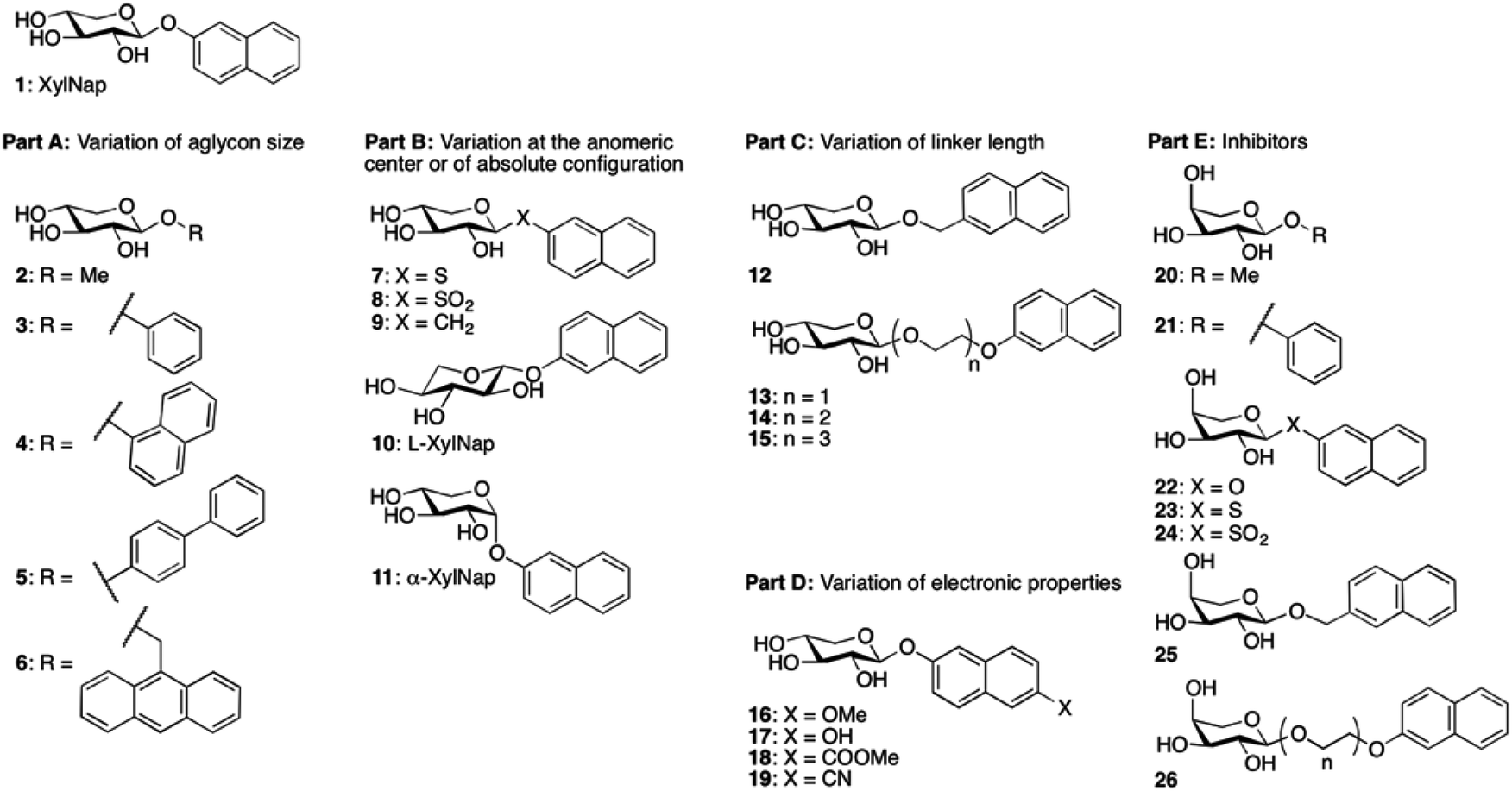 | ||
| Chart 1 Investigated glycosides. | ||
In our recent study4 we also observed strong inhibition of β4GalT7 by 2-naphthyl α-L-arabinoside, a xyloside analog epimerized at position 4. Thus, in addition to xylosides, we have included arabinosides to investigate inhibition of β4GalT7.
2 Results
2.1 Synthesis
Compounds 1,102,119,1212–15,1316,717,1418,1521,16 and 2217 have been reported previously, and compounds 2 and 20 are commercially available. Compounds 3, 4, 5, and 7 have been published before,10 however, full physical characterization has not been reported, hence, the syntheses and the analyses are described herein. The majority of the xylosides were synthesized through glycosylation of peracetylated β-D-xylopyranose 27 with different aromatic aglycons (phenol, 1-naphthol, 4-phenylphenol, 6-cyano-2-naphthol, and 2-naphthalenethiol) in CH2Cl2 with BF3·Et2O and Et3N (Scheme 1). The naphthyl thioxyloside 32 was smoothly oxidized to the corresponding β-linked sulfone 33 in excellent yield using mCPBA. To form the α-anomer of 1, the glycosylation reaction was performed in MeCN with BF3·Et2O and 2-naphthol without addition of Et3N. Subsequent de-O-acetylation using standard Zemplén conditions generated 3, 4, 5, 7, 8, 11, and 19 in good yields. 2-Naphthyl β-L-xylopyranoside 10 was synthesized according to the method described above for the xylosides with β-linkage, starting from peracetylated β-L-xylopyranose 34 (Scheme 1). (9-Anthracene)-methyl xyloside 6 could not be synthesized from peracetylated xylose 27, but was instead synthesized from peracetylated β-xylosyl bromide 36via a Koenigs–Knorr reaction using Ag2O followed by standard Zemplén deprotection (Scheme 1).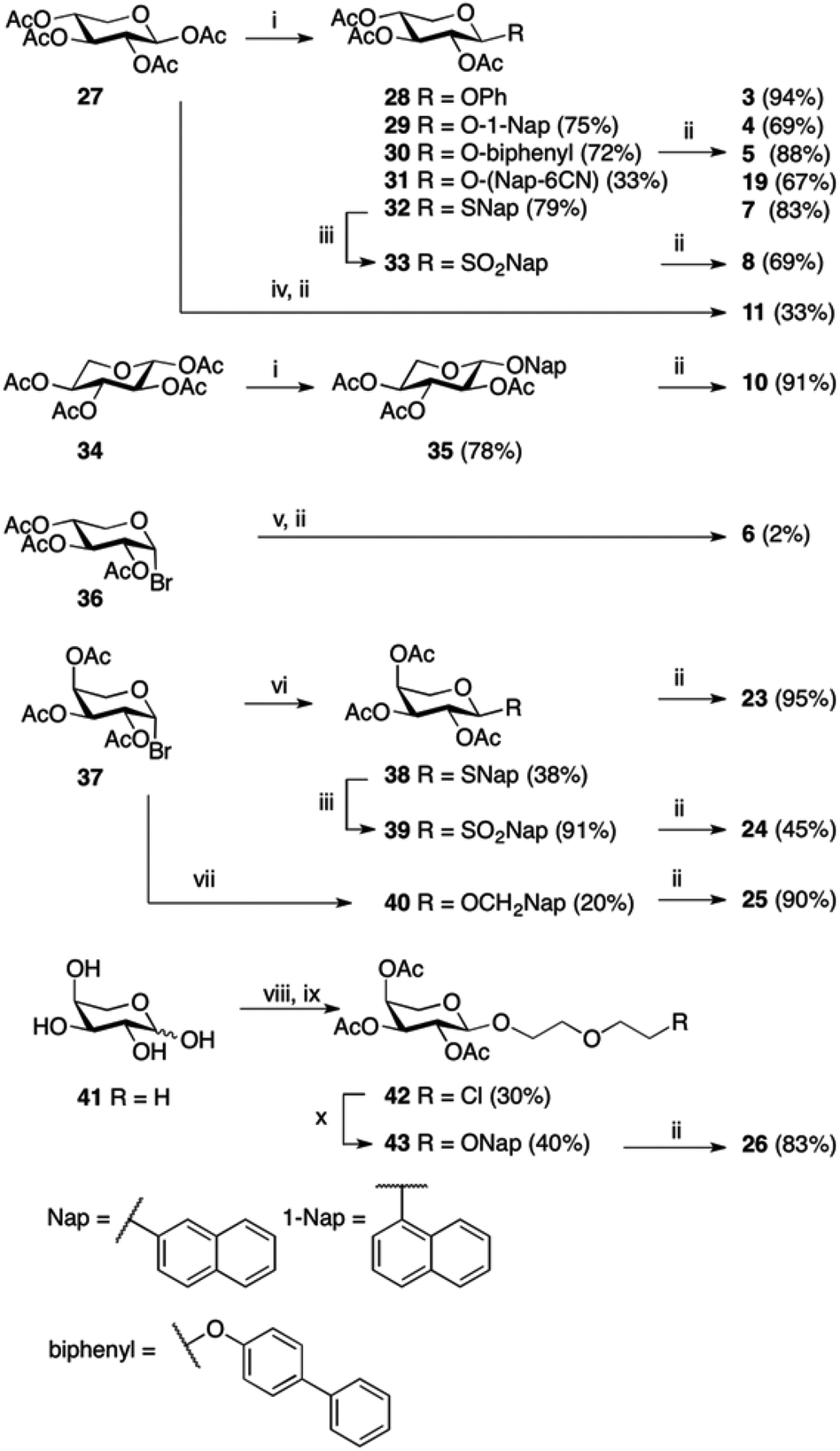 | ||
| Scheme 1 Reagents and conditions: (i) BF3·Et2O, 1-naphthol, Et3N, CH2Cl2; BF3·Et2O, 4-phenylphenol, Et3N, CH2Cl2; BF3·Et2O, 6-cyano-2-naphthol, Et3N, CH2Cl2; BF3·Et2O, 2-naphthalenethiol, CH2Cl2; BF3·Et2O, 2-naphthol, Et3N, CH2Cl2; (ii) NaOMe (1 M), MeOH; (iii) mCPBA, CH2Cl2, (iv) BF3·Et2O, 2-naphthol, MeCN; (v) 9-anthracenemethanol, Ag2O, 4 Å MS, CH2Cl2; (vi) 2-naphthalenethiol, TBAHS, Na2CO3 (1 M), EtOAc; (vii) 2-naphthalenemethanol, Ag2O, 4 Å MS, CH2Cl2; (viii) KOAc, Ac2O; (ix) BF3·Et2O, 2-(2-chloroethoxy)–ethanol, CH2Cl2; (x) 2-naphthol, K2CO3, 18-crown-6, DMF, 80 °C. | ||
Peracetylated β-arabinopyranosyl bromide 3718 was used as starting point for the synthesis of arabinosides. Phase transfer catalysis, a mild procedure with excellent anomeric stereoselectivity, was used to synthesize naphthyl thioarabinoside 38 (Scheme 1). To obtain the arabinosyl sulfone 24, thioarabinoside 38 was oxidized using the same procedure as for compound 8, generating the desired sulfone in 96% yield. Using arabinosyl bromide 37 as a donor in a Koenigs–Knorr reaction with 2-naphthalenemethanol, in the presence of Ag2O, generated 40. Deacetylation of 38–40 yielded the corresponding arabinosides 23–25, respectively. The Koenigs–Knorr glycosylation reaction could also be used for the synthesis of 26. However, the route via peracetylated L-arabinose increased the yield of 42 from 13% to 30%, calculated from L-arabinose 41 (Scheme 1). Thus, acetylation of L-arabinose 41 with KOAc in Ac2O generated an α/β-mixture (approximately 5.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and glycosylation of that mixture with 2-(2-chloroethoxy)–ethanol in the presence of BF3·Et2O formed the arabinoside 42. Treatment of 42 with 2-naphthol, K2CO3, and 18-crown-6 produced 43 in 40% and deacetylation generated arabinoside 26 in good yield.
:1), and glycosylation of that mixture with 2-(2-chloroethoxy)–ethanol in the presence of BF3·Et2O formed the arabinoside 42. Treatment of 42 with 2-naphthol, K2CO3, and 18-crown-6 produced 43 in 40% and deacetylation generated arabinoside 26 in good yield.
2.2 β4GalT7 assay
We have expressed a truncated version of β4GalT7 fused with glutathione S-transferase in E. coli and used it in enzymatic assays to measure the galactosylation by, and inhibition of, β4GalT7.4 To determine if the glycosides could serve as acceptor substrates for β4GalT7, various concentrations of the glycosides were incubated with the recombinant β4GalT7 and the donor substrate, UDP-Gal, for 30 minutes at 37 °C. The reaction progress was analyzed using HPLC with fluorescence detection. We found that none of the arabinosides served as substrate for β4GalT7 (data not shown), whereas all of the synthesized xylosides were galactosylated, except L-XylNap 10 and α-XylNap 11 (Table 1). The identities of galactosylated products were verified by LCMS analysis (ESI, Table S1†). The kinetic parameters, summarized in Table 1, were calculated using the nonlinear regression to the Michaelis–Menten model. We observed excess-substrate inhibition of the enzyme by 5–8, 16, and 18 (Fig. 2), at the studied substrate concentrations. For these compounds, the apparent kinetic parameters were calculated using the concentrations up to the highest observed reaction rate.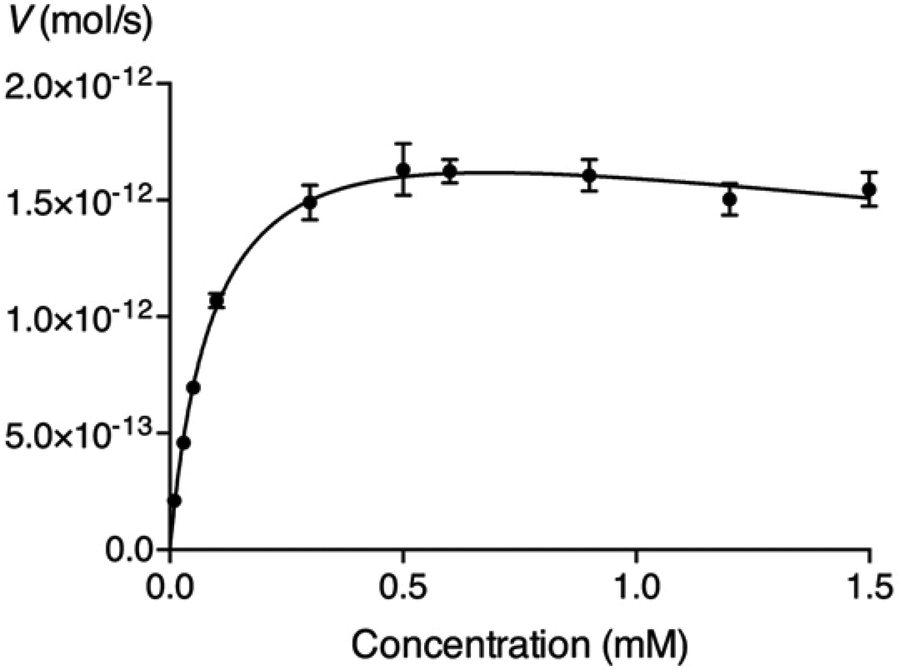 | ||
| Fig. 2 Michaelis–Menten representation of the activity of β4GalT7 (V) as a function of the concentration of compound 8. | ||
| Compound | K m (mM) | V max (pmol s−1) | k cat (s−1) | k cat/Km (mM−1 s−1) |
|---|---|---|---|---|
| a The assay was carried out with various batches of enzyme, and slightly different kinetic parameters for 1 were obtained. The compounds in each part were investigated with the same batch. Hence, comparisons can be made within the same part. b Apparent kinetic parameters were obtained for concentrations up to the highest observed reaction rate. | ||||
| Part A: variation of aglycon size | ||||
| 1 | 0.58 | 1.1 | 1.5 | 2.6 |
| 4 | 0.57 | 1.3 | 1.8 | 3.2 |
| 5 | 0.17 | 0.72 | 1.0 | 6.0 |
| 6 | 0.12 | 0.43 | 0.60 | 5.1 |
| Part B: variation at the anomeric center or of absolute configuration | ||||
| 1 | 0.58 | 1.1 | 1.5 | 2.6 |
| 7 | 0.25 | 1.7 | 2.3 | 9.4 |
| 8 | 0.066 | 1.7 | 2.4 | 37 |
| 9 | 2.4 | 1.8 | 2.5 | 1.0 |
| 10 | — | — | — | — |
| 11 | — | — | — | — |
| Part C: variation of linker length | ||||
| 1 | 0.77 | 1.2 | 1.7 | 2.2 |
| 12 | 0.48 | 1.9 | 2.6 | 5.5 |
| 13 | 0.60 | 2.0 | 2.7 | 4.6 |
| 14 | 0.091 | 0.93 | 1.3 | 14 |
| 15 | 0.24 | 2.4 | 3.3 | 13 |
| Part D: variation of electronic properties | ||||
| 1 | 0.58 | 1.1 | 1.5 | 2.6 |
| 16 | 0.58 | 1.6 | 2.2 | 3.8 |
| 17 | 0.31 | 1.2 | 1.6 | 5.2 |
| 18 | 0.31 | 1.1 | 1.5 | 4.8 |
| 19 | 0.60 | 2.0 | 2.8 | 4.7 |
To determine the potential inhibitory effect of selected glycosides, XylNap, which is known to be a substrate for β4GalT7, was used as a mimic of the natural substrate (i.e. the xylosylated core protein). A fixed amount of XylNap was added, together with different concentrations of glycosides, and the amount of formed GalXylNap was measured. A decrease in formation of GalXylNap, compared to control, indicates that the tested glycoside compete with XylNap for the binding site of β4GalT7. The decrease in formation of GalXylNap, was used to indirectly determine the galactosylation efficiency of the methyl and phenyl xylosides (compounds 2 and 3), since fluorescence could not be used for detection of these. As expected, compounds 2 and 3, showed less decrease of galactosylation of XylNap, than, for example, compound 7 (Table 2). Several arabinose analogs were investigated and found to inhibit β4GalT7 activity to various extents (Table 2), but none as efficiently as the previously reported compound 22.
| Compound | Inhibition (% of control) |
|---|---|
| Xylosides | |
| 2 | 21 ± 13 |
| 3 | 44 ± 3 |
| 7 | 80 ± 0.3 |
| 8 | 93 ± 1 |
| 10 | 0 |
| 11 | 0 |
| Arabinosides | |
| 20 | 11 ± 3 |
| 21 | 28 ± 8 |
| 22 | 63 ± 5 |
| 23 | 22 ± 3 |
| 24 | 38 ± 3 |
| 25 | 0 |
| 26 | 10 ± 0.3 |
2.3 GAG priming in cells
Compound 8 showed the highest activity (kcat/Km) according to the β4GalT7 assay (Table 1). To investigate if this result was transferable to the GAG priming ability in cell culture, the GAG priming of compound 8 in HCC70 cells was determined, using compound 1 and 7 as reference compounds.7,14 HCC70 cells were incubated with the xyloside analogs in low sulfate medium supplemented with [35S]sulfate. After 24 h of incubation at 37 °C, [35S]sulfate-labeled material was isolated from the culture media, separated based on size by HPLC, and the amount of radioactivity was measured to estimate the amount of GAG priming. Compounds 1 and 7 were found to initiate the formation of GAG priming to a similar extent, whereas compound 8 primed 3.4 times as much GAG chains as 1 (Fig. 3). This correlates well with data from the enzymatic assay.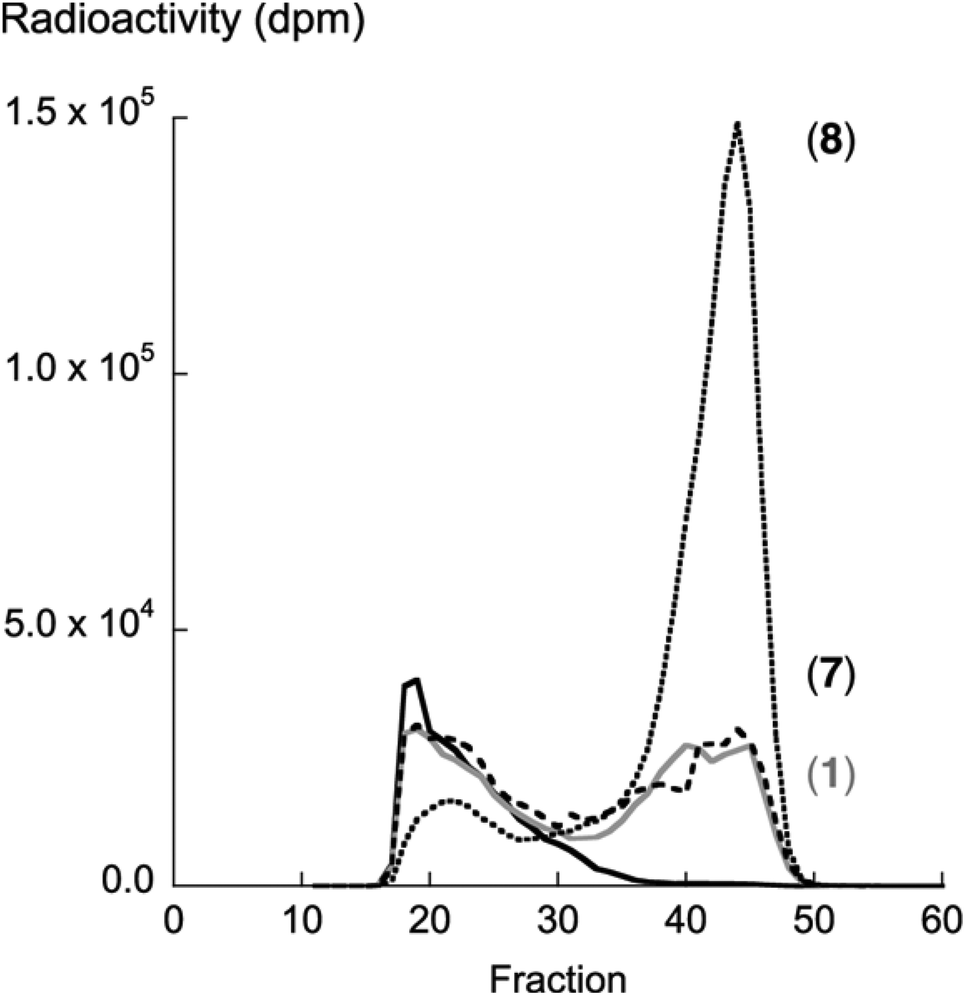 | ||
| Fig. 3 Xyloside-induced priming of GAG chains (approx. fractions 35–50) isolated from HCC70 cells. The cells were incubated with XylNap (1, solid grey line), 7 (dashed line), and 8 (dotted line) and secreted polyanionic material was subjected to size exclusion chromatography on a Superose 6 column. The solid black line shows the result for untreated cells. | ||
2.4 Molecular docking simulations of the Michaelis complex
To gain further insight into the galactosylation reaction, the xyloside acceptor substrates 1, 5, 7, 8, and 12–15 were studied in silico in complex with β4GalT7. Molecular docking simulations were performed with the Autodock VINA software and the acceptor ligands were docked to the protein in the closed conformation in the presence of a rigid UDP donor as well as manganese (based on coordinates from PDB IDs 4M4K and 4LW6). Dockings were performed in both presence and absence of water molecules19 in the crystal structure but it was concluded that additional insights were not gained when including water. In addition, protein interactions to the naphthyl moiety were hindered by water in the crystal structure. Thus, water molecules were excluded from the docking simulations.An initial docking was performed with acceptor ligand 1 in presence of UDP in the donor site (PDB ID 4LW6). The nine highest ranked poses showed a non-uniform distribution of the ligand throughout the binding pocket of the protein, including several cases with the xylose residues positioned in the donor substrate site. These findings indicate that the acceptor substrate competes for this site. It also highlights the importance of a galactose residue to form the acceptor substrate binding site (vide infra). Subsequent dockings were performed to mimic the Michaelis complex, i.e. with UDP-galactose as the donor (PDB ID 4M4K). From the generated binding poses of the docked ligands (1, 5, 7, 8, and 12–15), a similar positioning of the xylose moiety was observed. The acceptor binding site is a shallow pocket, which is defined by acidic residues as well as the donor substrate. The pocket encloses xylose whereas the aglycon moiety extends from the binding site, interacting with the exterior of the enzyme, consisting mainly of aromatic residues (Fig. 4a and b).
 | ||
| Fig. 4 Active site region of β4GalT7 mutant D211N complex with UDP-D-Gal, manganese and (a) ligand 12 or (b) ligand 8. Residues W207, G208, K259, and R263 have been hidden to increase visibility of the active site. | ||
One geometry optimized complex for each ligand, selected through cluster analyses of the docked poses, was analyzed in more detail. From these results we suggest that the xylose moiety is oriented in the binding site with a beneficial CH–π interaction between H5 and the aromatic ring of Y177. All xyloside hydroxyl groups act as hydrogen bond donors, whereas O3 also acts as a hydrogen bond acceptor for L209. The hydrogen-bonding pattern changes, depending on the coupling to the aromatic moiety. For ligand 1 and the sulfone containing ligand 8, the xylose moiety is tilted, compared to the other compounds, resulting in hydrogen bonds from both O2 and O3 to D212 and from O4 to N211 (Fig. 4b). Compounds 5, 7, and 12–15 show a more pronounced CH–π interaction from H5 to Y177 and O2, O3, and O4 hydrogen bond to D212, N211, and either O4 in UDP-Gal or N211, respectively (Fig. 4a). This latter orientation closely resembles that of xylobiose from the crystal structure.20 Furthermore, the naphthyl group generally adopts a face-to-face stacking to the aromatic ring of Y179.
3 Discussion
We have synthesized a number of glycosides and investigated their interactions with β4GalT7 using enzymatic assays, cell studies, and molecular docking simulations. From the computational studies, we observe that the acceptor binding site of β4GalT7 is a shallow pocket enclosing the xylose moiety where the aglycon extends out to the surface of the enzyme. The xyloside moiety displays only subtle differences in binding orientation between the different ligands. The naphthyl moiety tends to interact with a face-to-face π-stacking with Y179 on the outside of the pocket. Apart from compounds 10 and 11, all of the other studied xylosides (Chart 1) were galactosylated by β4GalT7, and several of the studied xylosides showed substrate inhibition. Even if the mechanism for substrate inhibition is unclear, it is possible that it originates in substrate binding to the enzyme without binding of UDP-Gal. Furthermore, our molecular modeling indicates that the acceptor substrate competes for the same site as the donor substrates, suggesting that the acceptor may act as an inhibitor by binding to the β4GalT7-UDP complex.3.1 Part A: variation of aglycon size
Several xylosides carrying relatively small aglycons have shown GAG priming ability in earlier cell studies and enzymatic assays. In an early study by Robinson et al.,21 it was shown that D-xylose initiated GAG priming to a small extent, whereas 2 was about four times, and 44 and 45 about twelve times, more efficient. In 1991, Lugemwa and Esko studied xylosides with different aglycon size.22 They found that xylosides carrying aromatic aglycons were more efficient GAG primers than aliphatic analogs. For example, GAG chains primed by 46, were shown to incorporate 14 times more [35S] sulfate compared to 47. Later, Esko and co-workers23 found that both 48 and 49 primed GAG chains in the same range as 46 and 1. The disaccharide 50 did not initiate GAG synthesis but when three of the hydroxyl groups were methylated, the priming of GAG chains was comparable to that by 1.24 The natural product 51 was also found to initiate GAG synthesis.25 In 2005, we synthesized compound 52, a fluorescently labeled analog to 17, which was taken up by cells but did not initiate priming of GAG chains.26 Apart from O-linked xylosides with large aglycons, Kuberan and co-workers27,28 have made triazole-linked xylosides of which some primed GAG chains, like 53, while others did not.The results from our β4GalT7 assay were in agreement with results from earlier investigations. According to the molecular modeling, there is a π–π interaction between the naphthyl moieties of 1 and 7 and the side-chain of Y179. It can be assumed that the π–π interaction between Y179 and phenyl xyloside 3 is weaker since the distance between the two residues is larger. Methyl xyloside 2, lacking an aromatic aglycon, cannot interact in such a favorable manner.
Compounds 1 and 4, which differ in the binding orientation of the naphthyl moiety, were galactosylated by β4GalT7 to a similar extent, whereas compound 5, with a biphenyl aglycon, showed a slightly increased galactosylation. These results correlate with the study by Esko and co-workers23 where 1, 4, and 5 were found to initiate GAG synthesis. The kinetic parameters for galactosylation of 6 were similar to 5. It is reasonable that the aglycon, which extends out of the binding pocket, provides stronger binding by favorable π–π interactions, but that the effect is limited and not favored by even larger aromatic moieties.
3.2 Part B: variation at the anomeric center or of absolute configuration
It has long been known that α-xylosides, such as 54, 55, 56, and 57 (Chart 2), do not initiate GAG synthesis, and these compounds have therefore often been included in studies as negative controls.21,29–32 However, there are studies in which some GAG priming by α-xylosides have been observed, e.g.5633 and some triazole-linked xylosides.34 | ||
| Chart 2 Discussed glycosides with various biological activities (literature data). | ||
Xylosides with S-xylosidic bonds generally initiate GAG priming comparable to the corresponding O-xylosides.21,32,33,35 For example, we have earlier investigated a series of hydroxynaphthyl 1-thio-β-D-xylopyranosides that all showed strong priming of relatively short GAG chains.36C- and N-xylosides have been reported to be less efficient as GAG primers compared to the corresponding O- or S-xylosides.33,35,37 Kuberan and co-workers27,28,34,38–40 have made several series of amide- and triazole-linked xylosides of which the GAG priming ability varied with variations in the aglycon moiety.
As expected, the L-xyloside 10 and the α-xyloside 11 were not found to be galactosylated by β4GalT7. The C-xyloside 9 was galactosylated to a significantly smaller extent than 1, in contrast to the sulfur analog 7 that was galactosylated more efficiently. Interestingly, the sulfone analog 8 was found to be galactosylated by β4GalT7 several times more efficiently than 1. The very efficient galactosylation of 8 by β4GalT7 was accompanied by efficient priming of GAG chains in cell studies.
The relationship between the nucleophile and the electrophile in a chemical reaction has been shown to be highly dependent on the distance and angle between the nucleophile and the electrophile.41 In this case one may speculate that the reaction kinetics of galactosylation by β4GalT7 is dependent on the orientation of O4 of the xylose moiety in relation to C1 of the galactose moiety, and that subtle differences in orientation might give rise to large effects in reaction efficiency.
3.3 Part C: variation of linker length
In 1994, Esko and co-workers23 noted that the GAG priming ability was not affected when the distance between the xylose residue and the naphthyl moiety in XylNap was increased by a linker. It has also been reported that a triazole-linker, compound 58, increased the GAG priming compared to 59.28Recently, we made a systematic investigation of the effects of the xylose–aglycon distance on GAG priming ability.13 A series of xylosides, where the xylose residue was separated from the naphthyl aglycon with linkers of different lengths, was synthesized (compounds 12–15), and the GAG priming abilities were investigated in cells. The results varied between different cell lines, but an enhancement of the GAG priming ability was generally observed when the xylose–naphthyl distance increased (Fig. 5).
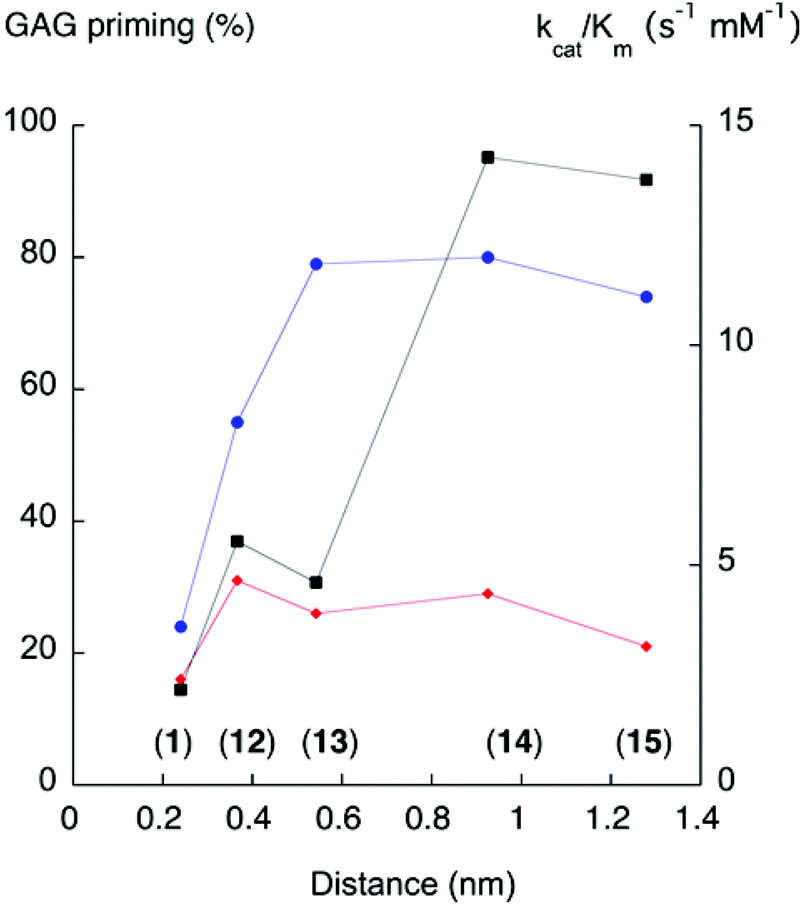 | ||
| Fig. 5 GAG priming and galactosylation plotted against xylose–aglycon distance (C1Xyl–C2Nap). The proportion of GAG priming is given as the integrated value of fractions from cells treated with xyloside, divided by the integrated values for fractions from untreated cells.13 The xylose-aglycon distances are measured in nm from the anomeric carbon to the aromatic carbon coupled to the linker. Circles (blue) indicate GAG priming in CCD-1095Sk cells, diamonds (red) indicate GAG priming in HCC70 cells, and squares (black) indicate kcat/Km values (s−1, mM−1). Compound numbers in parenthesis. | ||
In this study, the galactosylation of compounds 12–15 was investigated in the β4GalT7 assay. There is a trend towards increased galactosylation with increased xylose–aglycon distance (Fig. 5). However, the galactosylation does not perfectly correlate with the GAG priming for either cancer cells or normal cells. This leads us to speculate that the amount of xyloside-induced GAG priming is influenced not only by the galactosylation ability of the corresponding xylosides, but also other factors. For example, differences between cancer cells and normal cells regarding xyloside uptake, intracellular transport, or glycosyltransferases in the GAG biosynthesis.
3.4 Part D: variation of electronic properties
p-Nitrophenyl β-D-xylopyranoside 61 was one of the first xylosides for which GAG priming ability was observed, and it has been used as a control in several cell studies and enzymatic studies.29,42–44 Xylosides with a carboxyl group attached to the aromatic ring in the aglycon have been observed to be less efficient as GAG primers compared to the corresponding methyl ester or non-substituted compound.15,32,35 In solution, these xylosides are most probably charged, which limit the uptake and the cellular transportation. Furthermore, all 14 possible hydroxynaphthyl β-D-xylopyranosides have been evaluated and were shown to initiate synthesis of GAG chains with different proportions of HS chains.45 Moreover, the methoxy ether 16 was reported to initiate synthesis of GAG chains,7 and the methoxy ether of 62 was galactosylated in an enzymatic assay.46Since compound 17 has shown interesting properties, both in priming of GAG chains and in selective growth inhibition of tumor cells, we decided to investigate compounds modified in position 6 of the naphthyl moiety. Therefore, groups with electron donating or electron withdrawing properties were introduced in this position. The kinetic parameters for galactosylation by β4GalT7 of these compounds were found to be in the same range. Hence, the electron density of the aromatic residue does not seem to affect the binding to the active site to any greater extent.
3.5 Part E: inhibitors
Finally, to investigate if the galactosylation efficiency can be used for design of inhibitors of β4GalT7, we included a few arabinose analogs with various aglycons and investigated their ability to inhibit β4GalT7 activity. However, we observed that the corresponding arabinoside of an efficiently galactosylated xyloside is not necessarily an efficient inhibitor. Since the inhibitors do not participate in the reaction, the inhibitory effects shown by the arabinosides obviously reflect other steps in the binding sequence and thus deviate from the trends shown for galactosylation substrates.4 Conclusions
From structure–activity relationships combined with molecular docking simulations, we conclude that xylose should be conjugated to an aromatic aglycon in order to be efficiently galactosylated by β4GalT7, and subsequently initiate GAG synthesis in cells. Since the aglycons reside on the outside of the active site of the enzyme, quite bulky aglycons seem to be accepted. Furthermore, the aromatic aglycon can be separated from the xylose moiety by linkers and retain, or increase, the amount of galactosylation. Generally, only xylosides with β-glycosidic linkages are good substrates for β4GalT7. We also show that the galactosylation ability of a xyloside is increased by replacing the anomeric oxygen with sulfur, but decreased by replacing it with carbon. Substituents on the aromatic aglycon might alter the GAG priming ability depending on identity and position in the aromatic framework. Finally, we propose that reaction kinetics of galactosylation by β4GalT7 is dependent on subtle differences in orientation of the xylose moiety. Thus, a good substrate for galactosylation is not necessarily a good starting point for design of inhibitors.5 Experimental part
5.1 Synthesis
All moisture- and air-sensitive reactions were carried out under an atmosphere of dry nitrogen using oven-dried glassware. All solvents were dried prior to use unless otherwise stated. Purchased reagents were used without further purification. Organic phases were dried using Biotage ISOLUTE Phase separators. Chromatographic separations were performed on Matrex silica gel (25–70 μm), or on a Biotage Isolera One flash purification system using Biotage SNAP KP-Sil silica cartridges. Thin-layer chromatography was performed on precoated TLC alumina plates coated with silica gel 60 F254 0.25 mm (Merck). Spots were visualized with UV light or by staining with para-anisaldehyde. NMR spectra were recorded at ambient temperatures with a Bruker Avance II 400 MHz spectrometer at 400 MHz (1H) and at 100 MHz (13C). 1H NMR spectra were assigned using COSY (2D homonuclear shift correlation) with a gradient selection and NOESY (nuclear Overhauser effect spectroscopy). Chemical shifts are given in ppm, with reference to residual internals CDCl3 (δ 7.26), CD3OD (δ 3.31), (CD3)2SO (δ 2.50), or C6D6 (δ 7.16). Coupling constant values are given in Hz. Mass spectra were recorded on Micromass Q-Tof micro™. Optical rotations were measured on Perkin Elmer instrument, Model 341 polarimeter and are given in 10−1 deg cm2 g−1. A Waters 600 Series HPLC system equipped with a Waters Symmetry C18 column (5 μm, 19 × 100 mm) was used for purification. Synthesis and physical characterization of compounds 1,102,119,1212–15,1316,717,1418,1521,16 and 2217 have been reported previously and compounds 2, 20, 27, 28, 34, 36, and 41 are commercially available.5.1.4.1 Phenyl β-D-xylopyranoside (3). Compound 3 was obtained from 28 following the de-O-acetylation method as a white solid (94%). [α]20D −51 (c 1, MeOH), 1H-NMR (CD3OD) δ 7.25–7.30 (m, 2H, ArH), 7.03–7.07 (m, 2H, ArH), 6.98–7.02 (m, 1H, ArH), 4.85–4.87 (m, 1H, H-1), 3.92 (dd, 1H, J 5.4, 11.4 Hz, H-5e), 3.55–3.61 (m, 1H, H-4), 3.40–3.47 (m, 2H, H-2, H-3), 3.35 (dd, 1H, J 10.4, 11.2 Hz, H-5a); 13C-NMR (CD3OD) δ 158.9, 130.4, 123.4, 117.8, 102.8, 77.7, 74.7, 71.0, 66.9; HRMS calcd for C11H14O5Na+ [M + Na]: 249.0733; found: 249.0736.
5.1.4.2 1-Naphthyl β-D-xylopyranoside (4). Compound 4 was obtained from 29 following the de-O-acetylation method as a white solid (69%). [α]20D −73 (c 2, MeOH), 1H-NMR (CD3OD) δ 8.36–8.39 (m, 1H, ArH), 7.77–7.80 (m, 1H, ArH), 7.50 (d, 1H, J 8.4 Hz, ArH), 7.43–7.47 (m, 2H, ArH), 7.36 (t, 1H, J 8.0 Hz, ArH), 7.13 (d, 1H, J 8.0 Hz, ArH), 5.07 (d, 1H, J 7.6 Hz, H-1), 3.97 (dd, 1H, J 5.2, 11.6 Hz, H-5e), 3.63–3.69 (m, 2H, H-2, H-4), 3.51 (t, 1H, J 8.8 Hz, H-3), 3.40 (t, 1H, J 11.2 Hz, H-5a); 13C-NMR (CD3OD) δ 154.5, 136.1, 128.4, 127.5, 127.3, 126.8, 126.3, 123.3, 123.1, 110.5, 103.3, 77.8, 74.9, 71.1, 67.0; HRMS calcd for C15H16O5Na+ [M + Na]: 299.0895; found: 299.0895.
5.1.4.3 4-Phenylphenyl β-D-xylopyranoside (5). Compound 5 was obtained from 30 following the de-O-acetylation method as a white solid (88%). [α]20D −21 (c 0.7, MeOH), 1H-NMR (CD3OD) δ 7.53–7.58 (m, 4H, ArH), 7.38–7.42 (m, 2H, ArH), 7.27–7.31 (m, 1H, ArH), 7.11–7.15 (m, 2H, ArH), 4.91 (d, 1H, J 7.2 Hz, H-1), 3.94 (dd, 1H, J 5.6, 11.6 Hz, H-5e), 3.58–3.62 (m, 1H, H-4), 3.43–3.48 (m, 2H, H-2, H-3), 3.38 (dd, 1H, J 10.2, 11.5 Hz, H-5a); 13C-NMR (CD3OD) δ 158.5, 142.0, 136.8, 129.8, 129.0, 127.9, 127.7, 118.1, 102.9, 77.7, 74.8, 71.0, 67.0; HRMS calcd for C17H18O5Na+ [M + Na]: 325.1052; found: 325.1052.
5.1.4.4 (9-Anthracene)-methyl β-D-xylopyranoside (6). Compound 6 was obtained from 36 using glycosylation method B followed by de-O-acetylation of crude (9-anthracene)-methyl 2,3,4-tri-O-acetyl β-D-xylopyranoside as a light yellow solid (2%). [α]20D −7 (c 0.6, MeOH), 1H-NMR (CD3OD) δ 8.53 (s, 1H, ArH), 8.49 (d, 2H, J 8.8 Hz, ArH), 8.05 (d, 2H, J 8.4 Hz, ArH), 7.54–7.58 (m, 2H, ArH), 7.46–7.50 (m, 2H, ArH), 5.83 (d, 1H, J 11.2 Hz, CH2Ar), 5.67 (d, 1H, J 11.2 Hz, CH2Ar), 4.45 (d, 1H, J 7.6 Hz, H-1), 4.22 (dd, 1H, J 5.2, 11.2 Hz, H-5e), 3.49–3.60 (m, 1H, H-4), 3.20–3.29 (m, 3H, H-2, H-3, H-5a); 13C-NMR (CD3OD) δ 132.9, 132.6, 129.9, 129.6, 129.0, 127.3, 126.1, 125.6, 104.0, 77.9, 74.8, 71.3, 67.2, 63.8; HRMS calcd for C20H20O5Na+ [M + Na]: 363.1208; found: 363.1207.
5.1.4.5 2-Naphthyl 1-thio-β-D-xylopyranoside (7). Compound 7 was obtained from 32 following the de-O-acetylation method as a white solid (83%). [α]20D −50 (c 0.7, MeOH), 1H-NMR (CD3OD) δ 8.01 (d, 1H, J 0.8 Hz, ArH), 7.79–7.84 (m, 2H, ArH), 7.80 (d, 1H, J 8.8 Hz, ArH), 7.59 (dd, 1H, J 1.6, 8.8 Hz, ArH), 7.45–7.51 (m, 2H, ArH), 4.67 (d, 1H, J 9.2 Hz, H-1), 3.97 (dd, 1H, J 4.8, 11.2 Hz, H-5e), 3.46–3.52 (m, 1H, H-4), 3.37 (t, 1H, J 8.4 Hz, H-3), 3.23–3.31 (m, 2H, H-2, H-5a); 13C-NMR (CD3OD) δ 135.1, 134.0, 132.2, 131.8, 130.5, 129.3, 128.7, 128.5, 127.6, 127.3, 90.0, 79.2, 73.8, 70.9, 70.5; HRMS calcd for C15H16O4SNa+ [M + Na]: 315.0667; found: 315.0667.
5.1.4.6 2-Naphthyl β-D-xylopyranosyl sulfone (8). Compound 8 was obtained from 33 following the de-O-acetylation method as a white solid (69%). [α]21D −35 (c 0.5, MeOH); 1H NMR (CD3OD): δ 8.54 (s, 1H, ArH), 8.07–8.11 (m, 2H, ArH), 8.02 (d, 1H, J 8.4 Hz, ArH), 7.90 (dd, 1H, J 1.6, 8.8 Hz, ArH), 7.66–7.75 (m, 2H, ArH), 4.44 (d, 1H, J 9.2 Hz, H-1), 3.87 (dd, 1H, J 4.8, 11.2 Hz, H-5e), 3.63–3.67 (m, 1H, H-2), 3.35–3.39 (m, 2H, H-3, H-4), 3.16–3.20 (m, 1H, H-5a); 13C NMR (CD3OD): δ 137.0, 135.3, 133.5, 132.6, 130.63, 130.59, 130.0, 129.1, 128.8, 125.2, 93.7, 79.1, 71.2, 71.0, 70.2; HRMS calcd for C15H16O6SNa+ [M + Na]: 347.0565; found: 347.0557.
5.1.4.7 2-Naphthyl β-L-xylopyranoside (10). Compound 10 was obtained from 35 following the de-O-acetylation method as a white solid (91%). [α]21D +38 (c 0.7, MeOH); 1H NMR (CD3OD): δ 7.76–7.80 (m, 3H, ArH), 7.41–7.45 (m, 2H, ArH), 7.35–7.37 (m, 1H, ArH), 7.27 (dd, 1H, J 2.4, 8.8 Hz, ArH), 5.03 (d, 1H, J 7.2 Hz, H-1), 3.97 (dd, 1H, J 5.2, 11.2 Hz, H-5e), 3.58–3.64 (m, 1H, H-4), 4.43–4.53 (m, 3H, H-2, H-3, H-5a); 13C NMR (CD3OD): δ 156.7, 135.8, 131.3, 130.4, 128.6, 128.1, 127.4, 125.3, 120.0, 111.9, 102.9, 77.8, 74.8, 71.1, 67.0; HRMS calcd for C15H16O5Na+ [M + Na]: 299.0895; found: 299.0892.
5.1.4.8 2-Naphthyl α-D-xylopyranoside (11). Compound 11 was obtained from 27 and 2-naphthol using glycosylation method A followed by de-O-acetylation of crude 2-naphthyl 2,3,4-tri-O-acetyl α-D-xylopyranoside as a white solid (33%). [α]22D +181 (c 0.2, MeOH). 1H-NMR (CD3OD) δ 7.73–7.880 (m, 3H, ArH), 7.50 (d, 1H, J 2.4 Hz, ArH), 7.40–7.44 (m, 1H, ArH), 7.30–7.37 (m, 2H, ArH), 5.62 (d, 1H, J 3.6 Hz, H-1), 3.84–3.88 (m, 1H H-3), 3.59–3.67 (m, 4H, H-2, H-4, H-5a, H-5e); 13C NMR (CD3OD) δ 156.1, 135.9, 131.2, 130.4, 128.6, 128.1, 127.4, 125.2, 120.1, 111.9, 99.1, 75.2, 73.4, 71.4, 63.7; HRMS calcd for C15H16O5Na+ [M + Na]: 299.0895; found: 299.0887.
5.1.4.9 2-(6-Cyano-naphthyl) β-D-xylopyranoside (19). Compound 19 was obtained from 31 following the de-O-acetylation method as a white solid (67%). [α]20D −30 (c 0.9, MeOH), 1H-NMR (CD3OD) δ 8.28 (s, 1H, ArH), 7.93 (d, 1H, J 2.0 Hz, ArH), 7.91 (s, 1H, ArH), 7.60 (dd, 1H, J 1.6, 8.4 Hz, ArH), 7.50 (d, 1H, J 2.0 Hz, ArH), 7.39 (dd, 1H, J 2.4, 9.2 Hz, ArH), 5.10 (d, 1H, J 7.2 Hz, H-1), 3.98 (dd, 1H, J 5.2, 11.2 Hz, H-5e), 3.59–3.62 (m, 1H, H-4), 3.46–3.55 (m, 3H, H-2, H-3, H-5a); 13C-NMR (CD3OD) δ 159.1, 137.6, 135.0, 131.3, 129.9, 129.6, 127.9, 121.8, 120.3, 111.6, 108.3, 102.4, 77.6, 74.7, 71.0, 67.0; HRMS calcd for C16H15NO5Na+ [M + Na]: 324.0848; found: 324.0849.
5.1.4.10 2-Naphthyl 1-thio-α-L-arabinopyranoside (23). Compound 23 was obtained from 38 following the de-O-acetylation method as a white solid (95%). [α]21D −48 (c 1, MeOH); 1H NMR (CD3OD): δ 8.01 (d, 1H, J 1.2 Hz, ArH), 7.78–7.83 (m, 3H, ArH), 7.60 (dd, 1H, J 1.8, 8.6 Hz, ArH), 7.43–7.50 (m, 2H, ArH), 4.77 (d, 1H, J 8.4 Hz, H-1), 4.02 (dd, 1H, J 3.6, 12.4 Hz, H-5a), 3.90–3.92 (m, 1H, H-4), 3.75 (t, 1H, J 8.2 Hz, H-2), 3.60–3.65 (m, 2H, H-5e, H-3); 13C NMR (CD3OD): δ 135.1, 133.8, 133.6, 130.9, 130.0, 129.3, 128.7, 128.4, 127.5, 127.1, 90.2, 75.0, 71.6, 69.6, 69.5; HRMS calcd for C15H16O4SNa+ [M + Na]: 315.0667; found: 315.0659.
5.1.4.11 α-L-Arabinopyranosyl 2-naphthyl sulfone (24). Compound 24 was obtained from 39 following the de-O-acetylation method as a white solid (45%). [α]21D +2 (c 0.9, DMSO); 1H NMR ((CD3)2SO): δ 8.54 (s, 1H, ArH), 8.20 (d, 1H, J 8.0 Hz, ArH), 8.14 (d, 1H, J 8.8 Hz, ArH), 8.08 (d, 1H, J 8.0 Hz, ArH), 7.87 (dd, 1H, J 1.6, 8.4 Hz, ArH), 7.73–7.77 (m, 1H, ArH), 7.68–7.71 (m, 1H, ArH), 5.31 (d, 1H, J 6.0 Hz, OH), 4.97 (d, 1H, J 6.0 Hz, OH), 4.64 (d, 1H, J 2.0 Hz, OH), 4.44 (d, 1H, J 8.8 Hz, H-1), 3.76–3.82 (m, 1H, H-2), 3.72 (dd, 1H, J 2.0, 12 Hz, H-5a), 3.62 (s, 1H, H-4), 3.49 (d, 1H, J 11.6 Hz, H-5e) 3.41–3.43 (m, 1H, H-3); 13C NMR ((CD3)2SO): δ 135.0, 134.8, 131.6, 130.6, 129.5, 129.3, 128.7, 127.9, 127.6, 124.1, 92.6, 73.2, 70.5, 67.7, 66.7; HRMS calcd for C15H16O6SNa+ [M + Na]: 347.0565; found: 347.0557.
5.1.4.12 (2-Naphthyl)-methyl α-L-arabinopyranoside (25). Compound 25 was obtained from 40 following the de-O-acetylation method as a white solid (90%). [α]21D −20 (c 0.7, MeOH); 1H NMR (CD3OD): δ 7.88 (s, 1H, ArH), 7.82–7.85 (m, 3H, ArH), 7.54 (dd, 1H, J 1.6, 8.4 Hz, ArH), 7.44–7.49 (m, 2H, ArH), 5.02 (d, 1H, J 12.4 Hz, CH2), 4.79 (d, 1H, J 12.0 Hz, CH2), 4.35 (d, 1H, J 6.8 Hz, H-1), 3.92 (dd, 1H, J 3.2, 12.4 Hz, H-5a), 3.82–3.84 (m, 1H, H-4), 3.66 (dd, 1H, J 6.8, 8.8 Hz, H-2), 3.57 (dd, 1H, J 1.6, 12.4 Hz, H-5e), 3.53 (dd, 1H, J 3.4, 9 Hz, H-3); 13C NMR (CD3OD): δ 136.6, 134.8, 134.5, 129.0, 128.9, 128.7, 127.7, 127.13, 127.09, 126.9, 103.9, 74.3, 72.5, 71.6, 69.7, 66.9; HRMS calcd for C16H18O5Na+ [M + Na]: 313.1052; found: 313.1054.
5.1.4.13 5-(2-Naphthoxy)-3-oxo-1-pentyl α-L-arabinopyranoside (26). Compound 26 was obtained from 43 following the de-O-acetylation method as a white solid (83%). [α]21D +9 (c 0.9, MeOH); 1H NMR (CD3OD): δ 7.74–7.76 (m, 3H, ArH), 7.39–7.43 (m, 1H, ArH), 7.29–7.33 (m, 1H, ArH), 7.24 (d, 1H, J 2.8 Hz, ArH), 7.16 (dd, 1H, J 2.4, 8.8 Hz, ArH), 4.24–4.26 (m, 3H, H-1, CH2), 3.96–4.01 (m, 1H, CH2), 3.92–3.95 (m, 2H, CH2), 3.86 (dd, 1H, J 2.8, 12.4 Hz, H-5a), 3.72–3.81 (m, 4H, H-4, CH2), 3.58 (dd, 1H, J 6.8, 8.8 Hz, H-2), 4.50–4.54 (m, 2H, H-5e, H-3); 13C NMR (CD3OD): δ 158.1, 136.1, 130.6, 130.4, 128.6, 127.9, 127.3, 124.7, 119.8, 107.7, 105.0, 74.2, 72.4, 71.7, 70.9, 69.6, 69.4, 68.5, 66.9; HRMS calcd for C19H24O7Na+ [M + Na]: 387.1420; found: 387.1415.
5.1.4.14 1-Naphthyl 2,3,4-tri-O-acetyl-β-D-xylopyranoside (29). Compound 29 was obtained from 27 and 1-naphthol following glycosylation method A as a white solid (75%). 1H-NMR (CDCl3) δ 8.13–8.15 (m, 1H, ArH), 7.81–7.83 (m, 1H, ArH), 7.55 (d, 1H, J 8.0 Hz, ArH), 7.46–7.52 (m, 2H, ArH), 7.38 (t, 1H, J 7.6 Hz, ArH), 7.07 (d, 1H, J 7.6 Hz, ArH), 5.29–5.41 (m, 2H, H-1, H-2), 5.31 (t, 1H, J 7.2 Hz, H-3), 5.04–5.09 (m, 1H, H-4), 4.28 (dd, 1H, J 4.8, 12.4 Hz, H-5e), 3.60 (dd, 1H, J 7.6, 12.0 Hz, H-5a), 2.15, 2.11, 2.08 (s, 3H each, OAc); 13C-NMR (CDCl3) δ 170.0, 170.0, 169.7, 152.5, 134.6, 127.7, 126.7, 126.0, 125.9, 125.8, 122.8, 121.8, 108.9, 98.8, 70.6, 70.1, 68.6, 62.0, 21.0, 21.0, 20.9; HRMS calcd for C21H22O8Na+ [M + Na]: 425.1212; found: 425.1211.
5.1.4.15 4-Phenylphenyl 2,3,4-tri-O-acetyl-β-D-xylopyranoside (30). Compound 30 was obtained from 27 and 4-phenylphenol following glycosylation method A as a white solid (72%). 1H-NMR (CDCl3) δ 7.51–7.55 (m, 4H, ArH), 7.42 (t, 2H, J 7.2 Hz, ArH), 7.32 (t, 1H, J 7.4 Hz, ArH), 7.06–7.10 (m, 2H, ArH), 5.21–5.29 (m, 3H, H-1, H-2, H-3), 5.02–5.07 (m, 1H, H-4), 4.25 (dd, 1H, J 4.4, 12.4 Hz, H-5e), 3.55 (dd, 1H, J 7.6, 12.0 Hz, H-5a), 2.10, 2.10, 2.09 (s, 3H each, OAc); 13C-NMR (CDCl3) δ 170.0, 169.9, 169.4, 156.1, 140.5, 136.2, 128.8, 128.3, 127.1, 126.9, 117.1, 98.5, 70.8, 70.2, 68.5, 61.9, 20.8, 20.8, 20.7; HRMS calcd for C23H24O8Na+ [M + Na]: 451.1369; found: 451.1367.
5.1.4.16 2-(6-Cyano-naphthyl) 2,3,4-tri-O-acetyl-β-D-xylopyranoside (31). Compound 31 was obtained from 27 and 6-cyano-2-naphthol following glycosylation method A as a white solid (33%). 1H-NMR (CDCl3) δ 8.15 (s, 1H, ArH), 7.82 (d, 1H, J 13.6 Hz, ArH), 7.80 (d, 1H, J 13.2 Hz, ArH), 7.57 (dd, 1H, J 1.6, 8.4 Hz, ArH), 7.37 (d, 1H, J 2.4 Hz, ArH), 7.28 (dd, 1H, J 2.8, 9.2 Hz, ArH), 5.40 (d, 1H, J 5.2 Hz, H-1), 5.21–5.28 (m, 2H, H-2, H-3), 5.01–5.04 (m, 1H, H-4), 4.27 (dd, 1H, J 4.4, 12.0 Hz, H-5e), 3.49 (dd, 1H, J 6.8, 12.4 Hz, H-5a), 2.11, 2.10, 2.10 (s, 3H each, OAc); 13C-NMR (CDCl3) δ 169.9, 169.9, 169.5, 156.6, 136.0, 133.9, 130.5, 128.7, 128.4, 127.3, 120.6, 119.4, 110.9, 108.0, 97.9, 70.1, 69.7, 68.3, 61.9, 20.9, 20.9, 20.8; HRMS calcd for C22H21NO8Na+ [M + Na]: 450.1165; found: 450.1163.
5.1.4.17 2-Naphthyl 2,3,4-tri-O-acetyl-1-thio-β-D-xylopyranoside (32). BF3·OEt2 (1.0 mL, 7.9 mmol) was added to a stirred solution of 2-naphthalenethiol (766 mg, 4.78 mmol) and 1,2,3,4-tetra-O-acetyl-β-D-xylopyranose 27 (1.01 g, 3.19 mmol) in CH2Cl2 (3 mL) at rt. The reaction was performed as for glycosylation method A yielding 32 as a white solid (1.05 g, 79%). 1H-NMR (CDCl3) δ 7.98 (d, 1H, J 1.6 Hz, ArH), 7.78–7.83 (m, 3H, ArH), 7.54 (dd, 1H, J 2.0, 8.8 Hz, ArH), 7.47–7.51 (m, 2H, ArH), 5.20 (t, 1H, J 8.0 Hz, H-3), 5.00 (t, 1H, J 8.0 Hz, H-2), 4.90–4.96 (m, 2H, H-1, H-4), 4.32 (dd, 1H, J 5.2, 12.0 Hz, H-5e), 3.45 (dd, 1H, J 8.8, 12.0 Hz, H-5a), 2.12, 2.06, 2.05 (s, 3H each, OAc); 13C-NMR (CDCl3) δ 170.0, 169.9, 169.5, 133.6, 132.9, 132.2, 129.9, 129.6, 128.8, 127.8, 126.8, 86.4, 71.9, 70.0, 68.5, 65.2, 21.0, 20.9; HRMS calcd for C21H22O7SNa+ [M + Na]: 441.0984; found: 441.0980.
5.1.4.18 2-Naphthyl 2,3,4-tri-O-acetyl β-D-xylopyranosyl sulfone (33). Compound 33 was obtained from 32 following the oxidation method as a white solid (91%). [α]21D −81 (c 0.8, CHCl3); 1H NMR (CDCl3): δ 8.50 (s, 1H, ArH), 8.05 (d, 1H, J 8 Hz, ArH), 8.01 (d, 1H, J 8.8 Hz, ArH), 7.96 (d, 1H, J 8 Hz, ArH), 7.87 (dd, 1H, J 1.8, 8.6 Hz, ArH), 7.72 (dt, 1H, J 1.2, 7.6 Hz, ArH), 7.66 (dt, 1H, J 1.4, 7.6 Hz, ArH), 5.30 (m, 1H, H-2), 5.25 (t, 1H, J 8.8 Hz, H-3), 4.77–4.83 (m, 1H, H-4), 4.55 (d, 1H, J 9.2 Hz, H-1), 4.21 (dd, 1H, J 5.2, 11.6 Hz, H-5e), 3.37 (dd, 1H, J 9.6, 11.6 Hz, H-5a), 2.13, 2.01, 1.99 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.2, 169.8, 169.6, 135.9, 132.8, 132.1, 132.0, 130.0, 129.9, 129.3, 128.1, 127.8, 124.5, 90.0, 72.7, 68.3, 67.0, 66.9, 20.9, 20.7; HRMS calcd for C21H22O9SNa+ [M + Na]: 473.0882; found: 473.0879.
5.1.4.19 2-Naphthyl 2,3,4-tri-O-acetyl β-L-xylopyranoside (35). Compound 35 was obtained from 34 and 2-naphthol following glycosylation method A as a white solid (78%). [α]21D +28 (c 0.9, CHCl3); 1H NMR (CDCl3): δ 7.73–7.80 (m, 3H, ArH), 7.44–7.48 (m, 1H, ArH), 7.36–7.41 (m, 2H, ArH), 7.18 (dd, 1H, J 2.4, 8.8 Hz, ArH), 5.34 (d, 1H, J 6.0 Hz, H-1), 5.22–5.35 (m, 2H, H-3, H-2), 5.02–5.07 (m, 1H, H-4), 4.28 (dd, 1H, J 4.6, 12.2 Hz, H-5e), 3.60 (dd, 1H, J 7.6, 12 Hz, H-5a), 2.113, 2.105, 2.10 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.1, 170.0, 169.6, 154.5, 134.3, 130.2, 129.8, 127.8, 127.3, 126.7, 124.8, 118.9, 111.4, 98.7, 70.7, 70.2, 68.6, 62.0, 21.0, 20.93, 20.90; HRMS calcd for C21H22O8Na+ [M + Na]: 425.1212; found: 425.1202.
5.1.4.20 2-Naphthyl 2,3,4-tri-O-acetyl 1-thio-α-L-arabinopyranoside (38). Arabinopyranosyl bromide 3718 (330 mg) was dissolved in EtOAc (3.3 mL) and 2-naphthalenethiol (334 mg, 2.08 mmol), TBAHS (tetrabutylammonium hydrogen sulfate) (532 mg, 1.57 mmol), and aqueous Na2CO3 (1 M, 3.3 mL) were added. After 12 h, the reaction mixture was diluted with EtOAc (15 mL) and washed with satd aq. Na2CO3 (2 × 20 mL), brine (20 mL), and water (20 mL). The organic phase was dried before removal of solvent under reduced pressure. Column chromatography (SiO2, 10 → 30% EtOAc in heptane) gave 38 as a white solid (262 mg, 39% over 3 steps from L-arabinose). [α]21D +5 (c 1, CHCl3); 1H NMR (CDCl3): δ 8.01 (d, 1H, J 2 Hz, ArH), 7.78–7.83 (m, 3H, ArH), 7.56 (dd, 1H, J 1.8, 8.6 Hz, ArH), 7.47–7.52 (m, 2H, ArH), 5.28–5.32 (m, 2H, H-2, H-4), 5.14 (dd, 1H, J 3.4, 8.2 Hz, H-3), 4.92 (d, 1H, J 7.6 Hz, H-1), 4.20 (dd, 1H, J 4.4, 12.4 Hz, H-5a), 3.71 (dd, 1H, J 2.0, 12.8 Hz, H-5e), 2.13, 2.09, 2.07 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.4, 170.1, 169.6, 133.6, 132.8, 131.7, 130.6, 129.6, 128.7, 127.9, 127.8, 126.8, 126.7, 87.1, 70.6, 68.7, 67.6, 65.4, 21.0, 20.9; HRMS calcd for C21H22O7SNa+ [M + Na]: 441.0984; found: 441.0977.
5.1.4.21 2-Naphthyl 2,3,4-tri-O-acetyl α-L-arabinopyranosyl sulfone (39). Compound 39 was obtained from 38 following the oxidation method as a white solid (96%). [α]21D −44 (c 1, DMSO); 1H NMR (CDCl3): δ 8.56 (s, 1H, ArH), 8.06 (d, 1H, J 8.0 Hz, ArH), 8.01 (d, 1H, J 8.8 Hz, ArH), 7.93 (m, 2H, ArH), 7.72 (dt, 1H, J 1.3, 7.5 Hz, ArH), 7.64 (dt, 1H, J 1.3, 7.5 Hz, ArH), 5.53 (t, 1H, J 9.4 Hz, H-2), 5.13–5.14 (m, 1H, H-4), 5.05 (dd, 1H, J 3.2, 9.6 Hz, H-3), 4.55 (d, 1H, J 9.2 Hz, H-1), 4.10 (dd, 1H, J 2.4, 13.2 Hz, H-5a), 3.68 (dd, 1H, J 1.0, 13 Hz, H-5e), 2.53, 1.99, 1.64 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.2, 170.1, 169.6, 135.8, 132.8, 132.2, 132.1, 129.9, 129.8, 128.9, 128.0, 127.7, 124.9, 90.1, 71.0, 67.7, 67.6, 64.6, 21.0, 20.8, 20.4; HRMS calcd for C21H22O9SNa+ [M + Na]: 473.0882; found: 473.0877.
5.1.4.22 (2-Naphthyl)-methyl 2,3,4-tri-O-acetyl α-L-arabinopyranoside (40). Compound 40 was obtained from 3718 and 2-naphthalenemethanol following glycosylation method B as a white solid (20% over 3 steps from L-arabinose). 1H NMR (CDCl3): δ 7.79–7.83 (m, 3H, ArH), 7.75 (s, 1H, ArH), 7.44–7.49 (m, 2H, ArH), 7.41 (dd, 1H, J 1.6, 8.4 Hz, ArH), 5.25–5.31 (m, 2H, H-2, H-4), 4.99–5.05 (m, 2H, H-3, CH2), 4.75 (d, 1H, J 12.4 Hz, CH2), 4.52 (d, 1H, J 6.8 Hz, H-1), 4.06 (dd, 1H, J 3.4, 13.0 Hz, H-5a), 3.61 (dd, 1H, J 1.6, 12.8 Hz, H-5e), 2.13, 2.03, 2.00 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.3, 170.1, 169.4, 134.4, 133.1, 133.0, 128.2, 127.8, 127.7, 126.5, 126.3, 126.1, 125.5, 99.4, 70.4, 70.1, 69.2, 67.6, 63.0, 20.9, 20.8, 20.7; HRMS calcd for C22H24O8Na+ [M + Na]: 439.1369; found: 439.1361.
5.1.4.23 5-Chloro-3-oxo-1-pentyl 2,3,4-tri-O-acetyl-α-L-arabinopyranoside (42). KOAc (7.90 g, 80.5 mmol) was suspended in acetic anhydride (110 mL) and heated to 130 °C. L-Arabinose 41 (10.0 g, 66.9 mmol) was added in portions over 5 minutes and after 1.5 hours of heating at 130 °C, the reaction mixture was allowed to reach rt before poured onto ice. The aqueous phase was extracted with CH2Cl2 (3 × 500 mL) and the combined organic layers were washed with satd aq. NaHCO3 (1 L), dried, and stirred with activated charcoal. After 30 minutes, the mixture was filtered through silica and concentrated to yield 1,2,3,4-tetra-O-acetyl-L-arabinopyranose as a yellow oil (23.2 g) that was used without further purification. BF3·Et2O (1.10 mL, 8.68 mmol) was added to a stirred solution of crude peracetylated L-arabinose (1.09 g) and 2-(2-chloroethoxy)-ethanol (540 μL, 5.12 mmol) in CH2Cl2 (10 mL) at 0 °C. The reaction was performed as for glycosylation method A yielding 42 as a white solid (30% over 2 steps from L-arabinose). 1H NMR (CDCl3): δ 5.24–5.26 (m, 1H, H-4), 5.19 (dd, 1H, J 7.2, 9.2 Hz, H-2), 5.03 (dd, 1H, J 3.6, 9.2 Hz, H-3), 4.52 (d, 1H, J 7.2 Hz, H-1), 4.03 (dd, 1H, J 3.3, 13.2 Hz, H-5a), 3.92–3.97 (m, 1H, CH2), 3.59–3.75 (m, 8H, CH2, H-5e), 2.13, 2.07, 2.01 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.5, 170.3, 169.7, 101.1, 71.6, 70.5, 70.3, 69.2, 68.8, 67.8, 63.3, 43.0, 21.1, 21.0, 20.8; HRMS calcd for C15H23ClO9Na+ [M + Na]: 405.0928; found: 405.0919.
5.1.4.24 5-(2-Naphthoxy)-3-oxo-1-pentyl 2,3,4-tri-O-acetyl-α-L-arabinopyranoside (43). 2-Naphthol (127 mg, 0.879 mmol), K2CO3 (70 mg, 0.51 mmol), and 18-crown-6 (1.21 g, 4.57 mmol) were stirred in DMF (2 mL) for 1 hour before 42 (166 mg, 0.434 mmol), dissolved in DMF (3 mL), was added. The reaction mixture was heated to 80 °C for 40 hours before it was allowed to reach rt and water (10 mL) was added. The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic phases were washed with brine (50 mL) and dried before removal of solvent under reduced pressure. Column chromatography (SiO2, 20 → 40% EtOAc in heptane) gave 43 as a light brown solid (85 mg, 40%). 1H NMR (CDCl3): δ 7.71–7.77 (m, 3H, ArH), 7.41–7.45 (m, 1H, ArH), 7.31–7.35 (m, 1H, ArH), 7.17 (dd, 1H, J 2.4, 8.8 Hz, ArH), 7.14 (d, 1H, J 2.4 Hz, ArH), 5.23–5.25 (m, 1H, H-4), 5.21 (dd, 1H, J 7.0, 9.4 Hz, H-2), 5.02 (dd, 1H, J 3.6, 9.2 Hz, H-3), 4.50 (d, 1H, J 6.8 Hz, H-1), 4.21–4.23 (m, 2H, CH2), 3.96–4.03 (m, 2H, H-5a, CH2), 3.88–3.91 (m, 2H, CH2), 3.73–3.76 (m, 3H, CH2), 3.58 (dd, 1H, J 1.8, 13.0 Hz, H-5e), 2.11, 2.05, 2.01 (s, 3H each, OAc); 13C NMR (CDCl3): δ 170.4, 170.3, 169.6, 156.8, 134.5, 129.5, 129.1, 127.7, 126.8, 126.4, 123.8, 119.0, 106.7, 101.1, 70.6, 70.2, 69.9, 69.2, 68.9, 67.7, 67.5, 63.2, 21.0, 20.8, 20.7; HRMS calcd for C25H30O10Na+ [M + Na]: 513.1737; found: 513.1736.
5.2 β4GalT7 assay
The β4GalT7 assay has been described in detail previously.45.3 GAG priming in cells
The procedures for cell culture, radiolabeling, and isolation of xyloside-primed [35S]sulfate-labeled polyanionic material have been described in detail previously.145.4 Molecular docking simulations of the Michaelis complex
For molecular docking simulations, the protein crystal structure of Drosophila β-1,4-Galactosyltransferase 7 mutant D211N in complex with xylobiose, manganese, UDP-galactose, or UDP (PDB ID: 4M4K and 4LW6 respectively) was prepared using Autodock Tools (ADT).47 The xylobiose molecule was removed and water molecules were either included or excluded in the docking (vide supra). The ligands were built using the gview program (GaussView Version 5, Dennington et al., Semichem Inc., Shawnee Mission, KS, 2009.) and geometry-optimized using the ORCA 2.9.1 package48 with the B3LYP functional and a 6-31G* basis set.The docking simulations were performed using the Autodock VINA 1.1.2 package49 using a cubic 24 Å grid, an exhaustiveness of 64 and a random starting seed. Residues deemed to have possible interactions with the naphthyl group, K176, H178, and Y179, were treated as flexible residues. The nine top-scored poses from the docking simulations were analyzed and grouped based on interaction similarities. Subsequently, one representative structure from the major cluster was chosen for each ligand (among the three highest ranked poses in all cases). The selected structures were H-bond optimized and energy minimized employing a 0.6 Å heavy atom restraint (OPLS-2005 force field) with the Protein preparation wizard within the Maestro software.50 The resulting geometries were then evaluated.
Acknowledgements
This work was supported by grants from the Alfred Österlund Foundation, the Crafoord Foundation, the Evy and Gunnar Sandberg Foundation, Greta and John Kock, the Gunnar Nilsson Cancer Foundation, the Heart & Lung Foundation, the Knut and Alice Wallenberg Foundation, the Koch Foundation, Lars Hiertas Memorial Foundation, the Magnus Bergwall Foundation, the Medical Faculty of Lund University, Lund University, the Royal Physiographic Society in Lund, Syskonen Svenssons Foundation for Medical Research, the Swedish Cancer Society, the Swedish Medical Research Council (11550), and the Swedish Research Council.Notes and references
- R. Almeida, S. B. Levery, U. Mandel, H. Kresse, T. Schwientek, E. P. Bennett and H. Clausen, J. Biol. Chem., 1999, 274, 26165 CrossRef CAS PubMed.
- X. Bai, D. Zhou, J. R. Brown, B. E. Crawford, T. Hennet and J. D. Esko, J. Biol. Chem., 2001, 276, 48189 CAS.
- P. Carlsson and L. Kjellen, Handbook of Experimental Pharmacology, 2012, vol. 207, p. 23 Search PubMed.
- A. Siegbahn, S. Manner, A. Persson, E. Tykesson, K. Holmqvist, A. Ochocinska, J. Roennols, A. Sundin, K. Mani, G. Westergren-Thorsson, G. Widmalm and U. Ellervik, Chem. Sci., 2014, 5, 3501 RSC.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 5, 526 CrossRef CAS PubMed.
- M. Belting, L. Borsig, M. M. Fuster, J. R. Brown, L. Persson, L.-A. Fransson and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 371 CrossRef CAS PubMed.
- K. Mani, M. Belting, U. Ellervik, N. Falk, G. Svensson, S. Sandgren, F. Cheng and L.-A. Fransson, Glycobiology, 2004, 14, 387 CrossRef CAS PubMed.
- U. Nilsson, R. Johnsson, L.-A. Fransson, U. Ellervik and K. Mani, Cancer Res., 2010, 70, 3771 CrossRef CAS PubMed.
- K. Descroix and G. K. Wagner, Org. Biomol. Chem., 2011, 9, 1855 CAS.
- T. A. Fritz, F. N. Lugemwa, A. K. Sarkar and J. D. Esko, J. Biol. Chem., 1994, 269, 300 CAS.
- C. T. Bishop and F. P. Cooper, Can. J. Chem., 1962, 40, 224 CrossRef CAS.
- J. Malmberg, K. Mani, E. Saewen, A. Wiren and U. Ellervik, Bioorg. Med. Chem., 2006, 14, 6659 CrossRef CAS PubMed.
- K. Holmqvist, A. Persson, R. Johnsson, J. Lofgren, K. Mani and U. Ellervik, Bioorg. Med. Chem., 2013, 21, 3310 CrossRef CAS PubMed.
- K. Mani, B. Havsmark, S. Persson, Y. Kaneda, H. Yamamoto, K. Sakurai, S. Ashikari, H. Habuchi, S. Suzuki, K. Kimata, A. Malmstrom, G. Westergren-Thorsson and L.-A. Fransson, Cancer Res., 1998, 58, 1099 CAS.
- C.-O. Abrahamsson, U. Ellervik, J. Eriksson-Bajtner, M. Jacobsson and K. Mani, Carbohydr. Res., 2008, 343, 1473 CrossRef CAS PubMed.
- K. Higashi, H. Hori, T. Ishiyama, Y. Okimoto, I. Kusakabe and T. Yasui, Agric. Biol. Chem., 1983, 47, 1123 CrossRef CAS.
- A. Siegbahn, U. Aili, A. Ochocinska, M. Olofsson, J. Roennols, K. Mani, G. Widmalm and U. Ellervik, Bioorg. Med. Chem., 2011, 19, 4114 CrossRef CAS PubMed.
- S. C. Timmons and D. L. Jakeman, Carbohydr. Res., 2008, 343, 865 CrossRef CAS PubMed.
- M. Lundborg, E. Ali and G. Widmalm, Carbohydr. Res., 2013, 378, 133 CrossRef CAS PubMed.
- Y. Tsutsui, B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2013, 288, 31963 CrossRef CAS PubMed.
- H. C. Robinson, M. J. Brett, P. J. Tralaggan, D. A. Lowther and M. Okayama, Biochem. J., 1975, 148, 25 CAS.
- F. N. Lugemwa and J. D. Esko, J. Biol. Chem., 1991, 266, 6674 CAS.
- T. A. Fritz, F. N. Lugemwa, A. K. Sarkar and J. D. Esko, J. Biol. Chem., 1994, 269, 300 CAS.
- A. K. Sarkar, T. A. Fritz, W. H. Taylor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3323 CrossRef CAS.
- A. Sinha, W. H. Taylor, I. H. Khan, S. T. McDaniel and J. D. Esko, J. Nat. Prod., 1999, 62, 1036 CrossRef CAS PubMed.
- R. Johnsson, K. Mani, F. Cheng and U. Ellervik, J. Org. Chem., 2006, 71, 3444 CrossRef CAS PubMed.
- V. M. Tran, X. V. Victor, J. W. Yockman and B. Kuberan, Glycoconjugate J., 2010, 27, 625 CrossRef CAS PubMed.
- V. M. Tran and B. Kuberan, Bioconjugate Chem., 2014, 25, 262 CrossRef CAS PubMed.
- N. B. Schwartz, L. Galligani, P.-L. Ho and A. Dorfman, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4047 CrossRef CAS.
- N. B. Schwartz and L. Roden, J. Biol. Chem., 1975, 250, 5200 CAS.
- Y. Fukunaga, M. Sobue, N. Suzuki, H. Kushida and S. Suzuki, Biochim. Biophys. Acta, 1975, 381, 443 CrossRef CAS.
- J. A. Robinson and H. C. Robinson, Biochem. J., 1981, 194, 839 CAS.
- S. O. Kolset, K. Sakurai, I. Ivhed, A. Oevervatn and S. Suzuki, Biochem. J., 1990, 265, 637 CAS.
- B. Kuberan, M. Ethirajan, X. V. Victor, V. Tran, K. Nguyen and A. Do, ChemBioChem, 2008, 9, 198 CrossRef CAS PubMed.
- M. Sobue, H. Habuchi, K. Ito, H. Yonekura, K. Oguri, K. Sakurai, S. Kamohara, Y. Ueno, R. Noyori and S. Suzuki, Biochem. J., 1987, 241, 591 CAS.
- M. Jacobsson, K. Mani and U. Ellervik, Bioorg. Med. Chem., 2007, 15, 5283 CrossRef CAS PubMed.
- J. Malmberg, K. Mani, E. Sawen, A. Wiren and U. Ellervik, Bioorg. Med. Chem., 2006, 14, 6659 CrossRef CAS PubMed.
- X. V. Victor, T. K. N. Nguyen, M. Ethirajan, V. M. Tran, K. V. Nguyen and B. Kuberan, J. Biol. Chem., 2009, 284, 25842 CrossRef CAS PubMed.
- K. Raman and B. Kuberan, Mol. BioSyst., 2010, 6, 1800 RSC.
- T. K. N. Nguyen, V. M. Tran, V. Sorna, I. Eriksson, A. Kojima, M. Koketsu, D. Loganathan, L. Kjellen, R. I. Dorsky, C.-B. Chien and B. Kuberan, ACS Chem. Biol., 2013, 8, 939 CrossRef CAS PubMed.
- F. M. Menger, Adv. Chem. Ser., 1987, 215, 209 CrossRef CAS PubMed.
- M. Okayama, K. Kimata and S. Suzuki, J. Biochem., 1973, 74, 1069 CAS.
- F. Daligault, S. Rahuel-clermont, S. Gulberti, M.-T. Cung, G. Branlant, P. Netter, J. Magdalou and V. Lattard, Biochem. J., 2009, 418, 605 CrossRef CAS PubMed.
- R. L. Stevens and K. F. Austen, J. Biol. Chem., 1982, 257, 253 CAS.
- M. Jacobsson, U. Ellervik, M. Belting and K. Mani, J. Med. Chem., 2006, 49, 1932 CrossRef CAS PubMed.
- S. Gulberti, V. Lattard, M. Fondeur, J.-C. Jacquinet, G. Mulliert, P. Netter, J. Magdalou, M. Ouzzine and S. Fournel-Gigleux, J. Biol. Chem., 2005, 280, 1417 CrossRef CAS PubMed.
- M. Morris Garrett, R. Huey, W. Lindstrom, F. Sanner Michel, K. Belew Richard, S. Goodsell David and J. Olson Arthur, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455 CAS.
- Schrödinger Release 2014-3: Maestro, version 9.9, LLC, Schrödinger, New York, NY, 2014 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR, MS of disaccharides from β4GalT7 assay. See DOI: 10.1039/c4ob02632b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |