
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hemisynthesis of deuteriated adenosylhopane and conversion into bacteriohopanetetrol by a cell-free system from Methylobacterium organophilum†
Wenjun
Liu
a,
Anne
Bodlenner
b and
Michel
Rohmer
*a
aUniversité de Strasbourg/CNRS, Institut Le Bel, 4 rue Blaise Pascal, F 67070 Strasbourg, Cedex, France. E-mail: mirohmer@unistra.fr
bUniversité de Strasbourg/CNRS, ECPM, 25 rue Becquerel, F 67087 Strasbourg, Cedex 2, France
First published on 6th February 2015
Abstract
Adenosylhopane is a putative precursor of the widespread bacterial C35 biohopanoids. A concise and flexible hemisynthesis of adenosylhopane has been developed including as key steps a cross metathesis between two olefins containing either the hopane moiety or a protected adenosine derivative and a subsequent diimide reduction of the resulting olefin. Reduction by deuteriated diimide allowed deuterium labelling. This synthetic protocol represents a versatile tool to access to deuteriated composite bacterial hopanoids required for biosynthetic studies. Deuteriated adenosylhopane was thus converted into bacteriohopanetetrol by a crude cell-free system from Methylobacterium organophilum in the presence of NADPH, showing for the first time the precursor to product relationship between these two bacterial metabolites.
Introduction
(22R)-Adenosylhopane 2 (Scheme 1) has been found in fairly large amounts in a few species of bacteria1a–e and in trace amounts in many hopanoid producing bacteria.2 Adenosylhopane (and structurally tentatively related hopanoids) is one of the most common hopanoids in soils,3a–c and the presence of this biomarker in recent sediments is interpreted in terms of transport of soils into sediments.3b,4,5 | ||
| Scheme 1 Biogenetic scheme for the biosynthesis of C35 bacterial hopanoids. Identified genes in Methylobacterium extorquens, Rhodopseudomonas palustris and Streptomyces coelicolor: (i) hpnH/orf15; (ii) hpnG/orf14; hpnO/orf18.10–13 | ||
Adenosylhopane 2 is characterized by a unique carbon–carbon bond between C-30 of the hopane moiety and C-5′ of adenosine. The commonly occurring configuration of adenosylhopane was determined by circular dichroism and high-field NMR spectroscopy.1a Trace amounts of (22S)-adenosylhopane as the minor isomer was also reported from Rhodopseudomonas acidophila.1b Chemical conversion of adenosylhopane 2 into bacteriohopanetetrol 5 permitted the determination of the stereochemistry of all asymmetric centres of the side-chain of bacteriohopanetetrol as 22R, 32R, 33R and 34S.1b Later the synthesis of the eight side chain diastereomers of bacteriohopanetetrol confirmed that the absolute configuration of the C5 unit was identical to that of a D-ribitol linked via its C-5 carbon atom to the hopane moiety.6 The same configuration was also found for 35-aminobacteriohopanetriol 6via chemical correlation with bacteriohopanetetrol.7 This side-chain configuration is consistent with the hypothesis deduced from the labelling experiments: the additional C5 unit of elongated hopanoids originates from a D-ribose derivative.8,9 Adenosylhopane was consequently considered as a possible intermediate in the biosynthesis of C35 hopanoids, leading to bacteriohopanetetrol 5 or aminobacteriohopanetriol 6 (Scheme 1).8–10 This hypothesis has been corroborated by the identification of the hpnG gene in Methylobacterium extorquens11 and Rhodopseudomonas palustris12 and the orf14 gene in Streptomyces coelicolor:13 their deletion results in the accumulation of adenosylhopane 2 and in the absence of the major hopanoids, bacteriohopanetetrol 5 or aminobacteriohopanetriol 6.
The elucidation of the biosynthesis of the side-chain of C35 hopanoids requires sufficient amounts of adenosylhopane for future enzyme tests. Two distinct methods are available to produce complex hopanoids: fermentation or chemical synthesis. The amphiphilic character and the poor solubility in most organic solvents of biohopanoids represent severe limitations for their production by fermentation. Moreover, fermentation is only appropriate for a few major compounds as many complex hopanoids are produced by bacteria in only trace amounts. In addition, isolation is until now only performed on acetylated derivatives requiring a final deprotection to get the native free metabolites, which is either cumbersome or simply not available (e.g. for adenosylhopane or bacteriohopanetetrol derivatives with carbamoyl groups). Chemical synthesis, in contrast, allows an access to a broad range of naturally occurring hopanoids in sufficient amounts. Extensive work resulted in suitable syntheses of ribosylhopane, bacteriohopanetetrol and several other related structures.6,14–17 Some synthetic strategies are quite successful with excellent stereochemistry control and good overall yields.
No reliable synthesis is, however, available for adenosylhopane. The initial attempts towards the synthesis of adenosylhopane and related hopanoids were mainly based on two routes. The first strategy, inspired from the hypothetical biogenetic biosynthetic pathway, relies on a coupling between a C30 triterpene derivative and an adenosine derivative. Wittig-type coupling between a triterpene phosphonium salt with an adenosine-5′-aldehyde substrate led to the formation of adenosylhopane 2 in a very low yield,14 probably due to the steric hindrance induced by both the adenosine moiety and the pentacyclic triterpene skeleton in addition to the instability of the nucleoside-5′-aldehyde in the presence of the strong bases required for the generation of the non-stabilized phosphorane.18 Even the coupling strategy involving lithium (the smallest cation) activated hopane moiety, which was very efficient in ribosylhopane 3 synthesis, was disappointing to afford the bulkier 35-O-benzyl ribosylhopane analogue.17 The second strategy, which involves a glycosidic coupling between a ribosylhopane derivative and adenine in the presence of a Lewis acid was also rather unsuccessful, not only because of the poor solubility of adenine and the ribosylhopane derivative in acetonitrile, which promoted side reactions, but also because of the formation of both 35α and 35β isomers of adenosylhopane 2.14 Furthermore, the application of this methodology is also limited considering the number of synthetic steps.
This paper describes an efficient hemisynthesis of adenosylhopane including the possibility of deuterium labelling as well as the conversion of deuterium labelled adenosylhopane into bacteriohopanetetrol by a crude cell-free system form Methylobacterium organophilum, which represents the first direct and indisputable proof for a precursor to product relationship between adenosylhopane 2 and bacteriohopanetetrol 5.
Results and discussion
Hemisynthesis of adenosylhopane
The proposed strategy leading to adenosylhopane 2 is a convergent hemisynthesis with an excellent stereochemical control using commercially available starting materials. Moreover, in order to meet the need for isotopic labelled hopanoids for biosynthetic investigations, this strategy allows also the introduction of deuterium into the hopanoid structure in the last but one step. Our synthetic strategy relies on a cross-metathesis as the key coupling step between a methylene elongated hopane moiety 10 and a properly protected methylene elongated 5′-deoxy-5′-methyleneadenosine derivative 8 (Scheme 2). This approach has the advantage of preserving the stereogenic centres at C-22 on the hopane moiety and at C-4′ of the ribose derivative and avoids the inconvenient glycosylation between adenine and ribose,14 which must be regio- and diastereoselective. Metathesis19 seemed quite attractive because of its high tolerance towards functional groups and bulky structures,20a,b its mildness and impressive efficiency, and the possibility of late labelling by deuteriation of the double bond. Cross metathesis is known to be more difficult than dimerization, ring closing or ring opening metathesis due to the lost advantages from statistics, entropy and ring tension relief respectively. The trick is to use alkenes having strong reactivity differences or an excess of one counterpart.21a,b It was, however, encouraging to note that Andrei and Wnuk succeeded in coupling a protected 5′-deoxy-5′-methyleneadenosine with N-Boc-protected six-carbon amino acids bearing a terminal double bond,22 even if the methylenetriterpene 10 (Scheme 2) in our synthesis presents a steric hindrance closer to the terminal double bond than their cross-metathesis partner. Furthermore, both olefins 8 and 10 have the nice reactivity of terminal monosubstituted olefins21 to counteract the steric hindrance present one bond away from the olefin.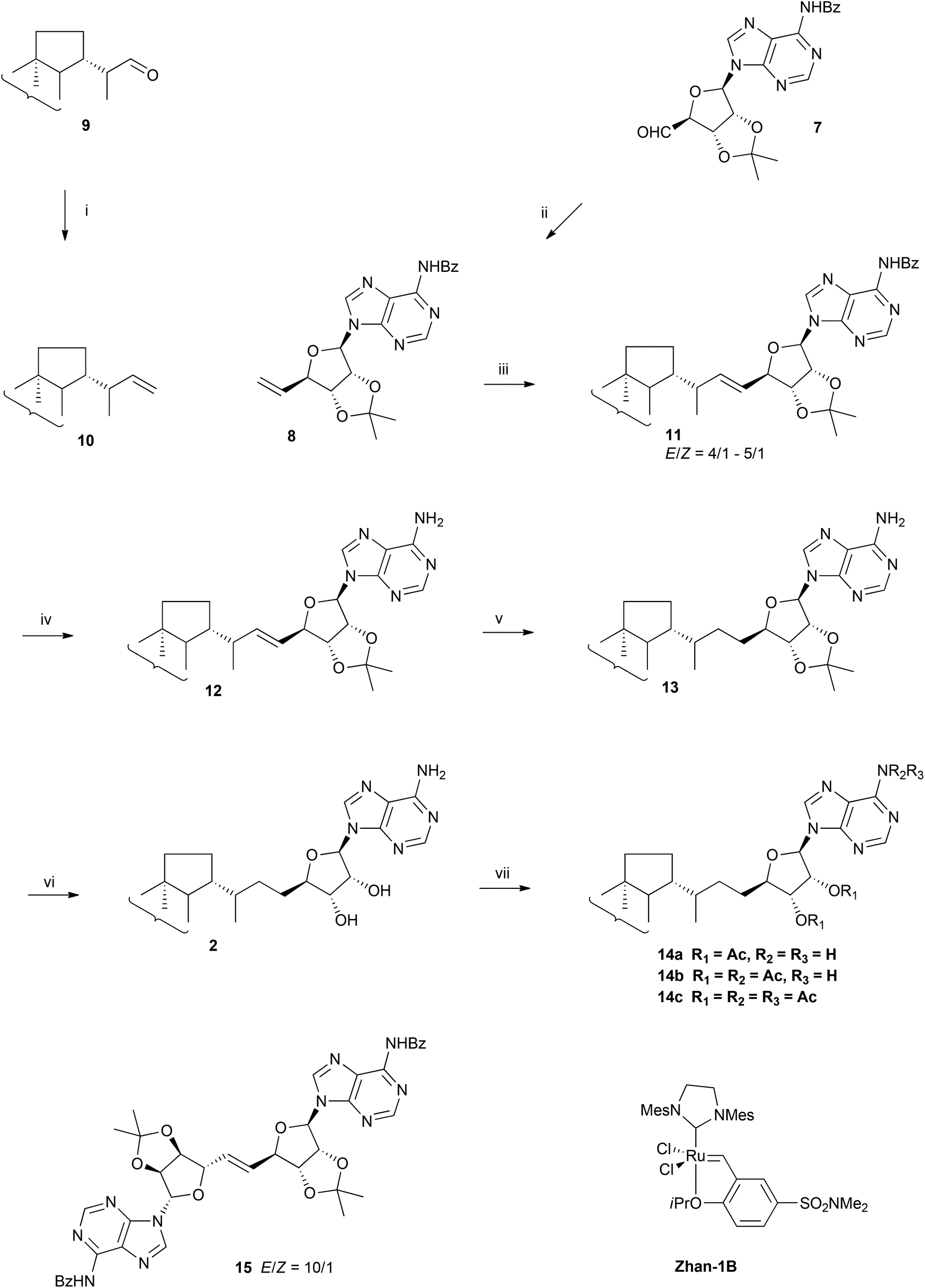 | ||
| Scheme 2 Hemisynthesis of adenosylhopane 2. (i) Ph3PCH3Br, n-BuLi, DMSO–THF, 50 °C, 98%; (ii) Ph3PCH3Br, n-BuLi, THF, −40 °C, 75%; (iii) metathesis; (iv) NH4OH, H2O, MeOH, CH2Cl2, 4 °C, 91%; (v) PADA, AcOH, Py, 90 °C, 73%; (vi) TFA, CHCl3–CH3OH, 0 °C, 92%; (vii) Ac2O, Py. | ||
Protected 5′-deoxy-5′-methyleneadenosine 8 (Scheme 2) was obtained by a Wittig methylene elongation of aldehyde 7, obtained in five steps from commercially available 2′,3′-O-isopropylideneadenosine by an adaptation of two published methods (cf. ESI, Scheme S1†).23a,b In parallel, homohop-30-ene 10 (Scheme 2), the triterpenic coupling partner, was synthesized in high yield via a Wittig reaction from (22S)-hopan-29-al 9, synthesized in four steps from hydroxyhopanone extracted from Dammar resin,15,24 and presenting the C-22 configuration of most bacterial hopanoids.25
Cross metathesis
Cross metathesis reactions between homohop-30-ene 10 and the 5′-methylene adenosine derivative 8 were tested under different conditions (Scheme 2 and Table 1). Given the high value of both olefins, and hoping that their reactivity are different enough,21a initial attempts of cross metathesis were performed with stoichiometric amounts. Reaction with alkene 8 and one equivalent of homohopene 10 in the presence of Hoveyda–Grubbs catalyst (HII), did not, however, give the cross coupling product 11 under reflux conditions (Table 1, entry 1). This lack of reactivity for those two terminal alkenes probably resulted from the high steric hindrance in the expected metallacyclobutanyl transition state, together with an activation energy barrier too high to be overcome under classical thermic conditions. Coupling product 11 was, however, isolated with an up to 13% yield when the same reaction was performed under microwave irradiation (Table 1, entry 2). Dimer 15 (Scheme 2) was also isolated in 43% yield, resulting from the self-metathesis of the nucleoside substrate 8. Unreacted starting materials 8 and 10, were also isolated (Table 1, entry 2). The product of self-metathesis of homohop-30-ene 10 was not detected. Ruthenium catalyst Grubbs II (GII) promoted cross metathesis between 8 and 10 in a lower yield, (Table 1, entry 4), suggesting a higher efficiency of HII over GII with those olefins. Finally, the N,N-dimethylaminosulfonyl group containing Hoveyda–Grubbs-type complex catalyst Zhan-1B, was found almost as active as HII for this cross metathesis reaction, and similar yields were obtained for the coupling product 11 and dimer 15 (Table 1, entries 2 and 3).| Catalysta | Conditions | 10/8 ratio | Products | |
|---|---|---|---|---|
| a 10 mol% of catalyst were used. b Reaction under reflux at ambient pressure. c MW is abbreviation of microwave irradiation. d Yield was not determined for 15. | ||||
| 1 | H II | 40 °C, DCM, 24 hb | 1/1 | No reaction |
| 2 | H II | MWc, 75 °C, DCM, 3 h | 1/1 | 11 (13%), 15 (43%) |
| 3 | Zhan-1B | MW, 75 °C, DCM, 3 h | 1/1 | 11 (12%), 15 (45%) |
| 4 | G II | MW, 75 °C, DCM, 3 h | 1/1 | 11 (6%), 15d |
| 5 | Zhan-1B | MW, 75 °C, DCM, 3 h | 10/1 | 11 (59%), 15 (40%) |
| 6 | H II | in sealed tubes, DCM, overnight | 10/1 | 11 (51%), 15 (46%) |
| 7 | Zhan-1B | in sealed tubes, DCM, overnight | 10/1 | 11 (52%), 15 (43%) |
| 8 | H II | MW, 75 °C, perfluorobenzene, 3 hb | 1/1 | 11 (6%), 15 (78%) |
Attempts to obtain olefin 11 from secondary cross metathesis between homohopene 10 and dimer 15 under microwave irradiation at 75 °C did not lead to the formation of the protected adenosylhopene 11, but allowed the recovery of the two starting reagents. The inability of dimer 15 to undergo secondary cross metathesis in addition to its formation from 8 under our conditions but its absence in the cross metathesis performed by Andrei and Wnuk with a type I olefin as cross metathesis partner,22 suggest that it belongs to type II olefins for cross metathesis reactions in Grubbs categorization of alkenes.21a Besides, the absence of the product of self-metathesis of homohop-30-ene 10 suggests that it is a type III olefin in spite of its terminal alkene.
Reacting two cross metathesis partners of different types using feedstock stoichiometries as low as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 normally allows selective cross metathesis, but in our case the yield remained low. Given these features, the crucial point to obtain higher yield for coupling compound 11 was to maintain a low concentration of adenosine derivative 8 compared to that of homohopene 10, minimizing thus the amount of dimerization. A large excess (10 eq.) of the less reactive homohopene 10 was thus introduced, and, as expected, the yield of protected adenosylhopene 11 rose significantly from 13% to 59% in the presence of Zhan-1B catalyst under microwave irradiation (Table 1, entry 5). However, the cross metathesis yield still remained moderate, probably due to the low reactivity of homohopene 10. Increasing more the stoichiometry ratio was not possible for solubility reasons.
:1 normally allows selective cross metathesis, but in our case the yield remained low. Given these features, the crucial point to obtain higher yield for coupling compound 11 was to maintain a low concentration of adenosine derivative 8 compared to that of homohopene 10, minimizing thus the amount of dimerization. A large excess (10 eq.) of the less reactive homohopene 10 was thus introduced, and, as expected, the yield of protected adenosylhopene 11 rose significantly from 13% to 59% in the presence of Zhan-1B catalyst under microwave irradiation (Table 1, entry 5). However, the cross metathesis yield still remained moderate, probably due to the low reactivity of homohopene 10. Increasing more the stoichiometry ratio was not possible for solubility reasons.
Under these conditions, the olefinic adenosine substrate 8 was fully consumed, resulting in the formation of 40% of dimer 15. A prolonged reaction time failed to further increase the yield in 11, supporting to a greater extent that dimer 15 was indeed unable to undergo a secondary hetero-metathesis even with homohop-30-ene 10 in excess. The inability of compound 10 to homodimerize, probably due to a strong steric hindrance induced by the hopane ring system and the pseudo-axial position of the side chain, allowed the recycling of the large excess of olefin 10.
The same reactions were performed in sealed tubes without microwave irradiation (Table 1, entries 6 and 7). A night long reaction time was required to reach a slightly lower cross metathesis yield, indicating that the highly beneficial effects of microwave irradiation did not only arise from the rapid heating and pressure increase allowed in the microwave oven (purely thermal/kinetic effect), but also from some specific thermal microwave effect, such as wall effect and selective heating of strongly microwave-absorbing heterogeneous catalysts in a less polar reaction medium.
Furthermore, a polar solvent (e.g. dichloromethane) is required to enhance the heating effect of microwave. Although perfluorobenzene was reported to be capable of increasing the activity of cross metathesis catalysts,26 its apolar character probably led to less efficient heating by microwave, resulting in a lower yield of 11 in comparison to the one obtained with the use of dichloromethane (Table 1, entries 2 and 8).
As expected, the cross-metathesis product 11 presented predominantly the E configuration as shown by the larger vicinal J30,31 coupling constant of the vinylic protons in the spectrum of the major E isomer (15.4 Hz) versus the smaller coupling constant in the spectrum of the minor Z isomer (11.0 Hz). Based on the integration of the 32-H and 33-H signals of both protected (E)- and (Z)-adenosylhop-32-ene 11 in the 1H-NMR spectrum, i.e. (E)-32-H at 4.74 ppm and (Z)-32-H at 5.03 ppm and (E)-33-H at 4.96 ppm and (Z)-33-H at 4.85 ppm, the relative amount of E isomer varied from 75% to 80%.
Reduction of the double bond of the protected adenosylhopene 11 turned out to be more challenging than expected. Catalytic hydrogenation was disappointing even under pressure, and gave very low yields in the presence of Pd/C (<10%), Adam's catalyst (<10%)14,27 or no reaction with Wilkinson's28 and Crabtree's29 catalysts. Again, the steric hindrance around the double bond probably allowed only restricted access or no access at all for the different catalysts to the double bond. This hindered access to the olefin was circumvented by the use of the small sized diimide, a short-lived reagent that can be implicated in the reduction of nonpolar multiple bonds. In a concerted mechanism, cis-diimide is converted into N2via a six-centre transition state corresponding formally to the syn addition of dihydrogen to the double bond.30a,b Taking into account that this reaction had to be extended to a deuterium labelled version, the acid promoted decarboxylation of potassium azodicarboxylate was chosen to generate diimide.31 Furthermore, the acid catalysis may speed up the equilibration of trans- and cis-diimide, favouring thus the hydrogen transfer from the cis-isomer to the alkene. Direct treatment of the N-protected adenosylhopene 11 with potassium azodicarboxylate and acetic acid, however, led to the partial loss of the 6′-N-benzoyl group. This protecting group was therefore removed before diimide reduction using a saturated methanolic ammonia solution in dichloromethane to afford the N-deprotected adenosylhopene 12. Diimide reduction of its double bond was then achieved by a continuous addition of potassium azodicarboxylate and acetic acid for 36 h and afforded the expected protected adenosylhopane 13 in 73% yield. The reaction required a large excess of diimide and a long reaction time, probably because of the steric hindrance around the double bond, resulting in a low reduction rate in comparison to the one of the disproportionation of diimide into nitrogen and hydrazine, the major competing reaction consuming the reducing agent. In order to minimize this disproportionation, potassium azodicarboxylate was added to the reaction mixture in small portions. Acetic acid was added only when the nitrogen evolution had ceased, keeping thus a low diimide concentration. Water was also avoided because it had been reported to be a powerful inhibitor of diimide reductions in aprotic solvents such as pyridine.31 Finally, acid catalysed deprotection of the acetonide of 13 with a methanolic TFA solution afforded adenosylhopane 2 in high yield. The reaction was conducted at 0 °C on a rotary evaporator to continuously remove 2,2-dimethoxypropane and shift the equilibrium. Purification of free adenosylhopane 2 also proved tricky, due to its amphiphilic character and its insolubility in polar solvents such as acetonitrile, methanol and water preventing purification by reversed phase HPLC. Purification was indeed successfully achieved by gravity column chromatography on silica gel with a polar ternary solvent (chloroform–methanol–ammonia).
Deuterium labelling of adenosylhopane
Only minor changes were required to obtain the deuterium labelled adenosylhopane 2-D (Scheme 3). Olefin 12 was first treated with CH3O2H to avoid the presence of extra protons coming from the exchangeable hydrogens of the amino group to yield 12-D. Monodeuteriated acetic acid CH3COO2H (98% isotope abundance) was adopted as deuterium source to generate dideuteriodiimide. Reduction of olefin 12-D was much slower with deuteriodiimide than that with natural abundance diimide due to a primary deuterium isotope kinetic effect. This relatively slow reaction resulted in a decreased yield (60%), but allowed to recover unreacted starting material 12 after 36 h treatment. The 1H-NMR spectrum of the recovered starting material 12 showed that the E/Z ratio decreased from 5:1 before diimide reduction to 1:1 after reduction, indicating a faster hydrogenation rate of the E olefin. This phenomenon has been frequently observed in diimide reductions and is attributed to the more cluttered transition state for a Z olefin than for an E olefin.30a
 | ||
| Scheme 3 Hemisynthesis of (30,31-2H2)adenosylhopane 2-D. (i) MeOD; (ii) PADA, AcOD, Py, 90 °C and (iii) H2O, 60%, 2 steps; (iv) TFA, CHCl3–CH3OH, 0 °C, 90%. | ||
The deuterium isotope abundance of the protected adenosylhopane 13-D was determined by mass spectrometry. In spite of all precautions against water contamination the deuterium content was not maximal, and the reaction resulted in a mixture of isotopomers: (30,31-2H2)-13-D (60%), (30-2H1)- and (31-2H1)-13-D (31%) and natural abundance 13 (9%). This incomplete labelling is probably due to the large excess of CH3COO2H (60 eq.) having only a 98% isotope abundance and to the primary deuterium kinetic effect, leading to a mixture of N22H2, N22H H and N2H2. Deuterium isotope abundance can be probably improved by using CH3COO2H with higher isotope abundance. The obtained labelling was, however, largely sufficient to perform the incorporation experiments.
Structure of adenosylhopane and of its bisdeuteriated isotopomer
To confirm the structure, synthetic adenosylhopane 2 was acetylated (Scheme 2). The spectroscopic data of adenosylhopane di-, tri- and tetraacetate 14a–c were compared with those of acetylated adenosylhopane isolated from bacteria.1a,b All the analytical data were consistent with those previously described. The assignments of the 1H and 13C-NMR signals were made with the help of 2D NMR (1H–1H COSY, HMBC and HSQC) and by comparison with those obtained on other hopanoids.16,32The structure of protected bisdeuteriated (31,32-2H2)adenosylhopane 13-D was confirmed by comparing its 1H- and 13C-NMR spectra recorded in (2H5)pyridine with those of the corresponding natural abundance adenosylhopane derivative 18. Four different configurations at C-30 and C-31 were observed after N22H2 reduction. Given that diimide reacts faster with the protected (E)-adenosylhopene 17, which is the dominant isomer, the major products resulting from syn addition of deuteriated diimide to the E double bond should be protected (30R,31R-2H2)- and (30S,31S-2H2)adenosylhopane, and the (30R,31S-2H2)- and (30S,31R-2H2)adenosylhopane diastereomers would be generated by the reduction of protected (Z)-adenosylhopene by diimide (cf. ESI and Scheme S2† for the discussion of the stereochemistry of those compounds and the interpretation of the NMR spectra).
Conversion of adenosylhopane into bacteriohopanetetrol by a cell free system from Methylobacterium organophilum
Methylobacterium organophilum has been chosen for its substantial production of hopanoids (up to 13 mg g−1, dry weight).33 It only synthesizes bacteriohopanetetrol derivatives, a glycoside and two ethers involving a carbapseudopentose moiety, free bacteriohopanetetrol being a minor hopanoid. This makes this bacterium a good candidate for incorporations with a crude cell-free system, since dilution of the deuterium labelled tetrol synthesized de novo from (2H)adenosylhopane by the natural abundance endogenous tetrol synthesized during the culture will be minimal. In addition, in order to diminish this dilution, unbroken cells and large cell debris containing most of the hopanoids were removed by centrifugation, and the incubations were only performed with the resulting supernatant.According to the hypothetical biogenetic scheme (Scheme 1), several steps are required for the conversion of adenosylhopane into bacteriohopanetetrol via ribosylhopane:10 a deadenylation into ribosylhopane, the hemiketal ring-opening of ribosylhopane and a reduction. To obtain ribosylhopane 3, the postulated precursor,10,13 two pathways have been proposed for the deadenylation: either a direct hydrolysis of adenosylhopane to ribosylhopane by a nucleosidase-like enzyme12 or a phosphorolysis of the nucleotide hemiaminal with the loss of adenine by a purine nucleoside phosphorylase-like enzyme, yielding ribosylhopane phosphate.11 In this case, ribosylhopane 35-phosphate cannot be reduced and has first to be converted by a phosphatase into ribosylhopane 3 prior reduction. The final step is in all cases the reduction of the open aldehyde form of ribosylhopane by an NADPH-dependent aldose reductase-type enzyme (Bodlenner and Rohmer, unpublished results). For those reasons, the incubations were performed in a phosphate buffer or in a phosphate free triethylamine buffer in the presence of NADPH. Adenosylhopane was introduced as a THF solution. Although the use of this solvent is not common in enzymatic processes, THF is a good solvent for this amphiphilic hopanoid and has the advantage of being water miscible. Fortunately, the presence of 10% THF in the incubation medium did not affect the enzymes activities. A lipid extraction followed by acetylation allowed isolation of bacteriohopanetetrol tetraacetate and analysis of the deuterium labelling by electron impact GC-MS (Fig. 1). Deuterium incorporation was evaluated from the relative abundances of the (m + 1)/z and (m + 2)/z versus those of the m/z signals (Table 2). Evidence that adenosylhopane was efficiently converted into BHT in a phosphate buffer was shown from intense 2H labelling for all ions containing the side chain [m/z 714 (M+), 699 (M+ − CH3), 654 (M+ − AcOH), 493 (fragment B, ring C cleavage)], but not for those without the side-chain: [m/z 369 (M+ − side chain), 191 (fragment A, ring C cleavage)] (Fig. 1),34 indicating that the 2H labelling was strictly localized in the C5 side chain as expected (Table 2, entry 2). In order to minimize the influence of the background noise, only the two most intense signals of the mass spectrum corresponding to fragments generated by ring C cleavage are shown and considered for evaluating the conversion of deuteriated adenosylhopane into bacteriohopanetetrol (Table 2).
 | ||
| Fig. 1 Fragmentation of the hopane skeleton by electron impact mass spectrometry. | ||
| Conditionsb | Fragment A (m/z = 191) | Fragment B (m/z = 493) | |||
|---|---|---|---|---|---|
| m + 1/m (%) | m + 2/m (%) | m + 1/m (%) | m + 2/m (%) | ||
| a Bacteriohopanetetrol was analysed as tetraacetate by GC/MS. Isotopic patterns of the fragments were measured according to the signal intensities observed for m/z (reference signal, 100%), m/z + 1 and m/z + 2. The standard deviation of the signal intensities of both fragments was calculated from the GC-MS analyses of seven samples of peracetylated bacteriohopanetetrol 5 isolated from seven different cell batches. A confidence interval of 95% can also be calculated by doubling the standard deviation value. The intensities if signals of fragments A and B have a 2% deviation. b All the incorporation experiments were performed at 30 °C for 24 h. A. Deuteriated adenosylhopane with cell-free extract in phosphate buffer. B. Deuteriated adenosylhopane with cell-free extract in TEA buffer. C. Decomposed deuteriated adenosylhopane after 24 h in phosphate buffer and then incubated with the cell-free extract in phosphate buffer. D. Deuteriated adenosylhopane with inactivated cell free system in phosphate buffer. c Reference mass spectrum of natural abundance bacteriohopanetetrol tetraacetate isolated from the unbroken cells and cell debris pellets recovered after centrifugation yielding the supernatant utilised for incubations. d High signal/noise ratio. | |||||
| 1 | Referencec | 20 ± 4 | 5 ± 2 | 28 ± 3 | 6 ± 1 |
| 2 | A | 19 | 4 | 27 | 47 |
| 3 | B | 21 | 4 | 36 | 19 |
| 4 | C | 24 | 1d | 29 | 6 |
| 5 | D | 19 | 2 | 28 | 6 |
Moreover, the ratio of bis- and monodeuteriated isotopomers (d2/d1 = 1.9/1) of adenosylhopane was found unchanged in the labelled bacteriohopanetetrol (d2/d1 = 1.9/1), indicating that incubated (2H)adenosylhopane is the only deuterium source and that there is no significant isotope effect in this enzymatic conversion, which is logical as no reaction affects the deuterium labelled carbon atoms in this biosynthetic pathway.
In order to confirm that the overall conversion of (2H)adenosylhopane 2-D into bacteriohopanetetrol 5 is purely enzymatic, (2H)adenosylhopane 2-D was incubated with a cell-free system inactivated by boiling. No deuterium labelling was found in the bacteriohopanetetrol isolated from this assay (Table 2, entry 5).
A non-enzymatic conversion of (2H)adenosylhopane 2-D into ribosylhopane 3 (or into ribosylhopane phosphate) was also excluded since a 3 days preincubation of (2H)adenosylhopane in a phosphate buffer with 10% THF at 30 °C did not lead to any detectable deuterium incorporation, even though a partial degradation of adenosylhopane was observed on TLC. Incubation of such a degraded sample resulted in no formation of 2H labelled bacteriohopanetetrol, indicating that even the decomposition products of (2H)adenosylhopane cannot be converted into bacteriohopanetetrol (Table 2, entry 4).
The present conversion of deuterium labelled adenosylhopane 2 into bacteriohopanetetrol 5 by a crude cell-free system of Methylobacterium organophilum in the presence of NADPH, indicates that the nucleoside side chain of adenosylhopane 2 is the precursor of the D-ribitol side chain of bacteriohopanetetrol 5. This biotransformation requires at least two reactions, depending on the nature of the nucleophile (phosphate or water) responsible of the cleavage of the glycosidic bond with adenine loss. Hoping to solve that question, the (2H)adenosylhopane incubations with a cell-free system of M. organophilum were performed using two different buffers. Although, the conversion was significantly higher in the phosphate buffer than in the phosphate free triethylamine buffer (Table 2, entries 2 and 3), there is no clear cut conclusion in favour of one of the two proposed deadenylation mechanism. A sufficient phosphate concentration originating from the cells might indeed be still present in the triethylamine buffer to achieve the phosphorolysis, and a slight inhibitory effect on the putative aldose reductase of triethylamine/triethylammonium cannot be excluded to explain the lower yields. In fact, ribosylhopane is effectively an intermediate in biohopanoid biosynthesis. Deletion of the orf18 gene in a S. coelicolor strain (corresponding to the hpnO gene in M. extorquens and R. palustris)11,12 allowed to identify for the first time in the Δorf18 mutant ribosylhopane 3 (Scheme 1), postulated twenty years ago as an intermediate in the biosynthesis of C35 hopanoids.10,15 It is accumulated in the absence of the putative transaminase encoded by the hpnO/orf18 gene catalysing the reductive transamination of the free aldehyde 4 into aminobacteriohopanetriol 6.13 Decisive proofs will be obtained later with tests performed with the isolated and purified enzymes.
In the final step, a reduction converts the open free aldehyde 4 of ribosylhopane 3 into bacteriohopanetetrol 5, which has been found to be NADPH-dependent (Bodlenner and Rohmer, unpublished results). The reaction resembles the reduction catalysed by an aldose reductase, but the gene encoding this enzyme is yet unknown in hopanoid producing bacteria.
Conclusion
A highly efficient synthesis of adenosylhopane and its deuterium labelled homologue has been achieved from the coupling of homohop-30-ene and a 5′-methylene adenosine derivative via a cross-metathesis. This approach was found to be the only one able of linking the two highly hindered hopane and adenine counterparts to date, affording thus a total control of all the stereogenic centres of this large molecule. Moreover the metathesis step allows further labelling through the reduction of the introduced double bond by diimide. The deuterium labelled adenosylhopane was thus obtained and incubated with a supernatant of a crude cell-free system of Methylobacterium organophilum yielding deuterium labelled bacteriohopanetetrol as shown by EI-MS of its tetraacetate. This allowed for the first time to demonstrate the substrate to product relationship between adenosylhopane and bacteriohopanetetrol. This work opens also the way for the efficient synthesis of other biohopanoids and for further enzymatic studies on isolated enzymes required for the full elucidation of hopanoid biosynthesis.Experimental
Reagents and solvents
Reagents were purchased from Sigma-Aldrich, Acros, Alfa Aesar or Carl Roth. Their purity was more than 98% in all cases. Dowex 50 (H+) resin was purchased from Aldrich and was activated right before use with a 10% HCl solution in water.All solvents were distilled before use. Dry solvents used in moisture sensitive reactions were obtained as follows: THF was freshly distilled from sodium benzophenone ketyl before use. CH2Cl2 was distilled over P2O5. Pyridine was distilled over KOH and then stored over molecular sieves (4 Å). DMSO was purchased from Aldrich (sure sealed) and used directly.
Purification techniques
The progress of reactions was monitored by analytical thin layer chromatography using aluminum-coated Merck 60 F254 silica plates. UV visible spots were directly visualized under UV light at λ = 254 nm. Hopanoids were revealed on TLC plate by UV light (λ = 366 nm) after spraying with a 0.1% berberine hydrochloride solution in EtOH.Most products were purified by flash column chromatography using Merck 60 (230–400 mesh) silica gel and appropriate eluents.35 Adenosylhopane 2 and 2-D were purified by gravity column chromatography using Merck 60 (63–200 μm) silica gel. Compounds in amounts below 10 mg were purified via preparative thin layer chromatography using Merck 60 F254 silica gel plates (0.25 mm layer thickness).
Spectroscopic analysis
1H-NMR and 13C-NMR were performed on BRUKER Avance 600 spectrometer (600 MHz for 1H, 150 MHz for 13C), Bruker Biospin 500 spectrometer (500 MHz for 1H, 125 MHz for 13C) and BRUKER Avance 300 spectrometer (300 MHz for 1H, 75 MHz for 13C). Most of the measurements were carried out using C2HCl3 as solvent and with CHCl3 (δ = 7.26 ppm) as internal standard for 1H-NMR and C2H Cl3 (δ = 77.0 ppm) for 13C-NMR. NMR spectra of adenosylhopane 2 and 2-D were recorded in (2H5)pyridine as solvent and with (2H4)pyridine (δ = 8.73, 7.58 and 7.21 ppm) as internal standard for 1H-NMR and (2H5)pyridine (δ = 149.9, 135.5 and 123.5 ppm) for 13C-NMR. The following abbreviations represent the multiplicity of the NMR signals: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; dd, doublet of doublet; ddd, doublet of doublet of doublet; dddd, doublet of doublet of doublet of doublet; arom., aromatic protons or carbon atoms; br, broad.1H- and 13C-NMR assignments of hopanoids were based on earlier assignments14,17,32 and were supported by additional experiments including DEPT, HSQC and HMBC techniques. 13C shift values of the carbon atoms in the hopanoid skeleton did not depend significantly on the nature of the side-chain and remained virtually unchanged after introduction of new carbon atoms or functional groups in the C5 side-chain.
GC-MS spectra were acquired on a Thermo TSQ Quantum mass spectrometer connected to a Thermo Trace GC ultra gas chromatograph (PTV injector, HP-5 MS column, 30 m, 0.25 mm internal diameter, 0.25 μm film thickness). Helium (constant flow 1 mL min−1) was used as carrier gas. The temperature program for GC analysis was: 3 min at 55 °C, from 55 °C to 320 °C at 10 °C min−1 and 50 min at 320 °C.
Mass spectra were obtained by direct inlet on a Thermo TSQ Quantum mass spectrometer at 70 eV in the electron impact positive ionization mode (EIMS) over a mass range of 50–800 Da (cycle time 0.5 s). The source temperature was set at 230 °C.
ESI-HRMS mass spectra were carried out on a Bruker MicroTOF spectrometer.
Melting points were measured with a BIBBY SMP3 apparatus and are corrected with benzophenone (m.p. 48 ± 1.5 °C). Optical rotations were measured on a Perkin Elmer 341 polarimeter at 20 ± 2 °C and 589 nm using 10 cm length and 1 mL volume cell.
(22S)-Homohop-30-ene (10)
A suspension of NaH (126 mg, 5 mmol) in DMSO (2.5 mL) was stirred at 75 °C for 50 min. The light green suspension was cooled down to room temperature, added into a solution of PPh3CH3Br (1.9 g, 5.4 mmol) in DMSO (27 mL) and stirred at room temperature for 20 min to afford a light green-grey suspension. The resulting mixture was then added into a solution of aldehyde 9 (297 mg, 0.70 mmol) in THF (20 mL) and stirred overnight at 50 °C. The reaction was quenched with water, and extracted three times with pentane. The combined organic phases were dried over anhydrous Na2SO4, filtered through cotton, and evaporated to dryness in vacuo to give an almost pure crystalline product, which was further purified by flash chromatography (petroleum ether) to yield the terminal olefin 10 (290 mg, 98%). Rf 0.67 (petroleum ether–EtOAc, 20:1).
M.p. = 203–205 °C.
1H-NMR (300 MHz, C2HCl3): δ/ppm = 5.53 (1H, ddd, J30,31a = 17.8 Hz, J30,31b = 9.5 Hz, J22,30 = 9.1 Hz, 30-H), 4.85 (1H, ddd, J30,31a = 17.8 Hz, Jgem = 2.0 Hz, J22–31a = 0.5 Hz, 31-Ha), 4.85 (1H, dd, J30,31b = 9.5 Hz, Jgem = 2.0 Hz, 31-Hb), 2.21 (1H, ddq, J21,22 = 9.7 Hz, J22,30 = 9.1 Hz, J22,29 = 6.5 Hz, 22-H), 1.8 (1H, tdd, J21,22 = 9.7 Hz, J20,21 = J17,21 = 4.8 Hz, 21-H), 1.01 (3H, d, J22,29 = 6.5 H, 22R-Me), 0.960 (6H, s, 8β-Me and 14α-Me), 0.851 (3H, s, 4α-Me), 0.818 (3H, s, 10β-Me), 0.796 (3H, s, 4β-Me), 0.710 (3H, s, 18α-Me).
13C-NMR (75 MHz, C2HCl3): δ/ppm = 145.3, 112.0, 56.1, 54.1, 50.4, 49.2, 45.0, 44.4, 42.2, 42.1, 41.9, 41.8, 41.7, 40.3, 37.4, 33.5, 33.4, 33.3, 33.2, 27.6, 24.0, 22.3, 22.0, 21.6, 20.9, 18.7, 16.6, 15.9.
EI-MS (direct inlet, positive mode 70 eV): m/z = 424 (M+, 19%), 409 (M+–CH3, 5%), 369 (M+–side chain, 20%), 203 (ring C cleavage, 100%), 191 (ring C cleavage, 88%).
N-Benzoyl-33,34-O-isopropylidene-adenosylhopene (11) and homodimer (15)
:3) to afford compound 11 (15 mg, 52%, E/Z ∼4:1) as a mixture of two isomers and dimer 15 (6 mg, 43%) as a by-product.
:3) to afford compound 11 (23 mg, 59%, E/Z ∼4:1) and dimer 15 (7.5 mg, 40%). Compound 11 was isolated as a colourless solid mixture of the E/Z isomers that were not separated. Rf 0.47 (CH2Cl2–MeOH, 100:3).
1H-NMR (300 MHz, C2HCl3) of (E)-11 and (Z)-11 (subscripts “E” and “Z” characterize respectively the 1H-NMR signals of (E)-11 and (Z)-11, which are respectively present in a 4:1 ratio): δ/ppm = 9.29 (1H, br. s, –NHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 8.82 (1H, s, 2′-H), 8.12 (0.2H, s, 8′-HZ), 8.10 (0.8H, s, 8′-HE), 8.04–8.01 (2H, m, Ar–H), 7.61–7.47 (3H, m, Ar–H), 6.14 (1H, d, J34,35 = 1.9 Hz, 35-HZ and 35-HE), 5.55 (0.8H, dd, J33,34 = 6.2 Hz, J34,35 = 1.9 Hz, 34-HE), 5.51 (0.2H, dd, J33,34 = 6.1 Hz, J34,35 = 1.8 Hz, 34-HZ), 5.46 (0.8H, dd, J30,31 = 15.4 Hz, J22,30 = 8.9 Hz, 30-HE), 5.37 (0.8H, dd, J30,31 = 15.4 Hz, J31,32 = 7.4 Hz, 31-HE), 5.28 (0.4H, dd, J30,31 = 11.0 Hz, J31,32 = 8.7 Hz, 30-HZ and 31-HZ), 5.03 (0.2H, dd, J31,32 = 8.7 Hz, J32,33 = 3.0 Hz, 32-HZ), 4.96 (0.8H, dd, J33,34 = 6.2 Hz, J32,33 = 3.0 Hz, 33-HE), 4.85 (0.2H, dd, J33,34 = 6.0 Hz, J32,33 = 3.0 Hz, 33-HZ), 4.74 (0.8H, dd, J31,32 = 7.3 Hz, J32,33 = 2.8 Hz, 32-HE), 2.71–2.65 (0.2H, m, 22-HZ), 2.17–2.11 (0.8H, m, 22-HE), 1.65 and 1.41 (6H, 2s, Me2C), 1.01 (0.6H, d, J22,29 = 6.2 Hz, 22R-MeZ), 0.97 and 0.95 (1.2H, 2s, 8β-MeZ and 14α-MeZ), 0.93 and 0.92 (4.8H, 2s, 8β-MeE and 14α-MeE), 0.89 (2.4H, d, J22,29 = 6.4 Hz, 22R-MeE), 0.84 (18H, s, 4α-Me), 0.80 (3H, s, 10β-Me), 0.781 (3H, s, 4β-Me), 0.64 (3H, s, 18α-Me).
O), 8.82 (1H, s, 2′-H), 8.12 (0.2H, s, 8′-HZ), 8.10 (0.8H, s, 8′-HE), 8.04–8.01 (2H, m, Ar–H), 7.61–7.47 (3H, m, Ar–H), 6.14 (1H, d, J34,35 = 1.9 Hz, 35-HZ and 35-HE), 5.55 (0.8H, dd, J33,34 = 6.2 Hz, J34,35 = 1.9 Hz, 34-HE), 5.51 (0.2H, dd, J33,34 = 6.1 Hz, J34,35 = 1.8 Hz, 34-HZ), 5.46 (0.8H, dd, J30,31 = 15.4 Hz, J22,30 = 8.9 Hz, 30-HE), 5.37 (0.8H, dd, J30,31 = 15.4 Hz, J31,32 = 7.4 Hz, 31-HE), 5.28 (0.4H, dd, J30,31 = 11.0 Hz, J31,32 = 8.7 Hz, 30-HZ and 31-HZ), 5.03 (0.2H, dd, J31,32 = 8.7 Hz, J32,33 = 3.0 Hz, 32-HZ), 4.96 (0.8H, dd, J33,34 = 6.2 Hz, J32,33 = 3.0 Hz, 33-HE), 4.85 (0.2H, dd, J33,34 = 6.0 Hz, J32,33 = 3.0 Hz, 33-HZ), 4.74 (0.8H, dd, J31,32 = 7.3 Hz, J32,33 = 2.8 Hz, 32-HE), 2.71–2.65 (0.2H, m, 22-HZ), 2.17–2.11 (0.8H, m, 22-HE), 1.65 and 1.41 (6H, 2s, Me2C), 1.01 (0.6H, d, J22,29 = 6.2 Hz, 22R-MeZ), 0.97 and 0.95 (1.2H, 2s, 8β-MeZ and 14α-MeZ), 0.93 and 0.92 (4.8H, 2s, 8β-MeE and 14α-MeE), 0.89 (2.4H, d, J22,29 = 6.4 Hz, 22R-MeE), 0.84 (18H, s, 4α-Me), 0.80 (3H, s, 10β-Me), 0.781 (3H, s, 4β-Me), 0.64 (3H, s, 18α-Me).
13C-NMR of the major isomer (E)-11 (75 MHz, C2HCl3): δ/ppm = 164.5, 152.6, 151.1, 149.6, 142.1, 133.6, 132.7, 128.7, 127.8, 124.5, 123.5, 114.3, 91.2, 88.6, 84.8, 84.4, 56.0, 54.0, 50.3, 49.1, 44.8, 44.3, 42.0, 41.8, 41.7, 41.5, 40.5, 40.2, 37.3, 33.4, 33.3, 33.2, 33.2, 27.5, 27.0, 25.4, 23.9, 21.9, 21.7, 21.5, 20.9, 18.6, 16.5, 15.9, 15.8.
HRMS (ESI): m/z [M + Na]+, calcd for C50H69N5NaO4+ 826.524, found 826.524.
Homodimer of 5′-methylene adenosine (15)
Dimer 15 was isolated as a solid mixture of two E/Z isomers (∼3:1). Rf = 0.16 (CH2Cl2–MeOH, 100:3).
1H-NMR (300 MHz, C2HCl3) (subscripts “E” and “Z” characterize respectively the 1H-NMR signals of (E)-15 and (Z)-15, which are respectively present in a 3:1 ratio): δ/ppm = 9.24 (1.5H, br. s, –NHBzE), 9.11 (0.5H, br. s, –NHBzZ), 8.79 (0.5H, br. s, 2-HZ), 8.63 (1.5H, br. s, 2-HE), 8.10 (0.5H, s, 8-HZ), 8.04–8.02 (4H, m, Ar-H), 7.95 (1.5H, s, 8-HE), 7.64–7.44 (6H, m, Ar-H), 6.15 (0.5H, d, J1′,2′ = 1.5 Hz, 1′-HZ), 6.06 (1.5H, d, J1′,2′ = 1.7 Hz, 1′-HE), 5.72 (1.5H, dd, J = 3.4, 1.5 Hz, 5′-HE), 5.63 (0.5H, dd, J = 5.5, 1.3 Hz, 5′-HZ), 5.56 (0.5H, dd, J2′,3′ = 6.1 Hz, J1′,2′ = 1.5 Hz, 2′-HZ), 5.44 (1.5H, dd, J2′,3′ = 6.3 Hz, J1′,2′ = 1.7 Hz, 2′-HE), 5.14–5.11 (0.5H, m, 3′-HZ), 4.94 (1.5H, dd, J2′,3′ = 6.3, J3′,4′ = 3.3 Hz, 3′-HE), 4.69–4.67 (0.5H, m, 4′-HZ), 4.63 (1.5H, m, 4′-HE), 1.63 and 1.38 (3H, 2s, Me2CZ), 1.59 and 1.36 (9H, 2s, Me2CE).
13C-NMR (75 MHz, C2HCl3): δ/ppm = 164.7E, 152.7Z, 152.4E, 150.9E, 149.8Z, 149.7E, 142.4Z, 142.3E, 133.5E, 133.4Z, 132.8Z, 132.7E, 131.2Z, 130.0E, 128.9Z, 128.7E, 128.0E, 127.9Z, 123.6E, 114.6Z, 114.5E, 91.1Z, 90.7E, 87.3E, 85.4Z, 84.5E, 84.3Z, 84.0E, 83.5Z, 27.0E, 27.0Z, 25.3Z, 25.2E.
HRMS (ESI): m/z [M + Na]+, calcd for C40H38N10NaO8+ 809.277, found 809.277.
(E)-33,34-O-Isopropylidene adenosylhop-30-ene ((E)-12) and (Z)-33,34-O-isopropylidene adenosylhop-30-ene ((Z)-12)
To a mixture of compounds (E)-11 and (Z)-11 (150 mg, 0.19 mmol) in CH2Cl2 (5 mL) was added a solution of ammonia in MeOH (7N, 20 mL) at 0 °C, and the mixture was stirred at 5 °C for 5 days. The volatiles were evaporated and the residue was purified via flash chromatography (CH2Cl2 to CH2Cl2–MeOH, 100:2) to give a mixture of (E)-12 and (Z)-12 (120 mg, 0.17 mmol, 91%) as a colourless solid, which was used in the next step without further purification. Rf = 0.36 (CH2Cl2–MeOH, 100:5).
1H-NMR (300 MHz, C2HCl3) for (E)-12 and (Z)-12 (subscripts “E” and “Z” characterize respectively the 1H-NMR signals of (E)-12 and (Z)-12, which are respectively present in a 4:1 ratio): δ/ppm = 8.36 (0.2H, s, 2′-HZ), 8.35 (0.8H, s, 2′-HE), 7.91 (0.2H, s, 8′-HZ), 7.89 (0.8H, s, 8′-HE), 6.07 (1H, d, J34,35 = 1.8 Hz, 35-HZ and 35-HE), 5.90 (2H, br. s, –NH2), 5.54 (0.8H, dd, J33,34 = 6.2 Hz, J34,35 = 1.9 Hz, 34-HE), 5.50 (0.2H, dd, J33,34 = 6.2 Hz, J34,35 = 1.9 Hz, 34-HZ), 5.45 (0.8H, dd, J30,31 = 15.3 Hz, J22,30 = 8.1 Hz, 30-HE), 5.36 (0.8H, dd, J30,31 = 15.3 Hz, J31,32 = 6.8 Hz, 31-HE), 5.31–5.29 (0.4H, m, 31-HZ and 30-HZ), 4.97 (0.2H, dd, J31,32 = 4.7 Hz, J32,33 = 2.9 Hz, 32-HZ), 4.94 (0.8H, dd, J33,34 = 6.3 Hz, J32,33 = 3.1 Hz, 33-HE), 4.83 (0.2H, dd, J33,34 = 6.3 Hz, J32,33 = 3.1 Hz, 33-HZ), 4.69 (0.8H, dd, J31,32 = 6.8 Hz, J32,33 = 2.9 Hz, 32-HE), 2.72–2.63 (0.2H, m, 22-HZ), 2.17–2.09 (0.8H, m, 22-HE), 1.63 (0.6H, s, Me2CZ), 1.61(2.4H, s, Me2CE), 1.40 (3H, s, Me2C), 1.00 (0.6H, d, J22,29 = 6.4 Hz, 22R-MeZ), 0.96 and 0.94 (1.2H, 2s, 8β-MeZ and 14α-MeZ), 0.93 (2.4H, s, 8β-MeE), 0.92 (2.4H, s, 14α-MeE), 0.89 (2.4H, d, J22,29 = 6.4 Hz, 22R-MeE), 0.834 (3H, s, 4α-Me), 0.799 (3H, s, 10β-Me), 0.779 (3H, s, 4β-Me), 0.640 (3H, s, 18α-Me).
13C-NMR (75 MHz, C2HCl3) for (E)-12: δ/ppm = 155.5, 153.0, 149.4, 141.7, 139.8, 124.8, 120.3, 114.2, 90.8, 88.4, 85.0, 84.4, 56.1, 54.1, 50.4, 49.1, 44.9, 44.3, 42.1, 41.8, 41.7, 41.6, 40.5, 40.3, 37.4, 33.5, 33.4, 33.3, 33.2, 27.5, 27.1, 25.4, 23.9, 22.1, 22.0, 21.7, 21.6, 20.9, 18.7, 16.6, 15.9.
HRMS (ESI): m/z [M + H]+, calcd for C43H65N5NaO3+ 722.498, found 722.497.
33,34-O-Isopropylidene adenosylhopane (13)
To a stirred slurry of potassium azodicarboxylate (90 mg, 0.46 mmol) in pyridine (1 mL) in a two-neck round bottom flask equipped with a condenser under N2 atmosphere was added a solution of 12 (32 mg, 0.046 mmol) in pyridine (3 mL). The mixture was heated to reflux, and a solution of anhydrous acetic acid (0.030 mL, 0.55 mmol) in pyridine (0.1 mL) was carefully added dropwise. Each drop of acetic acid solution was added after the end of N2 evolution. The mixture was left under reflux until the yellow colour vanished. Another five portions of potassium azodicarboxylate and subsequent acetic acid were necessary to increase the conversion. The reaction was quenched with water, and extracted with dichloromethane three times. The combined organic phases were washed with brine and dried over anhydrous Na2SO4, filtered through cotton, and evaporated to dryness in vacuo. The crude product was further purified by flash chromatography (CH2Cl2–MeOH 100:1 to 100:2) to yield the saturated product 13 (23 mg, 73%). Compound 13 was isolated as a colourless solid. Rf = 0.38 (CH2Cl2–MeOH, 100:5).
M.p. = 256–257 °C. [α]20D = +34 (c 0.35, CHCl3).
1H-NMR (C2HCl3, 300 MHz): δ/ppm = 8.35 (1H, s, 2′-H), 7.89 (1H, s, 8′-H), 6.03 (1H, dd, J34,35 = 2.4 Hz, 35-H), 5.85 (2H, s, –NH2), 5.51 (1H, J33,34 = 6.5 Hz, J34,35 = 2.4 Hz, 34-H), 4.80 (1H, dd, J33,34 = 6.5 Hz, J32,33 = 3.5 Hz, 33-H), 4.15 (1H, ddd, J31a,32 = 7.4 Hz, J31b,32 = 6.5 Hz, J32,33 = 3.5 Hz, 32-H), 1.60 and 1.38 (2 × 3H, 2s, Me2C), 0.928 (3H, s, 8β-Me), 0.921 (3H, S, 14α-Me), 0.83 (3H, d, J = 6.2 Hz, 22-Me), 0.834 (3H, s, 4α-Me), 0.800 (3H, s, 10β-Me), 0.779 (3H, s, 4β-Me), 0.656 (3H, s, 18α-Me).
13C-NMR data (C2HCl3, 75 MHz): δ/ppm = 155.4 (C-6′), 153.0 (C-2′), 139.8 (C-8′), 149.4 (C-4′), 120.3 (C-5′), 114.4 (CMe2), 90.5 (C-35), 87.7 (C-32), 84.3 (C-34), 84.0 (C-33), 56.1 (C-5), 54.4 (C-17), 50.4 (C-9), 49.3 (C-13), 45.7 (C-21), 44.3 (C-18), 42.1 (C-3), 41.8 (C-14), 41.7 (C-8), 41.5 (C-19), 40.3 (C-1), 37.4 (C-10), 36.4 (C-22), 33.6 (C-15), 33.4 (C-23), 33.2 and 33.1 (C-4 and C-7), 31.4 (C-30), 30.0 (C-31), 27.5 (C-20), 23.9 (C-12), 22.7 (C-16), 21.6 (C-24), 20.9 (C-11), 20.0 (C-29), 18.7 (C-2 and C-6), 16.6 and 16.5 (C-26 and C-27), 15.9 and 15.8 (C-25 and C-28).
HRMS (ESI): m/z [M + H]+, calcd for C43H67N5NaO3+ 724.514, found 724.512.
[30,31-2H2]-33,34-O-Isopropylidene adenosylhopane (13-D)
Compound 12 (51 mg, 0.073 mmol) was dissolved in a mixture of CH3O2H–C2HCl3 (1:2) under an atmosphere of N2 for H-2H exchange. After 5 min standing, the solvents were carefully evaporated under vacuum. The same procedure was repeated three times, and the N-deuteriated intermediate 12-D was carefully dried overnight under vacuum in the presence of P2O5. Compound 13-D was prepared following the procedure described for the synthesis of natural abundance 13 using potassium azodicarboxylate (850 mg, 43.8 mmol) and CH3COO2H (98% isotope abundance, 3.3 mL, 56.9 mmol) to generate deuteriated diimide in situ. Compound 13-D (31 mg, 60%) was isolated as a colourless solid with unreacted starting material 12 (8.6 mg, 17%) by TLC (CH2Cl2–MeOH, 100:5, Rf = 0.38). Deuterium content of 13-D was determined from HRMS (ESI) spectrum by evaluating the relative intensities of the m/z 724, 725 and 726 peaks corresponding to each isotopomer: natural abundance adenosylhopane (8%), monodeuteriated adenosylhopane (32%) and bisdeuteriated adenosylhopane (60%), trisdeuteriated adenosylhopane (0%).
1H-NMR (C2HCl3, 600 MHz): δ/ppm = 8.35 (1H, s, 2′-H), 7.90 (1H, s, 8′-H), 6.03 (1H, d, J34,35 = 2.4 Hz, 35-H), 5.92 (2H, s, –NH2), 5.51 (1H, ddd, J33,34 = 6.4 Hz, J34,35 = 2.4 Hz, J32,34 = 1.2 Hz, 34-H), 4.80 (1H, ddd, J33,34 = 6.4 Hz, J32,33 = 3.5 Hz, J31,33 = 1.2 Hz, 33-H), 4.15 (1H, dd for the major isotopomer, J31,32 = 7.2 Hz, J32,33 = 3.5 Hz, 32-H), 1.60 and 1.38 (2 × 3H, 2s, Me2C), 0.927 (3H, s, 8β-Me), 0.920 (3H, S, 14α-Me), 0.833 (3H, s, 4α-Me), 0.832 (3H, d, J = 6.3 Hz, 22-CH3), 0.798 (3H, s, 10β-Me), 0.778 (3H, s, 4β-Me), 0.655 (3H, s, 18α-Me).
13C-NMR data (C2HCl3, 150 MHz): δ/ppm = 155.5 (C-6′), 152.9 (C-2′), 139.8 (C-8′), 149.4 (C-4′), 120.3 (C-5′), 114.4 (CMe2), 90.5 (C-35), 87.63 and 87.60 (C-32, β-shift induced by deuterium at C-31), 84.3 (C-34), 84.0 (C-33), 56.1 (C-5), 54.3 (C-17), 50.4 (C-9), 49.2 (C-13), 45.7 (C-21), 44.3 (C-18), 42.1 (C-3), 41.8 (C-14), 41.6 (C-8), 41.5 (C-19), 40.3 (C-1), 37.4 (C-10), 36.35, 36.28 and 36.25 (C-22, β-shifts induced by deuterium at C-30), 33.6 (C-15), 33.4 (C-23), 33.2 and 33.1 (C-4 and C-7), missing signal (C-30 bearing a deuterium), missing signal (C-31 bearing a deuterium), 27.5 (C-20), 23.9 (C-12), 22.7 (C-16), 21.6 (C-24), 20.9 (C-11), 20.0 (C-29), 18.7 (C-2 and C-6), 16.6 and 16.5 (C-26 and C-27), 15.9 and 15.8 (C-25 and C-28).
HRMS (ESI): m/z [M + H]+, calcd for C43H65D2N5NaO3+ 726.526, found 726.523.
Adenosylhopane (2)
A solution of 13 (6.3 mg, 9 μmol) and TFA (2 mL) in CHCl3–MeOH (2:1, 5 mL) in a round bottomed flask was shaken under reduced pressure (450–500 mbar) at 0 °C for 2 h30 (rotary evaporator). When starting material could no more be detected by TLC, the reaction mixture was evaporated to dryness and the residue was purified by column chromatography over silica gel (63–200 μm) (CHCl3–MeOH–NH4OH, 100:2:0.5 to 100:5:0.5 to 100:7.5:0.5) to afford adenosylhopane 2 as a colourless solid (5.3 mg, 8 μmol 90%). Rf = 0.09 (CH2Cl2–MeOH 100:5). [α]20D = +34 (c 0.3, THF).
1H-NMR ((2H5)pyridine, 600 MHz): δ (ppm) = 8.77 (1H, s, 2′-H), 8.61 (1H, s, 8′-H), 8.29 (2H, s, –NH2), 7.74 (1H, d, J34,OH = 5.5 Hz, 34-OH), 7.04 (1H, d, J33,OH = 5.8 Hz, 33-OH), 6.72 (1H, d, J34,35 = 4.4 Hz, 35-H), 5.43 (1H, pseudo q, J = 4.7 Hz, 34-H), 4.77 (1H, pseudo q, J = 5.2 Hz, 33-H), 4.56 (1H, pseudo dt, J = 8.4 Hz, J = 4.9 Hz, 32-H), 2.13 (1H, m, 31-Ha), 1.96 (1H, m, 31-Hb), 1.79 (2H, m, 30-Ha and 20-Ha), 1.73 (1H, m, 21-H), 0.994 (3H, d, J = 6.3 Hz), 0.959 (3H, s, 8β-Me), 0.948 (3H, s, 14α-Me), 0.882 (3H, s, 4α-Me), 0.819 (3H, s, 4β-Me), 0.815 (3H, s, 10β-Me), 0.665 (3H, s, 18α-Me).
13C-NMR ((2H5)pyridine, 150 MHz): δ (ppm) = 157.6 (C-6′), 153.8 (C-2′), 150.6 (C-4′), 140.0 (C-8′), 121.3 (C-5′), 90.0 (C-35), 85.2 (C-32), 75.24 and 75.16 (C-33 and C-34), 54.6 (C-17), 56.4 (C-5), 50.7 (C-9), 49.6 (C-13), 46.4 (C-21), 44.5 (C-18), 42.3(C-3), 42.0 (C-14), 41.9 (C-8), 41.8 (C-19), 40.5 (C-1), 37.6 (C-10), 36.9 (C-22), 34.0 (C-15), 33.6 (C-7 and C-23), 33.4 (C-4), 32.3 (C-30), 31.0 (C-31), 27.9 (C-20), 24.2 (C-12), 23.0 (C-16), 21.8 (C-24), 21.2 (C-11), 20.4 (C-29), 19.0 (C-2 and C-6), 16.8 and 16.7 (C-26 and C-27), 16.1 and 16.0 (C-25 and C-28).
HRMS (ESI): m/z [M + H]+, calcd for C40H64N5O3+ 662.501, found 662.500.
(30,31-2H2)Adenosylhopane (2-D)
Deprotection of 13-D (9.3 mg, 13.2 μmol) by TFA (3 mL) in CHCl3–MeOH (2:1, 7 mL) following procedure D afforded deuteriated adenosylhopane 2-D (7.7 mg, 11.6 μmol, 90%). Rf = 0.09 (DCM–MeOH 100:5). Deuterium content of 2-D was calculated from HRMS (ES) spectrum: natural abundance adenosylhopane 8%; monodeuteriated adenosylhopane 32%; bisdeuteriated adenosylhopane 60%; trisdeuteriated adenosylhopane 0%.
1H-NMR ((2H5)pyridine, 600 MHz): δ (ppm) = 8.72 (1H, s, 2′-H), 8.60 (1H, s, 8′-H), 8.28 (2H, s, –NH2), 7.73 (1H, d, J34,OH = 5.1 Hz, 34-OH), 7.03 (1H, d, J33,OH = 5.6 Hz, 33-OH), 6.72 (1H, d, J34,35 = 4.4 Hz, 35-H), 5.43 (1H, pseudo q, J = 4.3 Hz, 34-H), 4.77 (1H, pseudo q, J = 5.0 Hz, 33-H), 4.57–4.54 (1H, m, 32-H), 2.11 (1H, dd, J31a, 32 = 8.3 Hz, J30b,31a = 4.6 Hz, 31-Ha), 1.93 (1H, pseudo t, J31b,32 = J30a,31b = 5.0 Hz, 31-Hb), 1.82–1.71 (2.7H, m, 30-Ha, 20-Ha and 21-H of isotopomer resulting from deuteriation of major isomer E-12), 1.75–1.72 (1H, m, 21-H), 0.993 (3H, d, J = 6.3 Hz), 0.961 (3H, s, 8β-Me), 0.949 (3H, s, 14α-Me), 0.883 (3H, s, 4α-Me), 0.820 (3H, s, 4β-Me), 0.816 (3H, s, 10β-Me), 0.667 (3H, s, 18α-Me).
13C-NMR ((2H5)pyridine, 150 MHz): δ (ppm) = 157.6 (C-6′), 153.8 (C-2′), 150.6 (C-4′), 140.0 (C-8′), 121.3 (C-5′), 90.0 (C-35), 85.20 and 85.15 (C-32, β-shift induced by deuterium at C-31), 75.21 and 75.15 (C-33 and C-34), 54.6 (C-17), 56.4 (C-5), 50.7 (C-9), 49.6 (C-13), 46.4 (C-21), 44.5 (C-18), 42.3(C-3), 42.0 (C-14), 41.9 (C-8), 41.8 (C-19), 40.5 (C-1), 37.6 (C-10), 36.90, 36.86 and 36.81 (C-22, β-shifts induced by deuterium at C-30), 33.9 (C-15), 33.6 (C-7 and C-23), 33.4 (C-4), missing signal at ca. 32.3 (C-30 bearing a deuterium), missing signal at ca. 31.0 (C-31 bearing a deuterium), 27.9 (C-20), 24.2 (C-12), 23.0 (C-16), 21.8 (C-24), 21.2 (C-11), 20.3 (C-29), 19.0 (C-2 and C-6), 16.8 and 16.7 (C-26 and C-27), 16.1 and 16.0 (C-25 and C-28).
HRMS (ESI): m/z [M + H]+, calcd for C40H622H2N5O3+ 664.513, found 664.513.
Adenosylhopane acetates (14a–c)
Adenosylhopane 2 (1.8 mg) was acetylated overnight at room temperature with a mixture of acetic anhydride and pyridine (0.3 mL, v/v, 1/2). Reagents were removed under vacuum. The resulting residue was purified by preparative thin layer chromatography (CH2Cl2–MeOH, 100:5) to afford adenosylhopane diacetate 14a (0.4 mg, Rf = 0.30), triacetate 14b (0.6 mg, Rf = 0.38)1a,b and tetraacetate 14c (0.8 mg, Rf = 0.66).
MS (EI, direct inlet, positive mode 70 eV): m/z = 746 (M+, 9%), 686 (M+ − AcOH, 6%), 626 (M+ − 2AcOH, 24%), 595 (6%), 538 (15%), 491 (5%), 389 (17%), 367 (M+ − CH2CO − side chain, 9%), 191 (ring C cleavage, 10%), 136 ([adenine + H]+, 100%).
MS (EI, direct inlet, positive mode 70 eV): m/z = 788 (M+, 2%), 610 (M+ − N,N-acetyladenine, 4%), 595 (ring C cleavage, 2%), 491 (1%), 389 (6%), 367 (M+ − CH2CO − side chain, 4%), 191 (ring C cleavage, 11%), 178 ([N-acetyladenine + H]+, 100%).
MS (EI, direct inlet, positive mode 70 eV): m/z = 830 (M+, 0.3%), 788 (M+ − CH2CO), 662 (1%), 610 (M+ − CH2CO − N,N-diacetyladenine, 8%), 595 (ring C cleavage − CH2CO, 5%), 491 (3%), 389 (16%), 367 (M+ − CH2CO − side chain, 8%), 191 (ring C cleavage, 20%), 178 (100%).
Culture of M. organophilum and preparation of the cell-free system
Methylobacterium organophilum DSM 760 was grown on a modified medium of Hestrin and Schramm36 (6 × 500 mL, yeast extract 5 g L−1, bactopeptone 5 g L−1, glucose 1 g L−1, anhydrous citric acid 1.04 g L−1, Na2HPO4 2.7 g L−1, pH = 6.8) in 2 L Erlenmeyer flasks on a rotatory shaker (200 rpm) at 30 °C. Optical densities of the bacterial cultures were measured at 595 nm on a Genesys 10UV spectrophotometer using 1 cm in length and 1 mL volume cuvettes. The cells were harvested before the end of the exponential growth phase (OD ∼ 0.6) by centrifugation (8 000 rpm, 10 min), and then washed with buffer either a sodium phosphate buffer (50 mM NaH2PO4–Na2HPO4, 0.5 mM, MgCl2, 1 mM dithiothreitol, pH 7.5) or a triethylamine buffer (0.1 M triethylamine, 0.5 mM MgCl2, 1 mM dithiothreitol, pH 7.5). After centrifugation (8 000 rpm, 10 min), the cell pellet (3 g) was suspended in the buffer (15 mL) and disrupted by sonication at 0 °C in a melting ice bath using a Branson SONIFIER B-30 (8 × 40 s with 3 min pause at 0 °C, 60% pulsed duty cycle, output control: 6). The crude cell-free system was centrifuged (10000 rpm, 10 min). The pellet was freeze-dried to recover bacteriohopanetetrol of natural abundance, and the supernatant was directly used for the incubation deuterium labelled adenosylhopane.
Conversion of deuteriated adenosylhopane into bacteriohopanetetrol by a crude cell-free system from M. organophilum
A solution of deuteriated adenosylhopane 2-D (200 μg) in THF (500 μL) was added to the supernatant (5 mL) described above in a glass screw cap vial. After addition of a solution of NADPH (1.5 mg) in water (100 μL), the sample was incubated at 30 °C by shaking (200 rpm). An additional portion of the cofactor solution (100 μL) was added after 4 h incubation and the sample was incubated for another 20 h. After lyophilisation, the residue was directly acetylated overnight at room temperature with Ac2O–pyridine (1:2 v/v, 3.9 mL). The reaction mixture was filtered over cotton and washed with toluene (3 × 2 mL) and CH2Cl2 (3 × 2 mL). The combined filtrates were concentrated in vacuo, and the residue was subjected to preparative TLC (cyclohexane–ethyl acetate, 3:7) to give an apolar fraction (Rf > 0.7) containing diploptene, diplopterol and acetylated bacteriohopanetetrol. This less polar fraction was further separated by TLC (cyclohexane–ethyl acetate, 5:1), yielding pure bacteriohopanetetrol tetraacetate (0.16 < Rf < 0.20), which was analysed by GC-EIMS.
Isolation of natural abundance bacteriohopanetetrol from M. organophilum or from cell-free system unbroken cells and cell debris pellet
The freeze-dried material (unbroken cells and large cell debris) recovered after the centrifugation yielding the supernatant used for the incubations was extracted with CHCl3–CH3OH (2:1, v/v) three times under reflux. The combined extracts were brought to dryness, acetylated with Ac2O–pyridine (1:2, v/v) and the excess of reagents was evaporated in vacuo. The residue was separated by preparative TLC (silica gel, cyclohexane–ethyl acetate, 3:7) to give an apolar fraction (Rf > 0.7) containing diploptene, diplopterol, and acetylated bacteriohopanetetrol. This mixture was further separated by preparative TLC (silica gel, cyclohexane–ethyl acetate, 5:1), yielding pure bacteriohopanetetrol tetraacetate (0.16 < Rf < 0.20).
Acknowledgements
This work was supported by a grant from the Foundation “Frontier Research in Chemistry” (Strasbourg, France). WL acknowledges financial support from the China Scholarship Council and the French embassy in Beijing. The authors express their gratitude to E. Motsch and Dr P. Schaeffer for EIMS and GC-EIMS measurements, Dr L. Allouche, B. Vincent and J.D. Sauer for NMR measurements and R. Carrière and Dr H. Nierengarten for ES-HRMS measurements.References
- (a) S. Neunlist and M. Rohmer, Biochem. J., 1985, 228, 769 CAS; (b) S. Neunlist, P. Bisseret and M. Rohmer, Eur. J. Biochem., 1988, 171, 245 CrossRef CAS PubMed; (c) M. Seemann, P. Bisseret, J. P. Tritz, A. B. Hooper and M. Rohmer, Tetrahedron Lett., 1999, 40, 1681 CrossRef CAS; (d) J. M. Bravo, M. Perzl, T. Härtner, E. L. Kannenberg and M. Rohmer, Eur. J. Biochem., 2001, 268, 1323 CrossRef CAS; (e) M. Eickhoff, D. Birgel, H. M. Talbot, J. Peckman and A. Kappler, Geobiology, 2013, 11, 268 CrossRef CAS PubMed.
- H. M. Talbot, M. Rohmer and P. Farrimond, Rapid Commun. Mass Spectrom., 2007, 21, 880 CrossRef CAS PubMed.
- (a) M. P. Cooke, H. M. Talbot and P. Farrimond, Org. Geochem., 2008, 39, 1347 CrossRef CAS PubMed; (b) M. P. Cooke, H. M. Talbot and T. Wagner, Org. Geochem., 2008, 39, 965 CrossRef CAS PubMed; (c) Y. Xu, M. P. Cooke, H. M. Talbot and M. J. Simpson, Org. Geochem., 2008, 40, 79 CrossRef PubMed.
- A. D. Selver, H. M. Talbot, O. Gustafsson, S. Boult and B. E. van Dongen, Org. Geochem., 2012, 51, 63 CrossRef PubMed.
- T. Wagner, W. Kallweit, H. M. Talbot, G. Mollenhauer, A. Boom and T. Zabel, Org. Geochem., 2014, 67, 85 CrossRef CAS PubMed.
- P. Bisseret and M. Rohmer, J. Org. Chem., 1989, 54, 2958 CrossRef CAS.
- S. Neunlist and M. Rohmer, J. Chem. Soc., Chem. Commun., 1988, 830 RSC.
- G. Flesch and M. Rohmer, Eur. J. Biochem., 1988, 175, 405 CrossRef CAS PubMed.
- M. Rohmer, B. Sutter and H. Sahm, J. Chem. Soc., Chem. Commun., 1989, 1471 RSC.
- M. Rohmer, Pure Appl. Chem., 1993, 65, 1293 CrossRef CAS.
- A. S. Bradley, A. Pearson, J. P. Sáenz and C. J. Marx, Org. Geochem., 2010, 41, 1075 CrossRef CAS PubMed.
- P. V. Welander, D. M. Doughty, C. H. Wu, S. Mehay, R. Summons and D. K. Newman, Geobiology, 2012, 10, 163 CrossRef CAS PubMed.
- W. Liu, E. Sakr, P. Schaeffer, H. M. Talbot, J. Donisi, T. Härtner, E. Kannenberg, E. Takano and M. Rohmer, ChemBioChem, 2014, 15, 2156 CrossRef CAS PubMed.
- P. Stampf, PhD Thesis, Université de Haute Alsace, Mulhouse, France, 1992.
- T. Duvold and M. Rohmer, Tetrahedron, 1999, 55, 9847 CrossRef CAS.
- W. D. Pan, PhD Thesis, Université de Paris VI, Paris, France, 2005.
- W. D. Pan, C. Sung, Y. M. Zhang, G. Y. Liang, P. Sinaÿ and S. P. Vincent, Chem. – Eur. J., 2007, 13, 1471 CrossRef CAS PubMed.
- S. F. Wnuk and M. J. Robins, Can. J. Chem., 1991, 69, 334 CrossRef CAS.
- R. H. Grubbs, in Handbook of metathesis, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS PubMed; (b) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed.
- (a) A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed; (b) S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900 CrossRef CAS PubMed.
- D. Andrei and S. F. Wnuk, Org. Lett., 2006, 8, 5093 CrossRef CAS PubMed.
- (a) R. S. Ranganathan, G. H. Jones and J. G. Moffatt, J. Org. Chem., 1974, 39, 290 CrossRef CAS; (b) S. Eppacher, P. K. Bhardwaj, B. Bernet, J. L. B. Gala, T. Knopfel and A. Vasella, Helv. Chim. Acta, 2004, 87, 2969 CrossRef CAS.
- W. J. Dunstan, H. Fazakerley, T. G. Halsall and E. R. H. Jones, Croat. Chem. Acta, 1957, 29, 173 CAS.
- A. Ensminger, PhD Thesis, Université Louis Pasteur, Strasbourg, France, 1974.
- C. Samoljowicz, M. Bienek, A. Zarecki, R. Kadyrov and K. Grela, J. Chem. Soc., Chem. Commun., 2008, 6282 RSC.
- V. Voorhees and R. Adams, J. Am. Chem. Soc., 1922, 44, 1397 CrossRef CAS.
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331 CrossRef CAS.
- (a) C. E. Miller, J. Chem. Educ., 1965, 42, 254 CrossRef CAS; (b) J. L. G. Spears and J. S. Hutchinson, J. Chem. Phys., 1988, 88, 240 CrossRef PubMed.
- J. W. Hamersma and E. I. Snyder, J. Org. Chem., 1965, 30, 3985 CrossRef CAS.
- J. Toulouse, PhD Thesis, Université de Strasbourg, Strasbourg, France, 2011.
- J. M. Renoux and M. Rohmer, Eur. J. Biochem., 1985, 151, 405 CrossRef CAS PubMed.
- H. Budzikiewicz, J. M. Wilson and C. Djerassi, J. Am. Chem. Soc., 1963, 85, 3688 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- S. Hestrin and M. Schramm, Biochem. J., 1954, 58, 345 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02560a |
| This journal is © The Royal Society of Chemistry 2015 |