
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational design of novel peptidomimetic inhibitors of cadherin homophilic interactions†
Fabio
Doro
a,
Cinzia
Colombo
a,
Chiara
Alberti
b,
Daniela
Arosio
c,
Laura
Belvisi
ac,
Cesare
Casagrande
a,
Roberto
Fanelli
d,
Leonardo
Manzoni
c,
Emilio
Parisini
e,
Umberto
Piarulli
d,
Elena
Luison
b,
Mariangela
Figini
b,
Antonella
Tomassetti
*b and
Monica
Civera
*a
aDipartimento di Chimica, Università degli Studi di Milano, Via C. Golgi 19, I-20133, Milan, Italy. E-mail: monica.civera@unimi.it; Fax: +39 02 50314072; Tel: +39 02 50314061
bDipartimento di Oncologia Sperimentale e Medicina Molecolare, Fondazione IRCCS Istituto Nazionale dei Tumori, Via G. Amadeo 42, I-20133, Milan, Italy. E-mail: Antonella.Tomassetti@istitutotumori.mi.it
cCNR – Istituto di Scienze e Tecnologie Molecolari (ISTM), Via C. Golgi 19, I-20133, Milan, Italy
dDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell'Insubria, Via Valleggio 11, I-22100, Como, Italy
eCenter for Nano Science and Technology @PoliMi, Istituto Italiano di Tecnologia, Via G. Pascoli 70/3, I-20133, Milan, Italy
First published on 14th January 2015
Abstract
We report a first set of peptidomimetic ligands mimicking the adhesive interface identified by recent crystallographic structures of E- and N-cadherin. Compounds 2 and 3 inhibit adhesion of epithelial ovarian cancer (EOC) cells with improved efficacy compared to the ADH-1 peptide, a N-cadherin antagonist that is in early clinical trials in EOC patients.
Cadherins are a large family of calcium-dependent cell adhesion molecules that are mostly expressed at the intercellular junctions. They are known to play a key role in important physiological processes such as tissue morphogenesis and stability, as well as in the immune system regulation. Over the past 20 years, dysregulation of the type I epithelial E-cadherin (cadh) and neuronal N-cadh has been shown to correlate with tumor progression and other pathological processes.1 The switch in expression from E-cadh to N-cadh is an important indicator of the epithelial–mesenchymal transition (EMT) occurring during cancer progression.1 Although E-cadh is considered a repressor for the majority of carcinomas, in epithelial ovarian cancer (EOC) E-cadh persists during tumor progression.2 We have shown that E- and N-cadh can be co-expressed in some advanced-stage EOCs and that E-cadh expression and homophilic interaction contribute to the proliferation of EOC cells.2
Cadherins from opposing cells interact by means of their extracellular regions, constituted by five Ig-like domains (EC1–EC5), and form multiple trans homophilic dimers arranged in a zipper-like fashion across the cellular adherens junctions.3,4 Several X-ray structures of type I and II cadherin ectodomains5 have shown that trans dimerization occurs through the opening of the highly conserved N-terminal six-residue portion of the EC1 (the “adhesion arm”) that contains a Trp residue in position 2 (Trp2), and its swapping into an acceptor pocket of the EC1 of the interacting protein, thus resulting in the so-called “strand dimer” formation. However, the complex and highly dynamical cadherin dimerization pathway has only been partially elucidated by the wealth of biochemical and biophysical studies performed to date.4 Ultimately, a variety of transient yet critical key intermolecular interactions involving different residues at different stages of the full cadherin dimerization trajectory play all a critical role in guiding the system through a number of intermediate intermolecular protein–protein arrangements during the recognition process that leads from monomeric cadherin to strand-dimer-formation and back.6
As a result of this highly dynamic behaviour and despite a growing interest in the field, the rational design of small ligands targeting cadherin protein–protein interactions is still in a very early stage. The first crystal structure of the N-cadh EC1 dimer revealed an association interface characterized by the His79-Ala80-Val81 (HAV) and the Ile53-Asn54-Pro55 (INP) sequences and not involving the strand swapping portion of the molecule.7 This triggered an interest in the synthesis of libraries of cyclic peptides based on HAV or INP sequences and of non-peptide HAV mimics for the inhibition of the cadherin dimerization process.8 Among them, the antagonist peptide N-Ac-CHAVC-NH2 (ADH-1 or Exherin™, Fig. 1) containing the HAV motif was promoted to phase I clinical trials in patients with advanced solid tumors which express N-cadh.9
 | ||
| Fig. 1 The N-cadherin antagonist peptide ADH-1 and peptidomimetic ligands synthesized 1–3. | ||
In this paper, we report the first small library of peptidomimetics based on the tetrapeptide sequence Asp1-Trp2-Val3-Ile4 (DWVI) of the N- and E-cadh N-terminal “adhesive arm”. To the best of our knowledge, this work represents the first attempt to rationally design small molecules targeting the strand dimer interfaces identified by recent crystallographic structures.5 Our compounds were tested in biochemical and functional assays on EOC cells for their ability to inhibit both N- and E-cadh homophilic interactions relative to ADH-1.
About 30 peptidomimetics of general formula NH3+-Asp-scaffold-Ile-NHCH3 were built in silico by replacing the central dipeptide Trp2-Val3 unit of the DWVI adhesive motif with several scaffolds developed in our and other laboratories (Fig. 2).10,11
 | ||
| Fig. 2 Scaffolds used for the generation of the virtual library of tetrapeptide mimics. | ||
All the peptidomimetics featured a conformationally constrained scaffold bearing a benzyl or phenyl ring to mimic the indole moiety of Trp2 in the native sequence. Based on the molecular architecture of the scaffolds, the aromatic ring was expected to be differently oriented into the binding pocket. To evaluate the ability of each of the compounds of our virtual library to reproduce the DWVI key interactions observed in the X-ray dimer structures (Fig. S1†) and during molecular dynamics simulations (Fig. S2 and S3†), we built a model of the EC1 fragment of E- and N-cadh starting from the corresponding X-ray structures and set up a docking protocol using the Glide V5.7 software12 (see ESI†).
Docking results were sorted according to the Glide score and filtered to match the two most important binding interactions: (i) the formation of an intermolecular salt bridge between the charged N-terminal amino group of Asp1 and the side chain carboxylate of Glu89 and (ii) the anchoring of the Trp2 side chain into a hydrophobic pocket described for the DWVI motif. Based on this analysis, three ligands were selected for synthesis and biological assays (Fig. 1).
In the docking poses, all the three ligands form the salt bridge (i) and insert the scaffold benzyl ring into the corresponding hydrophobic pocket of Trp2 (ii). In addition, in the N-cadh binding site an electrostatic interaction between ligand Asp1-NH3+ and the side chain of Asp27 is also observed, while in E-cadh there is the formation of a hydrogen bond with Asn27. The main difference in the binding mode of the three ligands is related to their 3D alignment with respect to the DWVI sequence of the crystallographic structures: while 2 and 3 are able to mimic the extended backbone arrangement of the reference tetrapeptide in both cadherin receptors (see Fig. 3 and S4† for docking results of 3), 1 orients the Ile residue back to the Asp1 amino acid and does not reproduce the experimental backbone conformation (Fig. S5†).
 | ||
| Fig. 3 Best pose of 3 (C in grey, N in blue and O in red) into the N-cadh binding site (blue), overlaid to DWVI (green) of the X-ray dimer. | ||
The three ligands were successfully synthesized using conventional high-yielding synthetic pathways in solution and in the solid phase (see ESI†).
The compounds were tested by ELISA for their ability to inhibit calcium-dependent cadherin binding using the N-cadh-expressing EOC cell line SKOV3 and N-cadh-Fc chimeric protein. Ligands 2 and 3 at 2 mM concentration inhibited N-cadh homophilic binding by 78% and 84%, respectively, and 50% and 65% at 1 mM concentration (Fig. 4A). Conversely, at 2 mM concentration ADH-1 and 1 provided about 50% inhibition of N-cadh-Fc/cells interactions and appeared nearly ineffective at 1 mM concentration (Fig. 4A). The compounds were then evaluated for their ability to inhibit EOC cells adhesion by observing the formation of cell monolayers in the presence of each ligand at 2 different concentrations (2 and 1 mM). As with ADH-1, at 2 mM concentration all compounds were able to inhibit the formation of cell monolayers of N-cadh-expressing cells (Fig. S6†). Notably, 2 and 3 were also active at 1 mM concentration, and 3 was able to inhibit cell–cell aggregation of N-cadh-expressing cells in suspension (Fig. 4B). When tested on the E-cadh-expressing EOC cell line OAW42, all compounds were found to be less efficient in inhibiting both E-cadh homophilic interactions and the formation of cell monolayers, with significant effects observed only at 2 mM concentration (Fig. S6 and S7†). In particular, by ELISA, at the concentration of 2 mM 1 and 3 gave about 50% inhibition of E-cadh-Fc binding to the cells while ADH-1 showed 30% inhibition (Fig. S7†), indicating a slightly better efficacy compared to ADH-1 in inhibiting also E-cadh homophilic interactions. Compound 2 was also able to inhibit the formation of cell monolayers at 1 mM concentration (Fig. S6†).
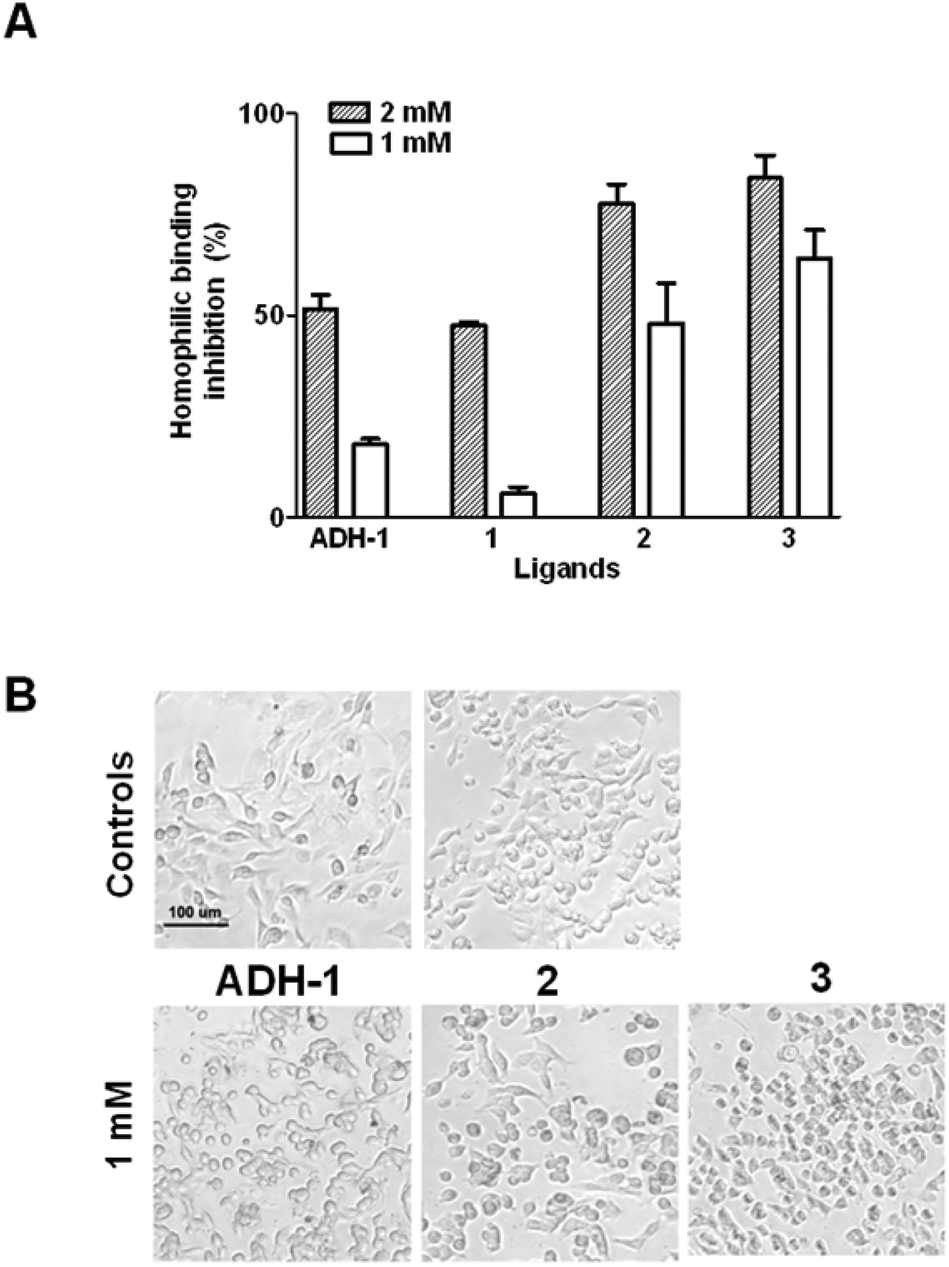 | ||
| Fig. 4 (A) Ligand inhibition of N-cadh-Fc binding to SKOV3 cells by ELISA. (B) Ligand inhibition of the SKOV3 cell monolayer formation by adhesion assay. Controls in the absence of ligand in water (left) and in 10% DMSO solution (right) are reported. | ||
Overall, compounds 2 and 3 resulted to be effective in inhibiting N-cadh homophilic and, to some extent, also E-cadh homophilic adhesion. Encouraged by the very promising results with N-cadh-expressing cells, we set out to evaluate by surface plasmon resonance (SPR) analysis the ability of the two compounds to specifically inhibit the N-cadh homo-dimerization in a cell-free experiment using the N-cadh-Fc recombinant protein. At 10 μM concentrations compounds 2 and 3 provided 98% and 55% inhibition of N-cadh homophilic binding, respectively, while ADH-1 showed only 28% inhibition (Fig. S8†). These data demonstrate the ability of both compounds 2 and 3 to specifically bind to N-cadh even in the μM range.
The combination of our computational investigation with the results of SPR assays has started to shed light on the ligand structural requirements for binding to N-cadh. For instance, it appears that the best inhibitors (2 and 3) of N-cadh homophilic binding maintain the key (i) and (ii) interactions and also align with the DWVI backbone (Fig. 3). Although N- and E-cadh share similar adhesive binding features for the DWVI sequence5 and no significant differences were observed in the interaction mode of our peptidomimetic ligands in the two cadherin docking models, the tests on the E-cadh-expressing EOC cell line OAW42 showed lower inhibition capability of E-cadh homophilic binding compared with those of N-cadh. Since a detailed picture of the complete adhesion pathway in the different cadherin family members is still elusive13 and a variety of factors can contribute to the formation and dissociation of cadherin dimers on cell membranes, our results can be interpreted in light of different considerations. First, two-dimensional affinities measured by micropipette adhesion assays on cell lines expressing E- or N-cadhs,4 have shown that E-cadh homophilic binding is stronger than that of N-cadh. For this reason, our compounds might be less efficient in inhibiting E-cadh than N-cadh interactions. Moreover, based on compelling structural data,4,5 we decided to focus exclusively on the inhibition of the swapped-dimer formation targeting the EC1 domain, although several extracellular domains are known to be involved in the formation of highly dynamic and transient protein–protein contacts at the adhesive interface.
It is worth stressing that targeting the interfaces between proteins has huge therapeutic potential, but it still remains a challenge to discover small drug-like molecules that are capable of modulating those protein–protein interactions that are by their nature highly dynamical. In fact, it is clear that the dynamic features of the cadherin dimerization process are likely to play against a traditional drug design approach. Accordingly, the design of a stable molecular interactor against the adhesion pocket would not just prove difficult but it would fail to achieve complete inhibition. However, peptidomimetic molecules that can transiently interfere with structurally validated intermediates in the cadherin dimerization pathway may successfully modulate cadherin-mediated adhesion, even in the absence of an unequivocal identification of the actual binding site and mode.
Conclusions
In this work, we report a first set of peptidomimetic ligands mimicking the adhesive E- and N-cadh DWVI sequence. Remarkably, two of our peptidomimetics inhibit the N-cadh-mediated adhesion process in EOC cells with somewhat improved efficacy compared to the ADH-1 cyclic peptide, which is being investigated in phase I clinical trials as a N-cadh antagonist in various tumors, including EOCs.9Thus, the small molecules generated in the present study are likely to represent new leads for the development of a novel class of modulators of cadherin-mediated adhesion. Such compounds may play an important role in the investigation of cellular processes, and in the design of novel diagnostic and therapeutic approaches against tumors, especially EOCs. Indeed, since the majority of EOCs grow and disseminate in the peritoneal cavity, the locoregional administration of drugs would allow the use of cadherin inhibitors without interfering with the cadherins expressed in normal epithelia.
Acknowledgements
We thank Università degli Studi di Milano for PhD Fellowship to FD and CINECA for computing facilities. We also gratefully acknowledge Ministero dell'Università e della Ricerca for financial support to MC (RBFR088ITV project), the AIRC for the IG13055 grant to AT and the European Union for the “Marie Curie” FP7-PEOPLE-2010-RG grant to EP (DETACH – N.268231). We thank Dr Silvana Canevari for the useful suggestions and support.Notes and references
- G. Berx and F. van Roy, Cold Spring Harbor Perspect. Biol., 2009, 1, a003129 CrossRef PubMed; O. W. Blaschuk and E. Devemy, Eur. J. Pharmacol, 2009, 625, 195 CrossRef PubMed.
- G. De Santis, S. Miotti, M. Mazzi, S. Canevari and A. Tomassetti, Oncogene, 2009, 28, 1206 CrossRef PubMed.
- J. Brasch, O. J. Harrison, B. Honig and L. Shapiro, Trends Cell. Biol., 2012, 22, 299 CrossRef PubMed.
- D. Leckband and S. Sivasankar, Curr. Opin. Cell Biol., 2012, 24, 620 CrossRef PubMed; J. Vendome, S. Posy, X. Jin, F. Bahna, G. Ahlsen, L. Shapiro and B. Honig, Nat. Struct. Mol. Biol., 2011, 18, 693 Search PubMed.
- O. J. Harrison, X. Jin, S. Hong, F. Bahna, G. Ahlsen, J. Brasch, Y. Wu, J. Vendome, K. Felsovalyi, C. M. Hampton, R. B. Troyanovsky, A. Ben-Shaul, J. Frank, S. M. Troyanovsky, L. Shapiro and B. Honig, Structure, 2011, 19, 244 CrossRef PubMed; E. Parisini, J. M. G. Higgins, J. Liu, M. B. Brenner and J. Wang, J. Mol. Biol., 2007, 373, 401 CrossRef PubMed; T. J. Boggon, J. Murray, S. Chappuis-Flament, E. Wong, B. M. Gumbiner and L. Shapiro, Science, 2002, 296, 1308 CrossRef PubMed; S. D. Patel, C. Ciatto, C. P. Chen, F. Bahna, M. Rajebhosale, N. Arkus, I. Schieren, T. M. Jessell, B. Honig, S. R. Price and L. Shapiro, Cell, 2006, 124, 1255 CrossRef PubMed.
- Y. Li, N. L. Altorelli, F. Bahna, B. Honig, L. Shapiro and A. G. Palmer 3rd, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16462 CrossRef PubMed.
- L. Shapiro, A. M. Fannon, P. D. Kwong, A. Thompson, M. S. Lehmann, G. Grübel, J. F. Legrand, J. Als-Nielsen, D. R. Colman and W. A. Hendrickson, Nature, 1995, 374, 327 CrossRef PubMed.
- E. Williams, G. Williams, B. J. Gour, O. W. Blaschuk and P. Doherty, J. Biol. Chem., 2000, 275, 4007 CrossRef PubMed; G. Williams, E.-J. Williams and P. Doherty, J. Biol. Chem., 2002, 277, 4361 CrossRef PubMed; S. M. Burden-Gulley, T. J. Gates, S. E. L. Craig, S. F. Lou, S. A. Oblander, S. Howell, M. Gupta and S. M. Brady-Kalnay, Peptides, 2009, 30, 2380 CrossRef PubMed.
- O. W. Blaschuk, Cell Tissue Res., 2012, 348, 309 CrossRef PubMed; A. Perotti, C. Sessa, A. Mancuso, C. Noberasco, S. Cresta, A. Locatelli, M. L. Carcangiu, K. Passera, A. Braghetti, D. Scaramuzza, F. Zanaboni, A. Fasolo, G. Capri, M. Miani, W. P. Peters and L. Gianni, Ann. Oncol., 2009, 20, 741 CrossRef PubMed; N. Yarom, D. Stewart, R. Malik, J. Wells, L. Avruch and D. J. Jonker, Curr. Clin. Pharmacol., 2013, 8, 81 Search PubMed.
- L. Manzoni, D. Arosio, L. Belvisi, A. Bracci, M. Colombo, D. Invernizzi and C. Scolastico, J. Org. Chem., 2005, 70, 4124 CrossRef PubMed; A. S. M. Ressurreição, A. Bordessa, M. Civera, L. Belvisi, C. Gennari and U. Piarulli, J. Org. Chem., 2008, 73, 652 CrossRef PubMed; D. Arosio, L. Belvisi, L. Colombo, M. Colombo, D. Invernizzi, L. Manzoni, D. Potenza, M. Serra, M. Castorina, C. Pisano and C. Scolastico, ChemMedChem, 2008, 3, 1589 CrossRef PubMed; L. Manzoni, L. Belvisi, D. Arosio, M. Civera, M. Pilkington-Miksa, D. Potenza, A. Caprini, E. M. V. Araldi, E. Monferini, M. Mancino, F. Podestà and C. Scolastico, ChemMedChem, 2009, 4, 615 CrossRef PubMed; M. Marchini, M. Mingozzi, R. Colombo, I. Guzzetti, L. Belvisi, F. Vasile, D. Potenza, U. Piarulli, D. Arosio and C. Gennari, Chem. – Eur. J., 2012, 18, 6195 CrossRef PubMed.
- Y. Peng, H. Sun and S. Wang, Tetrahedron Lett., 2006, 47, 4769 CrossRef PubMed; L. M. Beal, B. Liu, W. Chu and K. D. Moeller, Tetrahedron, 2000, 56, 10113 CrossRef.
- Glide, version 5.7, Schrödinger, LLC, New York, 2011 Search PubMed.
- P. Thiery, W. Engl, V. Viasnoff and S. Dufour, Curr. Opin. Cell Biol., 2012, 24, 614 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02538e |
| This journal is © The Royal Society of Chemistry 2015 |