
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn tandem or alone: a remarkably selective transfer hydrogenation of alkenes catalyzed by ruthenium olefin metathesis catalysts†‡
Grzegorz Krzysztof
Zieliński
,
Cezary
Samojłowicz
,
Tomasz
Wdowik
and
Karol
Grela
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, PO Box 58, 01-224, Warsaw, Poland. E-mail: karol.grela@gmail.com; Web: http://www.karolgrela.eu/ Fax: +48-22-632-66-81
First published on 9th December 2014
Abstract
A system for transfer hydrogenation of alkenes, composed of a ruthenium metathesis catalyst and HCOOH, is presented. This operationally simple system can be formed directly after a metathesis reaction to effect hydrogenation of the metathesis product in a single-pot. These hydrogenation conditions are applicable to a wide range of alkenes and offer remarkable selectivity.
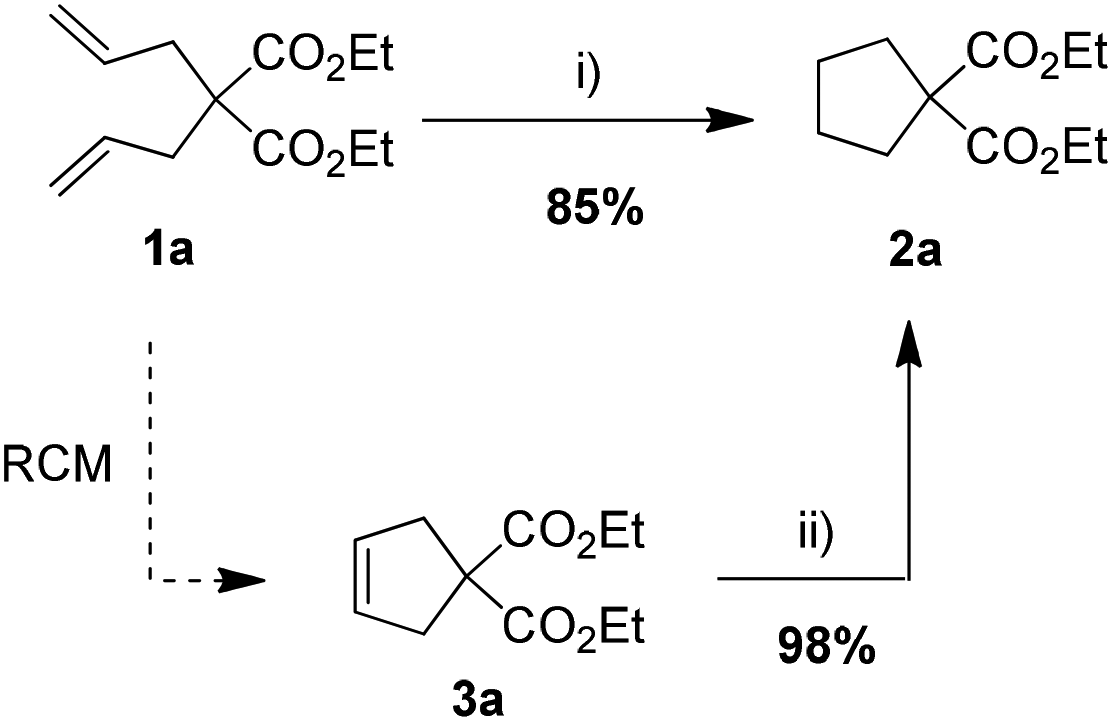
Catalytic hydrogenation olefins are one of the most important “classics” in organic chemistry.1 Due to high complexity of targets approached by contemporary organic synthesis, there is a very high demand for chemo- and regioselective methods to hydrogenate C–C multiple bonds in the presence of many other functionalities including other olefinic moieties. A difficulty associated with catalytic hydrogenation is the use of hydrogen gas, whose physical properties complicate its safe, efficient and economical storage. This creates a need for new selective, safe and environmentally friendly hydrogenation methodologies. Recently reduction of olefins has been carried out in a homogeneous fashion using transition metals (incl. Rh,2a Ir,2b Ru,2c–f Pd2g) and gaseous H2 or hydrogen sources such as alcohols,2h–j hydrazine,2k formic acid–triethylamine azeotrope,2l ammonia–borane,2m silanes,2n and others.2o,p
Over the past two decades olefin metathesis has transformed the design and practice of organic synthesis.3 It should be noted that this reaction can be easily incorporated in tandem processes, where the metathesis catalyst is triggered for a second transformation.4 One such sequence is metathesis–hydrogenation, typically conducted under high pressure of hydrogen, which is required to convert Ru alkylidene complexes into a hydrogenation catalyst.5 Recently a more convenient protocol, using NaBH4 as a hydrogen source has been reported.6
During our study on a new chelating ruthenium azinate complex (Az-II, Fig. 1), we have noticed its reactivity in numerous transformations.7 Depending on the conditions applied, the same complex promoted efficiently olefin metathesis, isomerization, cycloisomerization or reduction of a carbonyl group.7 Serendipitously, we have found recently that after the metathesis step is promoted by Az-II, adding sodium hydride and formic acid to the reaction mixture leads to a new catalytic system that is capable of efficient reduction of cycloolefins (Scheme 1).8 Intrigued by this preliminary observation we decided to study it in more detail.
 | ||
| Fig. 1 Selected ruthenium olefin metathesis catalysts (Cy = cyclohexyl). | ||
 | ||
| Scheme 1 Observed reactivity of azinate complex Az-II.8 Conditions: (i) 1. Az-II (1 mol%), C2Cl6 (4 mol%), THF, 80 °C, 3 h; 2. Az-II (1 mol%), NaH (0.1 equiv.), HCOOH (50 equiv.), THF, 80 °C, 20 h; (ii) Az-II (2 mol%), NaH (0.1 equiv.), HCOOH (50 equiv.), 80 °C, 20 h. | ||
Since the azinate complex Az-II is not commercially available, in the present study we decided to check if other, more standard Ru–alkylidene complexes, can also show a similar reactivity in the presence of HCOOH,9a so can be used in transfer hydrogenation or in olefin metathesis–hydrogenation sequences. To do so, three generations of representative Grubbs’ catalysts Gru-I, Gru-II and Gru-III have been selected (Fig. 1) and tested in a model reduction of cyclopentene 3a to cyclopentane 2a (Table 1). After some initial experiments, an optimized procedure was elaborated as follows: to a solution of olefin 3a a catalyst (2 mol%), and a base (0.2 equiv.) were introduced, followed immediately by 98% formic acid (50 equiv. relative to olefin). Then the reaction mixture was heated to 80 °C for 6 h in a sealed flask.9b After screening Ru-catalysts presented in Fig. 1, complex Gru-II, promoting hydrogenation at a comparable rate as the azinate complex Az-II, was chosen for further studies. The roles of the base (Table 1, entries 5–8) and the solvent (entries 9–11) were also investigated to find that sodium hydride can be replaced by sodium formate and the reaction can be conducted in solvents like THF, dimethyl carbonate, dimethoxyethane or dichloroethane.
| Entry | Catalyst | Base | Solventb | NMR yieldc [%] |
|---|---|---|---|---|
| a Conditions: catalyst (2 mol%), base (0.2 equiv.), HCO2H (50 equiv.), solvent, 80 °C, 6 h. b DMC = dimethyl carbonate; DME = 1,2-dimethoxyethane; DCE = 1,2-dichloroethane. c Yield determined by 1H NMR. | ||||
| 1 | Az-II | NaH | THF | 92 |
| 2 | Gru-I | NaH | THF | 27 |
| 3 | Gru-II | NaH | THF | >99 |
| 4 | Gru-III | NaH | THF | 95 |
| 5 | Gru-II | None | THF | 68 |
| 6 | Gru-II | t-BuOK | THF | 90 |
| 7 | Gru-II | HCOONa | THF | >99 |
| 8 | Gru-II | NaOH | THF | 78 |
| 9 | Gru-II | NaH | DMC | >99 |
| 10 | Gru-II | NaH | DME | 92 |
| 11 | Gru-II | NaH | DCE | >99 |
To check, if this reduction can be applied one-pot together with an olefin metathesis event, we conducted the RCM reaction of diene 1a in the presence of 2 mol% of Gru-II in THF at 40 °C. After 30 minutes, TLC revealed that the olefin metathesis step was complete. At this point 0.2 equiv. of HCOONa was added to the reaction mixture, followed by immediate addition of 50 equiv. of HCOOH. The reaction tube was closed and heated to 80 °C. After 7 hours the reduction was completed, as shown by NMR. Using this procedure, RCM-transfer hydrogenation experiments were conducted with a small set of dienes, producing, after aqueous work-up, corresponding cycloalkanes in good to excellent yield (Table 2).
| Entry | Substrate | Product | Timeb [h] | Yieldc [%] |
|---|---|---|---|---|
| a Conditions: (i) Gru-II (2 mol%), THF, 40 °C, 30 min, then (ii) HCOONa (0.2 equiv.), HCOOH (50 equiv.), 80 °C, THF. b Time of RCM (0.5 h) + time of reduction. c Yield of spectrally pure isolated products. In all cases full conversion was observed. d NaH was used instead of HCOONa. e Product isolated by extraction. Crude product was spectrally pure. f Product purified by silica gel column chromatography. | ||||
| 1d |
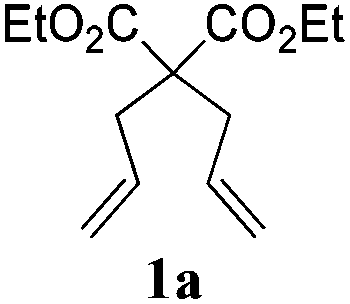
|

|
7.5 | 99e |
| 2 |
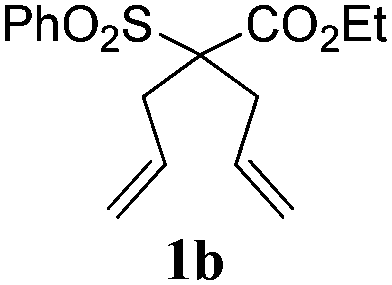
|

|
20.5 | 97f |
| 3 |

|
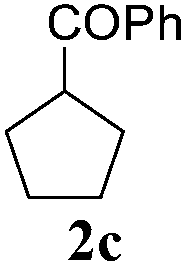
|
7.5 | 92f |
| 4d |

|

|
6.5 | 89e |
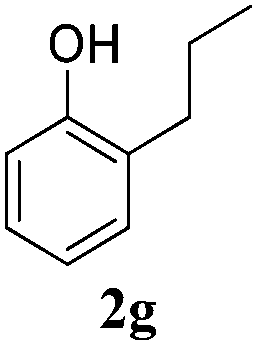
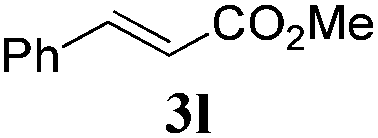
To examine deeper the substrate scope of this transfer hydrogenation reaction, a number of unsaturated substrates were subjected to the optimised reaction conditions. A range of substrates can be hydrogenated efficiently with this system (Table 3), proving excellent compatibility with a number of functional groups.
| Entry | Substrate | Product | Time [h] | Yieldb. [%] |
|---|---|---|---|---|
| a Conditions: Gru-II (2 mol%), HCOONa (0.2 equiv.), HCOOH (50 equiv.), 80 °C, THF. b Yields of spectrally pure isolated products. In parenthesis are conversions determined by 1H NMR, where not indicated full conversion was observed. c NaH was used instead of HCOONa. d Product isolated by extraction. Crude product was spectrally pure. e Product purified by silica gel column chromatography. f Product purified by bulb-to-bulb distillation. g Reaction with 3 mol% of Gru-II. h Product purified by crystallization. i Reaction with 4 mol% of Gru-II. | ||||
| 1 |

|

|
6 | 99c,d |
| 2 |

|

|
6 | 99d |
| 3 |

|

|
6 | 98e |
| 4 |

|

|
20 | 98c,f |
| 5 |

|

|
24 | 90f |
| 6g |

|
|
48 | 74e (100) |
| 7 |

|

|
6 | 83c |
| 8 |

|

|
6 | 91h |
| 9i |

|

|
336 | 43f (100) |
| 10i |

|

|
120 | (50) |
| 336 | (80) | |||
| 11i |
|

|
48 | 96e |
| 12 |

|

|
6 | 99d |
Interestingly, carbonyl functions in ketones10 and enones (Table 3, entries 3, 4, 7 and 8) do not undergo reduction under these conditions, which makes our system different from the previously described [Ru]/NaBH4 couple.6 As suggested by the experiment conducted with 3f, primary and secondary benzyl ethers and an allylic stereocenter survived the reaction untouched (Table 3, entry 5). Interestingly, compounds bearing unprotected OH group 3g (Table 3, entry 6) underwent transfer hydrogenation quantitatively and no esterification took place. The reaction of enones 3h, 3i and α,β-unsaturated ester 3l under our conditions lead to the reduction of conjugated C–C double bonds (Table 3, entries 7, 8 and 11). The second, more substituted C–C double bond present in β-damascone (3h) stayed untouched. Hydrogenation of the trisubstituted double bond in 3j proceeds much slower, requiring 336 h for completion (43% isolated yield, entry 9).11 However, we were surprised when we attempted the reduction of cyclohexene derivative 3k. Hydrogenation rates of small ring cycloalkenes depend on their strain energies,12a and in the case of reduction by diimide, it was shown that the relative rate of cyclopentene hydrogenation is 15.5 times faster than the rate of cyclohexene reduction.12b However, in the case of 3k the reaction was not complete even after 336 h (14 days) of heating at 80 °C, while both cyclopentene (entry 11) and cycloheptene rings (entry 12) were fully reduced in 6 hours. It is noteworthy that the cyclohexene ring is easily reduced by the newly published system composed of NaBH4 and Ru olefin metathesis catalysts.6
We speculated that the observed high sensitivity of the Gru-II/HCOOH catalytic system towards the ring strain and the substitution pattern can be utilised for selective reduction of one C–C double bond in the presence of others. To explore this interesting possibility, a competition experiment, shown in Scheme 2, was performed, to demonstrate the level of control offered by our transfer hydrogenation system. An equimolar mixture of 3a and 3k was subjected to the transfer hydrogenation process leading to complete reduction of the cyclopentene ring, with practically no reduction of a cyclohexene double bond under these conditions (Scheme 2).
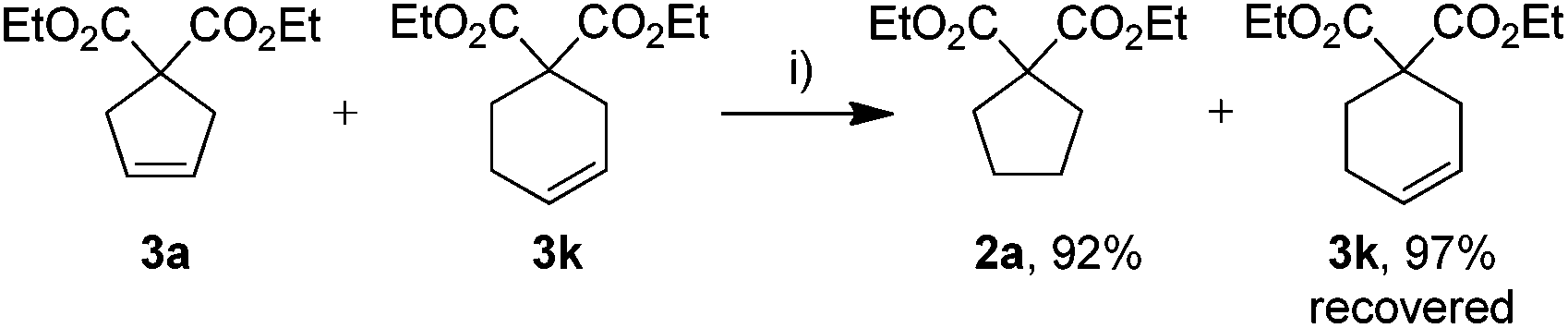 | ||
| Scheme 2 Competition study. Conditions: (i) Gru-II (2 mol%), HCOONa (0.2 equiv.), HCOOH (50 equiv.), THF, 4 h, 80 °C. Conversion determined by GC using an internal standard. | ||
While the system studied by us shows high level of selectivity, it offers also an additional possibility of being readily coupled with olefin metathesis reactions. An example shown in Scheme 3 starts with Ru-catalysed double-RCM of tetraene 1m forming selectively product 3m bearing one five- and one six-membered unsaturated ring. Then, only one of the two seemingly very similar C–C double bonds present in 3m was selectively reduced by Gru-II/HCOOH, yielding mono-saturated product 2m in good yield (Scheme 3, route i + ii). The same two-step sequence can be easily conducted in one-pot fashion, converting directly tetraene 1m into semihydrogenated cyclohexene derivative 2m in 90% yield (Scheme 3, route iii).
 | ||
| Scheme 3 Tandem double RCM-selective transfer hydrogenation of 1m. Conditions: (i) Gru-II (2 mol%), DCM, 1 h, 40 °C. (ii) Gru-II (2 mol%), HCOONa (0.2 equiv.), HCOOH (50 equiv.), THF, 4 h, 80 °C. (iii) Gru-II (2 mol%), THF, 0.5 h, 40 °C, then Gru-II (2 mol%), HCOONa (0.2 equiv.), HCOOH (50 equiv.), THF, 4 h, 80 °C. | ||
The precise details of the nature of catalytic species produced upon the action of HCOONa/HCOOH are unclear. It was reported that some Ru complexes can catalyse the decomposition of formic acid leading to the formation of CO2 and ruthenium hydride species.13 This suggests that entry into the catalytic cycle starts with the conversion of Az-II or Gru-II into Ru–hydride species, which act as the actual hydrogenation catalyst.14,15 A similar mechanism was also suggested for other Ru-catalysed transfer hydrogenation reactions.6 To prove the existence of Ru–H species in our system Grubbs’ second generation catalyst was placed in a dry NMR tube containing formic acid (20 equiv.) in THF-d8. Then the tube was closed and the reaction mixture was heated at 50 °C for 4 h. Then the tube was cooled down to room temperature and the NMR spectrum was recorded. A new signal with a chemical shift of −6.86 ppm appeared. This chemical shift is in the range characteristic for ruthenium hydride species.16 In line with this observation, in hydrogenations of 3a analysed before the end of reaction was reached, we observed some amounts of product 4a formed via the alkene isomerisation process (C–C double bond migration)17 that were decreasing with time (Scheme 4). Both these observations suggest the presence of Ru hydride species during reduction with the Ru/HCOOH system described by us.
 | ||
| Scheme 4 Incomplete isomerisation–hydrogenation of 3a. Conditions: (i) Gru-II (2 mol%), NaH (0.2 equiv.), HCOOH (50 equiv.), THF. Conversion determined by 1H NMR. | ||
Conclusions
In conclusion, a mild protocol for transfer hydrogenation of alkenes has been demonstrated that exhibits a highly functional group tolerance and surprising selectivity. For a number of alkenes, the Ru/HCOOH system can be seen as a safe alternative to dangerous pressurized hydrogen gas. This method can also be coupled with olefin metathesis events, allowing for efficient one-pot sequences. Unlike other Ru-catalysed hydrogenation protocols, Ru/HCOOH allows for selective reduction of a given C–C double bond in the presence of relatively similar ones and is compatible with keto and enone functionalities that can be reduced by NaBH4. Therefore the highly selective nature of this system provides a useful addition to the still expanding6 repository of hydrogenation methods.Experimental section
Procedure A. Reduction of C–C double bonds
Olefin (1 mmol) and 5 mL of anhydrous THF were placed under argon in a reaction tube. Next, catalyst Gru-II (2 mol%) was added to the resulting solution followed by the addition of HCOONa (0.2 mmol) and 98% HCOOH (50 mmol), and the reaction tube was closed. The reaction mixture was stirred for an appropriate period of time at 80 °C, then allowed to reach room temperature and poured into saturated aqueous solution of NaHCO3 (ca. 30 mL). The aqueous layer was extracted with an appropriate organic solvent and the combined organic phases were washed with brine, dried over MgSO4, filtered and the solvent was evaporated in vacuo to obtain the crude product, which was purified when necessary by column chromatography or by bulb-to-bulb distillation.1H NMR (400 MHz, CDCl3) δ = 4.20–4.12 (m, 4H), 2.21–2.11 (m, 4H), 1.71–1.62 (m, 4H), 1.23 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ = 172.7, 61.1, 60.4, 34.4, 25.4, 14.0.
1H NMR (400 MHz, CDCl3) δ = 2.50 (t, J = 7.4 Hz, 2H), 1.93 (t, J = 6.3 Hz, 2H), 1.69–1.59 (m, 4H), 1.53 (s, 3H), 1.45–1.39 (m, 2H), 1.04 (s, 6H), 0.93 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ = 212.0, 143.6, 128.7, 47.8, 39.0, 33.3, 31.2, 28.8, 20.95, 19.0, 16.7, 13.9.
Procedure B. Tandem olefin metathesis and C–C double bond reduction
Diene (1 mmol) in 5 mL of dry THF were placed in a reaction tube. Catalyst Gru-II (2 mol%) was added and the ring closing metathesis reaction was carried out for 0.5 h at 40 °C. Once the RCM reaction was complete according to TLC or GC, solid HCOONa (0.2 mmol) was added immediately followed by 98% HCOOH (50 mmol) and the reaction was continued for an appropriate period of time at 80 °C in a closed tube, then allowed to reach room temperature and poured into saturated aqueous solution of NaHCO3 (ca. 30 mL). The aqueous layer was extracted with an appropriate organic solvent and the combined organic phases were washed with brine, dried over MgSO4, filtered and the solvent was evaporated in vacuo to obtain the crude product, which was further purified when necessary by column chromatography or by bulb-to-bulb distillation.1H NMR (400 MHz, CDCl3) δ = 4.20–4.12 (m, 4H), 2.21–2.11 (m, 4H), 1.71–1.62 (m, 4H), 1.23 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ = 172.7, 61.1, 60.4, 34.4, 25.4, 14.0.
1H NMR (400 MHz, CDCl3) δ = 8.03–7.93 (m, 2H), 7.60–7.50 (m, 1H), 7.50–7.42 (m, 2H), 3.72 (quint, J = 7.88 Hz, 1H), 2.06–1.85 (m, 4H), 1.83–1.54 (m, 4H); 13C NMR (100 MHz, CDCl3) δ = 202.9, 137.0, 132.8, 128.6, 128.6, 46.5, 30.1, 26.4.
Acknowledgements
The authors are grateful to the National Science Centre (Poland) for the NCN Opus grant no. UMO-2013/09/B/ST5/03535.Notes and references
- (a) Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) S. Siegel, Heterogeneous Catalytic Hydrogenation of C=C and C≡C, in Comprehensive Organic Synthesis, Vol. 8 Reduction, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1993, pp. 417–443 Search PubMed; (c) H. Takaya, Homogeneous Catalytic Hydrogenation of C=C and C≡C, in Comprehensive Organic Synthesis Vol. 8 Reduction, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1993, pp. 443–471 Search PubMed.
- (a) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC; (b) R. Crabtree, Acc. Chem. Res., 1979, 12, 331 CrossRef CAS; (c) Q. Zhao, Y. Li, R. Liu, A. Chen, G. Zhang, F. Zhang and X. Fan, J. Mater. Chem. A, 2013, 1, 15039 RSC; (d) M. E. Vol'pin, V. P. Kukolev, V. O. Chernyshev and I. S. Kolomnikov, Tetrahedron Lett., 1971, 12, 4435 CrossRef; (e) I. S. Kolomnikov, V. P. Kukolev, V. O. Chernyshev and M. E. Vol'pin, Russ. Chem. Bull., Int. Ed., 1971, 21, 661 CrossRef; (f) I. S. Kolomnikov, Y. D. Koreshkov, V. P. Kukolev, V. A. Mosin and M. E. Vol'pin, Russ. Chem. Bull., Int. Ed., 1973, 22, 180 CrossRef; (g) V. Jurčík, S. P. Nolan and C. S. J. Cazin, Chem. – Eur. J., 2009, 15, 2509 CrossRef PubMed; (h) K. Ohkubo, T. Aoji, K. Hirata and K. Yoshinga, Inorg. Nucl. Chem. Lett., 1976, 12, 837 CrossRef CAS; (i) S. Horn and M. Albrecht, Chem. Commun., 2011, 47, 8802 RSC; (j) B. Schmidt, S. Krehl and V. Sotelo-Meza, Synthesis, 2012, 1603 CrossRef CAS PubMed; (k) M. Lamani, G. S. Ravikumara and K. R. Prabhu, Adv. Synth. Catal., 2012, 354, 1437 CrossRef CAS; (l) W. Leitner, J. M. Brown and H. Brunner, J. Am. Chem. Soc., 1993, 115, 152 CrossRef CAS; (m) C. E. Hartmann, V. Jurčík, O. Songis and C. S. J. Cazin, Chem. Commun., 2013, 49, 1005 RSC; (n) C. Menozzi, P. I. Dalko and J. Cossy, Synlett, 2005, 2449 CAS; (o) H. Imai, T. Nishiguchi and K. Fukuzumi, J. Org. Chem., 1977, 42, 431 CrossRef CAS; (p) T. Nishiguchi, A. Kurooka and K. Fukuzumi, J. Org. Chem., 1974, 39, 2403 CrossRef CAS.
- (a) Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003 Search PubMed; (b) Olefin Metathesis Theory and Practice, ed. K. Grela, John Wiley & Sons, Inc., Hoboken, NY, 2014 Search PubMed.
- For reviews, see: (a) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365 CrossRef CAS PubMed; (b) B. Schmidt and S. Krehl, Domino and Other Olefin Metathesis Sequences, in Olefin Metathesis Theory and Practice, ed. K. Grela, John Wiley & Sons, Inc., Hoboken, NY, 2014, pp. 187–232 Search PubMed.
- Selected examples: (a) J. Louie, C. W. Bielawski and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 11312 CrossRef CAS; (b) J. Cossy, F. C. Bargigga and S. BouzBouz, Tetrahedron Lett., 2002, 43, 6715 CrossRef CAS; (c) B. Schmidt and M. Pohler, Org. Biomol. Chem., 2003, 1, 2512 RSC; (d) X. Miao, C. Fischmeister, C. Bruneau, P. H. Dixneuf, J.-L. Dubois and J.-L. Couturier, ChemSusChem, 2012, 5, 1410 CrossRef PubMed.
- T. Connolly, Z. Wang, M. A. Walker, I. M. McDonald and K. M. Peese, Org. Lett., 2014, 16, 4444 CrossRef CAS PubMed.
- T. Wdowik, C. Samojłowicz, M. Jawiczuk, M. Malińska, K. Woźniak and K. Grela, Chem. Commun., 2013, 49, 674 RSC.
- T. Wdowik, C. Samojłowicz and K. Grela, unpublished results.
- (a) Formic acid is considered as viable and renewable feedstock for hydrogen. Ru(II) promoted catalytic formic acid dehydrogenation to a gaseous mixture of H2 and CO2 was reported: I. Mellone, M. Peruzzini, L. Rosi, D. Mellmann, H. Junge, M. Beller and L. Gonsalvi, Dalton Trans., 2013, 42, 2495 RSC; (b) In an open flask, the same catalytic system (2 mol% Gru-II, 50 equiv. HCOOH and 0.2 equiv. of NaH) leads to formation of 2a in 90% yield, however, after prolonged time of 15 hours. For detailed experimental procedures see ESI.‡.
- For representative examples of Ru-catalyzed transfer hydrogenation of ketones, see: (a) C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, J. Org. Chem., 2001, 66, 9020 CrossRef CAS; (b) M. Babin, R. Clément, J. Gagnon and F.-G. Fontaine, New J. Chem., 2012, 36, 1548 RSC; (c) O. O. Kovalenko, H. Lundberg, D. Hübner and H. Adolfsson, Eur. J. Org. Chem., 2014, 6639 CrossRef CAS; (d) ref. 13a.
- Interestingly, trisubstituted alkenes were reported to be inert to Ru-catalysed transfer hydrogenation, cf.ref. 6.
- (a) P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, J. Am. Chem. Soc., 1970, 92, 2377 CrossRef CAS; (b) The relative ratio of reduction with diimide is 15.5 for cyclopentene, 1.0 for cyclohexene, 12.1 for cycloheptene and 17.0 for cyclooctene, while for 1-methylcyclohexene is only 0.11. See: (c) E. W. Garbish Jr., S. M. Schildcrout, D. B. Patterson and C. M. Sprecher, J. Am. Chem. Soc., 1965, 87, 2932 CrossRef.
- (a) N. Manashe, E. Salant and Y. Shvo, J. Organomet. Chem., 1996, 514, 97 CrossRef; (b) T. Koike and T. Ikariya, Adv. Synth. Catal., 2004, 346, 37 CrossRef CAS.
- (a) B. Schmidt, Eur. J. Org. Chem., 2004, 1865 CrossRef CAS; (b) B. Alcaide, P. Almendros and A. Luna, Chem. Rev., 2009, 109, 3817 CrossRef CAS PubMed.
- It cannot be excluded that both, the initial ruthenium metathesis catalyst and its decomposition products after olefin metathesis step, are able to catalyse the hydrogen transfer reaction.
- (a) T. M. Trnka, J. P. Morgan, M. S. Sanford, T. E. Wilhelm, M. Scholl, T.-L. Choi, S. Ding, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 2546 CrossRef CAS PubMed; (b) M. B. Dinger and J. C. Mol, Eur. J. Inorg. Chem., 2003, 2827 CrossRef CAS; (c) D. Pingen, M. Lutz and D. Vogt, Organometallics, 2014, 33, 1623 CrossRef CAS.
- For decomposition pathways of ruthenium olefin metathesis catalysts and related processes, see: (a) S. H. Hong, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2004, 126, 7414 CrossRef CAS PubMed; (b) S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160 CrossRef CAS PubMed; (c) S. H. Hong, A. G. Wenzel, T. T. Salguero, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 7961 CrossRef CAS PubMed; (d) B. Schmidt and S. Hauke, Org. Biomol. Chem., 2013, 11, 4194 RSC.
- D. Domin, D. Benito-Garagorri, K. Mereiter, J. Fröhlich and K. Kirchner, Organometallics, 2005, 24, 3957 CrossRef CAS.
- E. Demole, P. Enggist, U. Säuberli and M. Stoll, Helv. Chim. Acta, 1970, 53, 56 CrossRef.
- L. Li, P. Cai, Q. Guo and S. Xue, J. Org. Chem., 2008, 73, 3516 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Mieczysław Mąkosza on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details and characterization data, including NMR spectra. See DOI: 10.1039/c4ob02480j |
| This journal is © The Royal Society of Chemistry 2015 |