
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopeptides containing the DEKS motif as conformationally restricted collagen telopeptide analogues: synthesis and conformational analysis†
Robert
Wodtke
ad,
Gloria
Ruiz-Gómez
b,
Manuela
Kuchar
ad,
M. Teresa
Pisabarro
b,
Pavlina
Novotná
c,
Marie
Urbanová
c,
Jörg
Steinbach
ad,
Jens
Pietzsch
ad and
Reik
Löser
*ad
aInstitute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, Bautzner Landstraße 400, 01328 Dresden, Germany. E-mail: r.loeser@hzdr.de; Fax: +49 351 260-2915; Tel: +49 351 250-3658
bStructural Bioinformatics, BIOTEC, Technische Universität Dresden, Tatzberg 47/49, 01307 Dresden, Germany
cDepartment of Physics and Measurements, Institute of Chemical Technology, 166 28 Prague, Czech Republic
dDepartment of Chemistry and Food Chemistry, Technische Universität Dresden, Bergstraße 66, 01069 Dresden, Germany
First published on 4th December 2014
Abstract
The collagen telopeptides play an important role for lysyl oxidase-mediated crosslinking, a process which is deregulated during tumour progression. The DEKS motif which is located within the N-terminal telopeptide of the α1 chain of type I collagen has been suggested to adopt a βI-turn conformation upon docking to its triple-helical receptor domain, which seems to be critical for lysyl oxidase-catalysed deamination and subsequent crosslinking by Schiff-base formation. Herein, the design and synthesis of cyclic peptides which constrain the DEKS sequence in a β-turn conformation will be described. Lysine-side chain attachment to 2-chlorotrityl chloride-modified polystyrene resin followed by microwave-assisted solid-phase peptide synthesis and on-resin cyclisation allowed for an efficient access to head-to-tail cyclised DEKS-derived cyclic penta- and hexapeptides. An Nε-(4-fluorobenzoyl)lysine residue was included in the cyclopeptides to allow their potential radiolabelling with fluorine-18 for PET imaging of lysyl oxidase. Conformational analysis by 1H NMR and chiroptical (electronic and vibrational CD) spectroscopy together with MD simulations demonstrated that the concomitant incorporation of a D-proline and an additional lysine for potential radiolabel attachment accounts for a reliable induction of the desired βI-turn structure in the DEKS motif in both DMSO and water as solvents. The stabilised conformation of the cyclohexapeptide is further reflected by its resistance to trypsin-mediated degradation. In addition, the deaminated analogue containing allysine in place of lysine has been synthesised via the corresponding ε-hydroxynorleucine containing cyclohexapeptide. Both ε-hydroxynorleucine and allysine containing cyclic hexapeptides have been subjected to conformational analysis in the same manner as the lysine-based parent structure. Thus, both a conformationally restricted lysyl oxidase substrate and product have been synthetically accessed, which will enable their potential use for molecular imaging of these important enzymes.
Introduction
Enzymes that act on peptides and proteins very often recognise their substrates in certain favoured conformations. This is especially well understood for proteases,1 while for other enzymes that participate in modifications of amino acid side chains the situation is less clear. Lysyl oxidase, an enzyme which catalyses the oxidative deamination of protein-bound lysine residues to the corresponding aldehydes called allysines (Scheme 1), is involved in the crosslinking of extracellular matrix proteins such as collagen and elastin.2,3 Lysyl oxidases have been identified as key players in the processes of hypoxia-induced tumour invasion and metastasis by remodelling the extracellular matrix mainly through crosslinking of type I collagen.6–8 Therefore, these enzymes represent a highly attractive target for functional molecular imaging of tumours. Among the different imaging modalities, positron emission tomography (PET) is unparalleled in terms of sensitivity and quantification and requires the development of target-tailored molecular probes labelled with positron-emitting radionuclides.9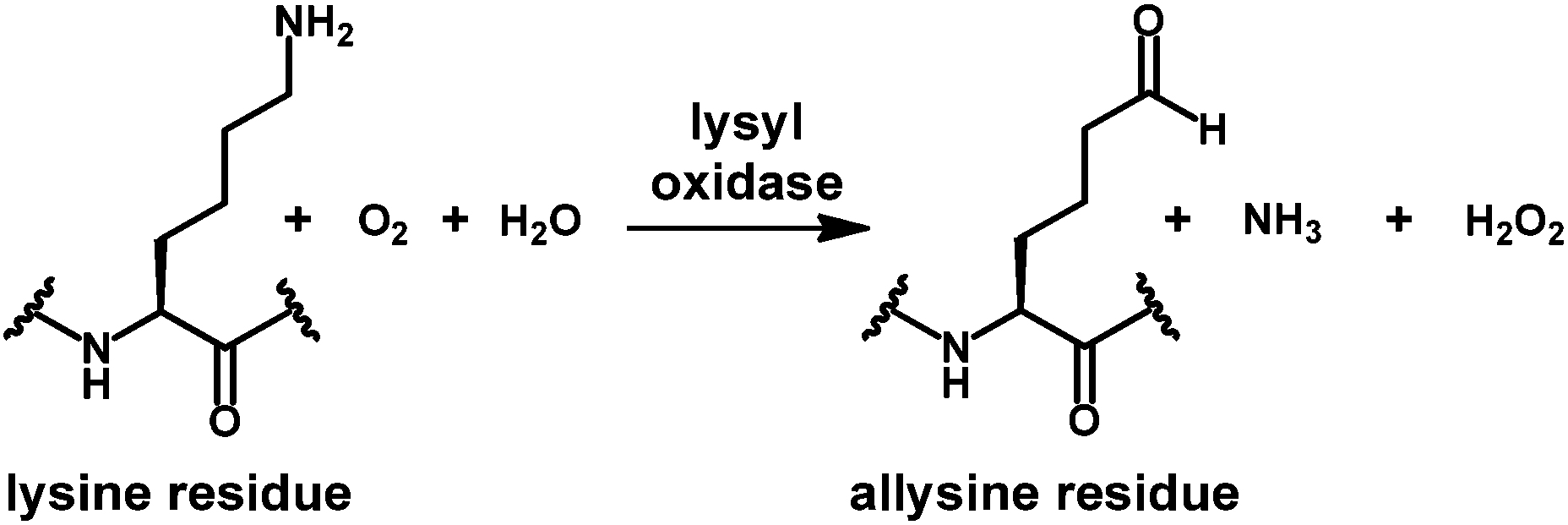 | ||
| Scheme 1 Lysyl oxidase-catalysed oxidative deamination of protein-bound lysine residues to allysine. | ||
For type I collagen, one major crosslink pathway involves the condensation of an allysine carbonyl with the amino group of an unmodified lysine or hydroxylysine residue to a Schiff base-type linkage.10 Notably, this process seems to occur spontaneously without enzymatic assistance. Especially well understood is the crosslinking process where the lysine residue located in the N-terminal telopeptide of the α1(I) chain undergoes oxidation to act as aldehyde donor towards a (hydroxy)lysine (930) residue located at the α1 chain in the C-terminal region of the triple-helical portion of another type I collagen molecule.5,11 On the basis of the Chou–Fasman algorithm and molecular modelling studies, it has been proposed that the N-terminal α1(I) telopeptide adopts a hairpin-like conformation with the central sequence Asp-Glu-Lys-Ser (DEKS) forming a β-turn.11 This model has been supported experimentally on the basis of 1H NMR and ECD12,13 as well as IR14 spectroscopic analysis. In detail, these investigations revealed a conformation for the DEKS motif that is in agreement with a type I β-turn. Extended molecular modelling studies made evident that the triple-helical region around lysine 930 constitutes a recognition site that is complementary to the hairpin-like shaped N-telopeptide of the α1 chain (Fig. 1).5 Detailed investigations to unravel the structural determinants contributing to the substrate properties of telopeptides concerning their conversion by lysyl oxidase have been performed by Nagan and Kagan. This study indicated the requirement of the interaction with the triple-helical region around Lys 930, as the negative charge of the Glu preceding the lysine residue at the telopeptide undergoing oxidation has to be compensated for efficient enzymatic conversion. Furthermore, the conformation of the peptide chain containing the Lys residue to be deaminated seems to be of importance for its recognition by the enzyme itself, as a telopeptide derivative containing Pro N-terminal before Lys exhibited the most favourable kinetic parameters.15
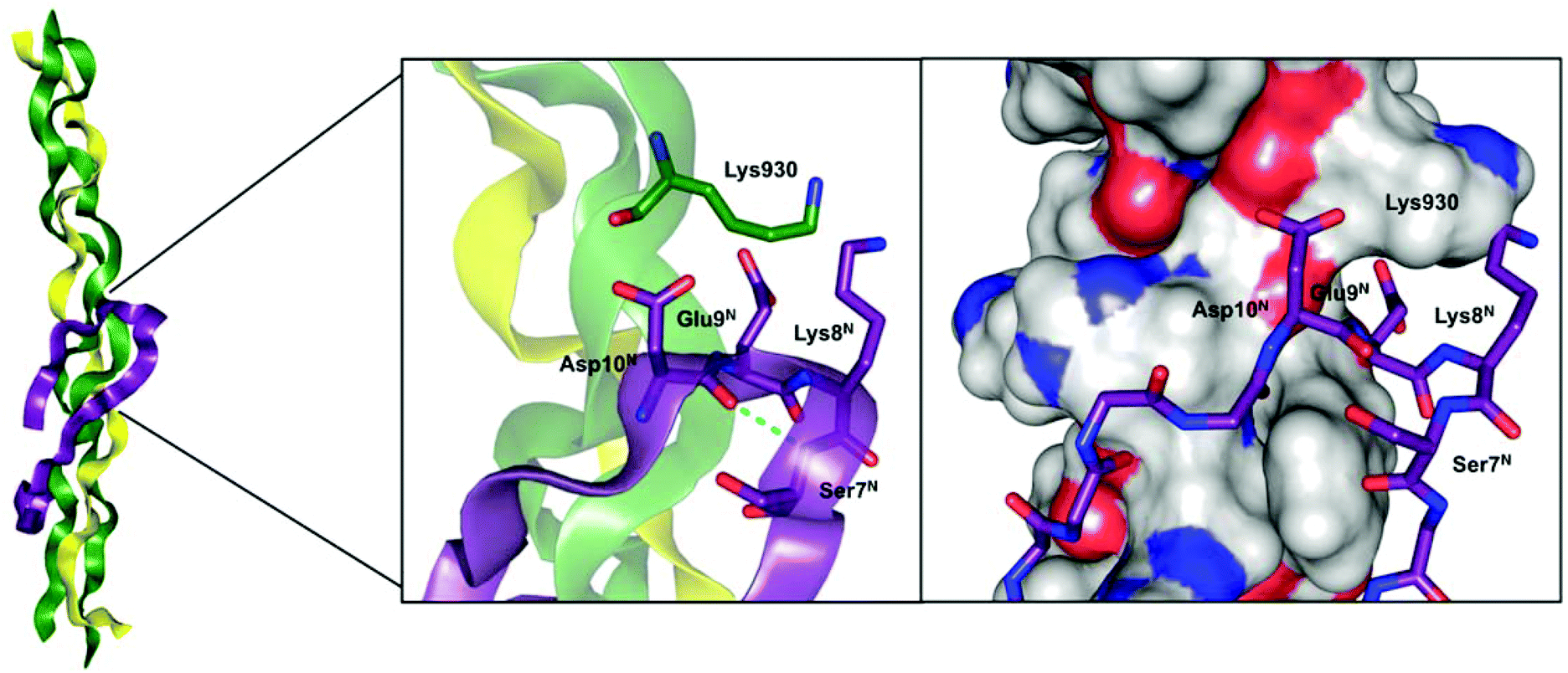 | ||
| Fig. 1 Model of the α1(I)-N-telopeptide docked to the triple-helical receptor region of bovine type I collagen. Left and Middle: The α1(I) chains are shown in green, the α2(I) chain in yellow and the α1(I)-N-telopeptide in purple each in ribbon plots. For the zoomed area, the βI-turn forming DEKS sequence in the α1(I)-N-telopeptide and the Lys930 in the collagen triple helix are represented in the stick model, the proposed hydrogen bond between Asp10N and Ser7N is depicted by the dotted line. Right: the α1(I) and α2(I) chains are shown in the surface model (grey = carbon, red = oxygen, blue = nitrogen) and the α1(I)-N-telopeptide is shown in stick representation (purple = carbon, red = oxygen, blue = nitrogen). All figures were prepared with PyMOL4 using the model developed by Malone et al.5 based on the file “Malone_etalFig1d3ce.pdb” included in the Supporting Information of that article. | ||
To image lysyl oxidase in vivo, the design of radiolabelled probes based on the α1(I)-N-telopeptide seems to be promising as their conversion will lead to aldehydes that can react with amino groups on collagen strands which may result in a signal enrichment on sites of enzymatic activity. As this will require the interaction of the probe molecule with collagen we envisaged stabilising a potential lysyl oxidase substrate derived from the α1(I)-N-telopeptide in a β-turn-like conformation.
The most promising approach to stabilise the DEKS sequence in the crucial turn seems to be its incorporation in head-to-tail cyclised peptides. This approach should be most feasible for cyclic penta- and hexapeptides, as their conformational properties are well understood.16–20 Nevertheless, the use of these macrocycles in biomedical applications is rather scarce as their synthesis can be challenging and cumbersome.21–24 Besides potentially enhanced target interaction, the cyclisation of peptides very often leads to improved pharmacokinetic properties over their linear counterparts,20,25 which was a further motivation of this work. In order to allow the attachment of the radiolabel, an additional amino acid had to be introduced. For the development of PET tracers based on small molecules, the positron emitter fluorine-18 represents the most advantageous radionuclide.26,27 Therefore, a 4-fluorobenzoyl group has been chosen to be attached to the side chain of a second lysine residue to allow the perspective labelling with fluorine-18 of the designed cyclopeptides.28
Herein we report on the synthesis of cyclic peptides containing the telopeptide-derived DEKS motif together with its allysine-containing analogue and their conformational analysis in both DMSO and water on the basis of NMR and chiroptical spectroscopy supported by molecular dynamics (MD) simulations. To assess the intrinsic turn-forming propensities of the DEKS motif, a linear analogue was synthesised and its enzymatic stability was investigated in comparison to the cyclohexapeptides.
Results and discussion
Synthesis
Initially, the conformational restriction of the DEKS motif was attempted within the cyclic pentapeptide 2 (Scheme 2). To this end, Fmoc-Lys-OAll (1) was prepared according to a published procedure29 and attached to the 2-chlorotrityl chloride resin (2-ClTrtCl resin) resulting in a loading degree of 0.26 mmol g−1. The corresponding resin-bound Fmoc-protected pentapeptide ester was sequentially deprotected at its C- and N-termini. This procedure ensures that the macrocycle-forming amide bond coupling will occur at a hydrogen-bonded peptide bond of the intended β-turn.30 Conditions for allyl ester cleavage were selected according to Horne et al. using Pd(PPh3)4 as catalyst and a solution of N-methylmorpholine and acetic acid in dichloromethane as allyl scavenger.31 To obtain the desired cyclopentapeptide, the resin-bound terminally deprotected pentapeptide was treated with HATU and DIPEA. Minicleavage followed by ESI-MS analysis indicated that the cyclisation procedure had to be repeated twice until no linear precursor was detectable. The amino group of the Dde-protected lysine was released by repetitive treatment with 2% hydrazine/DMF monitored spectrophotometrically followed by 4-fluorobenzoylation using 4-fluorobenzoyl chloride and triethylamine in dichloromethane. Cleavage from the resin and concomitant side chain deprotection yielded the desired DEKS and 4-fluorobenzoyl lysine containing cyclopentapeptide 2. HPLC analysis of the crude product indicated the presence of substantial amounts of side products that eluted at significantly greater retention times represented by a broad peak. These major impurities were assigned as oligomerisation/polymerisation products resulting from processes competing with cyclisation. The occurrence of these side products, despite the low initial loading of 0.26 mmol g−1, may reflect the poor tendency for cyclisation of the linear pentapeptide precursor. Isolation of the product from the crude mixture furnished the 4-fluorobenzoylated cyclopentapeptide 2 in 22% yield. 1H NMR analysis of 2 in DMSO at varying temperatures revealed chemical shift-temperature gradients for the amide protons <−2.5 ppb K−1, which indicates the absence of a defined secondary structure in this cyclic peptide (data not shown).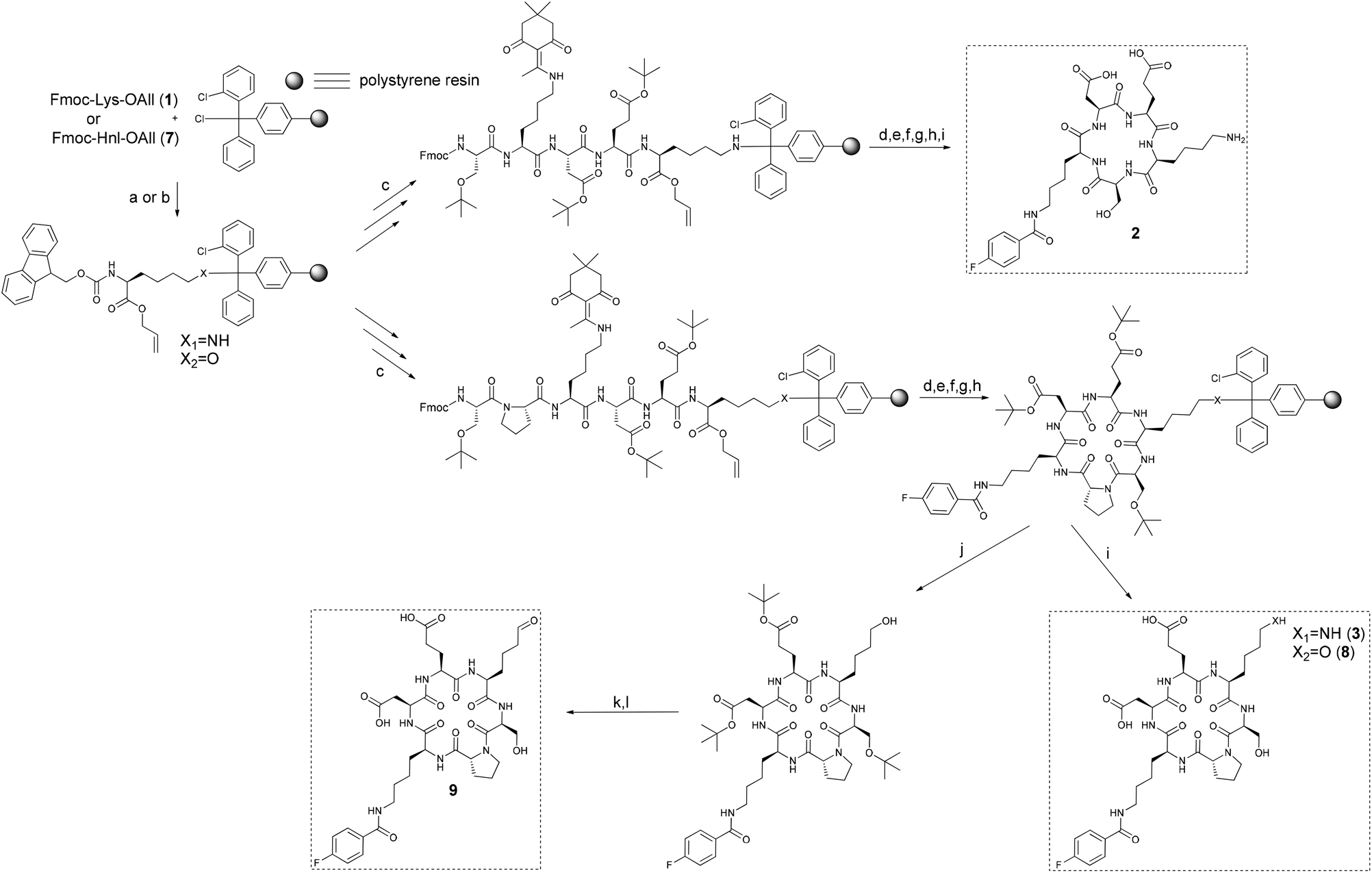 | ||
Scheme 2 Synthesis of cyclo(Ser-Lys(FBz)-Asp-Glu-Lys) (2) and cyclo(Ser-D-Pro-Lys(FBz)-Asp-Glu-Xaa) (3, 8, 9). Reagents and conditions: (a) 1. Fmoc-Lys-OAll × TFA, DIPEA, THF, 2 h, 2. CH2Cl2–CH3OH–DIPEA (17/1/2), 3 × 2 min; (b) Fmoc-Hnl-OAll, pyridine, CH2Cl2–DMF (1/1), 24 h or 65 h, 2. CH2Cl2–CH3OH–DIPEA (17/1/2), 3 × 2 min; (c) SPPS; (d) Pd(PPh3)4, CH2Cl2–NMM–CH3COOH (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1), Ar, 4 h; (e) 20% piperidine/DMF, 2 × 8 min; (f) HATU, DIPEA, DMF, 1–3 × 4 h; (g) 2% N2H4–DMF, 5–12 × 5 min; (h) 4-fluorobenzoyl chloride, TEA, CH2Cl2, 2 h; (i) TFA–TES–H2O (95:2.5/2.5), 3 h; (j) TFA–TES–CH2Cl2 (1/5/94), 30 min; (k) Dess-Martin periodinane, CH2Cl2, 3 h; (l) TFA–CH2Cl2 (9/1), 1 h. :2:1), Ar, 4 h; (e) 20% piperidine/DMF, 2 × 8 min; (f) HATU, DIPEA, DMF, 1–3 × 4 h; (g) 2% N2H4–DMF, 5–12 × 5 min; (h) 4-fluorobenzoyl chloride, TEA, CH2Cl2, 2 h; (i) TFA–TES–H2O (95:2.5/2.5), 3 h; (j) TFA–TES–CH2Cl2 (1/5/94), 30 min; (k) Dess-Martin periodinane, CH2Cl2, 3 h; (l) TFA–CH2Cl2 (9/1), 1 h. | ||
The synthesis of 2 resulted in low yields and no evidence for a stabilised structure was discernible. For this reason, it was envisaged to include a further amino acid acting as turn-inducing element. As both D-amino acids and proline are to be found frequently in position i + 1 of β-turns, the incorporation of one of these amino acids into the peptide chain can strongly support or induce turn formation.32,33D-Proline combines both structural features and has therefore the potential to induce turn-like conformations in cyclic hexapeptides as demonstrated by Matter and Kessler.34 Thus, it was decided to insert a D-proline between the N-terminal serine and the lysine to be fluorobenzoylated (Scheme 2). Assuming the added residue will adopt the i + 1 position of a β-turn, Ser, Lys(FBz), and Asp would be found in the positions i, i + 2, and i + 3, respectively. Potentially, this arrangement could lead to hydrogen bonding between the carbonyl oxygen of Ser and the amide NH of Asp, which would preorganise these residues for the formation of a second β-turn where they adopt the positions of i + 3 and i, respectively. Cyclisation of the linear hexapeptidic precursor was done as in the synthesis of 2. Notably, no repetition of the cyclisation procedure was necessary, which might reflect its stronger turn-forming propensity compared to the linear pentapeptide precursor. According to HPLC analysis, the content of 3 in the crude product was 80%, whereas that of 2 was only 22%.
As the oxidation reaction catalysed by lysyl oxidase leads to the transformation of a primary amine to its corresponding aldehyde, the synthesis of the allysine derivative 9 of cyclohexapeptide 3via its hydroxy analogue 8 was envisaged (Scheme 2). To conserve the preparative strategy, Fmoc-Hnl-OAll (7) (Hnl: ε-hydroxynorleucine) was intended to be attached to the 2-ClTrtCl resin. Subsequent construction of the hexapeptide, cyclisation, and cleavage under conservation of the tBu side chain protection groups should be followed by oxidation of the primary alcohol to the allysine-containing cyclohexapeptide and final deprotection. The synthesis of amino acid 7 was achieved in four steps (Scheme 3). Nα-Boc-protected L-ε-hydroxynorleucine (4) was accessible in satisfying yields by a chiral pool synthesis based on the diazotation of Nα-Boc-lysine with sodium nitroprusside and hydrolysis of the intermediate diazonium salt in a one-pot sequence published by Glenn et al.35 Treatment of 4 with allyl bromide furnished the Boc-protected L-ε-hydroxynorleucine allyl ester (5). Surprisingly, removal of the Boc group of 5 by treatment with trifluoroacetic acid was accompanied by trifluoroacetylation at the ε-hydroxyl group. As compound 6 was Fmoc-protected at its α-amino group under slightly basic aqueous conditions, the trifluoroacetic acid ester was quantitatively hydrolysed to the corresponding alcohol 7, which was the required building block for resin attachment.
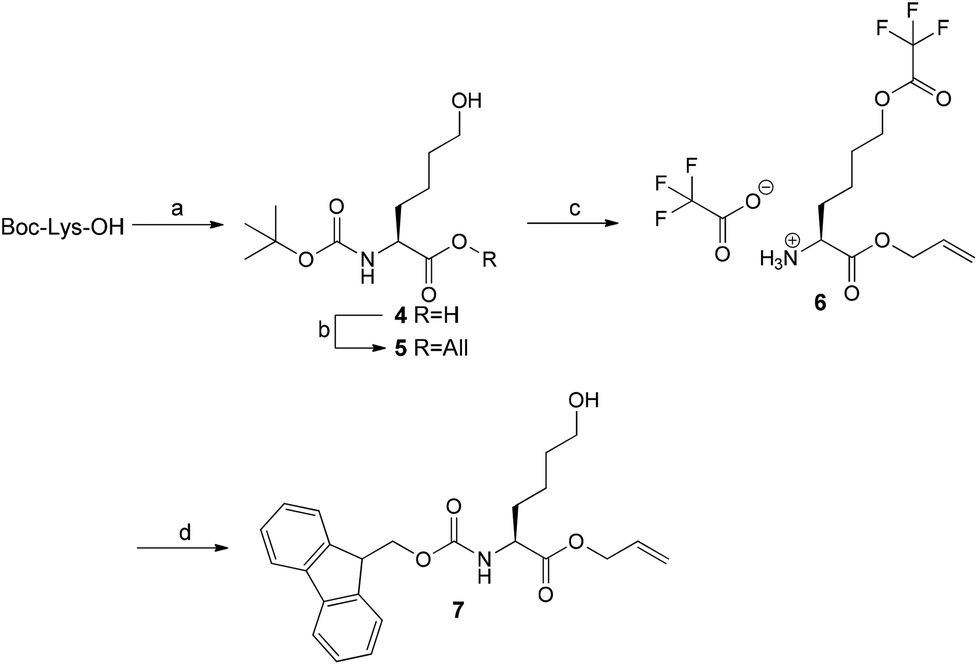 | ||
| Scheme 3 Synthesis of Fmoc-Hnl-OAll (7). Reagents and conditions: (a) sodium nitroprusside, 4 M NaOH, pH = 9–10, 6 h at 60 °C; (b) allyl bromide, DIPEA, DMF, 16 h at rt; (c) CH2Cl2–TFA (1:1, v/v), 2 h at rt; (d) Fmoc-OSu, 0.62 M Na2CO3, DME, 2 h at 4 °C to rt. | ||
Loading of 7 to the 2-ClTrtCl resin was tried under the same conditions as for the amino analogue Fmoc-Lys-OAll (1), but it resulted in no detectable resin attachment due to the low nucleophilicity of the hydroxyl group. Applying conditions developed for the functionalisation of the 2-ClTrtCl resin with Fmoc-protected amino alcohols, which employ pyridine in excess as base and prolonged reaction times,36 led to a loading degree of 0.86 mmol g−1 when one equivalent of 7 and a reaction time of 65 h was used. Construction of the hexapeptide sequence, cyclisation, and fluorobenzoylation was done according to the synthesis of the amino analogue. The increased resin loading of 0.86 mmol g−1 compared to 0.26 mmol g−1 in the synthesis of the lysine-containing cyclohexapeptide 3 did not have detrimental effects on the cyclisation, as confirmed by ESI-MS analysis of the product obtained by minicleavage of the Dde-protected cyclohexapeptide. Analysis of the fluorobenzoylated L-ε-hydroxynorleucine-containing cyclo-hexapeptide 8 released from the resin indicated the presence of an undesired component exhibiting a molecular mass that is 96 units higher than the product, which has been assigned to result from trifluoroacetylation at a free hydroxyl group. This esterification must occur at the hydroxyl group of L-ε-hydroxynorleucine rather than that of serine, as trifluoroacetylation has not been observed for cyclohexapeptide 3. Generally, trifluoroacetylation in solid-phase peptide synthesis during TFA-mediated cleavage has been reported only for N-terminal serine and threonine residues but not for internal ones.37 As trifluoroacetylation was also encountered along the route to building block 7 during TFA-mediated Boc deprotection of 5, it can be concluded that the nucleophilicity of the hydroxyl group of the L-ε-hydroxynorleucine must be increased due to sterical and/or electronical reasons. To hydrolyse the TFA ester, the crude material was dissolved in a 1:1 mixture of acetonitrile and water and kept at room temperature overnight. ESI-MS analysis clearly confirmed the complete hydrolysis of the TFA ester under these conditions. Purification by preparative HPLC revealed the existence of side products that eluted at greater retention times than the product 8, which was already observed for cyclopentapeptide 2 but not for cyclohexapeptide 3. Since the initial loading degree for the synthesis of 8 (0.86 mmol g−1) was significantly higher compared to 3 (0.23 mmol g−1), the generation of oligomerisation/polymerisation products was probably favoured and competed against the intramolecular cyclisation, even in the presence of D-proline. After purification by preparative HPLC and lyophilisation of the product-containing fractions, the desired cyclohexapeptide 8 was obtained in 30% yield.
For the synthesis of the allysine-containing cyclohexapeptide 9, the loading procedure of Fmoc-Hnl-OAll (7) to the 2-ClTrtCl resin was repeated with a reduced amount of the amino acid (1 eq. → 0.6 eq.) and shortened reaction time (65 h → 24 h). This variation resulted in a loading degree of 0.46 mmol g−1, which is still higher than that obtained with Fmoc-Lys-OAll (1; 0.26 mmol g−1) but should be sufficient to attenuate intermolecular reactions. To synthetically access the allysine-containing cyclohexapeptide 9, the selective oxidation of 8 was necessary. Oxidation of primary alcohols that stop at the aldehyde level is reported to work very well by employing the Dess-Martin periodinane as oxidising agent.38 As the deprotected cyclohexapeptide 8 also contains another hydroxyl group at its serine residue, this side chain should be protected to prevent oxidation there. Release of peptides linked to 2-ClTrtCl resin via their C-terminal carboxy group without affecting the acid-labile side-chain protecting groups can be conveniently accomplished by incubation with a 1:4 mixture of hexafluoroisopropanol and dichloromethane.39 However, when the resin containing fully protected 8 was subjected to these conditions, the yield of the released peptide was very low, as the 2-chlorotrityl ethers are obviously more acid-stable than the corresponding esters. A good yield for cleavage of the fully protected cyclohexapeptide (tri-tBu-8) was achieved with TFA–TES–CH2Cl2 (1:5:94). Oxidation of tri-tBu-8 with Dess-Martin periodinane and subsequent deprotection with TFA led to the desired allysine-containing cyclohexapeptide 9. Analysis by ESI-MS and 1H NMR clearly confirmed its identity as aldehyde. This is worth to note, as allysine-containing peptides have been reported to be unstable due to dehydrative cyclisation to tetrahydropyridine derivatives.40,41 Even over several months at room temperature in DMSO-d6, no changes were discernible for the aldehyde group from the 1H NMR spectrum. This result is in agreement with a more recent study in which no intrinsic instability of peptide-incorporated allysine residues was observed either.42
To gain insight into the intrinsic conformational properties of the DEKS motif, the linear version of cyclohexapeptide 3 formally resulting from a cut between the Lys(FBz) and D-Pro moieties of the corresponding cyclic peptide, the central residues of the intended β-turn, was synthesised. To mask the terminal charges, the required linear hexapeptide 10 (Fig. 2) was N- and C-terminally acetylated and amidated, respectively, and assembled by standard Fmoc solid-phase synthesis on Rink amide resin. The 4-fluorobenzoyl residue was introduced in analogy to the synthesis of the cyclic peptides.
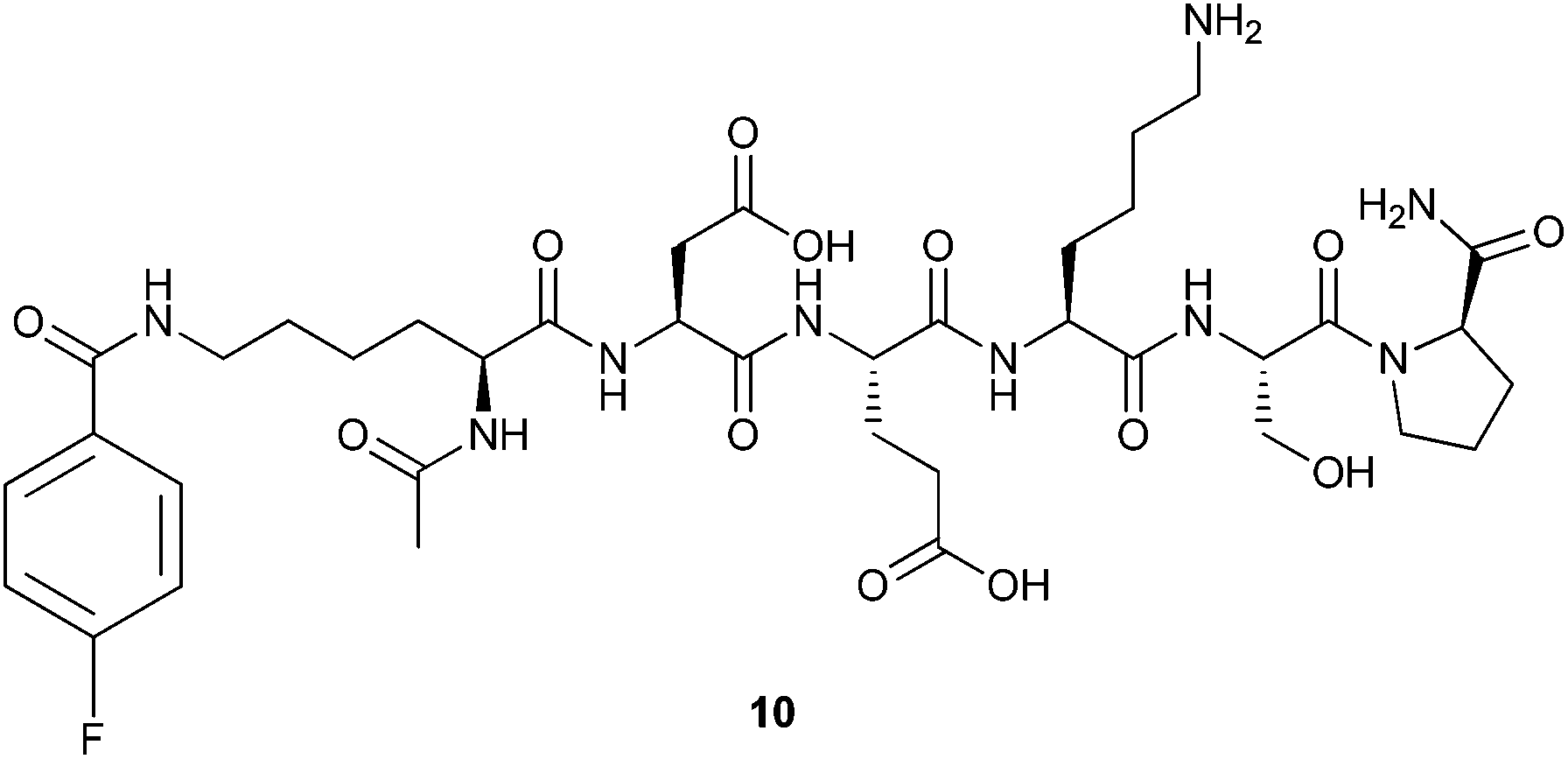 | ||
| Fig. 2 Structure of the linear hexapeptide 10. | ||
Conformational analyses
The successful synthesis of the three cyclohexapeptides 3, 8 and 9 allowed the investigation of their solution conformations by 1D and 2D 1H NMR experiments at 298 K in DMSO-d6. On the basis of TOCSY experiments, it was possible to assign all amide and CαH resonances in the 1H NMR spectra of these cyclic peptides unambiguously. Notably, no cis-amide bonds were detected. The larger dispersion in the chemical shifts of the NH and CαH protons of the constrained peptide 3 compared to the low chemical shift dispersion observed on its linear version 10 indicates a more defined structure for the cyclopeptide. This is particularly obvious for the region of the CαH chemical shifts, which in the case of cyclopeptide 3 shows distinctive signals for every proton whereas the signals of the Asp and Ser as well as the Glu, Lys and D-Pro Cα protons in the linear analogue 10 are overlapping with each other, respectively. Values greater than 8 Hz for the 3JNHCHα coupling constants of the residues Lys(FBz), Asp, and Lys, Aly or Hnl are consistent with a structure of restricted flexibility.43 This assumption is supported by the presence of medium to strong sequential dNN (i,i + 1) ROEs for the residues Glu, Lys/Aly/Hnl and Lys(FBz) each in position i, which lose the connectivity on the Asp residue. In addition, strong-medium and weak dαN (i,i + 1) ROEs were detected for D-Pro and Asp, and Lys/Aly/Hnl, Lys(FBz), Glu, respectively (Fig. 3). Values for the chemical shift temperature coefficients more positive than −4 ppb K−1 were observed for the amide protons of Asp, Glu, Lys/Hnl/Aly and Ser residues in all constrained hexapeptides (Fig. 4). Generally, temperature coefficients for amide protons in DMSO more positive than −2 ppb K−1 are considered indicative of intramolecular hydrogen bonds, whereas values more negative than −4 ppb K−1 allow to conclude solvent-exposed NH-groups.17,44 The most positive temperature coefficients are exhibited by the amide protons of the Asp and Ser residues, which indicates their involvement in intramolecular hydrogen bonds. This data combined with the information obtained from the ROESY spectra allows to conclude the presence of a β-turn formed by the residues Ser, D-Pro, Lys(FBz) and Asp stabilised by a i,i + 3 hydrogen bond between Ser CO (i) and Asp NH (i + 3). Generally, the criterion for the presence of a β-turn in a peptide sequence requires the Cα atoms of the first (i) and fourth (i + 3) residues to be less than 7 Å apart from each other.18,45,46 This feature implies for a cyclic hexapeptide that the presence of one β-turn accounts for the existence of a second β-turn for topological reasons.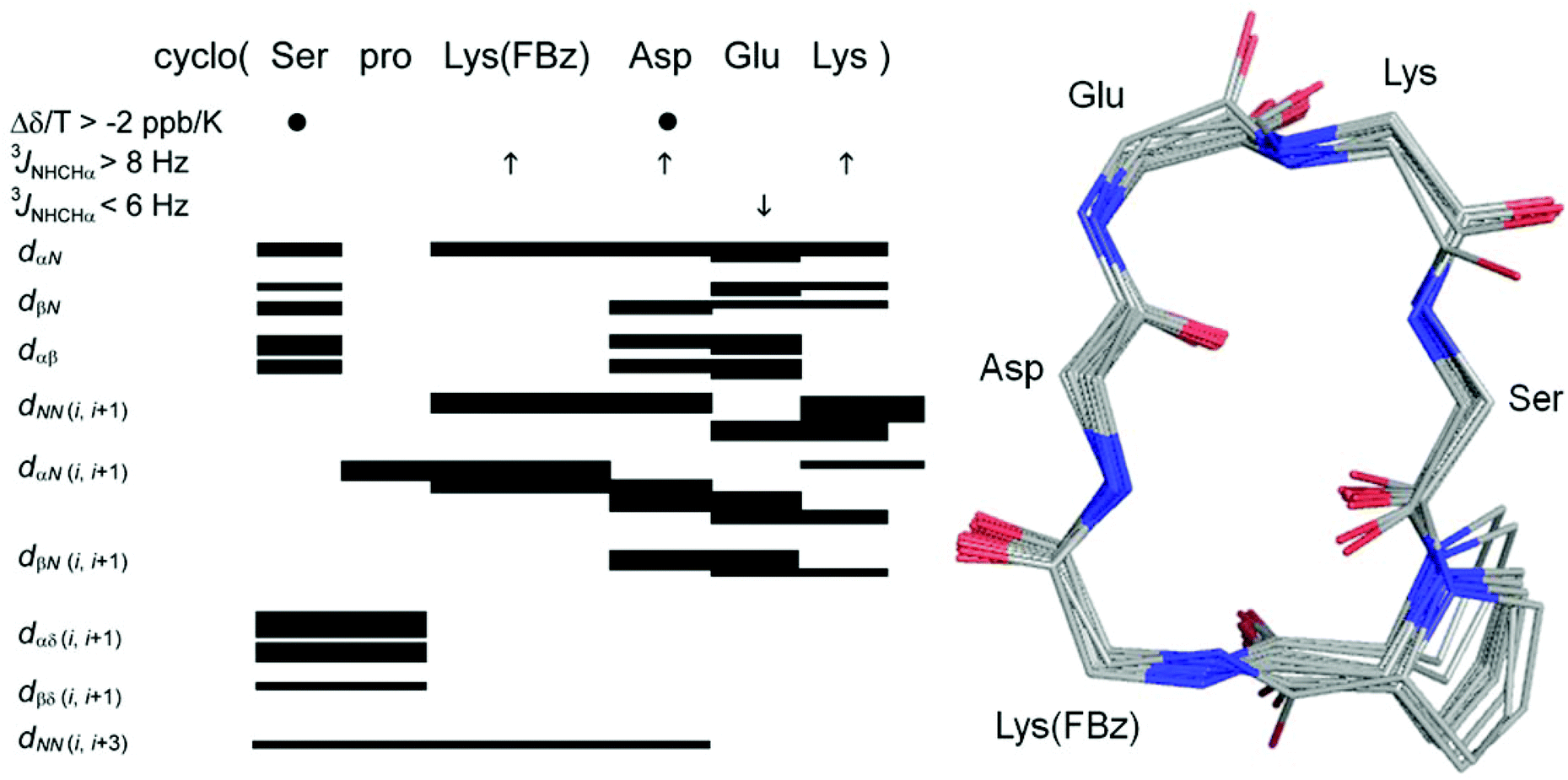 | ||
| Fig. 3 Left: summary of amide NH temperature coefficients (Δδ/T > −2 ppb K−1 shown by ●), 3JNHCHα coupling constants (>8 Hz represented by ↑ and <6 Hz shown by ↓) and ROEs correlations summary for cyclohexapeptide 3 in DMSO at 298 K. Right: 20-lowest energy structures of 3. For clarity only the side chain of pro is shown. For summary and structures of cyclohexapeptides 8 and 9 see ESI (Fig. S2†). For distance restraints of compounds 3, 8 and 9 see Tables S1–S3 in ESI.† | ||
 | ||
| Fig. 4 Amide temperature coefficients of the cyclohexapeptides 3, 3a, 8–9 and the linear hexapeptide 10 in DMSO or water (marked with *). Temperature coefficients more positive than −2.0 ppb K−1 (dashed red line) or −4.5 ppb K−1 (dashed blue line) indicate that the amide proton is part of an intramolecular hydrogen bond in DMSO and water, respectively. For the values of the amide temperature coefficients and plots of the temperature dependence of chemical shifts for the amide protons of 3, 3a, 8, 9 and 10 see ESI (Fig. S1†). | ||
Applied to the cyclohexapeptides 3, 8 and 9, this allows to infer that also the telopeptide-derived DEXS (X = Lys, Hnl, Aly) motif must form a β-turn-like structure, as Ser and Asp are the terminal residues of both the tetrapeptide sequences SpK(FBz)D and DEXS. This conclusion is in accordance with the observed temperature coefficient for the serine amide protons. Taking into account the threshold value for temperature coefficients in DMSO outlined above, the value of −2.2 ppb K−1 observed for the amide proton of the unmodified lysine residue is more ambiguous in regard to intramolecular H-bonding, but it is unlikely that this amide proton is exposed to the solvent. The chemical shift temperature coefficients exhibited by the amide protons of the linear DEKS-containing hexapeptide 10 were all more negative than −4 ppb K−1 except for the Glu NH (see MD section below), which allows to conclude their solvent exposure and thus the absence of turn-like structures in this compound (Fig. 4).
Solution structures were calculated from a total of 37 (15 inter-residue), 34 (11 inter-residue), and 29 (11 inter-residue) ROEs of 3, 8 and 9 in DMSO, respectively, four backbone ϕ-dihedral angle restraints derived from the amide coupling constants (−120 ± 30° for 3JNHCHα > 8 Hz and −65 ± 30° for 3JNHCHα < 6 Hz), and two H-bond restraints i,i + 3 involving the Asp and Ser residues (Fig. 3, S2 and Tables S1–S3 in ESI†). Structures were calculated in XPLOR-NIH47 using a dynamic simulated annealing (SA) protocol,48 followed by energy minimisation in the CHARMm force field.49 The 20 lowest-energy structures of 3, 8 and 9 did not have any distance (≥0.2 Å) or dihedral angle (≥2.4°) violations, converging with a backbone RMSD of 0.25, 0.21, and 0.24 Å, respectively. The presence of two β-turns flanked by Ser and Asp residues involving the sequences SpK(FBz)D and DEXS (X = Lys, Hnl, Aly) was consolidated in all three constrained peptides. The Cα distances between these flanking residues were 5.2 Å, which confirms the compliance with the criterion for β-turns as noted above.18,45,46 As expected, the turn involving D-Pro (i + 1) and Lys(FBz) (i + 2) residues exhibited ϕ and ψ angle values consistent with a type II reverse (II′) β-turn (Table 1). As previously observed,50 the geometrical characteristics of this turn impose short distances between the Cα atoms of the Ser and Asp flanking residues in the cyclic structures, which allows to nucleate a second β-turn around Glu (i + 1) and Xaa (Xaa = Lys, Hnl, Aly) (i + 2) (Table 1). Their ϕ and ψ angles showed values which are in accordance with a type I β-turn46 in 80–95% of the ensemble structures.
| Compound | ϕ(D-Pro) | ψ(D-Pro) | ϕ(Lys(FBz)) | ψ(Lys(FBz)) |
|---|---|---|---|---|
| 3 | 63.6 ± 3.0 | −108.0 ± 10.6 | −91.5 ± 6.2a | −5.9 ± 15.5 |
| 8 | 65.1 ± 2.3 | −113.7 ± 8.1 | −92.5 ± 6.9 | −3.3 ± 13.7 |
| 9 | 64.9 ± 2.5 | −110.4 ± 8.6 | −90.4 ± 4.5 | −10.7 ± 13.1 |
| Compound | ϕ(Glu) | ψ(Glu) | ϕ(Xaa) | ψ(Xaa) |
|---|---|---|---|---|
| a 90% of the 20 ensemble structures, 10% ϕ = −131.6 ± 9.1. b 80% of the 20 ensemble structures, 20% ϕ = −150.2 ± 0.1. c 95% of the 20 ensemble structures, 5% ϕ = −144.9. d 90% of the 20 ensemble structures, 10% ϕ = −139.9 ± 8.2. | ||||
| 3 | −57.4 ± 4.3 | −12.4 ± 15.3 | −90.5 ± 2.7b | −23.2 ± 21.6 |
| 8 | −58.1 ± 2.7 | −19.7 ± 3.9 | −89.9 ± 1.3c | −26.7 ± 28.2 |
| 9 | −53.5 ± 4.9 | −20.1 ± 9.2 | −88.7 ± 1.3d | −18.4 ± 28.5 |
DMSO is known to favour the formation of secondary structures in peptides.51 Given that the interaction of peptides with other biomolecules in vivo occurs in aqueous solution, the conformational behaviour of 3 was also exemplarily studied in water. NMR spectra of 3 at varying temperatures were acquired in water, and the signals were assigned based on the 2D TOCSY spectrum. As for 3 in DMSO, the dNN(i,i + 1) correlations in the 2D ROESY spectrum in water between Asp-Lys(FBz) and Lys-Ser clearly indicate the presence of the two β-turns (Fig. S3 in ESI†). Due to spectra acquisition with solvent suppression by water presaturation, it was not possible to obtain quantitative ROE signals for the Cα protons. Therefore, no solution structure could be calculated. Further evidence for structural order in the peptidic backbone of 3 in water is provided by considering the differences in the Cα proton chemical shifts between the values observed and the ones tabulated for random-coil conformations in water.52 As Fig. 5 shows, the secondary chemical shifts of the “inner residues” in the proposed turn (Glu/Lys and D-Pro/Lys(FBz)) are negative, whereas those of Asp and Ser, the residues bridging both turns, are positive. To evaluate whether amide protons in peptides and proteins participate in intramolecular hydrogen bonds a temperature chemical shift coefficient of −4.5 ppb K−1 is considered as threshold value in aqueous solution.53 Temperature coefficients more negative than this value indicate solvent-exposed amide protons. Interestingly, all amide protons of 3 in water display positive values, i.e. their chemical shifts increase with temperature, which is equivalent to greater deshielding of these protons at higher temperatures (Fig. 4). This result suggests that the changes in the chemical shifts of these protons over the observed temperature range is not exclusively determined by their hydrogen bond behaviour, as both intramolecular and intermolecular hydrogen bonds are weakened with increasing temperature.
 | ||
| Fig. 5 Secondary chemical shifts of Cα protons in cyclohexapeptide 3 in aqueous solution referenced to tabulated random coil values (Δδ = δCHα(3) − δCHα(random coil)).52 | ||
However, the NH protons of the residues that stabilise the two β-turns by acting presumably as H bond donors, Asp and Ser, exhibit the most positive values for Δδ/T. This finding is in accordance with the results obtained for DMSO as solvent, which indicates the similarity of the backbone conformations of 3 in DMSO and water. Generally, the chemical shift temperature coefficients should be interpreted cautiously to gain structural information, especially when the amide protons are in the vicinity to an aromatic moiety, as their chemical shift changes can be influenced by ring current effects.53 To investigate the influence of the 4-fluorobenzoyl moiety on the amide chemical shift temperature coefficients, the analogue of 3 lacking this group (compound 3a) was prepared. The NMR analysis of 3a in water revealed that its amide protons are slightly more deshielded than in 3 due to the lack of the aromatic ring current. Interestingly, the temperature coefficients for the chemical shifts of the amide protons in 3a did not differ significantly from those in 3, and the sequence of the values was the same in both derivatives, which suggests that the 4-fluorobenzoyl group has minor influence on the chemical shift temperature coefficients of the amide protons (Fig. 4).
The presence of a stabilised secondary structure within the cyclohexapeptides 3, 8 and 9 was further supported by electronic circular dichroism (ECD) spectra recorded in aqueous solution at different concentrations and in the absence and presence of 50% trifluoroethanol (TFE; Fig. 6). The spectra of 3 and 8 show a strong negative maximum at 201 nm and a weaker positive maximum at 185 nm. This ECD signature is in accordance with a type II reverse β-turn.54 Worth of note, no changes in the shape and intensity were observed upon addition of trifluoroethanol. The spectra of the allysine-containing analogue 9 are of similar shape even though lower absolute values for the molar ellipticities are exhibited. In contrast, the shape of the ECD spectrum of the linear hexapeptide 10 is of distinct character and resembles that of a type II poly(L-Pro) (PII) helix with a strong negative maximum at 197 nm and a weak positive maximum at 231 nm (Fig. 6).54 This motif of secondary structure has been very often observed in short peptides.55 Similar as for 3, 8 and 9, no major changes in the shape and intensity within the ECD spectrum of 10 were observable upon addition of TFE.
 | ||
| Fig. 6 ECD spectra of the cyclohexapeptides 3, 8, 9 and the linear analogue 10 in aqueous solution in the absence and presence of trifluoroethanol. The concentration of each peptide was 0.125 mg mL−1 The spectra for 0.5 mg mL−1 can be found in Fig. S4 in ESI.† | ||
Further evidence for the existence of a β-turn conformation in 3, 8 and 9 can be derived from their IR absorption and vibrational circular dichroism (VCD) spectra recorded in DMSO (Fig. 7). Compared to ECD spectra, which reflect the conformation of an oligopeptide as a whole, VCD spectra are more sensitive to local differences in conformation as they are derived from vibrational states and resemble an average conformation in the oligopeptide molecule.56 The absorption bands in the IR spectrum of 3 are 1691, 1660, 1635, 1540, and 1504 cm−1. The corresponding VCD spectrum exhibits a slightly positively biased negative couplet in the amide I region with the positive component at 1689 cm−1 and the negative component at 1658 cm−1. This signal is followed by a negative couplet in the amide II region consisting of a positive band at 1554 cm−1 and a negative band at 1523 cm−1. On the basis of VCD investigations on small peptides with a β-hairpin structure containing D-Pro in the position i + 1 of the central β-turn,57 the absorption bands at 1691 cm−1 and 1635 cm−1 in the IR spectrum and the VCD band at 1689 cm−1 can be attributed to the β-turn structure. The NMR-based structure calculations clearly indicated a high propensity for compound 3 to adopt a turn-extended-turn structure. Therefore, the absorption bands at 1660 cm−1 and the VCD band at 1658 cm−1 may result from the weak β-sheet character of this kind of structure. In this context, it should be noted that the IR and VCD bands of 3, which are attributed to the β-sheet and βII′-turn proportions, respectively, are of approximately equal intensities, in contrast to the β-hairpin structures investigated by Zhao et al. where the β-sheet proportion is dominating.57 The VCD spectrum of the allysine-containing cyclohexapeptide 9 is of similar signature in the amide I-region as the one of 3. Obviously, the β-sheet-related IR and VCD bands show stronger intensities in the case of the ε-hydroxynorleucine analogue 8, which otherwise exhibits the same band positions in the IR and VCD spectra as cyclohexapeptide 3. It is striking that the VCD spectrum of the linear analogue 10 is of similar shape as those of 3, 8 and 9. However, as deduced from the NMR results and the ECD spectra, the structure of 10 must be distinct from that of the cyclohexapeptides. The negatively biased negative amide I couplet consisting of a positive component at 1683 cm−1 and a negative component at 1660 cm−1 may indicate the presence of a PII helix,58 which is in agreement with the ECD results. The most significant hint for the absence of a β-turn-like structure in 10 is included in its IR spectrum, in which the band around 1635 cm−1 is missing, while the cyclic counterparts feature this vibrational transition (Fig. 7). Taken together, it can be concluded that in this case ECD provides more information regarding the secondary structure compared to VCD, as the VCD spectra of the cyclopeptides and the linear hexapeptide 10 differ only subtly from each other.
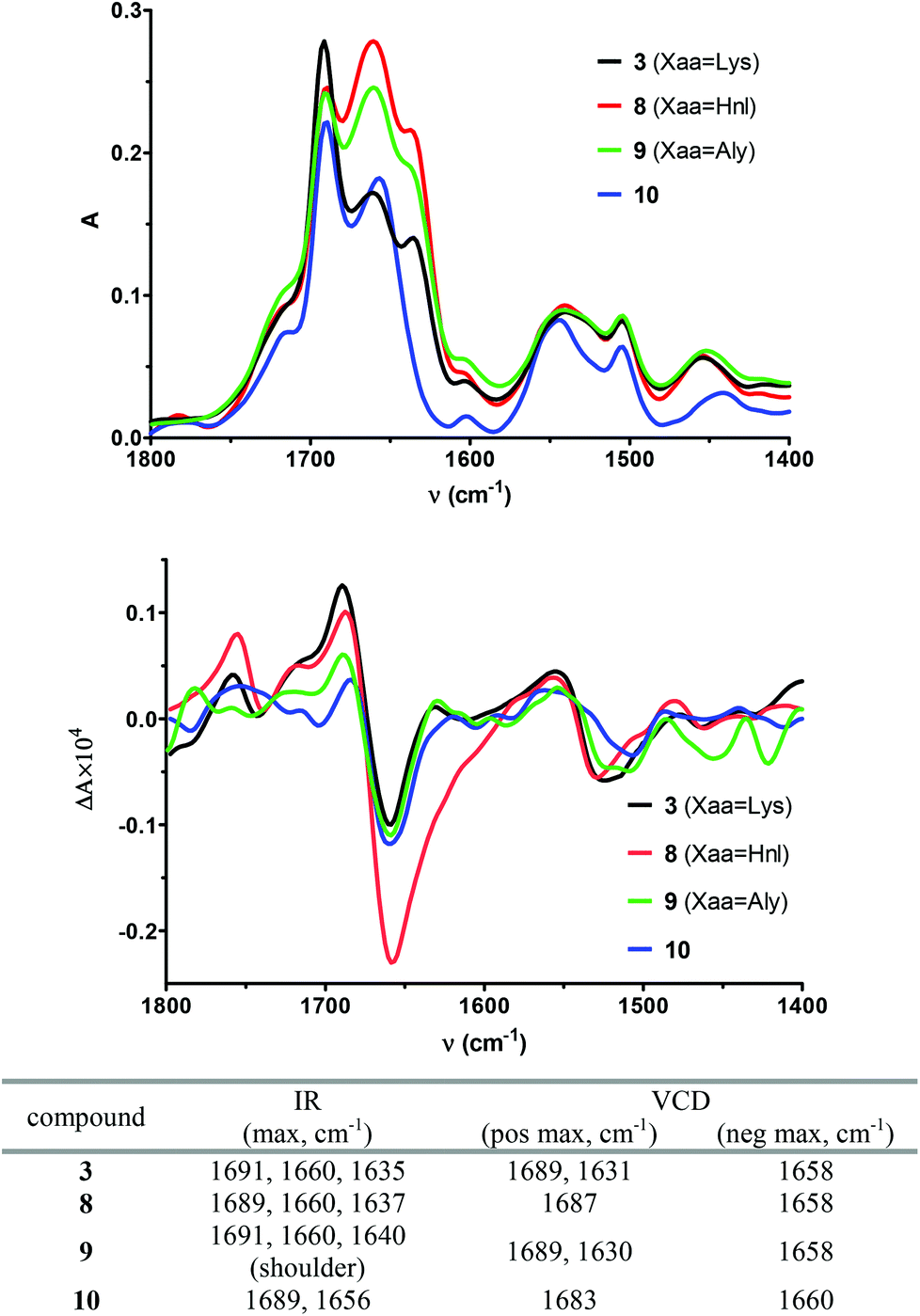 | ||
| Fig. 7 IR (top) and VCD (bottom) spectra of the cyclohexapeptides 3, 8, 9 and the linear analogue 10, and listing of characteristic signals in the amide I region (max = maximum, pos = positive, neg = negative). | ||
To gain further insight into the conformational behaviour of the constrained peptides 3 and 9, MD simulations were performed. Their conformational space was explored in DMSO and water as solvents. One of the best NMR-calculated structures was selected in each case and simulated without restraints for 100 ns using the ff99SB force field59 and standard protocols as implemented in the AMBER12 package.60 The trajectories were analysed in terms of backbone RMSD, backbone dihedral angles and H-bonding. The turn-extended-turn conformations observed from NMR-calculations were also dominant during the simulations in both DMSO and water (Table 2, Fig. 8 and 9).
 | ||
| Fig. 8 Backbone RMSD of 3 (black) and 9 (grey) in DMSO (top) and water (bottom). Backbone RMSD referred to the mass-weighted least-squares fitting of the average structures after removal of overall translational and rotational motions of 100 ns MD simulations. | ||
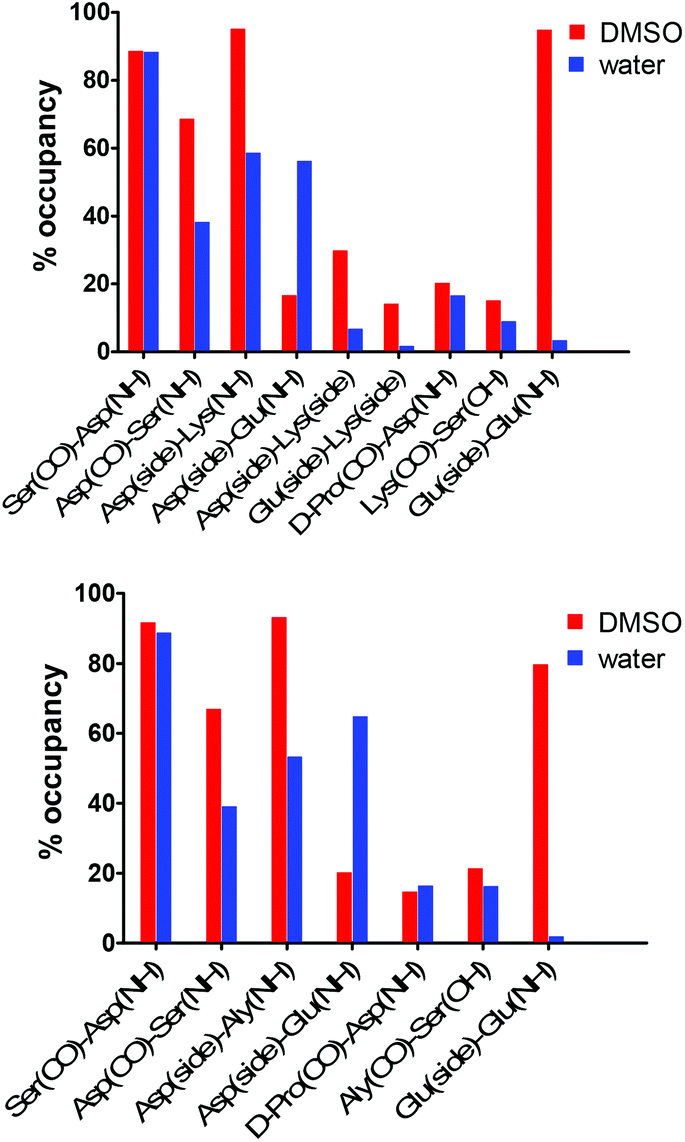 | ||
| Fig. 9 H-bond analysis derived from the 100 ns trajectories in DMSO (red) and water (blue). Top: cyclo(Ser-D-Pro-Lys(FBz)-Asp-Glu-Lys) (3), Bottom: cyclo(Ser-D-Pro-Lys(FBz)-Asp-Glu-Aly) (9). | ||
| ϕ(D-Pro) | ψ(D-Pro) | ϕ(Lys(FBz)) | ψ(Lys(FBz)) | Turnc (%) | |
|---|---|---|---|---|---|
| 3 | 66.3 ± 9.7 | −122.1 ± 11.9 | −83.3 ± 12.8 | 3.1 ± 17.2 | 97 |
| 3 | 63.5 ± 10.0 | −124.0 ± 13.4 | −82.3 ± 14.1 | 0.1 ± 19.3 | 98 |
| 9 | 66.1 ± 9.9 | −119.9 ± 12.3 | −84.4 ± 13.2 | 0.8 ± 16.3 | 96 |
| 9 | 63.7 ± 9.8 | −123.7 ± 13.5 | −81.8 ± 13.8 | −0.6 ± 19.2 | 98 |
| ϕ(Glu) | ψ(Glu) | ϕ(Xaa) | ψ(Xaa) | Turnc (%) | |
|---|---|---|---|---|---|
| a DMSO. b Water. c Calculated according to the angle deviations allowed around the ideal dihedral angles that define the classical type II reverse β-turn and type I β-turn, respectively.46 | |||||
| 3 | −59.9 ± 10.4 | −17.6 ± 11.4 | −92.0 ± 14.0 | −33.8 ± 12.3 | 82 |
| 3 | −60.9 ± 11.5 | −24.8 ± 16.8 | −103.2 ± 22.0 | −28.8 ± 18.8 | 87 |
| 9 | −65.9 ± 9.1 | −14.7 ± 10.6 | −93.6 ± 15.2 | −30.4 ± 13.5 | 86 |
| 9 | −62.5 ± 11.3 | −22.9 ± 15.4 | −103.7 ± 22.3 | −28.6 ± 19.0 | 86 |
Comparison of the backbone dihedral angles involving D-Pro, Lys(FBz), Glu, and Xaa (Xaa = Lys and Aly) in DMSO and water showed consistency with the two β-turn types previously identified (Table 2, ESI Fig. S5†). The type II reverse β-turn with D-Pro at position i + 1 was well maintained during at least 96% of the simulation time in each solvent. The type I β-turn involving Glu and Xaa (Xaa = Lys and Aly) residues was also consistent for 82–87% of the sampled conformations. Hydrogen bond analysis shed further light about the conformational space visited by the constrained cyclohexapeptides 3 and 9 in the different solvent environments (Fig. 9). The geometric criterion used to calculate the presence of intramolecular hydrogen bonds in 3 and 9 involved distance and angle cutoffs of 3.5 Å and 120°, respectively. The hydrogen bond involving the amide NH of Asp and the CO group of Ser showed similar occupancy (88–92%) in both compounds. These data are in good agreement with the experimentally determined amide temperature coefficients and provide evidence for the high stabilisation of the type II reverse β-turn in these cyclic hexapeptides. In addition, the H-bond involving the amide NH of Ser and the CO group of Asp was also observed, although with lower occupancy (68% and 67% for 3 and 9 in DMSO, respectively). The presence of this hydrogen bond dropped considerably in water (38% and 39% for 3 and 9, respectively). Consistent with the experimental data, the MD studies illustrate the presence of a highly populated H-bond in DMSO (>90%) between the side chain of Asp and the amide NH of Lys/Aly in 3 and 9, respectively (see Fig. S6 in ESI†). This result is in accordance with previous studies on β-turns in peptides and proteins, which pointed out that a hydrogen bond between an acceptor atom in the functional side chain of the residue at position i and the amide NH of residue at i + 2 is a common feature of type I β-turns.34,61–63 Moreover, the position i of type I β-turns in proteins is occupied by Asp in high statistical probability.63,64 Upon changing the solvent from DMSO to water the presence of the hydrogen bond between the Asp side chain and the amide NH of Lys/Aly was reduced by ca. 40%. To a similar extent (ca. 30%) dropped the occupancy of the SerNH–AspCO H-bond, which indicates the contribution of the i,i + 2 side chain-main chain interaction to the stabilisation of the βI-turn. The loss of this H-bond was compensated by an alternative contact between the Asp side chain and the amide NH of Glu. As opposed to the results obtained from NMR studies on the linear DEKS sequence, in which it has been concluded that a salt-bridge between the side chains of Glu and Lys could stabilise a type I β-turn (in CD3OH–H2O (60/40) as solvent),13 low H-bond occupancy was observed between these side chains in the constrained analogue 3 in DMSO. In fact, the Asp and Lys residues showed a tendency to form H-bond contacts between their side chains, which was twice as high compared to the side chains of Glu and Lys. The fact that the Glu-Lys side chain contact seems to be of negligible importance for the stabilisation of the βI-turn is furthermore supported by the results of H-bond analysis from the MD simulation for the allysine-derived cyclohexapeptide 9 (Fig. 9). These indicate that the H-bond contacts between Asp(CO) and Ser(NH), as well as between the Asp carboxylate and Aly(NH), have similar propensities as in the lysine analogue 3. The strong tendency for the formation of the Asp-Lys side chain contacts in DMSO can be explained by the highly populated H-bond between the Asp side chain and the amide NH of Lys, which may preorganise the Asp residue for the formation of that salt bridge while the side chain of Glu prefers to form H-bond contacts with its amide NH (see ESI Fig. S6†).65 In contrast, the occupancy of both H-bonds is significantly decreased in water. The intraresidual (backbone-side chain) H-bond in the Glu residue has been also observed for the linear hexapeptide 10, as indicated by the low temperature coefficient for the amide NH of Glu in DMSO (Fig. 4).
The increased structural flexibility of the cyclohexapeptides 3 and 9 in aqueous solution compared to DMSO as solvent is also reflected by the B factors for the backbone atoms of the individual residues (see Fig. S7 in ESI†).
Enzymatic stability
To obtain information on the biological stability of cyclohexapeptide 3, its susceptibility against enzymatic cleavage was investigated by HPLC in comparison to its linear counterpart 10. The presence of a lysine residue in these peptides renders the peptide bond between this residue and the following serine potentially prone towards proteolysis by the serine protease trypsin (EC 3.4.21.4). Compound 3 resisted degradation in the presence of bovine trypsin over a time of 30 min, even when stoichiometric amounts of the enzyme were employed (Fig. 10).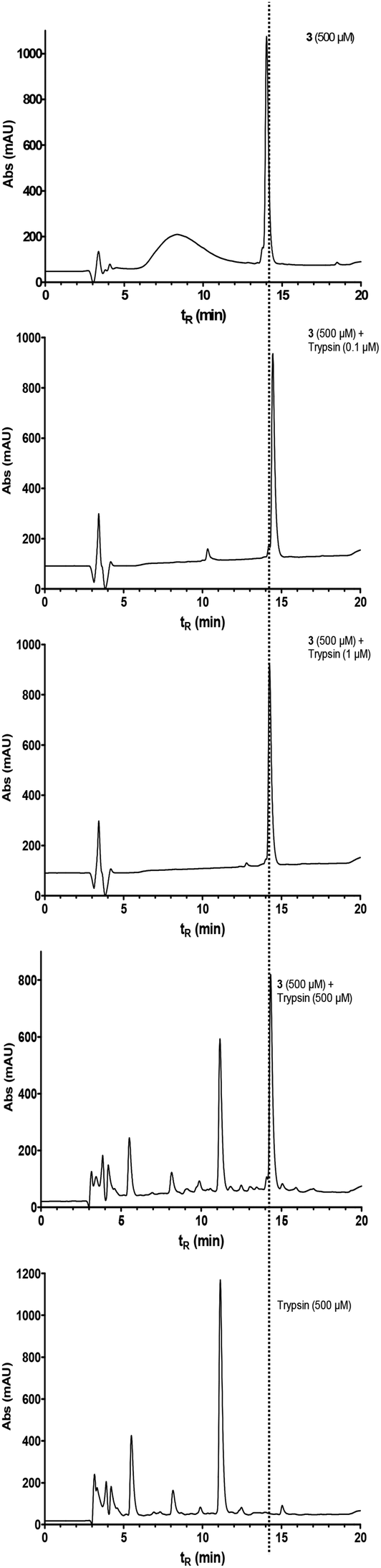 | ||
| Fig. 10 Stability of cyclohexapeptide 3 against cleavage by trypsin as determined by RP-HPLC. Compound 3 was incubated with bovine trypsin at varying concentrations over a time of 30 min. To distinguish the signals that originate from the enzyme, trypsin alone was analysed under the same conditions. | ||
In contrast, the linear analogue 10 was completely converted within the same time already at a trypsin concentration of 1 μM (see Fig. S8 in ESI†). These results provide further evidence for a stabilised structure of 3, as most proteases are unable to cleave peptide bonds within turn structures1 and, furthermore, demonstrate the advantages of small cyclic peptides with regard to metabolic stability.
Concluding remarks
The results of this study demonstrate that the collagen α1(I)-N-telopeptide-derived DEKS motif can be successfully stabilised in a type I β-turn as a part of a turn-extended-turn conformation in cyclic hexapeptides and thus preorganised to interact with its receptor region at type I collagen. D-Pro together with Lys(FBz) were introduced strategically in the cyclohexapeptide to favour a type II′ β-turn and to nucleate a type I β-turn in the DEKS collagen recognition sequence. Furthermore, the introduction of a second Lys residue allows the attachment of a reporter group to enable the convenient functionalisation with the radionuclide fluorine-18 for PET imaging studies. This together with the evaluation of the substrate properties towards lysyl oxidase-catalysed deamination is subject of ongoing investigations. The replacement of the DEKS-derived Lys residue by ε-hydroxynorleucine and allysine did not compromise the structural stabilisation of the βI-turn within this motif. The building block Fmoc-Hnl-OAll (7) required for the construction of these analogues was obtained by an efficient synthetic procedure which will account for the synthesis of further Hnl/Aly-containing cyclopeptides. Although the geometrical characteristics that define the type I β-turn around DEKS sequence is maintained in both DMSO and water, the occupancy of the H-bond involving the amide NH Ser residue and the CO group of Asp decreased considerably in the aqueous environment, which may have implications for the understanding of the collagen cross-linking process. Spectroscopic investigations of the linear analogue 10 did not reveal an inherent turn-forming propensity for the DEKS motif. Furthermore, the cyclic counterpart 3 was resistant towards tryptic cleavage, as opposed to 10. These results demonstrate the power of cyclic hexapeptides to constrain tetrapeptide sequences in β-turn-like conformations with concomitantly achieving stability against proteolytic degradation. Moreover, the combination of D-Pro as turn inducer together with an adjacent lysine for functionalisation with reporter groups might be useful as a general approach for the design of other imaging probes. Potentially, this approach may stimulate the design of other turn mimetic-based radiotracers, especially those targeting G-protein coupled receptors, as many ligands of peptide-activated GPCRs bind to their receptors in β-turn-like conformations.66,67Experimental section
General
All commercial reagents and solvents were used without further purification unless otherwise specified. Melting points were determined on a Galen III Boetius apparatus from Cambridge Instruments. Nuclear magnetic resonance spectra were recorded on a Varian Unity 400 MHz or an Agilent Technologies 400 MR spectrometer. Spectra were processed by using MestreNova (version 6.1.1-6384).68 NMR chemical shifts were referenced to the residual solvent resonances relative to tetramethylsilane (TMS). Mass spectra (ESI) were obtained on a Micromass Quattro LC or a Waters Xevo TQ-S mass spectrometer each driven by the Mass Lynx software. Elemental analysis was performed on a LECO CHNS-932 apparatus. Determination of resin loading was performed on a Thermo Scientific Helios α UV/Vis spectrophotometer.Chromatography
Thin-layer chromatography (TLC) was performed on Merck silica gel F-254 aluminium plates with visualisation under UV (254 nm) and/or staining with a 0.1% (m V−1) ninhydrin solution in ethanol. Preparative column chromatography was carried out using Merck silica gel (mesh size 230–400 ASTM) with solvent mixtures as specified for the particular compounds. Preparative HPLC for the purification of peptides 3, 3a, 8–10 was performed on a Varian Prepstar system equipped with UV detector (Prostar, Varian) and automatic fraction collector Foxy 200. A Microsorb C18 60-8 column (Varian Dynamax 250 × 21.4 mm) was used as the stationary phase and a binary gradient system of 0.1% CF3COOH–water (solvent A) and 0.1% CF3COOH–CH3CN (solvent B) at a flow rate of 10 mL min−1 served as the eluent. The conditions for the gradient elution are as follows: 0–3 min 90% A, 3–35 min gradient to 35% B, 35–40 min gradient to 100% B, 40–45 min 100% B, 45–50 min gradient back to 90% A.Analytical HPLC for both analyses of peptidic products as well as stability assays outlined below was performed on an Agilent Technologies 1200 LC system consisting of gradient pump G1311A combined with degasser G1322A, autosampler G1329A, column heater G1316A and diode array detector G1315D. The system was operated by the Agilent Chemstation 2008 software. A Nucleosil Standard C18 100-7 column (Macherey-Nagel EC 250 × 4.6) was used as stationary phase and a binary gradient system of 0.1% CF3COOH–water (solvent A) and 0.1% CF3COOH–CH3CN (solvent B) at a flow rate of 1 mL min−1 served as the eluent. The stationary phase was kept at 30 °C and the programme for elution was as follows: 0–3 min 90% A, 3–35 min gradient to 35% B, 35–40 min gradient to 100% B, 40–45 min 100% B, 45–50 min gradient back to 90% A.
Synthetic procedures
:1 v/v, 50 mL) and the resulting orange solution was stirred for 2 h at room temperature. The volatile components were removed in the N2 stream followed by repeated evaporations from diethyl ether (3 × 30 mL). The obtained brownish oily residue was purified via column chromatography (gradient from CH2Cl2–CH3OH 95:5 to CH2Cl2–CH3OH 80:20) to afford 1 (2.03 g, 91%) as an off-white, adhesive solid. Rf 0.55 (CHCl3–CH3OH–CH3COOH 75:25:2); 1H-NMR (DMSO-d6) δ: 7.89 (d, 3J = 7.5 Hz, 2H, 2 × CH of Fmoc), 7.83 (broad s, 3H, NH3+), 7.79 (d, 3J = 7.9 Hz, 1H, NH), 7.71 (dd, 3J = 7.4 Hz, 4J = 3.5 Hz, 2H, H-1,8 of Fmoc), 7.42 (t, 3J = 7.4 Hz, 2H, 2 × CH of Fmoc), 7.33 (t, 3J = 7.4 Hz, 2H, 2 × CH of Fmoc), 5.95–5.82 (m, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.30 (dd, 3J = 17.2 Hz, 2J = 1.5 Hz, 1H, CHCHH), 5.20 (dd, 3J = 10.5 Hz, 2J = 1.4 Hz, 1H, CHCHH), 4.58 (d, 3J = 5.3 Hz, 2H, CH2O of Fmoc), 4.38–4.26 (m, 2H, CH2O of allyl), 4.23 (t, 3J = 6.8 Hz, 1H, H-9 of Fmoc), 4.08–4.00 (m, 1H, CαH), 2.84–2.68 (m, 2H, CεH2), 1.78–1.46 (m, 4H, 2 × CH2), 1.45–1.26 (m, 2H, CH2); 13C-NMR (DMSO-d6) δ: 172.00, 157.98 (q, 2JC,F = 31.2 Hz, CO of TFA), 156.19, 143.75, 140.75, 132.39, 127.65, 127.07, 125.22, 120.14, 117.72, 65.63, 64.76, 53.76 (Cα), 46.65 (C-9 of Fmoc), 38.52, 30.04, 26.44, 22.44; 19F-NMR (DMSO-d6) δ: −74.13 (s, CF3); ESI-MS (ESI+) m/z: calc. for C24H29N2O4 [M + H]+, 409.21, found 409.6.
:1); m.p. 110–113 °C (Lit.72 112–113 °C); 1H-NMR (DMSO-d6) δ: 12.37 (s, 1H, COOH), 7.02 (d, 3J = 8.0 Hz, 1H, NH), 4.35 (broad s, 1H, OH), 3.87–3.78 (m, 1H, CαH), 3.39–3.33 (m, 2H, CεH2), 1.69–1.47 (m, 2H, CH2), 1.45–1.22 (s, 13H, 3 × CH3 of Boc, 2 × CH2); 13C-NMR (DMSO-d6) δ: 174.25, 155.57, 77.89 (quart. C of Boc), 60.48 (Cε), 53.48 (Cα), 32.04, 30.66, 28.21 (3 × CH3 of Boc), 22.25; ESI-MS (ESI+) m/z: calc. for C11H22NO5 [M + H]+, 248.15, found 248.4; Elemental analysis: calc. for C11H21NO5, C, 53.43; H, 8.56; N, 5.66; found C, 53.65; H, 8.61; N, 5.61.
:1 over 3:2 to 1:1). The product-containing fractions were combined and evaporated to afford 5 (1.22 g, 65%) as a yellow oil. Rf 0.16 (petroleum ether–ethyl acetate 3:2); 1H-NMR (CDCl3) δ: 5.97–5.84 (m, 1H, CHCH2), 5.33 (d, 3J = 17.2 Hz, 1H, CHCHH), 5.25 (d, 3J = 10.4 Hz, 1H, CHCHH), 5.08–5.00 (m, 1H, NH), 4.68–4.57 (m, 2H, CH2O of allyl), 4.33–4.28 (m, 1H, CαH), 3.64 (t, 3J = 6.3 Hz, 2H, CεH2), 1.90–1.39 (m, 15H, 3 × CH3 of Boc, 3 × CH2); 13C-NMR (CDCl3) δ: 172.67 (CO), 155.59 (CONH), 131.78 (CH of allyl), 118.94 (CH2 of allyl), 80.08 (quart. C of Boc), 65.98, 62.63, 53.48 (Cα), 32.76, 32.24, 28.47 (3 CH3 of Boc), 21.70; ESI-MS (ESI+) m/z: calc. for C14H26NO5 [M + H]+, 288.18, found 288.3; Elemental analysis: calc. for C14H25NO5, C, 58.52; H, 8.77; N, 4.87; found C, 58.01; H, 8.64; N, 4.78.
:1 v/v, 50 mL) and the resulting solution was stirred for 2 h at room temperature. The volatile components were removed in the N2 stream followed by repeated evaporations from diethyl ether (3 × 20 mL). The obtained residue was exposed to oil pump vacuum to remove remaining TFA. 1.84 g (quantitative yield containing 12% of residual TFA) of a brown oil were yielded. 1H-NMR (DMSO-d6) δ: 8.42 (s, 3H, NH3+), 5.99–5.87 (m, 1H, CHCH2), 5.38 (dd, 3J = 17.3 Hz, 2J = 1.5 Hz, 1H, CHCHH), 5.28 (dd, 3J = 10.5 Hz, 2J = 1.3 Hz, 1H, CHCHH), 4.70 (d, 3J = 5.0 Hz, 2H, CH2O of allyl), 4.38 (t, 3J = 6.3 Hz, 2H, CεH2), 4.16–4.07 (m, 1H, CαH), 1.91–1.64 (m, 4H, 3 × CH2), 1.56–1.31 (m, 2H, CH2); 19F-NMR (DMSO-d6) δ: −74.75 (s, CF3 of TFA), −74.79 (s, CF3 of Tfa); ESI-MS (ESI+) m/z: calc. for C11H17F3NO4 [M + H]+, 284.11, found 284.3.
:1 to 1:1). The product-containing fractions were combined to yield 7 (0.97 g, 58%) as colourless oil that solidified in the refrigerator. Rf 0.22 (petroleum ether–ethyl acetate 1:1); m.p. 100–103 °C; 1H-NMR (CDCl3) δ: 7.77 (d, 3J = 7.6 Hz, 2H, H-4,5 of Fmoc), 7.60 (d, 3J = 7.0 Hz, 2H, 2 × CH of Fmoc), 7.40 (t, 3J = 7.4 Hz, 2H, 2 × CH of Fmoc), 7.35–7.29 (m, 2H, 2 × CH of Fmoc), 5.98–5.84 (m, 1H, CHCH2), 5.40–5.23 (m, 2H, CHCHH, NH), 5.27 (dd, 3J = 10.4 Hz, 2J = 1.0 Hz, 1H, CHCHH), 4.65 (d, 3J = 5.5 Hz, 2H, CH2O of allyl) 4.45–4.35 (m, 3H, CH2O of Fmoc and CαH), 4.23 (t, 3J = 7.0 Hz, 1H, H-9 of Fmoc), 3.65 (t, 3J = 6.3 Hz, 2H, CεH2), 1.95–1.40 (m, 6H, 3 × CH2); 13C-NMR (CDCl3) δ: 172.34, 156.10, 143.96, 141.46, 131.64, 127.85, 127.20, 125.22, 120.13, 119.14, 67.16 (CH2O of Fmoc), 66.16, 62.58, 53.95 (Cα), 47.32 (C-9 of Fmoc), 32.63, 32.19, 21.64; ESI-MS (ESI+) m/z: calc. for C24H28NO5 [M + H]+, 410.20, found 410.2; Elemental analysis: calc. for C24H27NO5, C, 70.49; H, 6.65; N, 3.42; found C, 69.89; H, 6.67; N, 3.43.
CH2), 5.30 (dd, 3J = 17.2 Hz, 2J = 1.5 Hz, 1H, CHCHH), 5.20 (dd, 3J = 10.5 Hz, 2J = 1.4 Hz, 1H, CHCHH), 4.58 (d, 3J = 5.3 Hz, 2H, CH2O of Fmoc), 4.38–4.26 (m, 2H, CH2O of allyl), 4.23 (t, 3J = 6.8 Hz, 1H, H-9 of Fmoc), 4.08–4.00 (m, 1H, CαH), 2.84–2.68 (m, 2H, CεH2), 1.78–1.46 (m, 4H, 2 × CH2), 1.45–1.26 (m, 2H, CH2); 13C-NMR (DMSO-d6) δ: 172.00, 157.98 (q, 2JC,F = 31.2 Hz, CO of TFA), 156.19, 143.75, 140.75, 132.39, 127.65, 127.07, 125.22, 120.14, 117.72, 65.63, 64.76, 53.76 (Cα), 46.65 (C-9 of Fmoc), 38.52, 30.04, 26.44, 22.44; 19F-NMR (DMSO-d6) δ: −74.13 (s, CF3); ESI-MS (ESI+) m/z: calc. for C24H29N2O4 [M + H]+, 409.21, found 409.6.
:1); m.p. 110–113 °C (Lit.72 112–113 °C); 1H-NMR (DMSO-d6) δ: 12.37 (s, 1H, COOH), 7.02 (d, 3J = 8.0 Hz, 1H, NH), 4.35 (broad s, 1H, OH), 3.87–3.78 (m, 1H, CαH), 3.39–3.33 (m, 2H, CεH2), 1.69–1.47 (m, 2H, CH2), 1.45–1.22 (s, 13H, 3 × CH3 of Boc, 2 × CH2); 13C-NMR (DMSO-d6) δ: 174.25, 155.57, 77.89 (quart. C of Boc), 60.48 (Cε), 53.48 (Cα), 32.04, 30.66, 28.21 (3 × CH3 of Boc), 22.25; ESI-MS (ESI+) m/z: calc. for C11H22NO5 [M + H]+, 248.15, found 248.4; Elemental analysis: calc. for C11H21NO5, C, 53.43; H, 8.56; N, 5.66; found C, 53.65; H, 8.61; N, 5.61.
:1 over 3:2 to 1:1). The product-containing fractions were combined and evaporated to afford 5 (1.22 g, 65%) as a yellow oil. Rf 0.16 (petroleum ether–ethyl acetate 3:2); 1H-NMR (CDCl3) δ: 5.97–5.84 (m, 1H, CHCH2), 5.33 (d, 3J = 17.2 Hz, 1H, CHCHH), 5.25 (d, 3J = 10.4 Hz, 1H, CHCHH), 5.08–5.00 (m, 1H, NH), 4.68–4.57 (m, 2H, CH2O of allyl), 4.33–4.28 (m, 1H, CαH), 3.64 (t, 3J = 6.3 Hz, 2H, CεH2), 1.90–1.39 (m, 15H, 3 × CH3 of Boc, 3 × CH2); 13C-NMR (CDCl3) δ: 172.67 (CO), 155.59 (CONH), 131.78 (CH of allyl), 118.94 (CH2 of allyl), 80.08 (quart. C of Boc), 65.98, 62.63, 53.48 (Cα), 32.76, 32.24, 28.47 (3 CH3 of Boc), 21.70; ESI-MS (ESI+) m/z: calc. for C14H26NO5 [M + H]+, 288.18, found 288.3; Elemental analysis: calc. for C14H25NO5, C, 58.52; H, 8.77; N, 4.87; found C, 58.01; H, 8.64; N, 4.78.
:1 v/v, 50 mL) and the resulting solution was stirred for 2 h at room temperature. The volatile components were removed in the N2 stream followed by repeated evaporations from diethyl ether (3 × 20 mL). The obtained residue was exposed to oil pump vacuum to remove remaining TFA. 1.84 g (quantitative yield containing 12% of residual TFA) of a brown oil were yielded. 1H-NMR (DMSO-d6) δ: 8.42 (s, 3H, NH3+), 5.99–5.87 (m, 1H, CHCH2), 5.38 (dd, 3J = 17.3 Hz, 2J = 1.5 Hz, 1H, CHCHH), 5.28 (dd, 3J = 10.5 Hz, 2J = 1.3 Hz, 1H, CHCHH), 4.70 (d, 3J = 5.0 Hz, 2H, CH2O of allyl), 4.38 (t, 3J = 6.3 Hz, 2H, CεH2), 4.16–4.07 (m, 1H, CαH), 1.91–1.64 (m, 4H, 3 × CH2), 1.56–1.31 (m, 2H, CH2); 19F-NMR (DMSO-d6) δ: −74.75 (s, CF3 of TFA), −74.79 (s, CF3 of Tfa); ESI-MS (ESI+) m/z: calc. for C11H17F3NO4 [M + H]+, 284.11, found 284.3.
:1 to 1:1). The product-containing fractions were combined to yield 7 (0.97 g, 58%) as colourless oil that solidified in the refrigerator. Rf 0.22 (petroleum ether–ethyl acetate 1:1); m.p. 100–103 °C; 1H-NMR (CDCl3) δ: 7.77 (d, 3J = 7.6 Hz, 2H, H-4,5 of Fmoc), 7.60 (d, 3J = 7.0 Hz, 2H, 2 × CH of Fmoc), 7.40 (t, 3J = 7.4 Hz, 2H, 2 × CH of Fmoc), 7.35–7.29 (m, 2H, 2 × CH of Fmoc), 5.98–5.84 (m, 1H, CHCH2), 5.40–5.23 (m, 2H, CHCHH, NH), 5.27 (dd, 3J = 10.4 Hz, 2J = 1.0 Hz, 1H, CHCHH), 4.65 (d, 3J = 5.5 Hz, 2H, CH2O of allyl) 4.45–4.35 (m, 3H, CH2O of Fmoc and CαH), 4.23 (t, 3J = 7.0 Hz, 1H, H-9 of Fmoc), 3.65 (t, 3J = 6.3 Hz, 2H, CεH2), 1.95–1.40 (m, 6H, 3 × CH2); 13C-NMR (CDCl3) δ: 172.34, 156.10, 143.96, 141.46, 131.64, 127.85, 127.20, 125.22, 120.13, 119.14, 67.16 (CH2O of Fmoc), 66.16, 62.58, 53.95 (Cα), 47.32 (C-9 of Fmoc), 32.63, 32.19, 21.64; ESI-MS (ESI+) m/z: calc. for C24H28NO5 [M + H]+, 410.20, found 410.2; Elemental analysis: calc. for C24H27NO5, C, 70.49; H, 6.65; N, 3.42; found C, 69.89; H, 6.67; N, 3.43.
Procedures for loading of allyl esters 1 and 7 onto 2-ClTrtCl resin
High loading. The synthesis was accomplished according to ref. 36. A solution of 7 (634.7 mg, 1.55 mmol, 1 equiv.) in CH2Cl2–DMF (1
:1, 6 mL) was added to the preswollen (CH2Cl2, 6 mL, 30 min) 2-ClTrtCl resin (1.00 g, 1.55 mmol, 1 equiv.) in a PP filter vessel. Pyridine (0.5 mL, 6.2 mmol, 4 equiv.) was added subsequently and the mixture was sealed and agitated for 65 h at room temperature. The resin was successively washed with DMF (4 mL, 4 × 1 min), CH2Cl2 (4 mL, 4 × 1 min), CH2Cl2–CH3OH–DIPEA (17:1:2, 5 mL, 3 × 2 min) and finally with CH2Cl2 (4 mL, 4 × 1 min). The resin was dried in vacuo over night.
Low loading. A solution of 7 (270.3 mg, 0.66 mmol, 0.6 equiv.) in CH2Cl2–DMF (1
:1, 5 mL) was added to the preswollen (CH2Cl2, 6 mL, 30 min) 2-ClTrtCl resin (712 mg, 1.10 mmol, 1 equiv.) in a PP filter vessel. Pyridine (213 μL, 2.64 mmol, 2.4 equiv.) was added subsequently and the mixture was sealed and agitated for 24 h at room temperature. The resin was successively washed with DMF (4 mL, 4 × 1 min), CH2Cl2 (4 mL, 4 × 1 min), CH2Cl2–CH3OH–DIPEA (17:1:2, 5 mL, 3 × 2 min) and finally with CH2Cl2 (4 mL, 4 × 1 min). The resin was dried in vacuo over night.
Determination of the loading yield
The determination was accomplished according to ref. 75 with two aliquots of the resin. About 5 mg of the resin (exact value has to be noted) containing the Fmoc-protected amino acid allyl esters were placed into a PP reaction vessel and swollen in DMF (2 mL) for 30 min. The suspension was filtered and the resin was treated with 2% DBU in DMF (2 mL) and stirred for another 30 min. The suspension was filtered into a graduated 10 mL flask and the resin was washed with CH3CN (3 × 1 mL). Each filtrate was added to the flask and the solution was diluted to 10 mL with CH3CN. 2 mL of this solution were transferred to a graduated 25 ml flask and diluted with CH3CN to 25 mL. A reference solution was prepared the same way but without addition of the resin. The sample solutions were measured against the reference at 294 nm in the UV/Vis spectrometer. The loading yield was calculated by using the following equation.| Loading (mmol g−1)294 nm = (E×14214 μmol)/mresin |
Finally the average value was calculated.
Procedures for the syntheses of the peptides 2, 3, 3a, 8–10 on the 2-ClTrtCl resin
:1, 10 mL) for another 15 min. Removal of the Fmoc groups (except for N-terminal serine) was performed by using a solution of 20% piperidine in DMF with 0.1 M HOBt (1 × 7 mL at 35 W for 30 s and 1 × 7 mL at 44 W for 180 s, both at 75 °C). After each treatment, the resin was washed with DMF (5 + 28 mL). Coupling of each amino acid was performed with two solutions: 0.45 M HBTU in DMF (1 mL, 4.5 equiv.) and 2 M DIPEA in NMP (0.5 mL). The amino acids were applied as 0.2 M solutions in DMF. 2.5 mL (5 equiv.) of these solutions were taken for the coupling steps (300 s at 21 W and 75 °C). The peptidyl resin containing the protected linear precursor peptide was then washed with DMF (21 mL) and outside the peptide synthesizer with ethanol (4 × 5 mL) and CH2Cl2 (4 × 5 mL).
:2:1, 6 mL). After 2 min of degassing with Ar, Pd(PPh3)4 (57.8 mg, 0.05 mmol, kept under Ar atmosphere) was added to the mixture and the newly formed yellow suspension was degassed for another 2 min with Ar. The PP filter vessel was sealed and agitated for 4 h. The suspension was filtered and the peptidyl resin was successively washed with CH2Cl2 (5 mL, 4 × 1 min), DMF (5 mL, 4 × 1 min), 0.5% w/v sodium diethyldithiocarbamate in DMF (5 mL, 6 × 1 min) and finally with DMF (5 mL, 5 × 1 min) again. The removal of the Fmoc group was performed by treatment of the peptidyl resin with 20% piperidine in DMF (6 mL, 2 × 8 min). Then the peptidyl resin was washed with DMF (6 mL, 3 × 1 min), 5% DIPEA in DMF (6 mL, 3 × 1 min), DMF (6 mL, 1 × 1 min) and CH2Cl2 (6 mL, 5 × 1 min). The peptidyl resin was dried in vacuo over night.
Procedures for cleavage of the peptides from the 2-ClTrtCl resin and/or the protecting groups
The dry peptidyl resin was suspended in 1 or 3 mL of a solution of TFA–TES–H2O (95:2.5:2.5) for 1 or 3 h for small-scale analytical or complete cleavage, respectively. After filtering, the resin was washed with TFA (2 × 1 or 5 mL for analytical and complete cleavage, respectively) and the combined filtrates were evaporated in a N2 stream. Ice-cold diethyl ether (10 mL) was added to the oily residue and the mixture was cooled on ice for 30 min. After this, the newly formed white, voluminous precipitate was filtered off and dried in vacuo.
:5:94) for 30 min. After filtering and washing of the resin with CH2Cl2 (1 × 4 mL), NaHCO3 (52 mg) was added to the combined filtrates and the suspension was stirred for 10 min. After renewed filtration, the filtrate was evaporated in a N2 stream. The oily residue was dissolved in CH3CN–H2O (1 mL, 1:1) and stirred over night. Finally, the solution was evaporated in vacuo and lyophilised.
:1) and stirred for 1 h. The solution was evaporated in a N2 stream. Ice-cold diethyl ether (10 mL) was added to the oily residue and the mixture was kept on ice for 30 min. After this, the newly formed white, voluminous precipitate was filtered off and dried in vacuo.
Analytical data for the peptides
1H-NMR (water) δ: Ser: 7.46 (d, 3J = 6.5 Hz, NH), 3.87–3.76 (m, CβH2), CαH lies under the water signal D-Pro: 4.37–4.32 (m, CαH), 3.76–3.65 (m, CδH2), 2.21–1.55 (m, CβH2, CγH2) Lys (FBz): 8.72 (d, 3J = 7.4 Hz, CαNH), 8.46 (t, 3J = 5.8 Hz, CεNH), 7.79 (dd, 3JH,H = 8.7 Hz, 4JH,F = 5.4 Hz, H-2,6 FBz), 7.24 (t, 3JH,H = 3JH,F = 8.8 Hz, H-3,5 FBz), 4.32–4.21 (m, CαH), 3.45–3.34 (m, CεH2), 2.21–1.55 (m, CβH2, CδH2), 1.54–1.31 (m, CγH2) Asp: 8.24 (d, 3J = 9.1 Hz, NH), 3.07–2.89 (m, CβH2), CαH lies under the water signal Glu: 8.41 (d, 3J = 4.0 Hz, NH), 4.04–3.97 (m, CαH), 2.58–2.37 (m, CγH2), 2.21–1.55 (m, CβH2) Lys: 8.31 (d, 3J = 7.5 Hz, NH), 7.53 (broad s, NH3+), 4.32–4.21 (m, CαH), 3.07–2.89 (m, 2H, CεH2), 2.21–1.55 (m, CβH2 and CδH2), 1.54–1.31 (m, CγH2).
:3), NMR signals are only listed for the major isomer: 1H-NMR (DMSO-d6) δ: Lys (FBz): 8.46 (t, 3J = 5.5 Hz, CεNH), 8.09 (d, 3J = 6.9 Hz, CαNH), 7.90 (dd, 3JH,H = 8.8 Hz, 4JH,F = 5.6 Hz, H-2,6 FBz), 7.28 (t, 3JH,H = 3JH,F = 8.8 Hz, H-3,5 FBz), 4.16–4.07 (m, CαH), 3.27–3.18 (m, CεH2), 1.94–1.80 (m, CH3), 1.66–1.56 (m, CβHH), 1.56–1.42 (m, CβHH, CδH2), 1.41–1.20 (m, CγH2) Asp: 8.29 (d, 3J = 7.7 Hz, NH), 4.56–4.46 (m, CαH), 2.80–2.69 (m, CβHH), 2.59–2.49 (m, CβHH) Glu: 7.65 (d, 3J = 8.1 Hz, NH), 4.34–4.16 (m, CαH), 2.34–2.12 (m, CγH2), 1.94–1.80 (m, CβHH), 1.80–1.67 (m, CβHH) Lys: 7.85 (d, 3J = 7.8 Hz, NH), 7.72–7.57 (broad s, NH3+), 4.34–4.16 (m, CαH), 2.80–2.69 (m, CεH2), 1.66–1.56 (m, CβHH), 1.56–1.42 (m, CβHH, CδH2), 1.41–1.20 (m, CγH2) Ser: 8.20 (d, 3J = 6.8 Hz, NH), 4.56–4.46 (m, CαH), 3.67–3.55 (m, CβHH), 3.53–3.43 (m, CβHH) D-Pro: 7.05 (s, NHH), 6.91 (s, NHH), 4.34–4.16 (m, CαH), 3.75–3.67 (m, CδHH), 3.67–3.55 (m, CδHH), 2.08–1.95 (m, CβHH), 1.94–1.80 (m, CβHH, CγH2); 13C-NMR (DMSO-d6) δ: 174.06, 173.59, 172.16, 171.97, 171.55, 170.64, 170.41, 170.02, 169.11, 165.06, 163.74 (d, 1JC,F = 248.1 Hz, C-4 FBz), 158.08 (q, 2JC,F = 34.7 Hz, CO TFA), 131.10 (d, 4JC,F = 2.9 Hz, C-1 FBz), 129.73 (d, 3JC,F = 9.0 Hz, C-2,6 FBz), 115.11 (d, 2JC,F = 21.7 Hz, C-3,5 FBz), 61.06, 59.63, 53.15, 53.03, 51.85, 51.78, 49.55, 46.79, 38.75, 35.58, 31.31, 29.99, 29.28, 28.84, 27.22, 26.56, 23.99, 22.86, 22.40, 21.84, signal for one methylene group is missing; 19F-NMR (DMSO-d6) δ: −74.93 (s, CF3 TFA-Anion), −110.28 (tt, 3JFH = 8.8 Hz, 4JFH = 5.6 Hz, FBz); ESI-MS (ESI+) m/z: calc. for C38H57FN9O13, [M + H]+, 866.41, found 866.1.
NMR spectroscopy
The compounds 3, 8, 9 and 10 were dissolved in 600 μL of DMSO-d6. Additionally, compounds 3 and 3a were dissolved in 600 μL of H2O. In this case, an capillary insert containing D2O was placed into the sample tube to allow shiming. The H2O peak was suppressed by water presaturation. The 1D and 2D 1H NMR experiments (COSY, TOCSY (mixing time/spin-lock time 120 ms), and ROESY (mixing time 300 ms)) were acquired on a Varian 400 MHz or an Agilent Technologies 400 MR spectrometer at 298 K. VT-NMR experiments were performed to monitor amide exchange rates at varying temperatures up to 318 K in 5 K intervals. 3JNHCHα coupling constants were measured from 1H NMR spectra and ROE intensities were collected manually.ECD spectroscopy
Stock solutions of the peptides 3, 8, 9, and 10 were prepared in a concentration 0.5 mg mL−1 in 10 mM sodium phosphate pH 7.4. The ECD spectra were measured in a quartz cuvette with an optical path length of 1 mm (Starna, USA) using a J-810 spectropolarimeter (Jasco, Japan). The conditions of the measurements were as follows: a spectral region of 200 (180)–400 nm, a scanning speed of 20 nm min−1, a response time of 8 s, a resolution of 1 nm, a bandwidth of 1 nm and a sensitivity of 100 mdeg. The final spectrum was obtained as an average of 5 accumulations. The spectra were corrected for a baseline by subtracting the spectra of the corresponding polypeptide-free solution. The ECD measurements were conducted at room temperature.IR and VCD spectroscopy
Solutions of peptides 3, 8, 9, and 10 were prepared in DMSO to a concentration of 20 mg mL−1 in a total volume of 100 μL each. The VCD and infrared absorption spectra in the 1800–1400 cm−1 region were measured on a FTIR IFS 66/S spectrometer equipped with a PMA 37 VCD/IRRAS module (Bruker, Germany). The samples were placed in a demountable cell (A145, Bruker, Germany) composed of CaF2 windows separated by a 23 μm Teflon spacer. All of the VCD spectra were recorded at a spectral resolution of 8 cm−1 and are the average of the 15 blocks of 3686 scans (20 minutes). The OPUS 6.5 software (Bruker, Germany) was used for the VCD spectra calculations. The VCD spectra were corrected for a baseline, which was obtained as the VCD spectrum of the polypeptide-free solution measured under the same experimental conditions.Stability studies for trypsin-mediated degradation
A solution of trypsin from bovine pancreas (Serva 37290, lyophilised powder, 4 U mg−1) was freshly prepared in 0.3 M Tris-HCl pH 7.5 at a concentration of 1 mM and kept on ice. Stock solutions of 3 and 10 (2 mM each) were prepared in 0.3 M Tris-HCl pH 7.5. The peptide stock solutions were diluted with the trypsin solution and 0.3 M Tris-HCl to reach a final peptide concentration of 500 μM and enzyme concentrations of 0.1, 1.0, and 500 μM in a total volume of 100 μL. If necessary, the enzyme stock solution was prediluted with 0.3 M Tris-HCl pH 7.5. The reaction mixtures were incubated at 37 °C for 30 min. After this, an aliquot of 50 μL was withdrawn and added to 5 μL of 6 M HCl, vortexed and centrifuged at 1000g for 30 s. 40 μL of the supernatant was analysed by RP-HPLC as specified above.Structure calculations
Distance restraints used in calculating a solution structure for 3, 8, and 9 in DMSO-d6 were derived from ROESY spectra. ROE cross-peak volumes were classified manually as strong (upper distance constraint ≤2.7 Å), medium (≤3.5 Å), weak (≤5.0 Å) and very weak (≤6.0 Å). Standard pseudoatom distance corrections were applied for non-stereospecifically assigned protons.77 Conformational averaging was tackled by classifying the intensities conservatively. Only upper distance limits were included in the calculations to allow the largest possible number of conformers to fit the experimental data. 3JNHCHαcoupling constants measured from 1H NMR were used to apply backbone dihedral angle restraints as follows: ϕ was restrained to −120 ± 30° for 3JNHCHα ≥ 8 Hz and to −65 ± 30° for 3JNHCHα ≤ 6 Hz. Starting structures with randomised ϕ and ψ angles and extended side chains were generated using an ab initio simulated annealing protocol.48 The calculations were performed using the standard force field parameter set and topology file included in Xplor-NIH 2.34 package47,78 within house modifications to generate the backbone connection between the N-terminus of residue 1 and the C-terminus of residue 6 as well as the unnatural amino acids Lys(FBz), Hnl, and Aly. The final energy minimisation included 2000 steps of the conjugate gradient Powell algorithm and energy minimised using CHARMm force field.49 Final structures were visualised with PyMOL4 and had no distance (>0.2 Å) or angle (>2.4°) violations.Molecular dynamics (MD) simulations
MD simulations of cyclohexapeptides 3 and 9 were carried out using the ff99SB force field59 and standard protocols as implemented in the AMBER12 package (see below).60 Each system was solvated in a truncated octahedral box of DMSO or TIP3P water molecules and neutralised with Na+ counterions. In the case of DMSO, the solvent dielectric constant was set to 47. Molecular dynamic simulations were preceded by two energy-minimisation steps: (i) only the solvent and ions were relaxed and position restraints were applied to the cyclohexapeptide (500 kcal mol−1 Å−2) using 1000 steps of steepest descendent minimisation followed by 500 of conjugate gradient minimisation; (ii) the entire system was minimised without restraints by applying 3000 steps of steepest descendent and 3000 of conjugate gradient equilibration. Then the system was heated up from 200 K to 300 K in 20 ps with weak position restraints (10 kcal mol−1 Å−2) in the canonical ensemble (NVT). Langevin temperature coupling with a collision frequency γ = 1 ps−1 was used at this step. The system was equilibrated without restraints for 15 ns in the isothermal-isobaric ensemble (NPT) using the Langevin thermostat and Berendsen barostat under periodic boundary conditions. For simulations performed in DMSO, compressibility was set to 52.5 × 10−6 bar−1. A total of 100 ns unrestrained MD productions were carried out at 300 K in the canonical (NVT) or in the isothermal-isobaric (NPT) ensembles. Berendsen thermostat was used during NVT MD production. The SHAKE algorithm was used to constrain all bonds involving hydrogen atoms. A time step of 2 fs per step was used during the SHAKE algorithm. A cutoff of 8 Å was applied to treat the nonbonded interactions, and the Particle Mesh Ewald (PME) method was used to treat long-range electrostatic interactions. MD trajectories were recorded every 2 ps. Lys(FBz) and Aly were parametrised according to the ff99SB force field, and missed parameters were taken from the General Amber Force Field (GAFF).79 RESP charges were derived at the HF/6-31G* calculation level using Gaussian 09 version C.01.80–82 Trajectories were visualised with VMD.83 They were evaluated in terms of RMSD, distances, dihedral angles, and intramolecular H-bonds using the PTRAJ and CPPTRAJ modules implemented in AMBER. Data analysis and graphical representation were carried out with the R-package.84Acknowledgements
The authors wish to thank Mr Jerome Kretzschmar for advice in solvent suppression NMR experiments. This work was enabled by partial funding within the Helmholtz-Portfolio Topic “Technologie und Medizin – Multimodale Bildgebung zur Aufklärung des In vivo-Verhaltens von polymeren Biomaterialien” by the Helmholtz Association. Partial support by the Fonds der Chemischen Industrie to RL is cordially acknowledged. GRG is grateful for the award of a postdoctoral fellowship by the Alexander von Humboldt foundation. MTP group is funded by the German Research Council (DFG, SFB-TRR67; project A7).Notes and references
- P. K. Madala, J. D. A. Tyndall, T. Nall and D. P. Fairlie, Chem. Rev., 2010, 110, PR1–PR31 CrossRef CAS PubMed.
- H. A. Lucero and H. M. Kagan, Cell. Mol. Life Sci., 2006, 63, 2304–2316 CrossRef CAS PubMed.
- L. I. Smith-Mungo and H. M. Kagan, Matrix Biol., 1998, 16, 387–398 CrossRef CAS PubMed.
- W. DeLano, The PyMOL Molecular Graphics System, Version 1.5.0.3, Schrödinger, LLC, New York, 2009–2012 Search PubMed.
- J. P. Malone, A. George and A. Veis, Proteins, 2004, 54, 206–215 CrossRef CAS PubMed.
- J. T. Erler, K. L. Bennewith, M. Nicolau, N. Dornhöfer, C. Kong, Q. T. Le, J. T. Chi, S. S. Jeffrey and A. J. Giaccia, Nature, 2006, 440, 1222–1226 CrossRef CAS PubMed.
- K. R. Levental, H. Yu, L. Kass, J. N. Lakins, M. Egeblad, J. T. Erler, S. F. Fong, K. Csiszar, A. Giaccia, W. Weninger, M. Yamauchi, D. L. Gasser and V. M. Weaver, Cell, 2009, 139, 891–906 CrossRef CAS PubMed.
- J. Finney, H. J. Moon, T. Ronnebaum, M. Lantz and M. Mure, Arch. Biochem. Biophys., 2014, 546, 19–32 CrossRef CAS PubMed.
- L. Cunha, I. Horvath, S. Ferreira, J. Lemos, P. Costa, D. Vieira, D. S. Veres, K. Szigeti, T. Summavielle, D. Mathe and L. F. Metello, Mol. Diagn. Ther., 2014, 18, 153–173 CrossRef PubMed.
- D. R. Eyre and J.-J. Wu, Top. Curr. Chem., 2005, 247, 207–229 CAS.
- D. A. Helseth, J. H. Lechner and A. Veis, Biopolymers, 1979, 18, 3005–3014 CrossRef CAS.
- A. Otter, G. Kotovych and P. G. Scott, Biochemistry, 1989, 28, 8003–8010 CrossRef CAS PubMed.
- A. Otter, P. G. Scott, X. Liu and G. Kotovych, J. Biomol. Struct. Dyn., 1989, 7, 455–476 CAS.
- A. George, J. P. Malone and A. Veis, Proc. – Indian Acad. Sci., Chem. Sci., 1999, 111, 121–131 CAS.
- N. Nagan and H. M. Kagan, J. Biol. Chem., 1994, 269, 22366–22371 CAS.
- Y. A. Ovchinnikov and V. T. Ivanov, Tetrahedron, 1975, 31, 2177–2209 CrossRef CAS.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1982, 21, 512–523 CrossRef.
- D. P. Fairlie, G. Abbenante and D. R. March, Curr. Med. Chem., 1995, 2, 654–686 CAS.
- O. Ovadia, S. Greenberg, B. Laufer, C. Gilon, A. Hoffman and H. Kessler, Expert Opin. Drug Discovery, 2010, 5, 655–671 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- J. N. Lambert, J. P. Mitchell and K. D. Roberts, J. Chem. Soc., Perkin Trans. 1, 2001, 471–484 RSC.
- T. Jeremic, A. Linden and H. Heimgartner, Chem. Biodiversity, 2004, 1, 1730–1761 CAS.
- S. Royo Gracia, K. Gaus and N. Sewald, Future Med. Chem., 2009, 1, 1289–1310 CrossRef PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- A. Roxin and G. Zheng, Future Med. Chem., 2012, 4, 1601–1618 CrossRef CAS PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- P. Brust, J. van den Hoff and J. Steinbach, Neurosci. Bull., 2014, 30, 777–811 CrossRef CAS PubMed.
- M. Kuchar, M. Pretze, T. Kniess, J. Steinbach, J. Pietzsch and R. Löser, Amino Acids, 2012, 43, 1431–1443 CrossRef CAS PubMed.
- D. Seebach, E. Dubost, M. Löweneck, O. Flögel, J. Gardiner, S. Capone, R. I. Mathad, B. Jaun, M. Limbach, A. K. Beck, H. Widmer, D. Langenegger, D. Monna and D. Hoyer, Helv. Chim. Acta, 2008, 91, 1736–1786 CrossRef CAS.
- C. Gilon, C. Mang, E. Lohof, A. Friedler and H. Kessler, in Houben-Weyl Methods of Organic Chemistry, ed. M. Goodman, A. Felix and L. Moroder, G. Thieme, Stuttgart, 2003, vol. E22b, pp. 461–542 Search PubMed.
- W. S. Horne, C. M. Wiethoff, C. Cui, K. M. Wilcoxen, M. Amorin, M. R. Ghadiri and G. R. Nemerow, Bioorg. Med. Chem., 2005, 13, 5145–5153 CrossRef CAS PubMed.
- H. Kessler, R. Gratias, G. Hessler, M. Gurrath and G. Müller, Pure Appl. Chem., 1996, 68, 1201–1205 CrossRef CAS.
- M. G. Bomar, B. Song, P. Kibler, K. Kodukula and A. K. Galande, Org. Lett., 2011, 13, 5878–5881 CrossRef CAS PubMed.
- H. Matter and H. Kessler, J. Am. Chem. Soc., 1995, 117, 3347–3359 CrossRef CAS.
- M. P. Glenn, L. K. Pattenden, R. C. Reid, D. P. Tyssen, J. D. A. Tyndall, C. J. Birch and D. P. Fairlie, J. Med. Chem., 2002, 45, 371–381 CrossRef CAS PubMed.
- H. Wenschuh, M. Beyermann, H. Haber, J. K. Seydel, E. Krause, M. Bienert, L. A. Carpino, A. El-Faham and F. Albericio, J. Org. Chem., 1995, 60, 405–410 CrossRef CAS.
- G. Hübener, W. Göhring, H.-J. Musiol and L. Moroder, Pept. Res., 1992, 5, 287–292 Search PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, J. Chem. Soc., Chem. Commun., 1994, 2559 RSC.
- R. Dölz and E. Heidemann, Int. J. Pept. Protein Res., 1988, 32, 307–320 CrossRef.
- R. Dölz and E. Heidemann, Connect. Tissue Res., 1989, 18, 255–268 CrossRef.
- A. Heinz, C. K. Ruttkies, G. Jahreis, C. U. Schräder, K. Wichapong, W. Sippl, F. W. Keeley, R. H. H. Neubert and C. E. Schmelzer, Biochim. Biophys. Acta, 2013, 1830, 2994–3004 CrossRef CAS PubMed.
- D. F. Mierke, T. Huber and H. Kessler, J. Comput.-Aided Mol. Des., 1994, 8, 29–40 CrossRef CAS PubMed.
- Y. A. Bara, A. Friedrich, H. Kessler and M. Molter, Chem. Ber., 1978, 111, 1045–1057 CrossRef CAS.
- P. N. Lewis, F. A. Momany and H. A. Scheraga, Biochim. Biophys. Acta, 1973, 303, 211–229 CrossRef CAS.
- C. M. Wilmot and J. M. Thornton, Protein Eng., 1990, 3, 479–493 CrossRef CAS PubMed.
- C. D. Schwieters, J. J. Kuszewski and G. M. Clore, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62 CrossRef CAS.
- M. Nilges, A. M. Gronenborn, A. T. Brunger and G. M. Clore, Protein Eng., 1988, 2, 27–38 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- R. Haubner, D. Finsinger and H. Kessler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1375–1389 CrossRef.
- S. Ma, M. J. McGregor, F. E. Cohen and P. V. Pallai, Biopolymers, 1994, 34, 987–1000 CrossRef CAS PubMed.
- K. Tamiola, B. Acar and F. A. A. Mulder, J. Am. Chem. Soc., 2010, 132, 18000–18003 CrossRef CAS PubMed.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2000, 21, 249–261 CrossRef.
- C. Toniolo, F. Formaggio and R. W. Woody, in Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, John Wiley & Sons, Hoboken, 2012, vol. 2, pp. 499–544 Search PubMed.
- Z. Shi, R. W. Woody and N. R. Kallenbach, Adv. Protein Chem., 2002, 62, 163–240 CrossRef CAS PubMed.
- M. Urbanova and P. Malon, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2nd edn, 2012, pp. 337–369 Search PubMed.
- C. Zhao, P. L. Polavarapu, C. Das and P. Balaram, J. Am. Chem. Soc., 2000, 122, 8228–8231 CrossRef CAS.
- T. A. Keiderling and A. Lakhani, in Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, John Wiley & Sons, Hoboken, 2012, vol. 2, pp. 707–758 Search PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, I. T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- H. Matter, G. Gemmecker and H. Kessler, Int. J. Pept. Protein Res., 1995, 45, 430–440 CrossRef CAS PubMed.
- J. S. Richardson, Adv. Protein Chem., 1981, 34, 167–339 CrossRef CAS PubMed.
- B. Madan, S. Y. Seo and S. G. Lee, Proteins, 2014, 82, 1721–1733 CrossRef CAS PubMed.
- D. Pantoja-Uceda, C. M. Santiveri and M. A. Jiménez, Protein Design - Methods and Applications, in Methods in Molecular Biology, ed. R. Guerois and M. López de la Paz, Humana Press, Totowa, 2006, vol. 340, pp. 27–51 Search PubMed.
- M. P. Williamson and J. P. Waltho, Chem. Soc. Rev., 1992, 21, 227–236 RSC.
- G. Ruiz-Gómez, J. D. A. Tyndall, B. Pfeiffer, G. Abbenante and D. P. Fairlie, Chem. Rev., 2010, 110, PR1–PR41 CrossRef PubMed.
- M. Fani and H. R. Maecke, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39(Suppl 1), S11–S30 CrossRef PubMed.
- C. Cobas, S. Domínguez, N. Larin, I. Iglesias, C. Geada, F. Seoane, M. Sordo, P. Monje, S. Fraga, R. Cobas, C. Peng, J. A. García, M. Goebel and E. Vaz, MestReNova 6.1.1-6384, Mestrelab Research S.L., 2010 Search PubMed.
- D. Andreu, S. Ruiz, C. Carreno, J. Alsina, F. Albericio, M. A. Jimenez, N. de la Figuera, R. Herranz, M. T. Garcia-Lopez and R. Gonzalez-Muniz, J. Am. Chem. Soc., 1997, 119, 10579–10586 CrossRef CAS.
- J. Alsina, F. Rabanal, C. Chiva, E. Giralt and F. Albericio, Tetrahedron, 1998, 54, 10125–10152 CrossRef CAS.
- M. Adamczyk, D. D. Johnson and R. E. Reddy, Tetrahedron, 1999, 55, 63–88 CrossRef CAS.
- P. J. Maurer and M. J. Miller, J. Am. Chem. Soc., 1982, 104, 3096–3101 CrossRef CAS.
- S. Ficht, R. J. Payne, R. T. Guy and C. H. Wong, Chem. – Eur. J., 2008, 14, 3620–3629 CrossRef CAS PubMed.
- H. Nagase, C. G. Fields and G. B. Fields, J. Biol. Chem., 1994, 269, 20952–20957 CAS.
- M. Gude, J. Ryf and P. D. White, Lett. Pept. Sci., 2002, 9, 203–206 CAS.
- B. W. Bycroft, W. C. Chan, S. R. Chhabra and N. D. Hone, J. Chem. Soc., Chem. Commun., 1993, 778–779 RSC.
- K. Wüthrich, M. Billeter and W. Braun, J. Mol. Biol., 1983, 169, 949–961 CrossRef.
- C. D. Schwieters, J. J. Kuszewski, N. Tjandra and G. M. Clore, J. Magn. Reson., 2003, 160, 65–73 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821–7839 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- R Development Core Team, R. A language and environment for statistical computing, http://www.r-project.org., 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and additional Figures. See DOI: 10.1039/c4ob02348j |
| This journal is © The Royal Society of Chemistry 2015 |