
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric syntheses of three-membered heterocycles using chiral amide-based ammonium ylides†
Mathias
Pichler
a,
Johanna
Novacek
a,
Raphaël
Robiette
b,
Vanessa
Poscher
a,
Markus
Himmelsbach
c,
Uwe
Monkowius
d,
Norbert
Müller
a and
Mario
Waser
*a
aInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: Mario.waser@jku.at; Fax: +43 732 2468 8747; Tel: +43 732 2468 8748
bInstitute of Condensed Matter and Nanosciences, Université catholique de Louvain, Place Louis Pasteur 1 box L4.01.02, 1348 Louvain-la-Neuve, Belgium
cInstitute of Analytical Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria
dInstitute of Inorganic Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria
First published on 1st December 2014
Abstract
The use of carbonyl-stabilised ammonium ylides to access chiral glycidic amides and the corresponding aziridines has so far been limited to racemic trans-selective protocols. We herein report the development of an asymmetric approach to access such compounds with high levels of stereoselectivity using easily accessible chiral auxiliary-based ammonium ylides. The use of phenylglycinol as the chiral auxiliary was found to be superior to Evans or pseudoephedrine-based auxiliaries resulting in good to excellent stereoselectivities in both, epoxidation and aziridination reactions.
Introduction
The use of onium ylides has emerged as a powerful strategy for (dia)-stereoselective epoxide, aziridine, and cyclopropane syntheses.1,2 Whereas sulfonium ylides have been very frequently used for a variety of often very selective three-ring forming reactions,3,4 easily available ammonium ylides have been less routinely employed in the past.5–9 Especially the use of chiral amines to render such reactions enantioselective has been mainly limited to cyclopropanations5 so far. Indeed, their use in asymmetric epoxidation and aziridination reactions was found to be rather difficult, mainly due to the weaker leaving group ability of the amine group compared to the use of sulfonium ylides.6,7,9We have recently introduced a highly trans-selective protocol for the synthesis of glycidic amides and the corresponding aziridines by reacting amide-stabilised achiral ammonium ylide precursors 1 with aldehydes 2 or imines 3 (Scheme 1).7,9b Key to high yields was the use of trimethylamine as the amine leaving group, whereas the use of sterically more demanding chiral amines like Cinchona alkaloids did not result in any product formation. Due to this limitation, alternative strategies to control the absolute stereochemistry in these epoxide and aziridine forming reactions are necessary. The use of chiral auxiliary containing amides to control the face selectivity in enolate reactions is a very commonly employed and powerful strategy for numerous applications.10 A few examples about their use in sulfonium ylide-mediated reactions have been reported in the past11 but the use of chiral amide-based ammonium ylides has, to the best of our knowledge, not been reported yet. We have therefore undertaken a systematic study about the use of different classes of chiral auxiliaries to access chiral ammonium ylide precursors 6 and explore their potential in asymmetric three-membered ring heterocycle-forming reactions (Scheme 1). The main focus was on the development of a protocol for epoxidation reactions first, followed by a proof of concept for asymmetric aziridinations.
 | ||
| Scheme 1 Recently developed racemic trans-epoxidation and aziridination protocol and the targeted auxiliary-based stereoselective approach. | ||
Results and discussion
Our main focus was on three different classes of auxiliaries: α-amino acid based oxazolidinones (Evans auxiliaries)12 to obtain ammonium salts 6A, pseudoephedrine13-derived ammonium salts 6B, and α-amino acid based 1,3-oxazolidine-containing salt 6C. Synthesis of all three classes could be straightforwardly achieved by reacting bromoacetyl bromide 7 with the auxiliaries first, followed by treatment of the α-bromo acetamides 8 with trimethylamine (Scheme 2). | ||
| Scheme 2 Tested chiral ammonium salts 6. | ||
We started testing the applicability of these ammonium salts for the asymmetric epoxidation with benzaldehyde (2a) first. Our recently developed protocol for the synthesis of epoxides 4 (Scheme 1) required use of a large excess of aqueous NaOH (>100 eq.).7 Initial experiments with ammonium salts 6A resulted in hydrolysis of the auxiliary under these strongly basic conditions. Surprisingly, under relatively mild and weaker basic conditions, using Cs2CO3 as a solid base, full hydrolysis of the auxiliary also occurred (Scheme 3).14 Unfortunately, we were never able to suppress this hydrolysis of the amide bond and in no case formation of epoxide product could be observed. One possible mechanistic explanation for this extraordinary base-sensitivity of these usually rather base-stable amide motives10,12 may be an in situ ketene formation under the basic conditions. We could however not obtain experimental evidence supporting this hypothesis.
 | ||
| Scheme 3 Attempted use of the Evans auxiliary containing ammonium salts 6A. | ||
Next, we put our focus on the use of the pseudoephedrine based ammonium salts 6B for the targeted epoxidation. Pseudophedrine has recently been reported as a useful auxiliary in sulfonium ylide-mediated epoxidation reactions.11d First experiments using the free-OH containing ammonium ylide precursor 6B′ indicated formation of the target epoxide 10 as a single trans-diastereomer (as far as it could be judged by 1H NMR analysis of the crude product where 10 is present as a mixture of rotamers) and the two cyclization products 11 and 12 (Scheme 4a). Formation of 11 was also observed during the preparation of the ammonium acetamide 6B′. Unfortunately 11 turned out to be the main product under a variety of different conditions (always at least 50%). Formation of 12 on the other hand occurs via a base-mediated epoxide opening of 10 as indicated by the increasing amount of 12 formed under prolonged reaction conditions. This is a known transformation which was already described by Teran et al. when using sulfonium ylides.11d Interestingly, in their case no formation of 11 was observed during the epoxidation and the transformation of 10 to 12 required the use of sodium as a base in a distinct step, whereas in our experiments significant amounts of 12 were detected under either conditions (>20%). Compound 12 was always formed as a single diastereomer which fully matched the analytical data reported before.11d This also suggests that the initial epoxide formation occurs with a high level of face differentiation. However, due to the rather fast cyclization of the starting material to give 11 and the base-mediated epoxide opening, we were not able to obtain reasonable amounts (>30%) of epoxide 10 in any case. To overcome this obstacle, we tested the O-protected ammonium salts 6B′′ (Scheme 4b). Unfortunately this turned out to be non-satisfactory as the face selectivity was found to be rather low giving a mixture of diastereomers beside significant amounts of various decomposition products (due to the initial lack of selectivity the reaction conditions were not further optimized).
 | ||
| Scheme 4 Use of the pseudoephedrine containing ammonium salts 6B. | ||
Because of the limited applicability of the Evans oxazolidinones and pseudoephedrine as chiral auxiliaries for our ammonium ylide-mediated epoxidation we next turned our attention on the use of 1,3-oxazolidine-containing auxiliaries. These are less commonly employed in asymmetric transformations as compared to oxazolidinones or pseudoephedrine. However, we reasoned that the corresponding ammonium salts 6C may work well for our target reaction as they should be stable under the basic reaction conditions and the addition step should hopefully proceed with satisfying face-selectivity too. In addition, during the initial phase of this project, Teran et al. reported the use of chiral 1,3-oxazolidines (derived from the reaction of phenylglycinol (14) with different aldehydes) in sulfur ylide-mediated epoxidation reactions.11e Interestingly, they found that the absolute configuration of the product 16 depends exclusively on the configuration of the phenylglycinol moiety (C4) – and not on the configuration of the newly installed stereogenic center (C2) of the auxiliary. We opted for a slightly different approach by carrying out the oxazolidine formation by reacting (R)-14 with acetone, which proceeds very quickly giving the auxiliary in high yield and purity. We anticipated that this ketone-based 1,3-oxazolidine should show a higher stability under basic conditions than aldehyde-based ones and should therefore be suitable for our approach. The ylide precursor 6C could then easily be obtained in two more steps (Scheme 5).
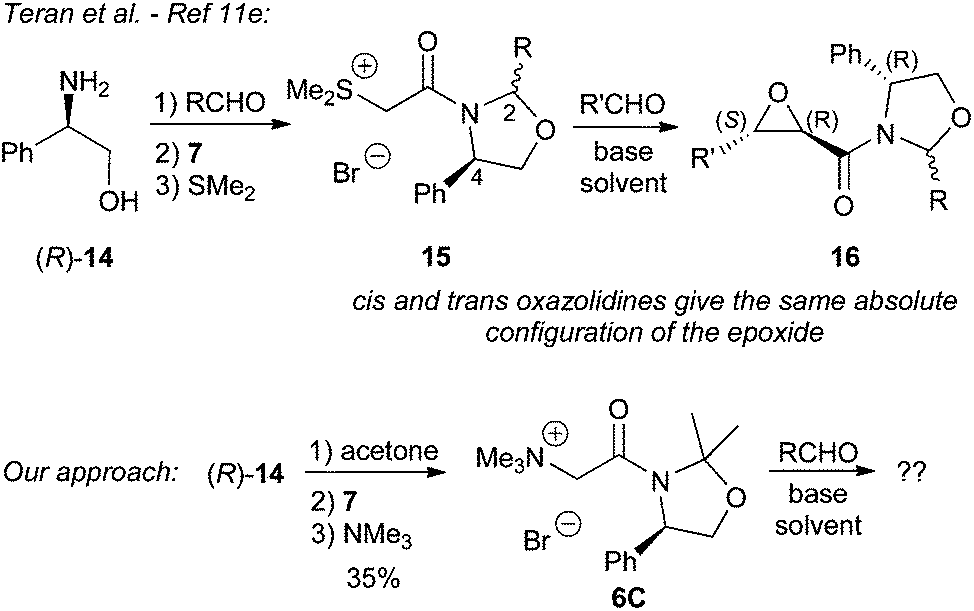 | ||
| Scheme 5 Use of phenylglycinol (14) to access chiral sulfonium11e and ammonium salts for asymmetric epoxidation reactions. | ||
In the initial epoxidation experiment of 6C with benzaldehyde (2a) in dichloromethane (using 6 eq. of Cs2CO3 as a solid base) epoxide 17a was formed as a single trans-diastereomer. Based on this promising result a systematic screening of different conditions was undertaken (Table 1 gives an overview about the most significant results obtained herein). In our attempts to determine the best-suited base and the optimum stoichiometric ratio we found that the strongly basic aqueous conditions that worked well in our racemic protocol7 did not give any product herein (entry 2). The use of K2CO3 (entry 3) or an equimolar amount of Cs2CO3 (entry 4) yielded also almost no product. However, an excess of Cs2CO3 and two equivalents of aldehyde allowed us to obtain trans-17a in 50% isolated yield after 24 h and in 74% after 72 h reaction time (entries 5 and 6). As expected from our previous experience using amide-stabilised ammonium ylides7 no cis-epoxide was formed under either conditions. In addition, also no second diastereoisomeric trans-epoxide could be observed in NMR spectra of the crude reaction mixture, illustrating that this chiral auxiliary leads to a high face-differentiation in the ylide addition step. Further screening of different solvents showed that toluene and i-PrOH can also be used as solvents for this reaction. Whereas the reaction was found to be slow in toluene, requiring either a prolonged reaction time (entry 9) or a higher reaction temperature (entry 10) to obtain 17a in good yield, reactions in i-PrOH were usually significantly faster (entry 12). However, hereby also, significant amounts of Cannizzaro disproportionation products of benzaldehyde could be detected whereas this side reaction is less pronounced in toluene. In addition, we also found that these reactions can be accelerated by ultrasonication, giving 17a in comparable quality and yield after 3 h (entries 11 and 13). Accordingly, this screening allowed us to identify different conditions which all gave the target epoxide 17a with very high diastereoselectivity and with isolated yields >70%.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2a (eq.) | Solv. | Base (eq.) | T [°C] | t [h] | Yielda [%] | drb [%] (trans)c |
| a Isolated yield. b Determined by 1H NMR of the crude reaction mixture. c In neither experiment any cis-diastereomer could be detected. d Incomplete conversion of 6C. e Carried out in an ultrasonic bath. | |||||||
| 1 | 4 | CH2Cl2 | Cs2CO3 (6×) | 25 | 24 | 35 | >98 |
| 2 | 2 | CH2Cl2 | NaOH (aq) (140×) | 25 | 24 | n.r. | n.d. |
| 3 | 2 | CH2Cl2 | K2CO3 (20×) | 25 | 24 | n.r. | n.d. |
| 4 | 1 | CH2Cl2 | Cs2CO3 (1×) | 25 | 24 | <10 | >98 |
| 5 | 2 | CH2Cl2 | Cs2CO3 (20×) | 25 | 24 | 50d | >98 |
| 6 | 2 | CH2Cl2 | Cs2CO3 (20×) | 25 | 72 | 74 | >98 |
| 7 | 2 | THF | Cs2CO3 (20×) | 25 | 24 | n.r. | n.d. |
| 8 | 2 | Toluene | Cs2CO3 (20×) | 25 | 24 | 50d | >98 |
| 9 | 2 | Toluene | Cs2CO3 (20×) | 25 | 72 | 72 | >98 |
| 10 | 2 | Toluene | Cs2CO3 (20×) | 60 | 24 | 73 | >98 |
| 11 | 2 | Toluene | Cs2CO3 (20×) | 60e | 3 | 75 | >98 |
| 12 | 2 | i-PrOH | Cs2CO3 (20×) | 25 | 24 | 78 | >98 |
| 13 | 2 | i-PrOH | Cs2CO3 (20×) | 60e | 3 | 84 | >98 |
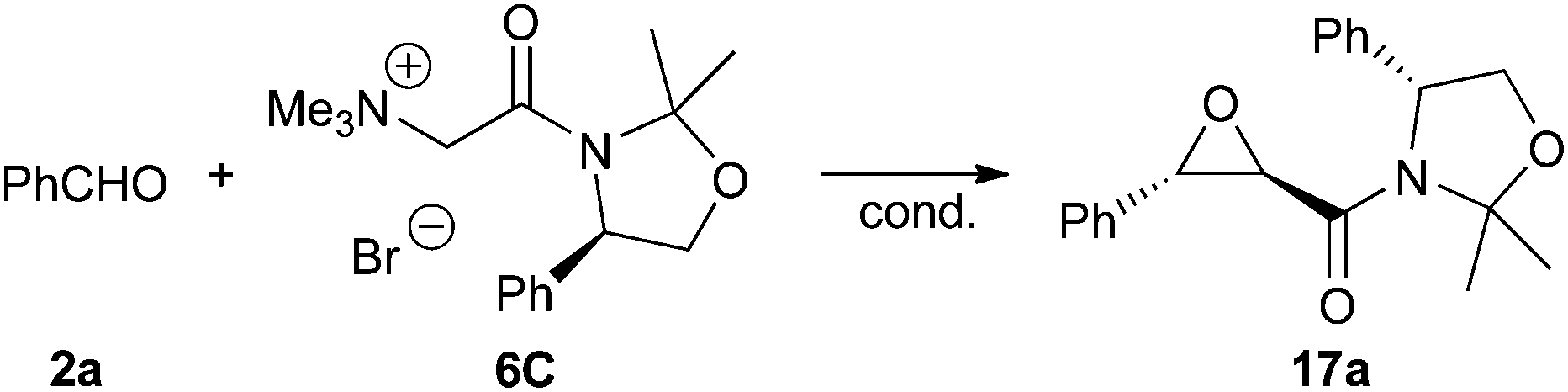
The absolute configuration of 17a was unambiguously proven by two different methods. First X-ray diffraction analysis of crystals of 17a proved the (2R,3S)-configuration of the trans-epoxyamide moiety.15 In addition 17a was also transferred into the known epoxyalcohol 18 upon treatment with LiBHEt3. Comparison of the specific optical rotation with literature values of 1811a,e also confirmed this absolute configuration of the epoxy moiety (Scheme 6). Unfortunately, this transformation did not allow us to recover the auxiliary.
 | ||
| Scheme 6 Molecular structure of 17a and conversion into the known epoxyalcohol 18. | ||
In order to get an insight into the interpretation of the observed high diastereoselectivity in all these epoxidation reactions, we undertook DFT calculations investigating the geometry of the most stable conformer of the ylide.16,17 It turned out that (Z)-ylides are significantly more stable than (E)-ylides (Scheme 7). This can most probably be accounted for by destabilizing steric interactions between the ammonium group and the auxiliary in the (E)-ylide and the presence of stabilizing electrostatic interactions between the ammonium group and the enolate oxygen in the (Z)-isomer.18 In addition, the (Z)-ylide in which the phenyl group blocks the Re-face of the α-carbon ((Z)-ylideRe) is thermodynamically more stable than the one where the Si-face is shielded ((Z)-ylideSi) (Scheme 7, upper part).
 | ||
| Scheme 7 DFT calculations of the ylide geometry (relative energies given in brackets) and proposed rationale for observed diastereoselectivity.16 | ||
Based on these results, the observed stereoselectivity could be explained by addition of the most stable ylide ((Z)-ylideRe) to benzaldehyde 2avia the Si-face (Scheme 7, lower part).17
With a set of high yielding reaction conditions available we next investigated the application scope of this epoxidation reaction. As shown in Table 2 a series of different aldehydes was tested. Notably, in neither case any cis-epoxide and no second trans-diastereomer could be detected. Due to the faster reaction rate of the parent benzaldehyde 2a in i-PrOH at room temperature the majority of the reactions were carried out under these conditions (cond. A). These worked reasonably fine for a series of aromatic aldehydes such as para- or ortho-methyl benzaldehydes (entries 3 and 4), halide-substituted ones (entries 5 and 6) and the biphenyl carbaldehyde 2f (entry 7). Also the more electron-rich para-methoxy benzaldehyde 2g could be reacted in good yield under these conditions, whereas the less active dimethylamino benzaldehyde 2h required harsher conditions (60 °C in toluene) to obtain the epoxide 17h in good crude yield. Remarkably, this was the first time that this aldehyde could be successfully used in any of our ammonium ylide-mediated epoxidation reactions.7 Unfortunately, epoxide 17h quickly decomposed during different purification methods. As expected, the more reactive cyano- and nitrobenzaldehydes 2i and 2j did not give any epoxide under the standard conditions (cond. A) but mainly the corresponding Cannizzaro disproportionation products. However, carrying out the reaction in toluene as a less polar solvent at room temperature allowed us to obtain the epoxides 17i and 17j in reasonable isolated yields. Notably, for the first time we have been able to use the aliphatic aldehyde 2k (entry 12) to obtain the corresponding epoxide in moderate yield. Such enolisable aldehydes had primarily undergone self aldol condensation reactions under the strongly basic conditions developed previously.7 Now the use of Cs2CO3 in toluene allowed us to overcome this limitation to some extent. Similar yield was obtained when reacting cyclohexanecarbaldehyde (2l) under these conditions (entry 13).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Ald. | Prod. | Cond.a | Yieldb [%] | drc [%] |
| a A: i-PrOH, 25 °C, 24 h, Cs2CO3 (s, 20 eq.); B: toluene, 60 °C, 24 h, Cs2CO3 (s, 20 eq.); C: toluene, 25 °C, 72 h, Cs2CO3 (s, 20 eq.); D: toluene, 25 °C, 24 h, Cs2CO3 (s, 20 eq.). b Isolated yield. c Determined by 1H NMR of the crude reaction mixture. d Full decomposition on silica gel, crude NMR yield given in brackets. | ||||||
| 1 | Ph | 2a | 17a | A | 78 | >98 |
| 2 | B | 73 | >98 | |||
| 3 | 4-MeC6H4– | 2b | 17b | A | 80 | >98 |
| 4 | 2-MeC6H4– | 2c | 17c | A | 79 | >98 |
| 5 | 4-ClC6H4– | 2d | 17d | A | 75 | >98 |
| 6 | 4-BrC6H4– | 2e | 17e | A | 85 | >98 |
| 7 | 4-PhC6H4– | 2f | 17f | A | 89 | >98 |
| 8 | 4-MeOC6H4– | 2g | 17g | A | 73 | >98 |
| 9 | 4-Me2NC6H4– | 2h | 17h | B | (60)d | >98 |
| 10 | 4-CNC6H4– | 2i | 17i | C | 62 | >98 |
| 11 | 3-NO2C6H4– | 2j | 17j | C | 68 | >98 |
| 12 | n-Decyl– | 2k | 17k | D | 42 | >98 |
| 13 | Cyclohexyl– | 2l | 17l | D | 39 | >98 |
Based on the detailed knowledge obtained on the use of the chiral amide 6C for asymmetric ylide-mediated epoxidations at hand, we carried out a short screening to test the potential of this concept for the related aziridination reaction (Table 3). In analogy to our previous experience using achiral amides 6 for racemic aziridinations9b the use of Cs2CO3 in CH2Cl2 was also found superior here compared to the use of i-PrOH or toluene as solvents. Interestingly, the use of N-tosyl imine 3a allowed the synthesis of the trans-aziridine 19a in high selectivity (entry 1). Unfortunately this compound was found to be relatively unstable and partially decomposed under a variety of purification conditions. This may be attributed to the rather strained nature of this trans-aziridine with the bulky tosyl group being cis to either the phenyl group or the chiral auxiliary.4c The use of N-Boc imine 3b gave an interesting result (entry 2). Formation of the expected major trans-aziridine 19b (isolated in 61% yield) was accompanied by minor amounts of the second trans-aziridine as well as around 10% of cis-19b. Besides this reduced stereoselectivity in the aziridine formation (compared to epoxidation) also notable amounts of the α,β-unsaturated β-amino amide 20b were obtained. Formation of analogous olefins was already observed during our previous racemic studies but only when using electron-poor N-Boc benzaldimines (e.g. NO2-substituted) or heteroaromatic N-Boc aldimines, whereas not even traces thereof could be detected using more electron-rich imines like 3b.9b Unfortunately we were not able to suppress formation of 20 by changing the reaction conditions (in contrast: using less base gave a larger amount of 20). When using more electron-rich imines 3c and 3d the formation of olefins 20c and 20d was reduced, but still not totally suppressed (entries 3 and 4). Again, formation of the minor trans-aziridine and cis-diastereomer was observed too. While the isolated yield of 19c may be a bit lower because of a reduced rate of addition due to the ortho-substituent, the more electron-rich p-methoxy containing 19d was found to be totally unstable on silica gel and also partially decomposed during alumina column chromatography.19 The naphthyl-based imine 3e performed similarly to imine 3b (compare entries 5 and 2) giving the major trans-aziridine 19e in a reasonable isolated yield of 59%. By contrast, the less electron-rich bromine-substituted 3f gave the olefin 20f as the main product (entry 6), while, as expected, the electron-poor 3g did not allow us to obtain any aziridine at all (entry 7).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | PG | Ar | 3 | 19 (dr)a,b [%] | 20 [%] | Yield [%] (trans-19)c |
| a Determined by 1H NMR of the crude reaction mixture. b Values in brackets give the diastereomeric ratios of aziridines (transmajor/transminor/cis) – only one cis-isomer could be detected. c Isolated yield of the major trans-aziridine. d Only one trans-aziridine detected. e Partial decomposition on silica gel, crude NMR yield given in brackets. f Full decomposition on silica gel and partial on alumina, crude NMR yield given in brackets. | ||||||
| 1 | Tos | Ph | 3a | >98 transd | n.d. | 39 (70)e |
| 2 | Boc | 3b | 82 (87/3/10)b | 18 | 62 | |
| 3 | 2-MeC6H4– | 3c | 96 (85/3/12)b | 4 | 56 | |
| 4 | 4-MeOC6H4– | 3d | 95 (88/3/9)b | 5 | 57 (75)f | |
| 5 | Napht-2-yl | 3e | 77 (85/3/12)b | 23 | 58 | |
| 6 | 4-BrC6H4– | 3f | 39 (86/0/14)b | 61 | 32 | |
| 7 | 3-NO2C6H4– | 3g | n.d. | >99 | n.d. | |

The absolute configuration of the major trans-aziridine was determined by anomalous X-ray diffraction analysis of crystals of bromine-containing 19f. Again, in analogy to the epoxidation reaction, the (2R,3S)-configuration of the trans-heterocyclic moiety was found for the major isomer (Fig. 1).15
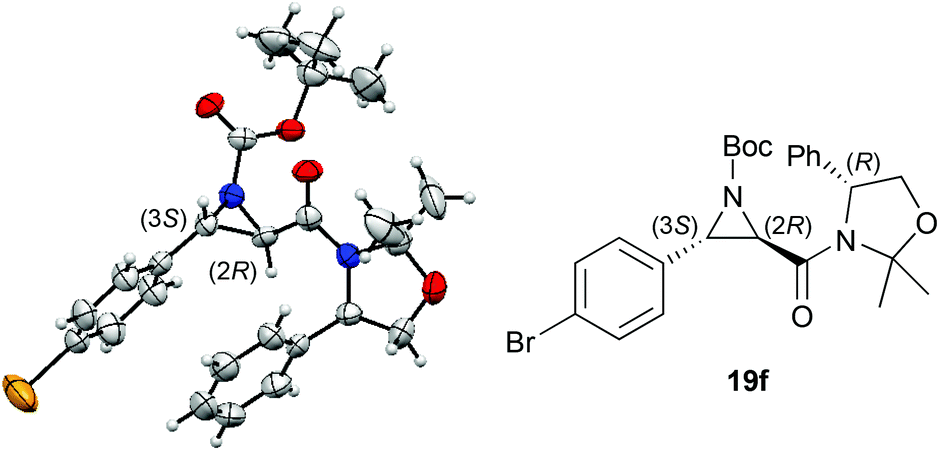 | ||
| Fig. 1 Molecular structure of 19f. | ||
Experimental
General
1H- and 13C-NMR spectra were recorded on a Bruker Avance DRX 500 MHz spectrometer, a Bruker Avance III 300 MHz spectrometer, and on a Bruker Avance III 700 MHz spectrometer with TCI cryoprobe. All NMR spectra were referenced using the residual 1H solvent peak as secondary reference. High resolution mass spectra were obtained using a Thermo Fisher Scientific LTQ Orbitrap XL with an Ion Max API Source. All analyses were made in the positive ionisation mode. IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with ATR unit. All chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. All reactions were performed under an Ar-atmosphere. CH2Cl2 was distilled over P2O5 and stored under Ar (it was not necessary to dry CH2Cl2 prior to every experiment and usually this quality could be used successfully in these reactions over the course of 3–4 weeks after distillation). Single-crystal structure analyses were carried out on a Bruker Smart X2S diffractometer operating with Mo-Kα radiation (λ = 0.71073 Å).Geometry optimization has been performed using the Jaguar 8.0 pseudospectral program package using the well-established B3LYP hybrid density functional with the D3 dispersion correction and the standard split valence polarized 6-31G* basis as implemented in Jaguar. All the optimization calculations were carried out using the Poisson–Boltzmann polarizable continuum method as incorporated in Jaguar, and parameters for CH2Cl2. Energies were obtained by single point energy calculations at the B3LYP-D3/6-311+G**(CH2Cl2) level. The correct nature of each stationary point has been checked by performing frequency calculations at the B3LYP/6-31G*(CH2Cl2) level of theory. Thermal and entropic contributions to free energy (at 298.15 K) and zero-point energy have been obtained from these frequency calculations. We have made a systematic attempt to locate all possible local minima, with the data presented referring to the lowest energy form.
Ammonium amide 6C
(R)-14 (3.00 g, 21.9 mmol) was dissolved in 50 mL acetone and 3.2 g anhydrous MgSO4 were added and the reaction mixture was stirred for 3 h at 20 °C. After filtration and evaporation to dryness the product 21 was obtained in 95% (3.68 g, 20.7 mmol) and used without further purification. The 1H-NMR-spectrum is in full accordance to literature.201H-NMR (300 MHz, δ, CDCl3, 298 K): 1.45 (s, 3H), 1.52 (s, 3H), 2.04 (b, 1H), 3.71 (t, 1H, J = 7.8 Hz), 4.29 (t, 1H, J = 7.8 Hz), 4.54 (t, 1H, J = 7.8 Hz), 7.26–7.41 (m, 5H, Ar–H) ppm.Compound 21 (3.68 g, 20.7 mmol) was dissolved in 25 mL CH2Cl2 and 83 mL aqueous saturated Na2CO3 solution were added. Then bromide 7 (2.9 mL, 21.7 mmol, 1.05 eq.) was added and the mixture was vigorously stirred for 4 h. After addition of aqueous saturated NaHCO3 the aqueous layer was separated and washed three times with 20 mL CH2Cl2. The combined organic phases were dried over anhydrous MgSO4, filtrated and the solvent removed under reduced pressure. The product was purified by column chromatography (silica gel, heptanes–EtOAc = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 8C (2.57 g, 8.6 mmol, 41% yield) as a light-brown solid. The 1H-NMR-spectrum was in accordance to literature.211H-NMR (300 MHz, δ, CDCl3, 298 K): 1.64 (s, 3H), 1.87 (s, 3H), 3.45 (d, 1H, J = 11.0 Hz), 3.52 (d, 1H, J = 11.0 Hz), 3.94 (dd, 1H, J1 = 9.0 Hz, J2 = 2.7 Hz), 4.41 (dd, 1H, J1 = 9.0 Hz, J2 = 6.5 Hz), 5.07 (dd, 1H, J1 = 6.5 Hz, J2 = 2.7 Hz), 7.25–7.45 (m, 5H) ppm.
:1) to give 8C (2.57 g, 8.6 mmol, 41% yield) as a light-brown solid. The 1H-NMR-spectrum was in accordance to literature.211H-NMR (300 MHz, δ, CDCl3, 298 K): 1.64 (s, 3H), 1.87 (s, 3H), 3.45 (d, 1H, J = 11.0 Hz), 3.52 (d, 1H, J = 11.0 Hz), 3.94 (dd, 1H, J1 = 9.0 Hz, J2 = 2.7 Hz), 4.41 (dd, 1H, J1 = 9.0 Hz, J2 = 6.5 Hz), 5.07 (dd, 1H, J1 = 6.5 Hz, J2 = 2.7 Hz), 7.25–7.45 (m, 5H) ppm.
Compound 8C (2.57 g, 8.6 mmol) was dissolved in THF (26 mL) and NMe3 (2.50 mL, 20.4 mmol, 1.2 eq., 33% solution in EtOH) was added. After stirring for 24 h at r.t. the solvent was removed with vacuum distillation. The crude product (purity >90%) was purified by column chromatography (heptanes → CH2Cl2–MeOH = 5:1) to give the ammonium salt 6C in 90% yield (2.90 g, 8.1 mmol) as a white foam. [α]22D (c = 0.6, CH2Cl2) = −92; 1H-NMR (700 MHz, δ, CDCl3, 298 K): 1.65 (s, 3H), 1.86 (s, 3H), 2.98 (d, 1H, J = 16.3 Hz), 3.44 (s, 9H), 3.91 (dd, 1H, J1 = 9.2 Hz, J2 = 1.7 Hz), 4.46 (dd, 1H, J1 = 9.2 Hz, J2 = 6.5 Hz), 5.83 (dd, 1H, J1 = 6.5 Hz, J2 = 1.7 Hz), 6.07 (d, 1H, J = 16.3 Hz), 7.28–7.51 (m, 5H) ppm; 13C NMR (176 MHz, δ, CDCl3, 298 K): 23.5, 25.5, 54.7, 60.1, 64.8, 71.8, 97.5, 126.8, 128.5, 129.5, 140.5, 161.0 ppm; IR (film): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3011, 2987, 2937, 2882, 1654, 1434, 1412, 1378, 1351, 1237, 1204, 1133, 1064, 1048, 923, 896, 843, 703, 664, 604, 579, 563, 517, 501 cm−1; HRMS (ESI): m/z calcd for C16H25N2O2+: 277.1910 [M]+; found: 277.1904.
= 3011, 2987, 2937, 2882, 1654, 1434, 1412, 1378, 1351, 1237, 1204, 1133, 1064, 1048, 923, 896, 843, 703, 664, 604, 579, 563, 517, 501 cm−1; HRMS (ESI): m/z calcd for C16H25N2O2+: 277.1910 [M]+; found: 277.1904.
General epoxidation procedure using ammonium amide 6C
Ammonium salt 6C was dissolved in the appropriate solvent (20 mL mmol−1 ammonium salt) and Cs2CO3 (20 eq.) was added to the reaction mixture. After 5 min the aldehyde (2 eq.) was added and the suspension was stirred for the indicated time at the given temperature. The reaction was quenched with water and extracted with toluene. The organic phase was washed with brine and dried with anhydrous Na2SO4, filtrated and the solvent was removed under reduced pressure. The epoxide was purified by column chromatography (silica gel, heptanes–EtOAc = 7:3).
= 3032, 2989, 2933, 2873, 1658, 1458, 1437, 1387, 1363, 1252, 1205, 1081, 1066, 894, 849, 772, 749, 695, 660, 596, 552, 513 cm−1; HRMS (ESI): m/z calcd for C20H21NO3: 324.1594 [M + H]+; found: 324.1595.
General procedure for the preparation of aziridines
A mixture of 6C (0.4 mmol), aldimine 3 (2 eq.), and Cs2CO3 (20 eq.) in CH2Cl2 (8 mL) was vigorously stirred for 24 h at room temperature. CH2Cl2 and brine were added and the phases separated. The aqueous layer was extracted twice with CH2Cl2, the combined organic layers were extracted with brine and the aqueous layer was re-extracted twice with CH2Cl2. The combined organic layers were dried over Na2SO4, filtrated, evaporated, and dried in vacuo. Column chromatography (silica gel, heptanes–EtOAc = 20:1–2:1) gave the aziridines 19 in the reported yields. In most cases the minor cis-isomers and the olefins 20 could not be obtained in pure form.
= 3009, 2984, 2935, 2868, 1715, 1652, 1433, 1395, 1364, 1333, 1253, 1223, 1204, 1074, 1053, 819, 755, 746, 705, 691, 635 cm−1; HRMS (ESI): m/z calcd for C25H30N2O4: 423.2278 [M + H]+; found: 423.2287.
Conclusions
The use of easily available phenylglycinol as a chiral auxiliary in ammonium ylide-mediated reactions was found to be a promising strategy to obtain chiral three-membered ring heterocycles with good to excellent stereoselectivities and in good yields. In general it was found that the epoxidation reaction is rather broad in its application scope giving glycidic amides as single stereoisomers in all cases. The aziridination is slightly less selective giving the major trans-isomer with at least 85% diastereoselectivity but, depending on the electronic nature of the starting imine, with varying amounts of an α,β-unsaturated β-amino amide side-product.Acknowledgements
This work was supported by the Austrian Science Funds (FWF): Project no. P26387-N28. Computational resources have been provided by the supercomputing facilities of the Université catholique de Louvain (CISM/UCL) and the Consortium des Équipements de Calcul Intensif en Fédération Wallonie Bruxelles (CÉCI) funded by the Fond de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under convention 2.5020.11. The NMR spectrometers used were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”). RR is a Chercheur qualifié of the F.R.S.-FNRS.Notes and references
- For reviews see: (a) V. K. Aggarwal, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999, vol. 2, p. 679 Search PubMed; (b) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841–5883 CrossRef CAS PubMed.
- For seminal contributions about sulfur ylides see: (a) A. W. Johnson and R. B. Lacount, Chem. Ind., 1958, 1440–1441 CAS; (b) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867–868 CrossRef CAS; (c) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353–1364 CrossRef CAS.
- For selected examples highlighting the use of sulfur ylides in epoxidation reactions see: (a) O. Illa, M. Arshad, A. Ros, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 1828–1830 CrossRef CAS PubMed; (b) J. Zanardi, C. Leriverend, D. Aubert, K. Julienne and P. Metzner, J. Org. Chem., 2001, 66, 5620–5623 CrossRef CAS PubMed; (c) M. Davoust, J.-F. Briere, P.-A. Jaffres and P. Metzner, J. Org. Chem., 2005, 70, 4166–4169 CrossRef CAS PubMed; (d) V. K. Aggarwal and J. Richardson, Chem. Commun., 2003, 2644–2651 RSC; (e) Y.-G. Zhou, X.-L. Hou, L.-X. Dai, L.-J. Xia and M.-H. Tang, J. Chem. Soc., Perkin Trans. 1, 1999, 77–80 RSC; (f) V. K. Aggarwal, J. P. H. Charmant, D. Fuentes, J. N. Harvey, G. Hynd, D. Ohara, W. Picoul, R. Robiette, C. Smith, J.-L. Vasse and C. L. Winn, J. Am. Chem. Soc., 2006, 128, 2105–2114 CrossRef CAS PubMed; (g) V. K. Aggarwal, G. Hynd, W. Picoul and J.-L. Vasse, J. Am. Chem. Soc., 2002, 124, 9964–9965 CrossRef CAS PubMed; (h) O. Illa, M. Namutebi, C. Saha, M. Ostovar, C. C. Chen, M. F. Haddow, S. Nocquet-Thibault, M. Lusi, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2003, 135, 11951–11966 CrossRef PubMed; (i) A. Solladie-Cavallo and A. Diep-Vohuule, J. Org. Chem., 1995, 60, 3494–3498 CrossRef CAS.
- For the use of sulfur ylides in aziridine synthesis see: (a) A.-H. Li, L.-X. Dai and X.-L. Hou, J. Chem. Soc., Perkin Trans. 1, 1996, 867–869 RSC; (b) V. K. Aggarwal, M. Ferrara, C. J. O'Brien, A. Thompson, R. V. H. Jones and R. Fieldhouse, J. Chem. Soc., Perkin Trans. 1, 2001, 1635–1643 RSC; (c) R. Robiette, J. Org. Chem., 2006, 71, 2726–2734 CrossRef CAS PubMed; (d) T. Saito, M. Sakairi and D. Akiba, Tetrahedron Lett., 2001, 42, 5451–5454 CrossRef CAS; (e) V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcelloni, Angew. Chem., Int. Ed., 2001, 40, 1433–1436 CrossRef CAS; (f) V. K. Aggarwal and J.-L. Vasse, Org. Lett., 2003, 5, 3987–3990 CrossRef CAS PubMed; (g) A. Solladie-Cavallo, M. Roje, R. Welter and V. Sunjiç, J. Org. Chem., 2004, 69, 1409–1412 CrossRef CAS PubMed.
- For ammonium ylide mediated cyclopropanations see: (a) M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS PubMed; (b) C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024–6028 CrossRef CAS PubMed; (c) C. D. Papageorgiou, M. A. Cubillo de Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641–4644 CrossRef CAS PubMed; (d) N. Bremeyer, S. C. Smith, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 2681–2684 CrossRef CAS; (e) C. D. Papageorgiou, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2003, 42, 828–831 CrossRef CAS PubMed; (f) S. Kojima, K. Hiroike and K. Ohkata, Tetrahedron Lett., 2004, 45, 3565–3568 CrossRef CAS; (g) S. Yamada, J. Yamamoto and E. Ohta, Tetrahedron Lett., 2007, 48, 855–858 CrossRef CAS; (h) N. Kanomata, R. Sakaguchi, K. Sekine, S. Yamashita and H. Tanaka, Adv. Synth. Catal., 2010, 352, 2966–2978 CrossRef CAS.
- For benzylic ammonium ylide mediated epoxidations see: (a) T. Kimachi, H. Kinoshita, K. Kusaka, Y. Takeuchi, M. Aoe and M. Ju-ichi, Synlett, 2005, 842–844 CrossRef CAS; (b) H. Kinoshita, A. Ihoriya, M. Ju-ichi and T. Kimachi, Synlett, 2010, 2330–2334 CAS; (c) R. Robiette, M. Conza and V. K. Aggarwal, Org. Biomol. Chem., 2006, 4, 621–623 RSC.
- For amide-stabilised ammonium ylide mediated epoxidations see: (a) M. Waser, R. Herchl and N. Müller, Chem. Commun., 2011, 47, 2170–2172 RSC; (b) R. Herchl, M. Stiftinger and M. Waser, Org. Biomol. Chem., 2011, 9, 7023–7027 RSC.
- For cyano-stabilised ammonium ylide mediated epoxidations see: (a) A. Kowalkowska, D. Sucholbiak and A. Jonczyk, Eur. J. Org. Chem., 2005, 925–933 CrossRef CAS; (b) A. Jonczyk and A. Konarska, Synlett, 1999, 1085–1087 CrossRef CAS; (c) A. Alex, B. Larmanjat, J. Marrot, F. Couty and O. David, Chem. Commun., 2007, 2500–2502 RSC.
- For ammonium ylide-mediated aziridinations see: (a) L. D. S. Yadav, R. Kapoor and Garima, Synlett, 2009, 3123–3126 CrossRef CAS; (b) S. Aichhorn, G. N. Gururaja, M. Reisinger and M. Waser, RSC Adv., 2013, 3, 4552–4557 RSC.
- For selected reviews see: (a) A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307–1370 CrossRef CAS; (b) P. Arya and H. Qin, Tetrahedron, 2000, 56, 917–947 CrossRef CAS; (c) D. A. Evans and J. T. Shaw, Actual. Chim., 2003, 35–38 CAS; (d) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS PubMed.
- For chiral amide containing sulfonium ylides in epoxidations see: (a) F. Sarabia, S. Chammaa, M. Garica-Castro and F. Martin-Galvez, Chem. Commun., 2009, 5763–5765 RSC; (b) D. M. Aparicio, J. L. Teran, D. Gnecco, A. Galindo, J. R. Juarez, M. L. Orea and A. Mendoza, Tetrahedron: Asymmetry, 2009, 20, 2764–2768 CrossRef CAS; (c) F. Sarabia, C. Vivar-Garcia, M. Garica-Castro and J. Martin-Ortiz, J. Org. Chem., 2011, 76, 3139–3150 CrossRef CAS PubMed; (d) D. M. Aparicio, D. Gnecco, J. R. Juarez, M. L. Orea, A. Mendoza, N. Waksman, R. Salazar, M. Fores-Alamo and J. L. Teran, Tetrahedron, 2012, 68, 10252–10256 CrossRef CAS; (e) P. G. Gordillo, D. M. Aparicio, M. Flores, A. Mendoza, L. Orea, J. R. Juarez, G. Huelgas, D. Gnecco and J. L. Teran, Eur. J. Org. Chem., 2013, 5561–5565 CrossRef CAS.
- Seminal report describing the use of these auxiliaries: D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- Seminal reports about the use as a chiral auxiliary: (a) M. Larcheveque, E. Ignatova and T. Cuvigny, Tetrahedron Lett., 1978, 19, 3961–3964 CrossRef; (b) A. G. Myers, B. H. Yang, H. Chen and J. L. Gleason, J. Am. Chem. Soc., 1994, 116, 9361–9362 CrossRef CAS.
- These conditions were found to be useful for the parent epoxidation of ammonium salts 1 with different aldehydes: S. Aichhorn, On the Diastereoselective Synthesis of Aziridines and Epoxides using Nitrogen Ylides, Diploma Thesis, Institute of Organic Chemistry, Johannes Kepler University Linz, 2012 Search PubMed.
- (a) CCDC 1023711 (Cambridge Crystallographic Data Centre, http://www.ccdc.cam.ac.uk) contains the supplementary crystallographic data for 17a; (b) CCDC 1030561 (Cambridge Crystallographic Data Centre, http://www.ccdc.cam.ac.uk) contains the supplementary crystallographic data for 19f.
- Calculations were carried out at the B3LYP-D3/6-311+G**(CH2Cl2)//B3LYP-D3/6-31G*(CH2Cl2) level using Jaguar, version 8.0, Schrodinger, LLC, New York, NY, 2011. Reported energies are free energies (kcal mol−1) relative to reactants in their most stable conformation.
- Systematic more general computational studies delineating ammonium ylide mediated epoxidation reactions will be an integral part of our future research in this field.
- For computational studies showing preference of (Z)-configuration in sulfonium and phosphonium ylides see ref. 3f and: R. Robiette, J. Richardson, V. K. Aggarwal and J. N. Harvey, J. Am. Chem. Soc., 2005, 127, 13468–13469 CrossRef CAS PubMed.
- The aziridines were generally less stable than the epoxides especially at higher temperature or under acidic conditions.
- S. Kanemasa and K. Onimura, Tetrahedron, 1992, 48, 8631–8644 CrossRef CAS.
- R. J. R. Lumby, P. M. Joensuu and H. W. Lam, Tetrahedron, 2008, 64, 7729–7740 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, characterization of new compounds, and copies of NMR spectra. CCDC 1023711 and 1030561. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02318h |
| This journal is © The Royal Society of Chemistry 2015 |