
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A unified lead-oriented synthesis of over fifty molecular scaffolds†
Richard G.
Doveston
a,
Paolo
Tosatti
a,
Mark
Dow
a,
Daniel J.
Foley
a,
Ho Yin
Li
a,
Amanda J.
Campbell
b,
David
House
b,
Ian
Churcher
b,
Stephen P.
Marsden
*a and
Adam
Nelson
*ac
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: s.p.marsden@leeds.ac.uk; a.s.nelson@leeds.ac.uk
bGlaxoSmithKline Medicines Research Centre, Stevenage, SG1 2NY, UK
cAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
First published on 18th November 2014
Abstract
Controlling the properties of lead molecules is critical in drug discovery, but sourcing large numbers of lead-like compounds for screening collections is a major challenge. A unified synthetic approach is described that enabled the synthesis of 52 diverse lead-like molecular scaffolds from a minimal set of 13 precursors. The divergent approach exploited a suite of robust, functional group-tolerant transformations. Crucially, after derivatisation, these scaffolds would target significant lead-like chemical space, and complement commercially-available compounds.
Introduction
Control of molecular properties is crucial in drug discovery, and experience has provided sets of guidelines that can steer medicinal chemists towards drug-like chemical space. For example, Lipinski's rule-of-five was formulated to predict oral bioavailability,1,2 and, in the case of central nervous system (CNS) drug discovery, more restricted guidelines can assist the design of compounds able to penetrate the blood-brain barrier.3,4 Conversely, relaxation of molecular property guidelines is generally required to enable the discovery of small molecule inhibitors of protein–protein interactions.‡5,6 The molecular properties of clinical candidates strongly influence the probability of successful progression towards marketed drugs.7–11 Key parameters that correlate strongly with success in drug discovery include molecular weight,7,8 lipophilicity8–10 and the fraction of sp3-hybridised carbons,11 some of which have been captured within a single metric that estimates drug-likeness.12The identification of high quality lead compounds is a key challenge in early stage drug discovery. Lead optimisation tends to increase both molecular weight and lipophilicity, as well as molecular complexity.13–16 It is advisable, therefore, to control the properties of lead compounds to facilitate the emergence of clinical candidates with desirable drug-like characteristics. As a result, approaches have been developed to define chemical space that is populated by molecules with good lead-like properties typically by considering factors such as lipophilicity (e.g.17 −1 < clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P < 3), molecular size (e.g.17 14 ≤ heavy atoms ≤ 26) and removal of undesirable sub-structures.17
P < 3), molecular size (e.g.17 14 ≤ heavy atoms ≤ 26) and removal of undesirable sub-structures.17
High-throughput screening provides a significant source of starting points for drug discovery.14,18 Control of the molecular properties of screening compounds can both aid subsequent development into high-quality lead molecules, and increase the efficiency of the exploration of chemical space.19 However, sourcing large numbers of diverse compounds with appropriate molecular properties has been identified as a major challenge.17 In a recent analysis, just 2.6% of 4.9 M commercially-available compounds were found to survive filtering by molecular weight (200 ≤ MR ≤ 360), lipophilicity (−1 < clogP < 3) and various structural features known to be undesirable in drug candidates.17 In addition, the ability of emerging synthetic methods to deliver lead-like compounds has been very poorly demonstrated17 despite significant development of diversity-oriented synthetic approaches.20–22 The problem is in fact exacerbated when diversity is also considered because chemists have historically explored chemical space unsystematically and in an uneven manner;23 indeed, metrics have been developed to capture the scaffold diversity of screening collections.24 The realisation of lead-oriented synthesis has recently been framed as a major academic and practical challenge17 which has prompted the development of approaches25,26 to the synthesis of specific classes of lead-like molecules.27–30
We have established a programme focussed on developing general synthetic approaches to diverse and novel lead-like molecular scaffolds i.e. scaffolds with the potential, following diversification, to yield large numbers of compounds with lead-like molecular properties. In this case, our approach to exemplifying the strategy exploited a single connective reaction in combination with just six distinct cyclisations (Scheme 1). Ir-catalysed amination31–36 was selected as the connective reaction because we had previously retooled it for compatibility with polar, sp3-rich, functionalised substrates35 that would have particular value for targeting lead-like chemical space. Thus, reaction between allylic carbonates 1 and amines 2 would provide cyclisation precursors 3. Importantly, the building blocks would be armed with functional groups to enable subsequent cyclisation – either once (e.g. → 4 or 5) or twice (e.g. → 6) – to yield product scaffolds. In addition, more complex scaffolds might be accessible by exploitation of a third building block (e.g. → 7–9). Remaining functionality would then be available for late-stage scaffold decoration. Our aim was to prepare, in a synthetically concise and efficient manner, a wide range of diverse and novel molecular scaffolds that, following decoration, would target broad regions of lead-like chemical space and thus demonstrate the potential of the strategy to underpin early-stage drug discovery.
 | ||
| Scheme 1 Overview of the unified approach to the synthesis of lead-like molecular scaffolds. Reaction between an allylic carbonate (1; blue) and an amine (2; green) would yield a cyclisation precursor 3. Reactive sites that may enable cyclisation or functionalisation (filled circles), third building blocks (red) and new bonds (bold) are indicated. | ||
Results
Synthesis of cyclisation precursors
We selected a number of densely functionalised cyclisation precursors that could facilitate the synthesis of a wide range of novel and diverse lead-like molecular scaffolds. Thirteen cyclisation precursors were prepared from combinations of an amine (10 alternatives) and an allylic carbonate (2 alternatives) building block in good to excellent yield, and with high enantio- or diastereoselectivity (Scheme 2). The building blocks were either commercially available or were prepared on a multi-gram scale using well established methods. In most cases, our previously reported protocol, which enables efficient coupling of unprotected polar amines, was used.35 Thus, the active catalyst was generated from 2 mol% [Ir(dbcot)Cl]2, 4 mol% chiral ligand [either (R,R,aR)- or (S,S,aS)-10] and 4 mol% BuNH2 in DMSO at 55 °C before the building blocks were added. In some cases, the use of DMSO was not required, and THF was used as the reaction solvent.31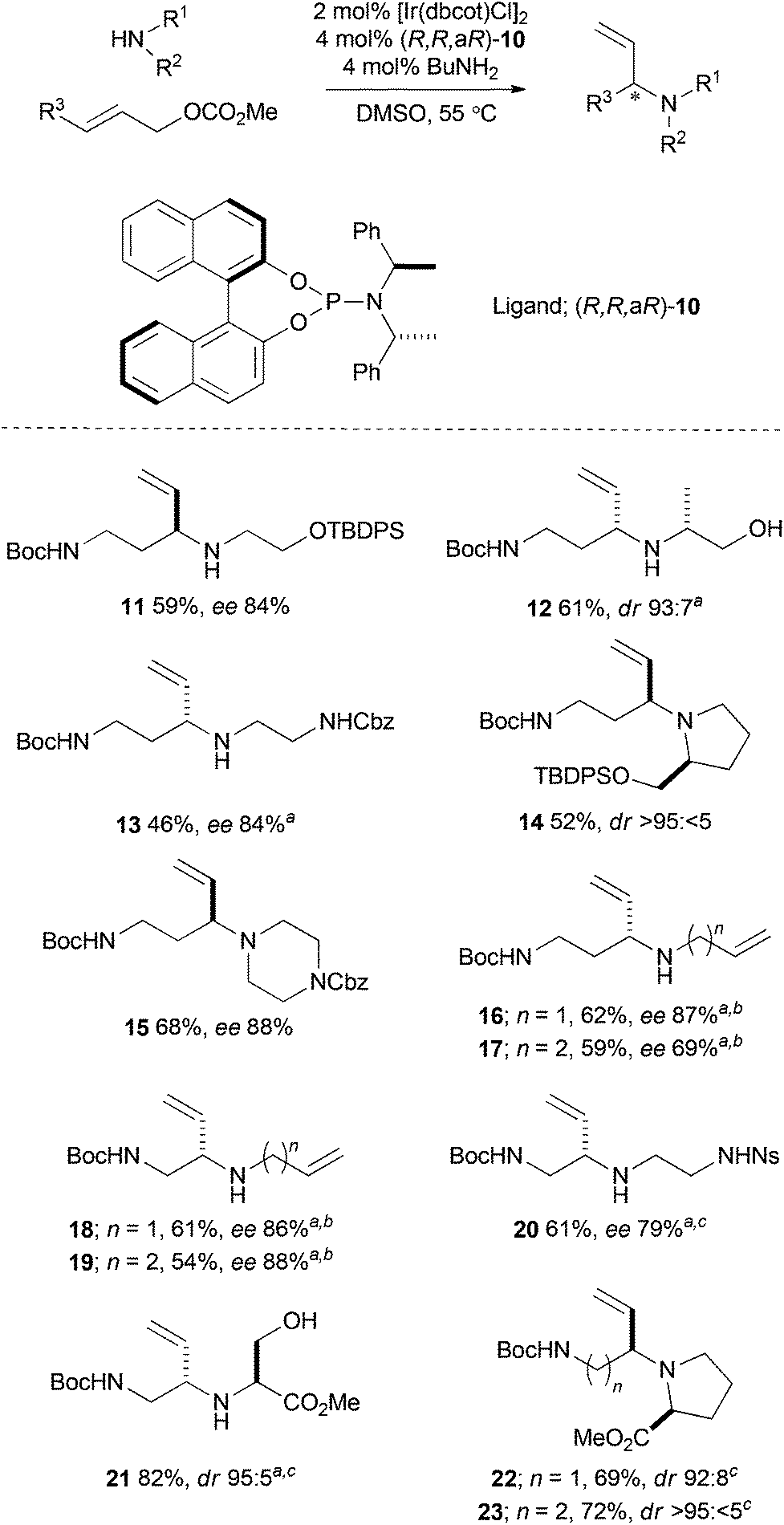 | ||
| Scheme 2 Synthesis of cyclisation precursors by Ir-catalysed reaction between amine and allylic carbonate building blocks. a(S,S,aS)-10 was used. bPrNH2 and THF were used. cThe amine HCl salt and 1.3 eq. K3PO4 were used. dbcot = dibenzo[a,e]cyclooctatetraene; TBDPS = tert-butyldiphenylsilyl; Ns = 2-nitrobenzenesulfonyl. | ||
Synthesis of lead-like molecular scaffolds
A toolkit of six cyclisation reactions was then exploited to convert the thirteen cyclisation precursors (11–23) into lead-like molecular scaffolds. Selected syntheses of scaffolds are presented in Schemes 3 and 4 (see ESI† for full details). In order to maximise the number of scaffolds prepared, a divergent synthetic approach was adopted that exploited each of the six methods in the first cyclisation step (Methods A–F; Scheme 3). | ||
| Scheme 3 Selected syntheses of lead-like scaffolds prepared using a single cyclisation step (indicated by colour: A, yellow, Pd-catalysed aminoarylation; B, purple, iodocyclisation/displacement; C, pink, reaction with CDI; D, green, reaction with α-halo acetyl halide; E, light blue, ring-closing metathesis; F, peach, lactamisation). Typical methods (see ESI† for full details): A: Aryl bromide (1.2 eq.), 5 mol% Pd(OAc)2, 10 mol% DPE-Phos, Cs2CO3 (2.5 eq.), 1,4-dioxane, 105 °C; B: (i) NsCl (1.2 eq.), NEt3 (2.0 eq.), DMAP (0.1 eq.), rt, then TBAF (1.2 eq.), AcOH (1.2 eq.), THF, rt; (ii) NIS (1.5 eq.), MeCN, 65 °C; (iii) ArSH (1.5 eq.), DBU (2.5 eq.), MeCN, rt; (iv) mCPBA (4.0 eq.), CH2Cl2, rt; (v) PhSH (1.2 eq.), DBU (1.5 eq.), MeCN, rt; C1: CH2Cl2–TFA, 0 °C → rt, then CDI (1.5 eq.), DBU (4.0 eq.), THF, 50 °C; C2: CDI (4.5 eq.), DMF, 110 °C; C3: CDI (1.5 eq.), DBU (2.5 eq.), THF, 50 °C; D1: Chloroacetyl chloride (1.5 eq.), NEt3 (5.0 eq.), CH2Cl2, 0 °C → rt, then NaH (2.0 eq.), NaI (1.0 eq.), THF, rt; D2: (i) TMSCl (1.1 eq.), NEt3 (3.0 eq.),CH2Cl2, 0 °C → rt, then bromoacetyl bromide (1.5 eq.), then 20% AcOH (aq.), rt; (ii) 35% NaOH (aq.) (5.0 eq.), Bu4NSO4 (0.5 eq.), CH2Cl2, 0 °C → rt; E1: 5 mol% Grubbs II, CH2Cl2, reflux; F1: CH2Cl2–TFA, 0 °C → rt, then K2CO3 (6.0 eq.), CH2Cl2, H2O, rt; F2: CH2Cl2–TFA, 0 °C → rt, then NaOtBu (2.0 eq.), THF, reflux; TBDPS = tert-butyldiphenylsilyl; Ns = 2- or 4-nitrobenzenesulfonyl; DMAP = 4-dimethylaminopyridine; TBAF = tetra-n-butylammonium fluoride; DBU = 1,8-diazabicycloundec-7-ene; mCPBA = m-chloroperoxybenzoic acid; DPE-Phos = bis-[2-(diphenylphosphino)phenyl]ether; TFA = trifluoroacetic acid; CDI = carbonyl diimidazole. | ||
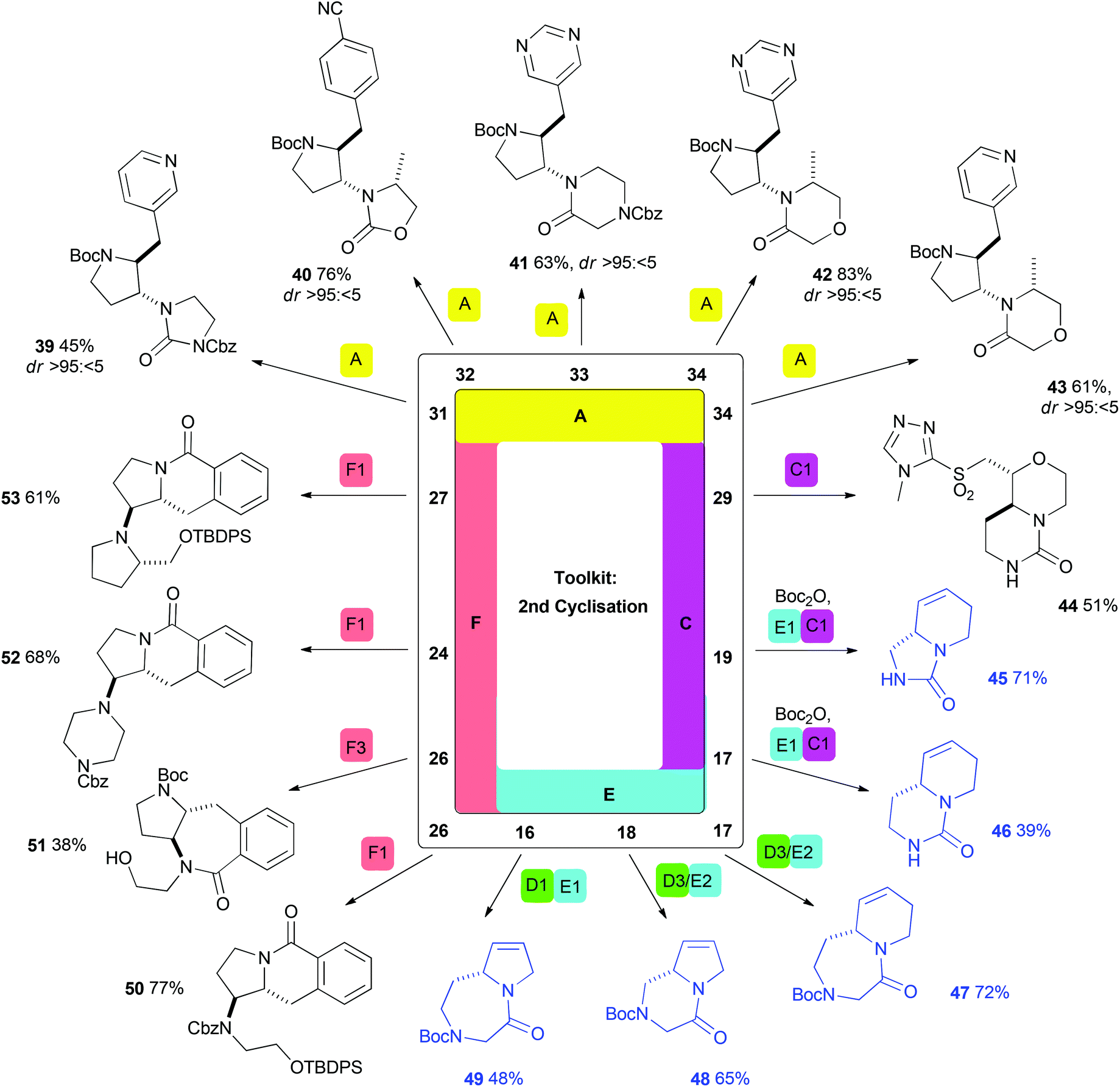 | ||
| Scheme 4 Selected syntheses of lead-like scaffolds prepared using a second cyclisation step (including telescoped procedures; shown in blue). Methods are consistent with those in Scheme 3 (indicated by colour: A, yellow, Pd-catalysed aminoarylation; B, purple, iodocyclisation/displacement; C, pink, reaction with CDI; D, green, reaction with α-halo acetyl halide; E, light blue, ring-closing metathesis; F, peach, lactamisation). Additional methods (see ESI† for full details): D3/E2: (i) Bromoacetyl bromide (1.1 eq.), DIPEA (1.2 eq.), CH2Cl2, 0 °C → rt; (ii) 5 mol% Grubbs II, CH2Cl2, 45 °C; (iii) NaH (2.0 eq.), THF, rt; F3: 10% Pd/C (20 mol% Pd), ethylenediamine (1.0 eq.), MeOH, rt, then Cs2CO3 (10.0 eq.), DMF, 110 °C; TBDPS = tert-butyldiphenylsilyl. | ||
For example, Pd-catalysed aminoarylation (Method A),37,38 if necessary after protection of the secondary amine, enabled conversion of the cyclisation precursors 11, 14 and 15 into the pyrrolidines 24–28. In each case, the reaction proceeded efficiently and with high diastereoselectivity.§ Iodocylisation39 (Method B) enabled the cyclisation precursor 11 to be converted, after protecting group manipulation, into the corresponding morpholine (dr 56:44); subsequent treatment with 4-methyl-4H-1,2,4-triazole-3-thiol and DBU gave separable sulfides which were oxidised and then deprotected to give the morpholine 29. Methods A and B both exploit a potentially variable building block – a (het)aryl bromide or a thiol respectively – and may therefore enable variation of the specific scaffold prepared. The application of alternative electron-deficient (het)aryl bromides in an initial cyclisation by aminoarylation was exemplified in the synthesis of nine scaffolds, five of which are shown in Scheme 3.
Two cyclisation reactions (Methods C and D) exploited simple bifunctional reagents that enabled the formation of a range of five-, six- and seven-membered ring systems. Thus, reaction with CDI (Methods C), if necessary after a telescoped protecting group removal, gave either cyclic ureas (e.g.30 and 31) or oxazolidinones (e.g.32). Similarly, reaction with either chloroacetyl chloride or bromoacetyl bromide, followed by alkylation, (Methods D) enabled the synthesis of ketopiperazines (e.g.33) and ketomorpholines (e.g.34). In addition, 2-keto-1,4-diazepane formation was also possible from the homologous substrates, although generally telescoped with a second cyclisation step (see below). Ring-closing metathesis (Methods E) was often used as a single cyclisation to form tetrahydropyridines (e.g.35) or dihydropyrroles (e.g.36) but could also be telescoped with a second cyclisation step (see below). Finally, lactamisation (Methods F) enabled the formation of either six- (e.g.37) or seven- (e.g.38) membered lactams.
In many cases, a second cyclisation step was possible using one of the complementary cyclisation reactions (Scheme 4). The robustness of Pd-catalysed aminoarylation (Method A) enabled the formation of pyrrolidines from a wide range of substrates. For example, the substrates 31–34 were converted into the pyrrolidines 39–43 with consistently high diastereoselectivity. Again, the ability to vary the scaffold prepared through careful choice of (het)aryl bromide was widely demonstrated. Method C also had utility as a second cyclisation: Boc deprotection of 29, followed by reaction with CDI, enabled the synthesis of the bicyclic scaffold 44.
As already noted, telescoped procedures incorporating a second cyclisation step were often used to great effect (Scheme 4; structures shown in blue). For example, ring-closing metathesis (Methods E) followed by urea formation (Methods C) enabled the synthesis of bicyclic compounds 45 and 46 directly from acyclic cyclisation precursors (19 and 17). Alternatively, telescoped diketopiperazine or 2-keto-1,4-diazepane formation (Methods D) and ring-closing metathesis procedures enabled the facile synthesis of compounds 47–49.
Lactamisation (Method F) had particular value for substrates prepared by aminoarylation with 2-methoxycarbonylphenyl bromide in the first cyclisation. For example, deprotection of 24, 26 and 27 triggered cyclisation to yield either six- (e.g.50, 52 or 53) or seven- (e.g.51) membered lactams. In the case of pyrrolidine 26, the selective formation of two distinct scaffolds (50 and 51) was possible using orthogonal deprotection reactions.
Discussion
To assess the value of the 52 scaffolds prepared, a virtual library of functionalised compounds was enumerated (see ESI† for full details). The deprotected scaffolds were combined with 59 exemplar medicinal chemistry capping groups. For most scaffolds, up to two capping groups were used. However, for scaffolds whose synthesis had already involved a variable reactant (e.g. those prepared by aminoarylation), only one capping group was exploited. The resulting virtual library comprised 19530 likely synthetically-accessible small molecules. To confirm the validity of this analysis, we demonstrated experimentally that scaffold decoration was possible to yield exemplar lead-like compounds from the virtual library (see ESI†).
First, the lead-likeness of the members of the virtual library was assessed (Fig. 1, Panel A). Compounds were successively filtered by molecular size (14 ≤ number of heavy atoms ≤ 26), lipophilicity (−1 ≤ AlogP ≤ 3) and undesirable structural features (see ESI† for specific structural filters). About 59% of the compounds in the virtual library had lead-like molecular properties, and the majority of the outlying compounds only narrowly failed the molecular property filters (heavy atoms: μ = 23.8, σ = 4.0; AlogP: μ = 0.3, σ = 1.3). By comparison, with these specific filters, just 23% of ∼9 M commercially-available compounds from the entire ZINC database40 were lead-like, with the majority of compounds lying well outside lead-like chemical space (heavy atoms: μ = 25.9, σ = 5.4; AlogP: μ = 1.7, σ = 2.9) (Fig. 1, Panel B).¶41 Remarkably, we also showed that, with this set of capping groups, each one of the 52 scaffolds allowed significant regions within lead-like chemical space to be targeted (see ESI†). Our unified synthetic approach thus specifically targeted lead-like chemical space.
 | ||
| Fig. 1 Analysis of the molecular properties of a virtual library of 19530 compounds derived from the 52 molecular scaffolds and 1% of the ZINC database (90911 randomly-selected compounds).|| Panel A: Distribution of the molecular properties of the virtual library. 59% of the compounds (green) survive successive filtering by molecular size (14 ≤ number of heavy atoms ≤ 26; failures shown in red) and lipophilicity (−1 ≤ AlogP ≤ 3; failures shown in orange) and various structural filters; 0.27% of the compounds (shown in black) failed the structural filters. Panel B: Distribution of the molecular properties of the compounds from the ZINC database. Using the same approach, 23% of the compounds survive the iterative filtering process, and 9% of the compounds fail a structural filter. Panel C: Mean Fsp3 of the compounds from the ZINC database (red) and our virtual library (overall mean, black; and mean for the compounds based on each of the 52 scaffolds, green). | ||
Second, we determined the fraction of sp3-hybridised carbon atoms (Fsp3) in the virtual compounds (Fig. 1, Panel C). It has previously been shown that Fsp3 correlates strongly with success because compounds in the discovery phase (mean Fsp3: 0.36) have lower Fsp3 than marketed drugs (mean Fsp3: 0.47).11 It has thus been stated that accessing more three-dimensional lead compounds is a desirable goal.11,17 The mean Fsp3 of the virtual compounds (0.58) compared very favourably with that of the random sample of compounds from the ZINC database (0.33). Thus, our synthetic approach can yield compounds with significantly greater sp3 character than most commercially-available compounds, thereby expanding the range of molecular architectures available within lead-like chemical space and offering more flexibility in lead optimisation.
Third, the novelty and diversity of the 52 scaffolds was assessed. A substructure search was performed in which the ZINC database (9046036 compounds) was interrogated with each of the deprotected scaffolds. In 43 cases (82%), the deprotected scaffold was not found as a substructure in any compound in the database. Even in the remaining 9 cases, the deprotected compound was not known in the CAS registry. The diversity of, and relationship between, the scaffolds was assessed using an hierarchical analysis (see ESI†).42 It was found that 42 frameworks were represented at the graph-node-bond level, which were related hierarchically to 13 “parent” frameworks. There is significant scaffold diversity at each level of hierarchy, meaning that the scaffolds are not simply closely related derivatives.
In total, 52 diverse molecular scaffolds were prepared from just thirteen different cyclisation precursors. Initially, pairs of building blocks were combined using a single connective reaction – Ir-catalysed allylic amination – before a divergent synthetic approach was used to convert these cyclisation precursors into 52 molecular scaffolds. This approach exploited a toolkit of just six cyclisation reactions, and required on average just 3.0 operations** for the synthesis of the scaffolds from the constituent building blocks. Furthermore, the unified and modular nature of the strategy means that it has the potential to deliver many additional scaffolds through expansion of the range of building blocks used (e.g. by use of homologated, and stereo- or regioisomerically substituted variants).
Conclusions
Our unified synthetic approach yielded molecular scaffolds that were novel, diverse and lead-like. It was shown that functionalisation of the scaffolds would allow significant lead-like chemical space to be targeted that complements that occupied by commercially-available molecules. A key challenge in lead-oriented synthesis is the identification of complementary and robust reactions with broad functional group compatibility, particularly convergent reactions that may be used to link building blocks.††43 The success of our unified lead-oriented synthetic approach was founded in the identification of a robust convergent reaction that, together with a range of cyclisation reactions, could be exploited in the synthesis of a wide range of novel, diverse and lead-like molecular scaffolds. An increased armoury of such robust convergent reactions would crucially expand the relevant chemical space accessible to drug discovery programmes, and may help to address the grand challenge of increasing productivity in the pharmaceutical sector.44,45Acknowledgements
We thank EPSRC (EP/J00894X/1) and GSK for funding, Stephen Pickett for computational analyses, George Burslem, Stuart Warriner and Alan Nadin for useful discussions.Notes and references
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- C. A. Lipinski, Drug Discovery Today, 2004, 1, 337 CrossRef CAS PubMed.
- H. Pajouhesh and G. R. Lenz, NeuroRx, 2005, 2, 541 CrossRef PubMed.
- T. T. Wager, R. Y. Chandrasekaran, X. Hou, M. D. Troutman, P. R. Verhoest, A. Villalobos and Y. Will, ACS Chem. Neurosci., 2010, 1, 420 CrossRef CAS PubMed.
- O. Sperandio, C. H. Reynès, A.-C. Camproux and B. O. Villoutreix, Drug Discovery Today, 2010, 15, 220 CrossRef CAS PubMed.
- T. L. Nero, C. J. Morton, J. K. Holien, J. Wielens and M. W. Parker, Nat. Rev. Cancer, 2014, 14, 248 CrossRef CAS PubMed.
- M. C. Wenlock, R. P. Austin, P. Barton, A. M. Davis and P. D. Leeson, J. Med. Chem., 2003, 46, 1250 CrossRef CAS PubMed.
- M. J. Waring, Bioorg. Med. Chem. Lett., 2009, 19, 2844 CrossRef CAS PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS PubMed.
- T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90 CrossRef CAS PubMed.
- T. I. Oprea, A. M. Davis, S. J. Teague and P. D. Leeson, J. Chem. Inf. Comput. Sci., 2001, 41, 1308 CrossRef CAS PubMed.
- E. Perola, J. Med. Chem., 2010, 53, 2986 CrossRef CAS PubMed.
- M. M. Hann, Med. Chem. Commun., 2011, 2, 349 RSC.
- G. M. Keserü and G. M. Makara, Nat. Rev. Drug Discovery, 2009, 8, 203 CrossRef PubMed.
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188 CrossRef CAS PubMed.
- M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856 CrossRef CAS PubMed.
- D. Robbins, A. F. Newton, C. Gignoux, J.-C. Legeay, A. Sinclair, M. Rejzek, C. A. Laxon, S. K. Yalamanchili, W. Lewis, M. A. O'Connell and R. A. Stockman, Chem. Sci., 2011, 2, 2232 RSC.
- D. Morton, S. Leach, C. Cordier, S. Warriner and A. Nelson, Angew. Chem., Int. Ed., 2009, 48, 104 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed.
- M. Krier, G. Bret and D. Rognan, J. Chem. Inf. Model., 2006, 46, 512 CrossRef CAS PubMed.
- A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt, R. J. Schenck and A. J. Trippe, J. Org. Chem., 2008, 73, 4443 CrossRef CAS PubMed.
- P. MacLellan and A. Nelson, Chem. Commun., 2013, 49, 2383 RSC.
- R. Doveston, S. P. Marsden and A. Nelson, Drug Discovery Today, 2014, 19, 813 CrossRef CAS PubMed.
- S. V. Ryabukhin, D. M. Panov, D. S. Granat, E. N. Ostapchuk, D. V. Kryvoruchko and O. O. Grygorenko, ACS Comb. Sci., 2014, 16, 146 CrossRef CAS PubMed.
- A. Borisov, V. Voloshchuk, M. Nechayev and O. Grygorenko, Synthesis, 2013, 2413 CAS.
- T. James, I. Simpson, J. A. Grant, V. Sridharan and A. Nelson, Org. Lett., 2013, 15, 6094 CrossRef CAS PubMed.
- T. James, P. MacLellan, G. M. Burslem, I. Simpson, J. A. Grant, S. Warriner, V. Sridharan and A. Nelson, Org. Biomol. Chem., 2014, 12, 2584 CAS.
- S. Spiess, C. Welter, G. Franck, J.-P. Taquet and G. Helmchen, Angew. Chem., Int. Ed., 2008, 47, 7652 CrossRef CAS PubMed.
- J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS PubMed.
- W.-B. Liu, J.-B. Xia and S.-L. You, Top. Organomet. Chem., 2012, 38, 155 CrossRef.
- P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147 CAS.
- P. Tosatti, J. Horn, A. J. Campbell, D. House, A. Nelson and S. P. Marsden, Adv. Synth. Catal., 2010, 352, 3153 CrossRef CAS.
- G. Helmchen, in Molecular Catalysts: Structure and Functional Design, ed. H. Gade and P. Hofmann, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, DOI:10.1002/9783527673278.ch11.
- D. N. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157 CrossRef CAS PubMed.
- J. S. Nakhla and J. P. Wolfe, Org. Lett., 2007, 9, 3279 CrossRef CAS PubMed.
- S. Bera and G. Panda, ACS Comb. Sci., 2012, 14, 1 CrossRef CAS PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757 CrossRef CAS PubMed . The “available now” library was used.
- A. Chuprina, O. Lukin, R. Demoiseaux, A. Buzko and A. Shivanyuk, J. Chem. Inf. Model., 2010, 50, 470 CrossRef CAS PubMed.
- A. Schuffenhauer, P. Ertl, S. Roggo, S. Wetzel, M. A. Koch and H. Waldmann, J. Chem. Inf. Model., 2007, 47, 47 CrossRef CAS PubMed.
- K. D. Collins and F. Glorious, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed.
- K. C. Nicolaou, Angew. Chem., Int. Ed., 2014, 53, 9128 CrossRef CAS PubMed.
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02287d |
| ‡ It has been suggested that new approaches may be required to translate such molecules into marketed drugs (see ref. 5). |
| § Substrates bearing a remote o-nitrosulfonyl-protected amine did not undergo clean aminoarylation (see ESI†). |
| ¶ Similar analyses of the molecular properties of commercially-available compounds have previously been reported. With similar molecular property filters and different structural filters, it was concluded that just 2.6% of 4.9 M compounds from various suppliers were lead-like (see ref. 7). In an analysis of compound collections from 29 suppliers using less constrained filters (200 < MR < 460, −4 < clogP < 4.2), 32% of ∼5.2 M compounds were found to be lead-like (see ref. 41). |
| || The properties of these compounds are representative of the entire ZINC database of ∼9 M commercially-available compounds (see ESI†). |
| ** For the purposes of this analysis, a synthetic operation is defined as a process conducted in a single reaction vessel. |
| †† An approach to identify such highly robust reactions has recently been reported (ref. 43). |
| This journal is © The Royal Society of Chemistry 2015 |