
Highly regio- and diastereo-selective synthesis of novel tri- and tetra-cyclic perhydroquinoline architectures via an intramolecular [3 + 2] cycloaddition reaction†
M.
Bakthadoss
*ab,
D.
Kannan
b,
J.
Srinivasan
b and
V.
Vinayagam
b
aDepartment of Chemistry, Pondicherry University, Pondicherry – 605 014, India. E-mail: bhakthadoss@yahoo.com; Fax: (+91) 44 22352494; Tel: (+91) 22202812
bDepartment of Organic Chemistry, University of Madras, Guindy Campus, Chennai-600025, Tamil Nadu, India
First published on 5th January 2015
Abstract
A facile and efficient synthetic protocol was established for the construction of novel tri- and tetra-cyclic pyrrolo/pyrrolizinoquinoline architectures via the in situ formation of azomethine ylide followed by an intramolecular [3 + 2] cycloaddition reaction strategy. This protocol leads to the creation of two/three new rings and three/four contiguous stereocentres, in which one of them is a tetra-substituted carbon center, in a highly diastereoselective fashion with excellent yields.
Introduction
In the domain of heterocyclic compounds, tetrahydroquinoline derivatives are very important and are recognized as small molecule transformers with hepatic micro RNA function, reducing the replication of Hepatitis C virus, and they are also used as apoptosis inducers.1 They are well known for their potential utility in medicinal chemistry such as antioxidant, antimalarial, anticoagulant, anti-HIV and antitubercular properties. Among various aza-heterocyclic compounds containing pyrrolo[3,2-c]quinoline2 frameworks, martinelline alkaloids isolated from the roots of Martinella iquitosensis are known to exhibit antibiotic properties and have also been reported as the first non-peptide antagonists for B2 receptors and bradykinin B1.3 Because of the interesting medicinal applications of these entities in drug discovery, the development of new and novel synthetic strategies towards the efficient construction of tetrahydroquinoline derivatives remains an important endeavour in this area of chemical research.4 A representative example of tetrahydroquinoline, pyrroloquinoline based molecules5 are Torcetrapib, (+)-Melonine, (±)-Heliotridane, (±)-Isoretronecanol, martinelline and Antitrypanosomal,1e which are also shown in Fig. 1.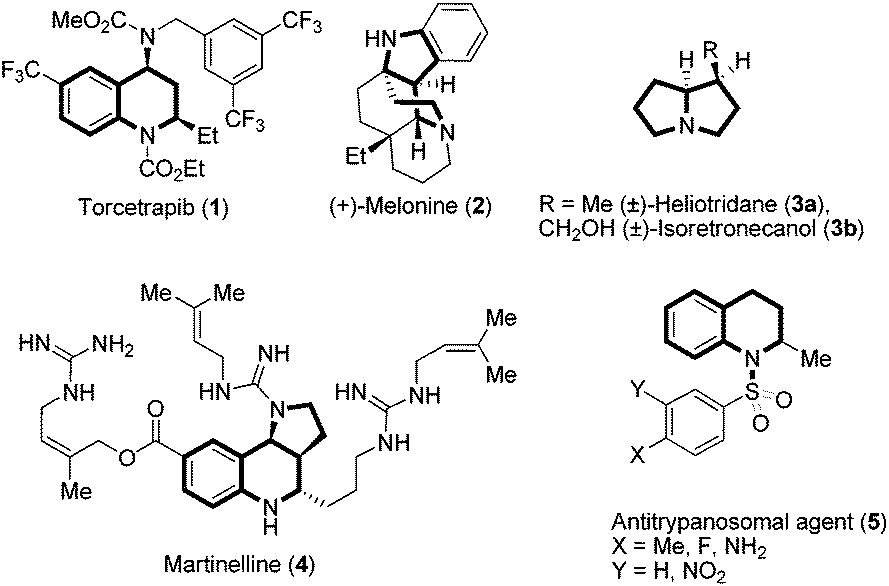 | ||
| Fig. 1 Some of the bioactive molecules and natural products embodying tetrahydroquinoline and other privileged motifs. | ||
The 1,3-dipolar cycloaddition reaction is one of the prominent and efficient protocol towards the synthesis of five membered heterocyclic compounds, particularly pyrrolidines.6 Even though an array of methods7 are available in the previously reported studies, the 1,3-dipolar cycloaddition, encompassing azomethine ylides trapped with alkenes, is the most expedient synthetic strategy for the construction of modified pyrrolidine and pyrrolizidine architectures.
The Baylis–Hillman reaction is one of the versatile classes of reactions in the field of modern organic synthesis. Precisely, the derivatives of Baylis–Hillman adducts are widely employed as crucial synthons for various intermediates leading towards the syntheses of several natural products and bio-active compounds.8 To the best of our knowledge, Baylis–Hillman derivatives9 have not been utilized for the syntheses of fused tri- and tetra-cyclic pyrrolo/pyrrolizinoquinoline derivatives via 1,3-dipolar cycloaddition reaction to date.
Therefore, we envisaged that the Baylis–Hillman derivatives (6a–k & 10a–g) could be effectively employed as a key starting material for the synthesis of tri- and tetra-cyclic perhydroquinolines as other Baylis–Hillman derivatives have been successfully utilized in our laboratory for the synthesis of a wide variety of polyheterocyclic frameworks via a cycloaddition reaction.6f–h The requisite precursors can be prepared by the treatment of various Baylis–Hillman bromides with N-tosylated aminoaldehyde.9c,10 Further treatment of various N-tosyl-N-allyl-2-aminobenzaldehydes bearing conjugated ester or nitrile moieties with N-methyl glycine/L-proline will afford the tricyclic pyrroloquinolines and tetracyclic pyrrolizinoquinolines with ester/nitrile functionality in the ring junction as shown in the retrosynthetic analysis (Scheme 1).
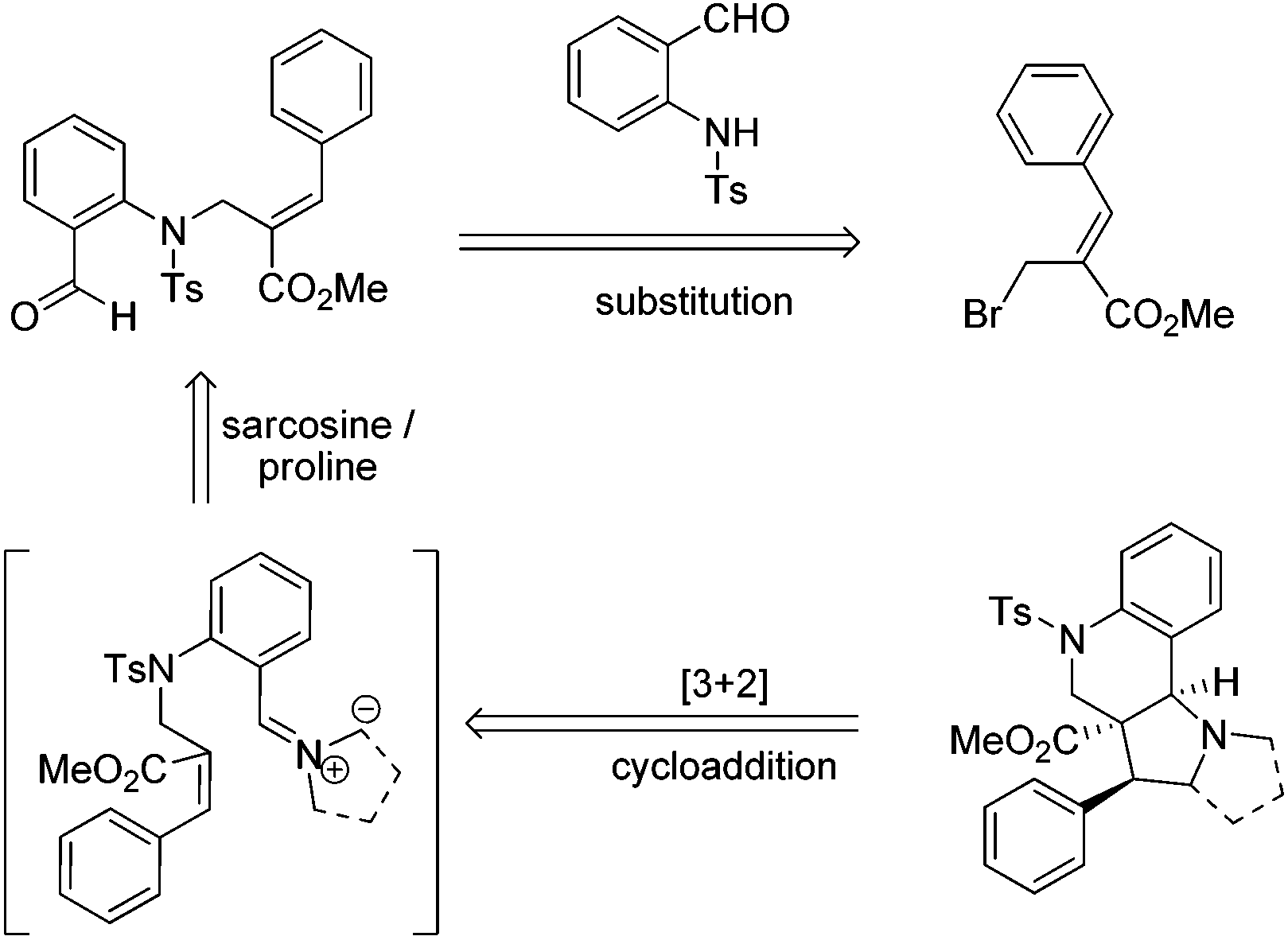 | ||
| Scheme 1 Retrosynthetic strategy for the synthesis of tri- and tetra-cyclic pyrroloquinoline architectures. | ||
To execute our idea, we treated N-allylated aldehyde (6a) and N-methyl glycine (7) in acetonitrile under reflux conditions, which successfully led to the formation of desired tricyclic pyrrolo[3,2-c]quinoline (9a) containing an ester functionality at the angular position in excellent yield (94%) via 1,3-dipolar cycloaddition reaction comprising an imine formation, decarboxylation, and [3 + 2] cycloaddition reaction, as shown in Table 1.
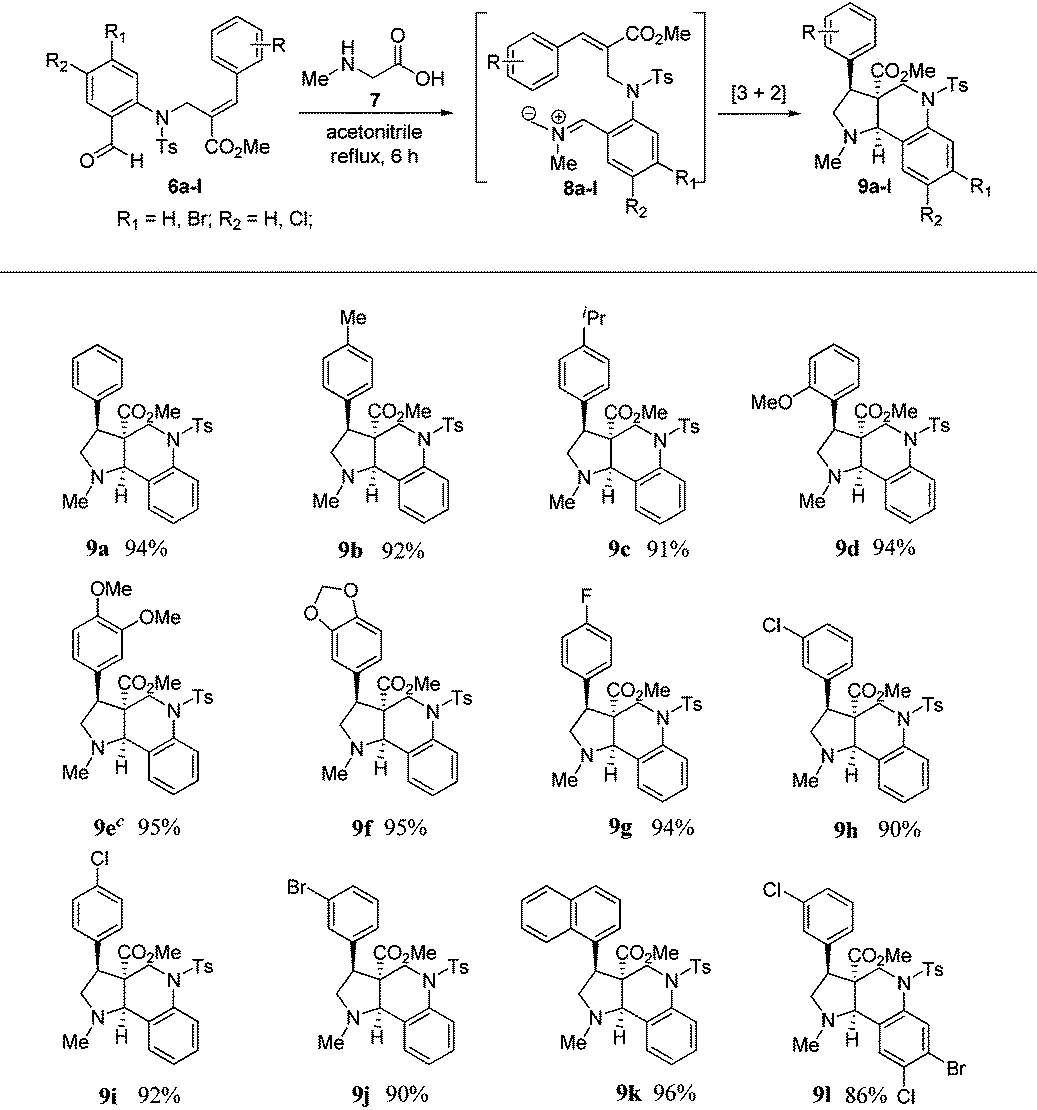
Encouraged by this result, we subjected a variety of N-allylated aldehydes (6b–l) with N-methyl glycine under the aforementioned reaction conditions, which afforded the anticipated tricyclic pyrrolo[3,2-c]quinolines (9b–l) containing an ester functionality at the angular position in the racemic form with excellent yields (86%–96%), and the results are shown in Table 1. It is important to note that this is the first report for the efficient construction of tricyclic pyrrolo[3,2-c] quinolines from Baylis–Hillman derivatives.
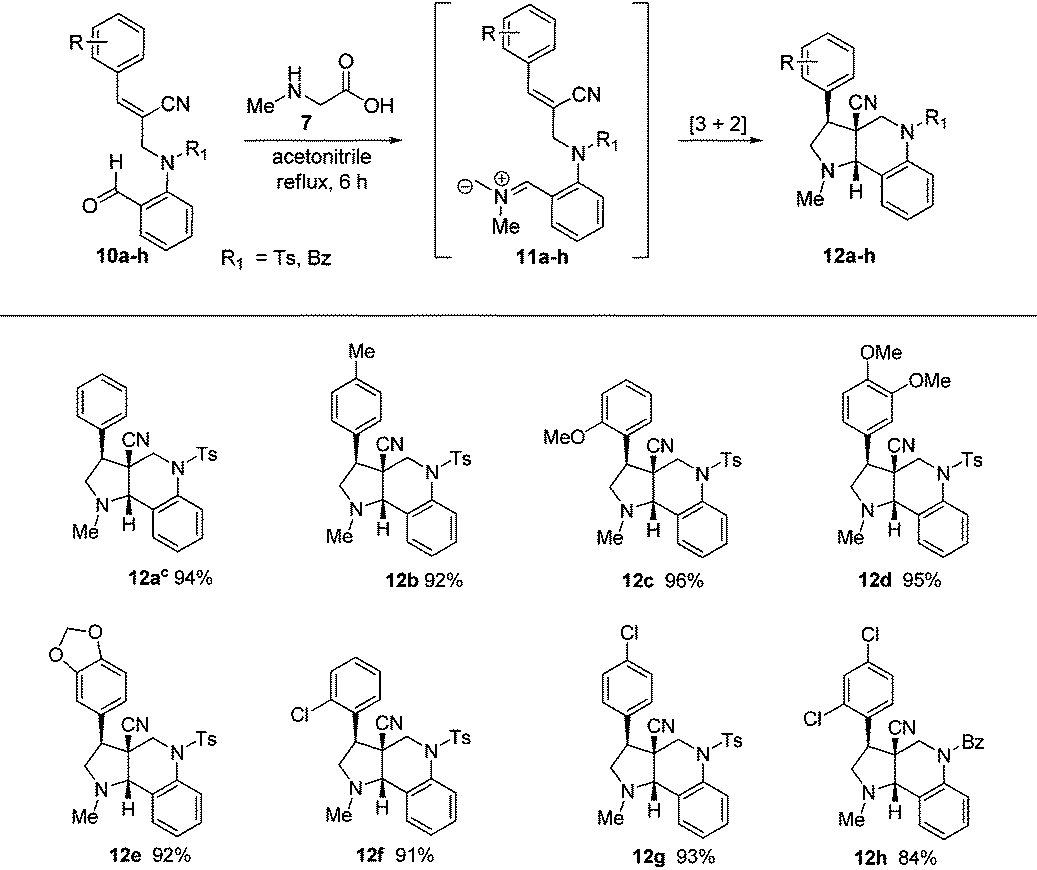
After obtaining successful results, we decided to extend the methodology further to N-allylated aldehydes (10a–h) containing a nitrile functionality. Therefore, a similar reaction of N-allylated aldehydes (10a–h) with N-methyl glycine (7) in acetonitrile at reflux temperature for 6 h successfully produced the desired tricyclic pyrroloquinolines (12a–h) as racemates in excellent yields (84%–96%), as shown in Table 2.
| a All reactions were carried out on a 1 mmol scale with N-allylated aldehydes (10a–h) and 1.1 mmol of N-methyl glycine (7), which were allowed to stir in acetonitrile at reflux for 6 h. b Isolated yields of the pure product. c The structure of the molecule was further confirmed by single-crystal X-ray data (see ESI). |
|---|
|
It can be noted that the tricyclic martinelline core structure was achieved using B–H derivatives through [3 + 2] cycloaddition for the first time. Delighted by these results, we decided to utilize this methodology towards the synthesis of tetracyclic pyrrolizinoquinolines.
In order to construct pyrrolizinoquinolines, we treated various N-allylated aldehydes (6a, 6c, 6f & 6g) with L-proline (13) in acetonitrile at reflux temperature for 10 h, which successfully produced the anticipated tetracyclic pyrrolizinoquinolines (15a–d) possessing the angular ester functionality in very good yields (84%–88%), as shown in Table 3. To probe the reaction further, we also treated various N-allylated aldehydes (10a–b, 10e, 10g) with L-proline (13) in acetonitrile at reflux temperature for 10 h, which successfully afforded the desired tetracyclic pyrrolizinoquinolines (17a–d) containing a nitrile functionality at the angular position in excellent yields (82%–86%), as shown in Table 4.
| a All reactions were carried out on a 1 mmol scale with N-allylated aldehydes (6a, 6c, 6f, 6g) and 1.1 mmol of L-proline (13) in acetonitrile at reflux temperature for 10 h. b Isolated yields of the pure product. c The structure of the molecule was further confirmed by single-crystal X-ray data. (see ESI). |
|---|
|
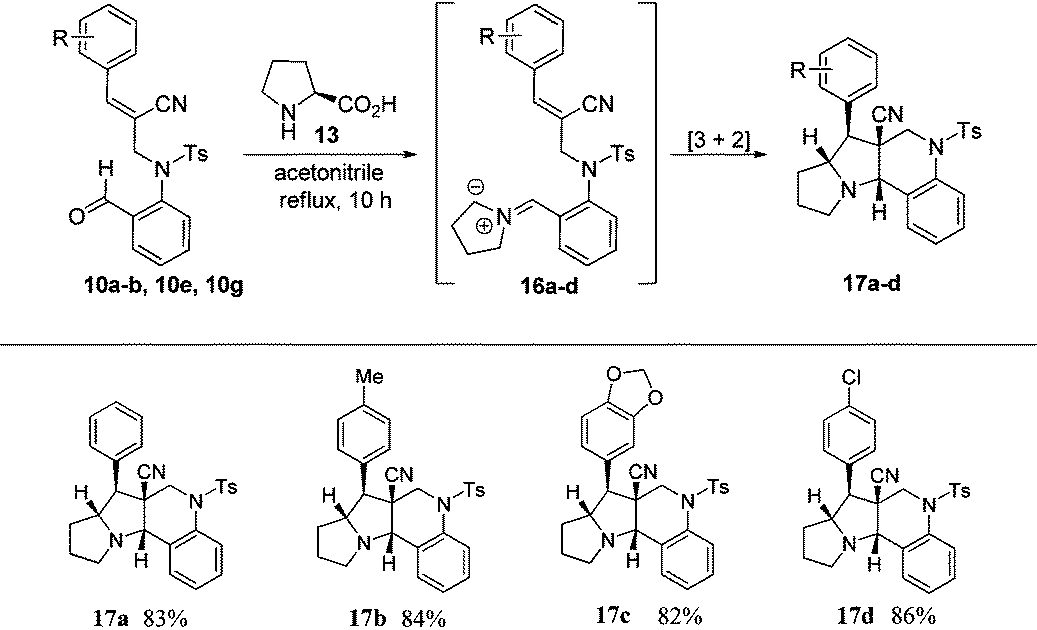
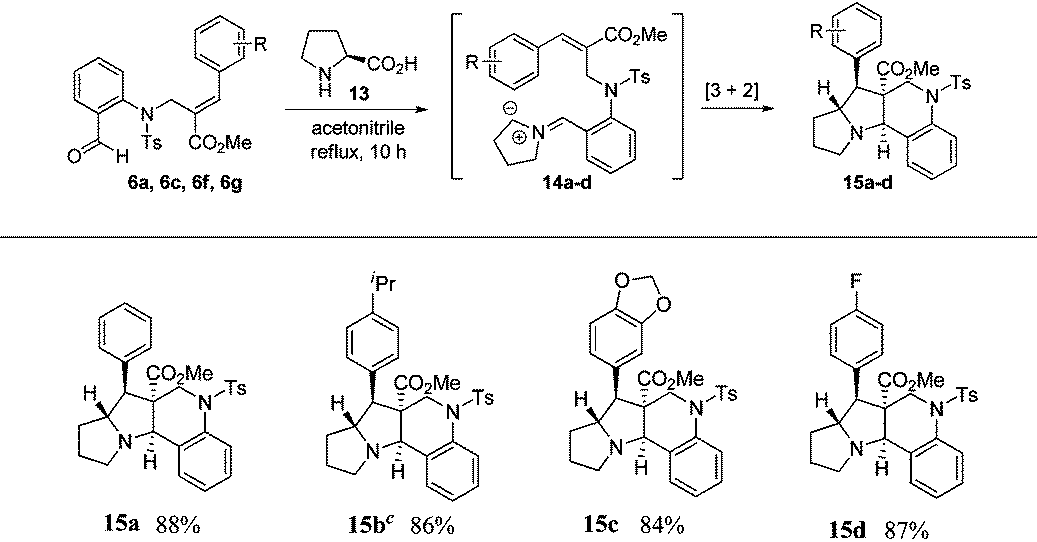
Based on the ORTEP diagram of the pyrroloquinoline 9e, which is shown in Fig. 2, the relative stereochemistry of the phenyl group and the adjacent ester moiety are in trans orientation, while, the ester moiety and the ring junction proton exist in a cis orientation. In addition, the X-ray crystal structure of tricyclic pyrroloquinoline 12a reveals that the phenyl group, which is adjacent to the nitrile moiety, and ring junction proton are in cis orientation (Fig. 2). It is quite obvious to note that the trans product was formed, due to olefin 6a having a trans geometry (aryl and ester groups present in the vicinal positions of compound 6). Similarly, olefin 10a, having a cis geometry (aryl and nitrile moieties present in the vicinal positions of compound 10), led to the formation of the cis product, which clearly shows the stereospecificity of the [3 + 2] cycloaddition reaction.
 | ||
| Fig. 2 ORTEP diagram of compound 9e and 12a. | ||
Furthermore, in the relative stereochemistry of tetracyclic pyrrolizinoquinoline compound 15b, the aryl moiety and the adjacent ester moiety are in trans-orientation, whereas the ester unit and the ring junction proton are in cis orientation based on the ORTEP diagram shown in Fig. 3.
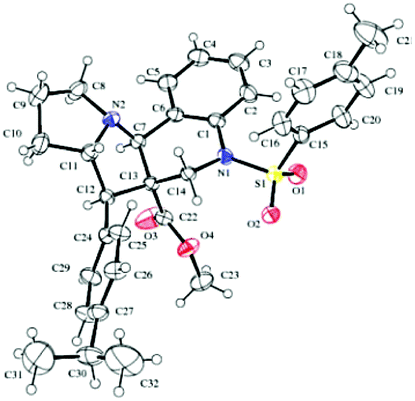 | ||
| Fig. 3 ORTEP diagram of compound 15b. | ||
The plausible pathways for the formation of tricyclic pyrroloquinoline ring system can be explained based on the transition state depicted in Scheme 2. It is important to mention here that both endo (path A & C) and exo (path B and D) transition states are possible for the formation of pyrroloquinoline frameworks. However, the endo transition state (path A and C) is the more favorable and predominant due to attractive π-interactions and repulsive steric interactions in the 1,3-dipolar cycloaddition reactions.
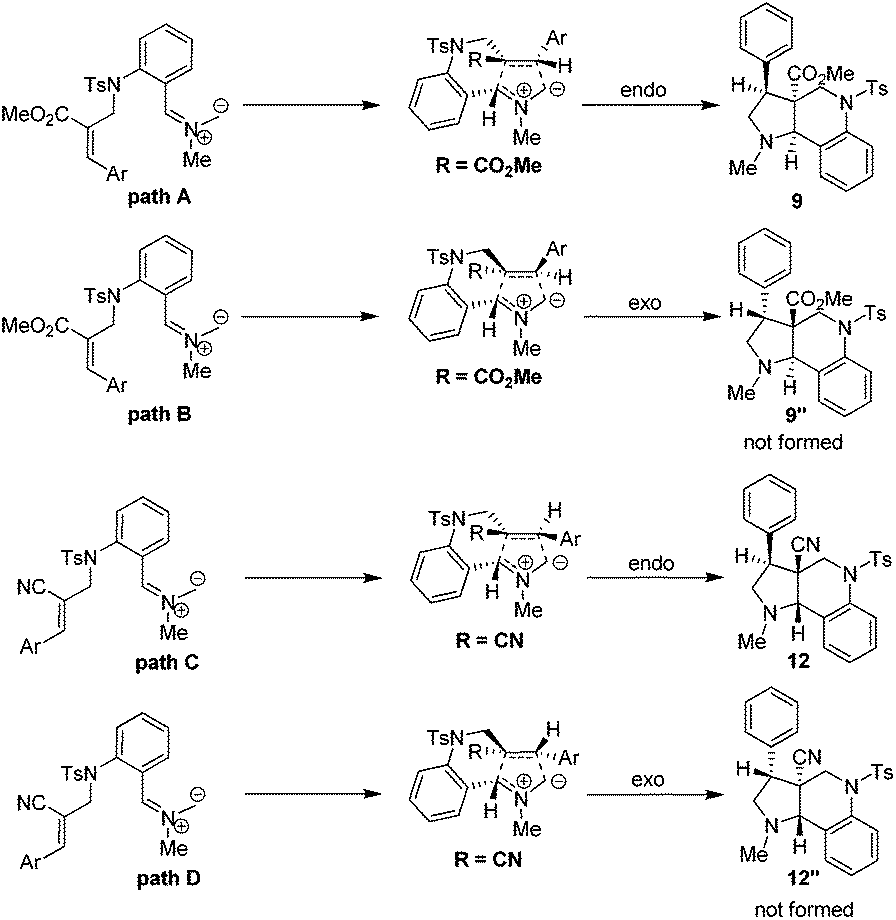 | ||
| Scheme 2 Plausible pathway for the formation of pyrrolo quinolines. | ||
Conclusions
We have successfully developed a simple and efficient protocol for the facile synthesis of complex angularly substituted tri- and tetra-cyclic frameworks containing a pyrrolo/pyrrolizinoquinoline ring system via an intramolecular 1,3-dipolar cycloaddition reaction using Baylis–Hillman derivatives. Angularly substituted tri- and tetra-cyclic compounds were obtained in a highly diastereoselective fashion with high yields. This approach also opens up new opportunities for the preparation of a library of pyrroloquinoline compounds for further biological screening.Acknowledgements
We thank DST-SERB, New Delhi for the financial support. We also thank DST-FIST for the NMR facility. D.K and J.S would like to thank CSIR for their award of SRF.Notes and references
- (a) D. Butler, Nature, 2007, 449, 158 CrossRef CAS PubMed; (b) A. A. Bowers, M. G. Acker, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2010, 132, 7519 CrossRef CAS PubMed; (c) A. Dastan, A. Kulkarni and B. Torok, Green Chem., 2012, 14, 17 RSC; (d) F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS PubMed; (e) V. Sridharan, P. A. Suryavanshi and J. C. Menendez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed; (f) M. Bakthadoss, A. Devaraj and D. Kannan, Eur. J. Org. Chem., 2014, 1505 CrossRef CAS.
- (a) J. A. Nieman and M. D. Ennis, Org. Lett., 2000, 2, 1395 CrossRef CAS PubMed; (b) S. G. Davies, A. M. Fletcher, J. A. Lee, T. J. A. Lorkin, P. M. Roberts and J. E. Thomson, Tetrahedron, 2013, 69, 9779 CrossRef CAS PubMed; (c) K. M. Witherup, R. W. Ransom, A. C. Graham, A. M. Bernard, M. J. Salvatore, W. C. Lumma, P. S. Anderson, S. M. Pitzenberger and S. L. Varga, J. Am. Chem. Soc., 1995, 117, 6682 CrossRef CAS; (d) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC.
- (a) Y. Takeda, T. Nakabayashi, A. Shirai, D. Fukumoto, T. Kiguchi and T. Naito, Tetrahedron Lett., 2004, 45, 3481 CrossRef CAS PubMed; (b) M. K. Gurjar, S. Pal and A. V. R. Rao, Heterocycles, 1997, 45, 231 CrossRef CAS; (c) T. C. T. Ho and K. Jones, Tetrahedron, 1997, 53, 8287 CrossRef CAS.
- (a) Z. Rong, Q. Li, W. Lin and Y. Jia, Tetrahedron Lett., 2013, 54, 4432 CrossRef CAS PubMed; (b) H. R. Tan, H. F. Ng, J. Chang and J. Wang, Chem. – Eur. J., 2012, 18, 3865 CrossRef CAS PubMed; (c) S. Kitamura, T. Harada, H. Hiramatsu, R. Shimizu, H. Miyagawa and Y. Nakagawa, Bioorg. Med. Chem. Lett., 2014, 24, 1715 CrossRef CAS PubMed; (d) Y. Wanga, M. Yu, J. Zhu, J. K. Zhang, F. Kayser, J. C. Medina, K. Siegler, M. Conn, B. Shan, M. P. Grillo, J. J. Liu and P. Coward, Bioorg. Med. Chem. Lett., 2014, 24, 1133 CrossRef PubMed.
- (a) D. B. Damon, R. W. Dugger and R. W. Scott, U.S. Patent, 6, 689, 897, 2004 Search PubMed; (b) S. Baassou, H. M. Mehri, A. Rabaron and M. Plat, Tetrahedron Lett., 1983, 24, 761 CrossRef CAS; (c) J. Robertson, M. A. Peplow and J. Piilai, Tetrahedron Lett., 1996, 37, 5825 CrossRef CAS; (d) R. K. Dieter and R. Watson, Tetrahedron Lett., 2002, 43, 7725 CrossRef CAS; (e) D. A. Powell and R. A. Batey, Org. Lett., 2002, 4, 2913 CrossRef CAS PubMed.
- (a) J. W. Lown, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, NY, 1984, vol. 1, p. 2 Search PubMed; (b) I. Coldham, A. J. M. Burrell, H. D. S. Guerrand and N. Oram, Org. Lett., 2011, 13, 1267 CrossRef CAS PubMed; (c) K. V. Gothelf, in Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and S. K. A. Jorgensen, Wiley-VCH, Weinheim, 2002, p. 211 Search PubMed; (d) K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed; (e) G. Pandey, N. R. Gupta and S. R. Gadre, Eur. J. Org. Chem., 2011, 740 CrossRef CAS; (f) M. Bakthadoss and G. Murugan, Eur. J. Org. Chem., 2010, 5825 CrossRef CAS; (g) M. Bakthadoss, N. Sivakumar, G. Sivakumar and G. Murugan, Tetrahedron Lett., 2008, 49, 820 CrossRef CAS PubMed; (h) M. Bakthadoss and N. Sivakumar, Synlett, 2011, 1296 CrossRef CAS PubMed.
- (a) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765 CrossRef CAS PubMed; (b) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484 CrossRef CAS PubMed; (c) M. S. T. Morin and B. A. Arndtsen, Org. Lett., 2014, 4, 1056 CrossRef PubMed; (d) E. H. Krenske, A. Patel and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 17638 CrossRef CAS PubMed; (e) W. Sun, G. Zhu, C. Wu, G. Li, L. Hong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 8633 CrossRef CAS PubMed; (f) J. M. Aracil, C. Nájera and J. M. Sansano, Chem. Commun., 2013, 49, 11218 RSC.
- (a) D. Basavaiah and G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68 RSC; (b) T. Y. Liu, M. Xie and Y. C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC; (c) D. Basavaiah, G. C. Reddy and K. C. Bharadwaj, Tetrahedron, 2014, 70, 7991 CrossRef CAS PubMed; (d) D. Basavaiah and D. M. Reddy, RSC Adv., 2014, 4, 23966 RSC; (e) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed; (f) S. Watanabe, A. Tada, Y. Tokoro and S.-i. Fukuzawa, Tetrahedron Lett., 2014, 55, 1306 CrossRef CAS PubMed; (g) R. Lee, F. Zhong, B. Zheng, Y. Meng, Y. Lu and K.-W. Huang, Org. Biomol. Chem., 2013, 11, 4818 RSC.
- (a) M. Bakthadoss, D. Kannan and R. Selvakumar, Chem. Commun., 2013, 49, 10947 RSC; (b) M. Bakthadoss, D. Kannan and G. Sivakumar, Synthesis, 2012, 793 CrossRef CAS PubMed; (c) M. Bakthadoss and D. Kannan, RSC Adv., 2014, 4, 11723 RSC; (d) M. Bakthadoss, G. Sivakumar and D. Kannan, Org. Lett., 2009, 11, 4466 CrossRef CAS PubMed; (e) M. Bakthadoss and G. Sivakumar, Tetrahedron Lett., 2014, 55, 1765 CrossRef CAS PubMed.
- Starting materials 6 and 10 were prepared from O-aminobenzyl alcohol according to a previously reported procedure.9c.
Footnote |
| † Electronic supplementary information (ESI) available: Representative experimental procedures, with all spectral data of 9a–k, 12a–g, 15a–d, 17a–d, crystal data, ORTEP diagram. CCDC 764210 for 9e, 833470 for 12a and 976506 for 15b. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02203c |
| This journal is © The Royal Society of Chemistry 2015 |