
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic flow cascade for the synthesis of protected mandelonitrile derivatives†
Mariëlle M. E.
Delville
a,
Kaspar
Koch
b,
Jan C. M.
van Hest
a and
Floris P. J. T.
Rutjes
*a
aRadboud University Nijmegen, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: F.Rutjes@science.ru.nl
bFutureChemistry B.V., Toernooiveld 100, 6525 EC Nijmegen, The Netherlands
First published on 8th December 2014
Abstract
A chemoenzymatic two-step cascade process, with both steps having incompatible reaction conditions, was successfully performed in continuous flow. The chemoenzymatic aqueous formation of cyanohydrins was integrated with a subsequent organic phase protection step in a single flow process utilising a membrane-based phase separation module. The wider applicability of our setup was demonstrated with the synthesis of nine protected cyanohydrin derivatives, all obtained in good yields and high to excellent enantioselectivity.
Since the beginning of this century, microreactors have become a well-established tool in the field of organic synthesis. As a result, the number of reactions that is carried out in continuous flow rather than in batch, both in academic and industrial labs, is continuously growing.1 Integration of two or more single-step flow reactions to establish multistep continuous flow processes is a logical step forward to optimally profit from the advantages of flow chemistry. Although this seems a straightforward extension of existing technology, genuine applications (not including inline collection of the intermediate product, followed by telescoping into the next flow reactor) so far have been limited.2 Challenges involved when setting up integrated multistep flow processes include the number of reaction steps, flow rate control, solvent compatibility of the individual steps, need for intermediate workup, and dilution effects.3
In conjunction with previous flow chemistry research in our group,4 and with supramolecular approaches developed by us to combine incompatible reaction conditions,5 we focused on developing a two-step sequence by integrating a chemoenzymatic (aqueous) step with a regular organic reaction. In such an approach, the issues of solvent compatibility and intermediate workup need to be addressed. Concerning the latter, over the last few years several continuous flow phase separation methods have been developed. Probably most thoroughly explored are solid-supported scavengers entrapped in a glass column.6 Excess reagents and side-products are scavenged on solid supports resulting in a product solution, which is then sufficiently pure for the next transformation. These solid phase workup modules, however, have generally limited capacity and need to be replaced or regenerated on a regular basis. A second approach of inline workup proceeds through liquid–liquid extraction, which is realised by dedicated phase separation modules. Several separation strategies have been reported in literature such as utilising different materials or coatings,7 gravity,8 or wetting properties of a membrane surface.9,10 In our case, we chose to work with a commercially available phase separation module which utilises a hydrophobic membrane.
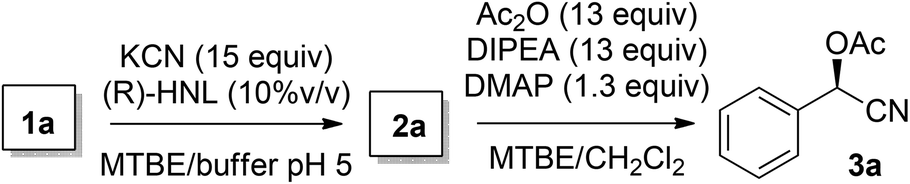
The chemoenzymatic continuous flow synthesis of cyanohydrins 2, versatile building blocks that have found widespread use in organic synthesis,11,12 was previously reported by us (Scheme 1).13 Microreactor technology enables the safe handling of in situ generated and toxic HCN for the enzyme-catalysed addition to aldehydes 1. Since free cyanohydrins tend to racemise, in particular under slightly basic conditions,4d we aimed to combine the aqueous cyanohydrin formation with protection of the hydroxyl function in a single flow system. Formation of protected cyanohydrins 3 generally takes place in the organic phase, which is incompatible with the aqueous conditions of the chemoenzymatic transformation.
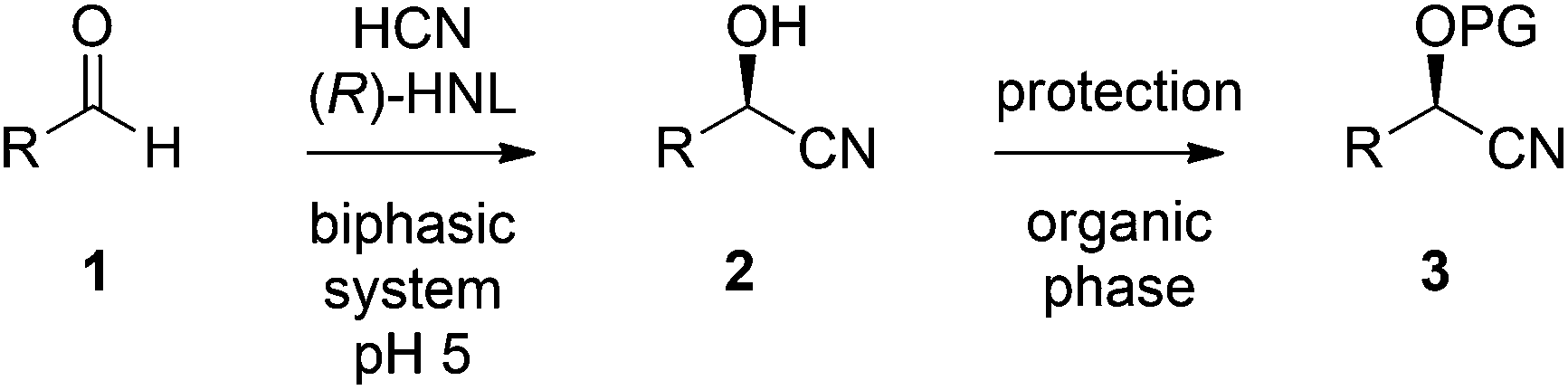 | ||
| Scheme 1 Two-step synthesis of protected cyanohydrins 3. | ||
Introduction of a liquid–liquid phase separation module would enable us to perform this chemoenzymatic cascade in a single continuous flow process. Thus, we report the first two-step chemoenzymatic flow synthesis of which the incompatible reaction steps are efficiently integrated by utilisation of an inline separation module.
Initially the separate reactions steps were optimised, starting from previously identified flow conditions using benzaldehyde 1a (R = Ph).13 The reactants were 10% (v/v) of a crude cell lysate containing an (R)-selective hydroxynitrile lyase (HNL)14 in a biphasic mixture of methyl tert-butyl ether (MTBE), containing the substrate (0.23 M), and a citrate buffer of pH 5, containing KCN (0.69 M) to in situ generate HCN for 5 minutes at room temperature. The flow rates of the aqueous and organic solutions were set to 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively, and such that a residence time (reaction time) of 5 minutes was obtained. Analysis was performed by chiral HPLC.
:1, respectively, and such that a residence time (reaction time) of 5 minutes was obtained. Analysis was performed by chiral HPLC.
Direct removal of the aqueous phase would eliminate an additional quenching step necessary to accurately determine reaction times. We chose to connect the microreactor to a separation device based on membrane technology, because of its robustness, wide applicability, high chemical resistance and ease of scaling up.10 More specifically, a commercially available FLLEX (Flow Liquid–Liquid Extraction)15 module utilizing a PTFE membrane was applied to separate the two liquid phases in flow. Dichloromethane (ratio CH2Cl2 with respect to biphasic buffer 1:4) was added to the biphasic reaction mixture to improve phase separation and for solubility reasons in the subsequent protection step. We found that, despite the fact that the aqueous phase contained a crude cell lysate, by applying a pressure difference of 0.2 bar over the separation module, both phases could be efficiently separated without emulsions remaining or clogging of the membrane.
Under the aforementioned reaction conditions, 57% conversion into mandelonitrile (2a) was observed (Table 1, entry 1). Raising the temperature to 40 °C gave an increase in conversion to 74% (entry 2). Additionally, applying a longer reaction time of 12 minutes resulted in a conversion of 83%. Higher conversions were hard to achieve due to the equilibrium of the chemoenzymatic step.16
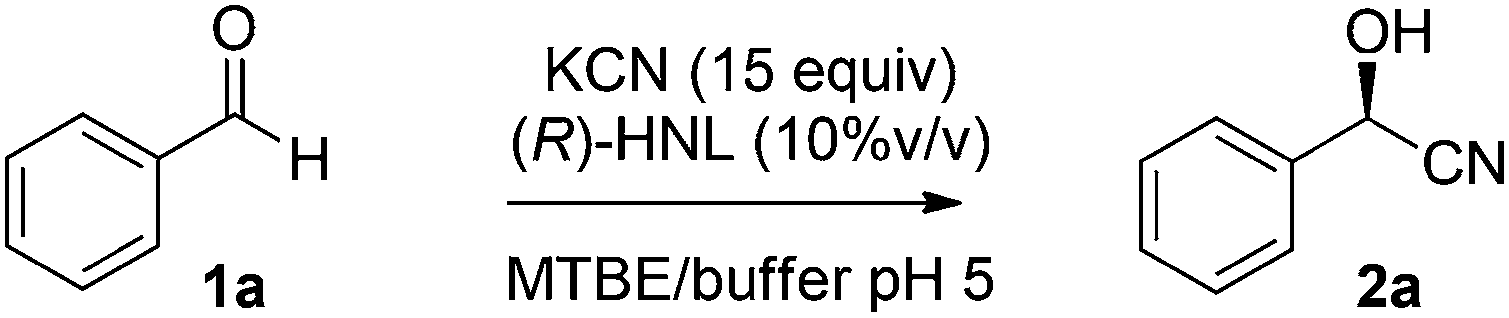
Having established optimal reaction conditions for the chemoenzymatic reaction, the subsequent step was investigated being protection through acetylation of the hydroxyl group11,12g based on a batchwise procedure from Bühler et al. (Ac2O, pyridine, 50 °C, 2 h).17 In flow, Ac2O was added to the mandelonitrile solution prior to addition of the base in order to prevent instant racemisation. Water was used to quench the reaction. Unfortunately, pyridine caused clogging of the microreactor due to the formation of insoluble pyridine salts; this was circumvented by the usage of DIPEA instead. Initially the acetylation was performed in dry MTBE using Ac2O from a commercial source (Table 2, entry 1) providing acetylated mandelonitrile (rac-3a) in a moderate isolated yield of 53%.
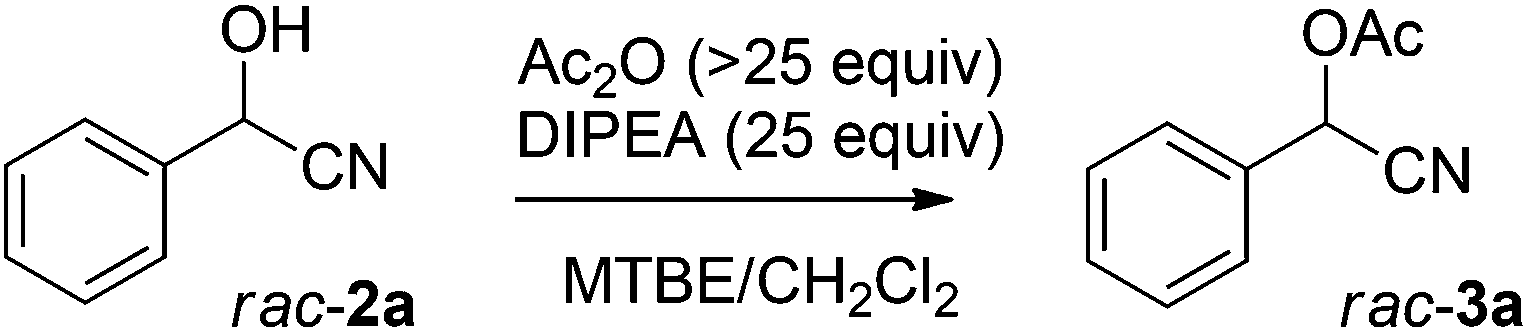
Purification of Ac2O18 before use increased the yield to 90% (Table 2, entry 2). Since the protection will be performed after the chemoenzymatic step, the separated MTBE/CH2Cl2 phase will still be saturated with water. Therefore, we also conducted the acetylation in water-saturated MTBE yielding racemic product 3a in 79% yield (entry 3). Karl-Fischer titration experiments showed that the water concentration in water-saturated MTBE was 460 mM, which explains the lower yield for the acetylation reaction under these conditions. Attempts to remove water from MTBE by inline use of a column filled with crushed 4 Å molecular sieves, or one with Na2SO4 as a drying agent, were unsuccessful due to an insufficient drying capacity. It was also not possible to increase the Ac2O molar ratio, since a maximum Ac2O concentration was reached by employing neat acetic anhydride (10.4 M) was used in combination with the required flow rates.
With the different components of the two-step process in place, integration of the two reactions in one single flow process was investigated (Scheme 2). First, the flow process for acetylated cyanohydrins (3) was investigated. In order to keep the solvents in the liquid phase and therefore maintaining control over the flow rates, a 40 psi backpressure regulator (BPR) was introduced. This additionally led to an increased backpressure necessary for the FLLEX module (80 psi) in order to create a pressure drop over the system to prevent back flushing of the reaction mixture.
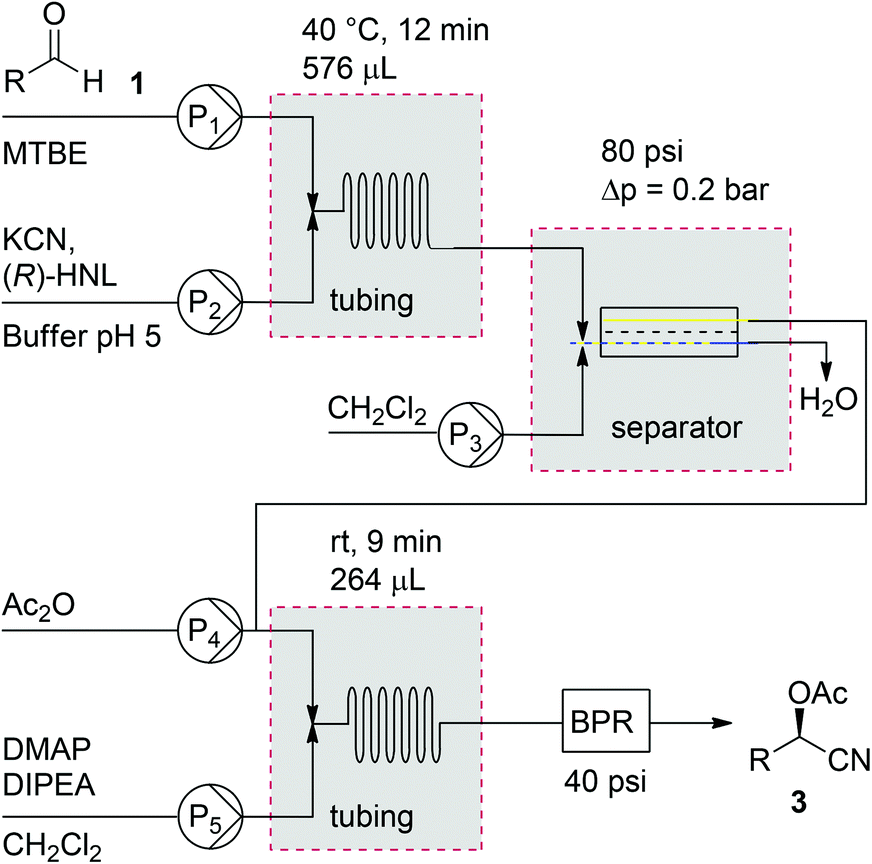 | ||
| Scheme 2 Schematic representation of the flow setup. | ||
Using the flow conditions from entries 3 in Tables 1 and 2, acetylated mandelonitrile (3a) was obtained from this integrated process in an isolated yield of 61% (Table 3, entry 1). This is in line with the expected outcome by combining the yields of the individual reaction steps. The ee of the product, however, appeared 90% while complete retention of 98% ee was expected. We hypothesised that this was caused in the second step due to partial racemisation under the basic conditions. Upon lowering of the acetylation temperature (entry 2), product 3a was obtained in 60% yield and 95% ee, most likely due to slower racemisation. Simultaneous acceleration of the acetylation was achieved by addition of 10% DMAP to the DIPEA flow, leading to a further increase of the enantioselectivity to 98% ee (entry 3).
After successful integration of the two-step chemoenzymatic cascade with benzaldehyde (1a), the setup was evaluated for a broader range of mandelonitrile derivatives (Table 4). The overall yields were in the same range as for acetylated mandelonitrile (3a) except for the more electron-donating substituents shown in entries 2 and 3, which is in line with previously reported results.19 The same holds for the ee's reported in Table 4, which are all high to excellent except for the aliphatic substrate 1e, which again is in agreement with precedent from literature.13
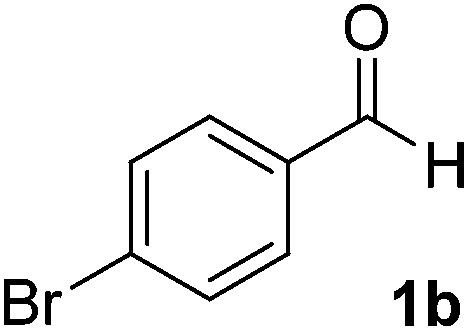
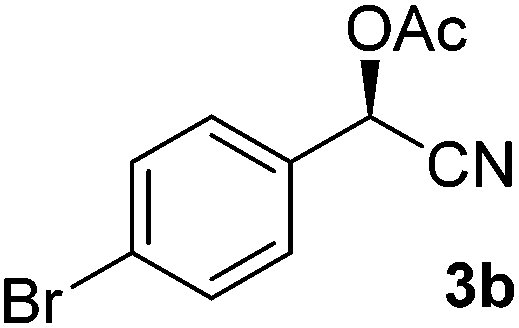
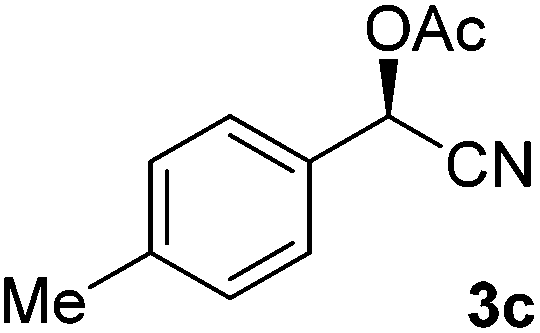
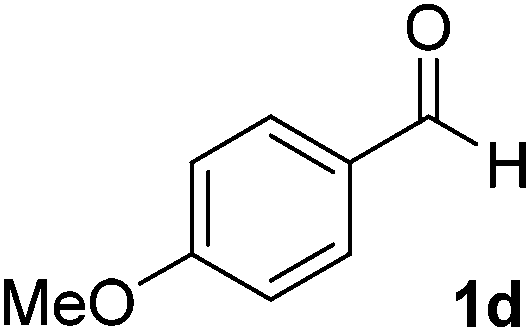
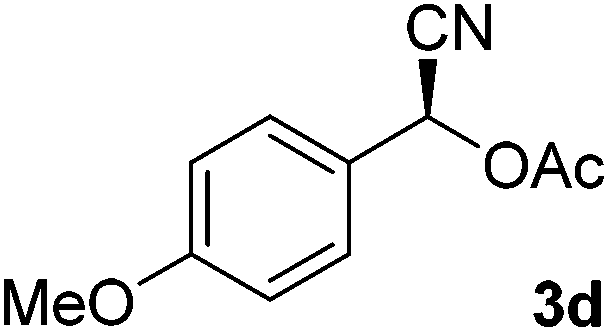
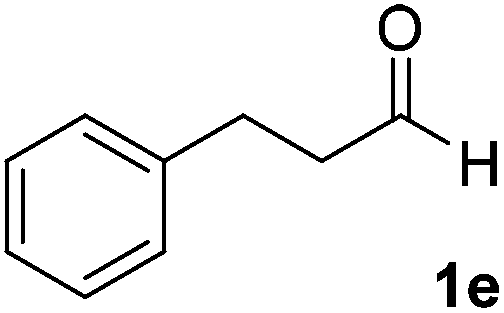
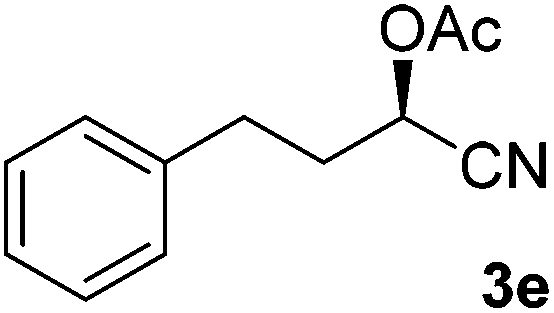
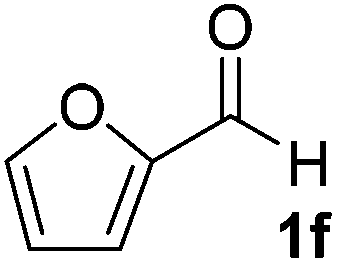
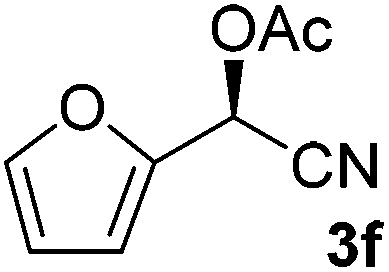
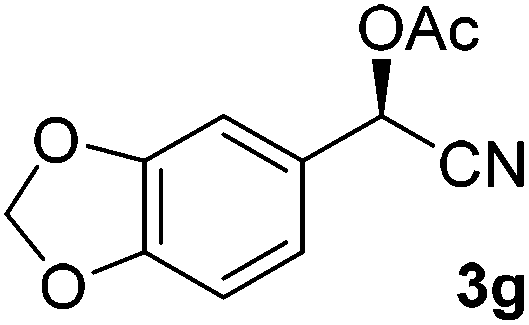
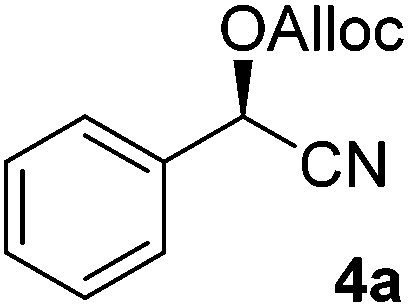
To enlarge the scope of the cyanohydrin functionalisation, we extended the chemoenzymatic flow cascade to other protecting groups as well (Table 5). First, allyloxycarbonyl (Alloc) protection was readily achieved (entry 1) by using the same flow scheme, but replacing Ac2O with neat AllocCl (9.4 M). The chemoenzymatic reaction was performed under the optimised conditions, but upon performing the protection at rt in the presence of DMAP the system was clogged. Therefore, the inline protection reaction was performed at 50 °C without addition of DMAP. This gave rise to Alloc-protected cyanohydrin 4a in 62% yield, but with a somewhat lower ee of 87% as probably caused by the elevated temperature. To minimise waste production and to recover the enzyme solution, the water phase collected from the latter experiment was also directly reused in a second flow cascade. Without the addition of fresh reagents and enzyme, Alloc-protected mandelonitrile 4a was now obtained in 52% overall yield and 80% ee. Secondly, the 2-methoxyisopropyl (MIP)-group was successfully introduced. Unlike the acetyl moiety, this protecting group is introduced under acidic conditions, is stable to base, and its use in flow chemistry has recently been described by us.4d
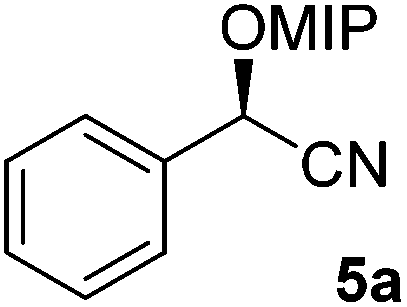
In the flow synthesis Ac2O was replaced by 2-methoxypropene (2.7 M in MTBE) and DMAP/DIPEA by camphorsulfonic acid (2.4 mM in MTBE). Flows were set to realise a reaction time of 200 seconds at a reaction temperature of 60 °C. Quenching of the reaction mixture was achieved by the addition of DIPEA. In this way, cyanohydrin 5a was synthesised in 68% yield and 97% ee.
Conclusions
In this article we describe the first continuous flow cascade of an aqueous chemoenzymatic reaction integrated with an organic phase protection step. The combination of both incompatible reaction steps into a single flow system was enabled by using a membrane-based phase separation module. We showed that our flow set-up can be used for the direct synthesis of acetylated cyanohydrins, which are formed in similar overall yields and ee's as in the separate reaction steps, but saves one workup and extraction procedure. We also demonstrated that this approach can be extended to carbonate (Alloc) and acetal (MIP)-protection of the intermediate cyanohydrins.Acknowledgements
We thank Dr Martin Schürmann (DSM, Geleen, The Netherlands) for providing the HNL enzyme. The NWO-ACTS Process-on-a-Chip (PoaC) programme is kindly acknowledged for financial support.Notes and references
- For recent reviews, see e.g.: (a) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384 CrossRef CAS PubMed; (b) C. Wiles and P. Watts, Green Chem., 2014, 16, 55 RSC; (c) J. Yoshida, Y. Takahasi and A. Nagaki, Chem. Commun., 2013, 49, 9896 RSC; (d) B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300 CrossRef CAS PubMed.
- For selected examples, see: (a) H. Usutani, Y. Tomida, A. Nagaki, H. Okamoto, T. Nokami and J. Yoshida, J. Am. Chem. Soc., 2007, 129, 3046 CrossRef CAS PubMed; (b) R. B. Andrew, L. P. Sarah, C. K. Daniel, J. B. Steven and D. T. McQuade, Angew. Chem., Int. Ed., 2009, 48, 8547 CrossRef PubMed; (c) A. Herath, R. Dahl and N. D. P. Cosford, Org. Lett., 2010, 12, 412 CrossRef CAS PubMed; (d) R. A. Maurya, K.-I. Min and D.-P. Kim, Green Chem., 2014, 16, 116 RSC; (e) P. Poechlauer, S. Braune, B. Dielemans, B. Kaptein, R. Obermüller and M. Thatagar, Chim. Oggi, 2012, 30, 51–54 CAS.
- J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17 CrossRef CAS.
- (a) M. M. E. Delville, J. C. M. van Hest and F. P. J. T. Rutjes, Beilstein J. Org. Chem., 2013, 9, 1813 CrossRef PubMed; (b) L. Babich, A. F. Hartog, L. J. C. van Hemert, F. P. J. T. Rutjes and R. Wever, ChemSusChem, 2012, 5, 2348 CrossRef CAS PubMed; (c) M. M. E. Delville, P. J. Nieuwland, P. Janssen, K. Koch, J. C. M. van Hest and F. P. J. T. Rutjes, Chem. Eng. J., 2011, 167, 556 CrossRef CAS PubMed; (d) M. M. E. Delville, J. J. F. van Gool, I. M. van Wijk, J. C. M. van Hest and F. P. J. T. Rutjes, J. Flow Chem., 2012, 4, 124 CrossRef; (e) R. Becker, K. Koch, P. J. Nieuwland and F. P. J. T. Rutjes, Chim. Oggi, 2011, 29, 47 CAS.
- For selected examples, see: (a) M. C. M. van Oers, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Commun., 2014, 50, 4040 RSC; (b) J. S. Willemsen, J. C. M. van Hest and F. P. J. T. Rutjes, Chem. Commun., 2013, 49, 3143 RSC; (c) Z. Wang, M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Angew. Chem., Int. Ed., 2012, 51, 10746 CrossRef CAS PubMed.
- (a) S. V. Ley, Chem. Rec., 2012, 12, 378 CrossRef CAS PubMed; (b) R. M. Myers, K. A. Roper, I. R. Baxendale and S. V. Ley, in From Modern Tools for the Synthesis of Complex Bioactive Molecules, eds. J. Cossy and S. Arseniyadis, Wiley & Sons, New Jersey, 2012, pp. 359–394 Search PubMed; (c) T. P. Petersen, S. Mirsharghi, P. C. Rummel, S. Thiele, M. M. Rosenkilde, A. Ritzén and T. Ulven, Chem. – Eur. J., 2013, 19, 9343 CrossRef CAS PubMed.
- (a) W. A. Gaakeer, M. H. J. M. Croon, J. van der Schaaf and J. C. Schouten, Chem. Eng. J., 2012, 207–208, 440 CrossRef CAS PubMed; (b) C. Kositanont, T. Tagawa, Y. Yamada, S. Putrivisutisak and S. Assabumrungrat, Chem. Eng. J., 2013, 215–216, 404 CrossRef CAS PubMed; (c) O. K. Castell, C. J. Allender and D. A. Barrow, Lab Chip, 2009, 9, 388 RSC.
- (a) D. X. Hu, M. O'Brien and S. V. Ley, Org. Lett., 2012, 14, 4246 CrossRef CAS PubMed; (b) B. Li, D. Widlicka, S. Boucher, C. Hayward, J. Lucas, J. C. Murray, L. Samp, J. VanAlsten, Y. Xiang and J. Young, Org. Process Res. Dev., 2012, 16, 2031 CrossRef CAS.
- (a) H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704 CrossRef CAS PubMed; (b) T. Noël, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 5943 CrossRef PubMed; (c) S. Sharma, R. A. Maurya, K.-I. Min, G.-Y. Joeng and D.-P. Kim, Angew. Chem., Int. Ed., 2013, 125, 7712 CrossRef.
- A. E. Cervera-Padrell, S. T. Morthensen, D. J. Lewandowski, T. Skovby, S. Kiil and K. V. Gernaey, Org. Process Res. Dev., 2012, 16, 888 CrossRef CAS.
- G. A. Tolstikov, L. M. Khaliliv, F. Z. Galin, E. V. Vasil'eva, D. B. Amirkhanov, M. G. Migranov and A. A. Panasenko, Chem. Nat. Compd., 1988, 24, 246 CrossRef.
- (a) R. J. H. Gregory, Chem. Rev., 1999, 99, 3649 CrossRef CAS PubMed; (b) J. Holt and U. Hanefeld, Curr. Org. Synth., 2009, 6, 15 CrossRef CAS; (c) W. Wang, X. Liu, L. Lin and X. Feng, Eur. J. Org. Chem., 2010, 4751 CrossRef CAS; (d) A. Baeza, J. M. Sansano, J. M. Saá and C. Nájera, Pure Appl. Chem., 2007, 79, 213 CrossRef CAS; (e) M. North, Tetrahedron: Asymmetry, 2003, 14, 147 CrossRef CAS; (f) T. Purkarthofer, W. Skranc, C. Schuster and H. Griengl, Appl. Microbiol. Biotechnol., 2007, 76, 309 CrossRef PubMed; (g) D. Ebbinghaus, A. Hinterhuber, W. Thielert, H. Hungenberg and H. Weckwert, U.S. Patent, 0276013, 2007 Search PubMed.
- (a) K. Koch, R. J. F. van den Berg, P. J. Nieuwland, R. Wijtmans, H. E. Schoemaker, J. C. M. van Hest and F. P. J. T. Rutjes, Biotechnol. Bioeng., 2008, 99, 1028 CrossRef CAS PubMed; (b) K. Koch, R. J. F. van den Berg, P. J. Nieuwland, R. Wijtmans, M. G. Wubbolts, H. E. Schoemaker, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Eng. J., 2008, 135S, S89 CrossRef PubMed.
- For a review on HNL-enzymes see e.g.: U. Hanefeld, Chem. Soc. Rev., 2013, 12, 6308 RSC.
- http://www.syrris.com .
- W. F. Willeman, P. J. Gerrits, U. Hanefeld, J. Brussee, A. J. J. Straathof, A. van der Gen and J. J. Heijnen, Biotechnol. Bioeng., 2002, 77, 239 CrossRef CAS PubMed.
- H. Bühler, F. Effenberger, S. Förster, J. Roos and H. Wajant, ChemBioChem, 2003, 4, 211 CrossRef PubMed.
- W. K. F. Armarego and C. L. L. Chai, in Purification of Laboratory Chemicals, Elsevier, Cornwall, 5th edn, 2003, pp. 83 Search PubMed.
- (a) G. Lin, S. Han and Z. Li, Tetrahedron, 1999, 55, 3531 CrossRef CAS; (b) A. Solís, H. Luna, H. I. Pérez, N. Manjarrez, R. Sánchez, M. Albores-Velasco and R. Castillo, Biotechnol. Lett., 1998, 20, 1183 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02128b |
| This journal is © The Royal Society of Chemistry 2015 |